

Abstract
The first influenza pandemic of the 21st century was caused by novel H1N1 viruses that emerged in early 2009. Molecular evolutionary analyses of the 2009 pandemic influenza A H1N1 [A(H1N1)pdm09] virus revealed two major clusters, cluster I and cluster II. Although the pathogenicity of viruses belonging to cluster I, which became extinct by the end of 2009, has been examined in a nonhuman primate model, the pathogenic potential of viruses belonging to cluster II, which has spread more widely in the world, has not been studied in this animal model. Here, we characterized two Norwegian isolates belonging to cluster II, namely, A/Norway/3568/2009 (Norway3568) and A/Norway/3487-2/2009 (Norway3487), which caused distinct clinical symptoms, despite their genetic similarity. We observed more efficient replication in cultured cells and delayed virus clearance from ferret respiratory organs for Norway3487 virus, which was isolated from a severe case, compared with the efficiency of replication and time of clearance of Norway3568 virus, which was isolated from a mild case. Moreover, Norway3487 virus to some extent caused more severe lung damage in nonhuman primates than did Norway3568 virus. Our data suggest that the distinct replicative and pathogenic potentials of these two viruses may result from differences in their biological properties (e.g., the receptor-binding specificity of hemagglutinin and viral polymerase activity).
INTRODUCTION
A novel H1N1 influenza virus emerged in Mexico and the United States in spring 2009 and quickly spread among humans worldwide, causing the first influenza pandemic of the 21st century. Infection with the 2009 pandemic influenza A H1N1 [A(H1N1)pdm09] virus caused mostly mild clinical symptoms, but an appreciable number of severe cases were also reported in healthy individuals who had no other underlying health problems (2). Whether these differences in clinical severity were due to virulence factors of A(H1N1)pdm09 virus or to host factors remains unclear.
The A(H1N1)pdm09 virus resulted from the reassortment of human, swine, and avian influenza viruses (9, 10, 32, 34). The PB2 and PA genes of A(H1N1)pdm09 virus originated from a North American avian virus, the PB1 gene came from a human H3N2 virus, the hemagglutinin (HA), NP, and NS genes came from a classical swine virus, and the neuraminidase (NA) and M genes came from a Eurasian avian-like swine virus (9, 10, 32, 34). The mean evolutionary rate for the viral genome of A(H1N1)pdm09 virus was high (32), and molecular evolutionary analyses of viral genome sequences revealed two major clusters, cluster I and cluster II, which originated from Mexico, Texas, and California and from New York, respectively (4, 7, 30). Cluster I viruses appeared in early April 2009 and became extinct by the end of 2009, whereas cluster II viruses emerged in late April 2009 and have been circulating ever since (4). The pathogenicity of A(H1N1)pdm09 viruses belonging to cluster I was evaluated in a nonhuman primate model (12, 13, 28). However, the pathogenic potential of currently circulating cluster II viruses in nonhuman primates and ferrets remains poorly understood.
Here, we characterized two Norwegian isolates that belong to the previously uncharacterized cluster II, which were clinically different from each other, in vitro and in vivo. Our data suggest that these viruses exhibit different replication properties in cells and ferrets, and moreover, they possess distinct pathogenic potentials in a nonhuman primate model, which likely result from differences in their biological properties, such as the receptor-binding specificity of HA and their viral polymerase activity.
MATERIALS AND METHODS
Cells and viruses.
Madin-Darby canine kidney (MDCK) cells were maintained in Eagle's minimal essential medium (MEM) containing 5% newborn calf serum. Human lung tissue samples were prepared and maintained as described previously (31). Normal human alveolar epithelial (HPAEpi) cells were obtained from ScienCell Research Laboratories (San Diego, CA) and maintained according to the manufacturer's manual.
A/Norway/3487-2/2009 (Norway3487) and A/Norway/3568/2009 (Norway3568) viruses were isolated from a fatal and a mild case occurring in late July and late September 2009, respectively. The viruses used in this study were generated by using reverse genetics (23) and were completely sequenced to rule out unwanted mutations.
Plasmid construction.
Reverse genetics systems were established by using the method described by Neumann et al. (23). Briefly, viral cDNAs were synthesized by reverse transcription of viral RNAs with an oligonucleotide, Uni-12 (5′-AGCAAAAGCAGG-3′), complementary to the conserved 3′ end of the viral RNA, as previously described (15). The cDNAs were amplified by using PCR with gene-specific oligonucleotides and then inserted into the pHH21 vector.
Glycan array analyses.
Viruses were grown in MDCK cells, clarified by low-speed centrifugation, laid over a cushion of 30% sucrose in phosphate-buffered saline (PBS), and ultracentrifuged at 25,000 rpm for 2 h at 4°C in a Beckman SW28 rotor. Virus stocks were aliquoted and stored at −80°C. Virus concentrations were determined by use of an HA assay with 0.5% (vol/vol) turkey red blood cells. Custom microarray slides were printed for the Centers for Disease Control and Prevention (CDC; Atlanta, GA) by using the Consortium for Functional Glycomics glycan library (CDC version 1 slides; see Table S1 in the supplemental material for the glycans) as described previously (3). Virus preparations were thawed and suspended in PBS supplemented with 3% (wt/vol) bovine serum albumin to an HA titer of 128, previously established to be optimal for glycan array analyses. Virus suspensions were supplemented with 300 nM zanamivir, overlaid on the printed region of the slides, and then incubated with gentle agitation in a closed container for 1 h at room temperature. Unbound virus was then eluted with brief rinses in PBS. Slides were immediately incubated with hyperimmune sheep or ferret serum to A/California/07/2009 (H1N1) HA (30 min), a biotinylated anti-sheep or -ferret IgG antibody (30 min), and a streptavidin-Alexa Fluor 635 conjugate (30 min) (Invitrogen, Carlsbad, CA) with brief PBS washes between incubations. After the final PBS wash, slides were briefly rinsed in deionized water, dried under a gentle stream of air, and immediately subjected to imaging. Fluorescence intensities were captured by using a ProScanArray HT scanner (PerkinElmer, Waltham, MA). Image analyses were carried out with ImaGene (version 8) software (BioDiscovery, El Segundo, CA). Data were processed in the Microsoft Excel program to group similar sialoglycans and generate a simplified chart.
Experimental infection of ferrets.
We used 5- to 8-month-old female ferrets (Triple F Farms, Sayre, PA), which were serologically negative by hemagglutination inhibition (HI) assay for currently circulating human influenza viruses. Baseline body temperatures and body weights were established by two measurements prior to infection. Six ferrets per group were intramuscularly anesthetized with ketamine and xylazine (5 mg and 0.5 mg, respectively, per kg of body weight) and intranasally inoculated with 106 PFU of virus (in 500 μl). On days 3 and 6 postinfection, three ferrets per group were euthanized for virus titration. Virus titers in organs were determined by use of plaque assays in MDCK cells.
Experimental infection of nonhuman primates.
Two- to 4-year-old cynomolgus macaques, which were obtained from Harlan Laboratories (Madison, WI), were used according to protocols approved by the University of Wisconsin Graduate School Animal Care and Use Committee. As described elsewhere (13, 36), five animals per group were anesthetized with ketamine via intramuscular injection and inoculated with a suspension containing a total of 6.7 × 107 PFU of the respective virus through a combination of intratracheal (4.5 ml), intranasal (0.5 ml per nostril), ocular (0.1 ml per eye), and oral (1 ml) routes. Using implanted chips, macaques were monitored every day for changes in body temperature. On days 1, 3, 5, and 7 postinfection, nasal washes and bronchoalveolar lavage (BAL) fluid samples were collected from animals. The BAL procedures were performed by first introducing a red rubber feeding tube into the tracheal lumen. Up to 3 ml of 1% lidocaine was instilled as needed to control bronchospasm. The tip of the feeding tube was then gently wedged into a subsidiary bronchus and lavage was performed by infusion of four 10-ml aliquots of sterile, pyrogen-free saline into the bronchus, followed by aspiration using a 10-ml syringe. Typically, 25 to 35 ml of lavage fluid was recovered.
At the indicated time points postinfection, two or three macaques per group were euthanized for virologic and pathological examinations. The virus titers in various organs, nasal washes, and BAL fluid were determined by using plaque assays in MDCK cells.
Pathological examination.
Tissues of animals were preserved in 10% phosphate-buffered formalin for pathological examination. They were then processed for routine paraffin embedding and cut into 5-μm-thick sections. One section from each tissue sample was subjected to standard hematoxylin-and-eosin staining, while another was processed for immunohistological staining with an anti-influenza virus rabbit antibody (R309) that reacts comparably with all test viruses. Specific antigen-antibody reactions were visualized by use of 3,3′-diaminobenzidine tetrahydrochloride staining and a Dako EnVision system (Dako Co. Ltd., Tokyo, Japan).
Cytokine and chemokine measurement.
Cytokines and chemokines in the BAL fluid of macaques were measured by using a Milliplex MAP nonhuman primate cytokine/chemokine panel (Millipore, Bedford, MA) with a Bio-Plex 200 system (Bio-Rad Laboratories, Hercules, CA).
Luciferase assay.
A luciferase assay was performed to examine viral polymerase activity as described previously (25). Briefly, 293T cells were transfected with plasmids for the expression of viral proteins PA, PB1, PB2, and NP derived from Norway3487 or Norway3568 viruses and pPolI-WNA-Flu expressing the NA gene encoding the firefly luciferase gene. Plasmid pGL4.74[hRuc/TK] (Promega, Madison, WI) served as an internal control for the dual-luciferase assay. After transfection, the cells were incubated at 37°C for 48 h, and then luciferase activity was measured with a dual-luciferase reporter system (Promega, Madison, WI) on a Glomax microplate luminometer (Promega, Madison, WI) according to the manufacturer's instructions.
Statistical analyses.
Statistical analyses, except for the pathological severity scores, were performed by using analysis of variance (ANOVA) in GraphPad Prism (version 5.0) software (GraphPad Software Inc., La Jolla, CA); P values of <0.05 were considered significant. Pathological severity scores were analyzed by using the Student t test; to control for the multiplicity effect, P values were adjusted using Benjamini-Hochberg's procedure, keeping the false discovery ratio <0.05.
RESULTS
Identification of amino acid differences between two clinically distinct Norwegian virus isolates.
Here, we used two A(H1N1)pdm09 viruses, A/Norway/3487-2/2009 (Norway3487) and A/Norway/3568/2009 (Norway3568), which were isolated from a fatal and a mild case occurring in late July and late September 2009, respectively. Sequencing analysis revealed that these Norwegian viruses belong to cluster II, which has been circulating worldwide, because these viruses possess several characteristic sequences found in cluster II viruses (Table 1). In contrast, A/California/04/2009, A/Netherlands/602/2009, A/Mexico/4108/2009, and A/Mexico/InDRE4487/2009 viruses, whose pathogenicity has been tested in nonhuman primates (12, 13, 28), belong to cluster I, which has been extinct since the end of 2009 (4). Norway3487 and Norway3568 differ by only 10 amino acids (4 in the polymerase subunit PB2 and 1 in the polymerase subunit PB1, 2 in the interferon-antagonist protein NS1, 1 in NA, and 2 in HA [Table 2]). However, none of the amino acid changes known to affect virulence were found in PB2, PB1-F2, HA, or NS1 (6, 8, 11, 14, 16, 18, 24, 29, 33), except for an Asp-to-Gly amino acid change at position 222 of the HA of Norway3487 virus, which has recently been identified as a potential virulence marker of A(H1N1)pdm09 viruses (35, 37).
Table 1.
Nucleotide and amino acid residues located in six segments of A(H1N1)pdm09 viruses specific for the two clusters
| Virus | Residue at the indicated positionsa |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| HA |
NA 742 (248) | M1 |
NP |
NS1 367 (123) | PB2 2163 (721) | ||||
| 658 (220) | 1480 (470) | 492 (164) | 600 (200) | 298 (100) | 1143 (381) | ||||
| Cluster I | T (S) | C (L) | A (N) | G (Q) | G (A) | G (V) | G (A) | A (I) | G (K) |
| Cluster II | A (T) | T (L) | G (D) | A (Q) | A (A) | A (I) | A (A) | G (V) | A (K) |
| Norway3487 | A (T) | T (L) | T (D) | A (Q) | T (A) | G (G) | T (D) | C (V) | A (K) |
| Norway3568 | A (T) | T (L) | T (D) | A (Q) | T (A) | G (G) | T (D) | C (V) | A (K) |
Fereidouni et al. (7) analyzed all complete genome sequences of A(H1N1)pdm09 viruses available as of 10 September 2009. Nucleotide positions and amino acid positions (in parentheses) for all genes are counted from the start codon. Sequences of Norway3487 and Norway3568 viruses are also shown.
Table 2.
Amino acid differences between Norway3487 and Norway3568 virusesa
| Virus | Amino acid |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PB1 720b | PB2 |
HA |
NA 257 | NS1 |
||||||
| 4 | 191 | 340 | 410 | 126 | 222 | 103 | 127 | |||
| Norway3487 | S | I | E | N | M | D | G | R | L | N |
| Norway3568 | Y | T | K | K | V | N | D | K | F | D |
Comparison of the amino acid sequences of Norway3487 and Norway3568 viruses. There were no differences in the sequences of the PA, NP, M1, M2, NS2, and PB1-F2 proteins between these viruses.
Numbers represent amino acid positions.
Growth properties of Norwegian viruses in vitro and in vivo.
To compare the replicative ability of Norway3487 and Norway3568 viruses in vitro and in vivo, we generated these viruses by reverse genetics (23) and sequenced their genomes completely to rule out unwanted mutations. We then tested their growth properties in MDCK and normal human alveolar epithelial (HPAEpi) cells at 35°C (Fig. 1). Cells were infected with viruses at a multiplicity of infection (MOI) of 0.001, and culture supernatants were harvested at the indicated time points for virus titration. Norway3568 virus exhibited a delay in growth in MDCK cells at 24 h postinfection, with titers being 2 log units lower than those of Norway3487 virus (P < 0.001; Fig. 1A). In HPAEpi cells, Norway3487 virus replicated more efficiently than did Norway3568 virus; titers of Norway3487 virus were 1 log unit higher than those of Norway3568 virus (P < 0.001 at 24, 48, 60, and 72 h postinfection; P < 0.01 at 36 h postinfection; Fig. 1B).
Fig 1.
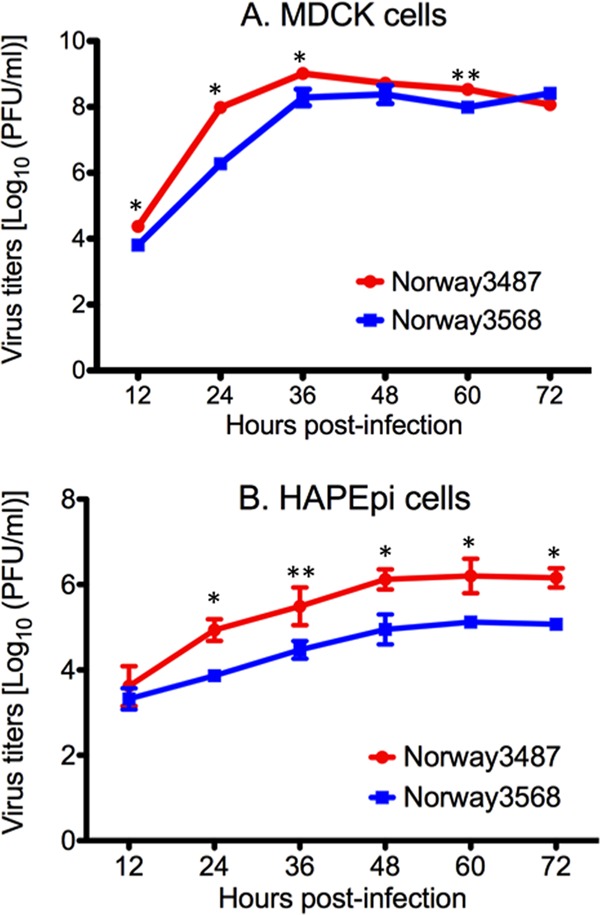
Viral growth kinetics in MDCK cells (A) and normal human alveolar epithelial cells (B). Cells were infected with Norway3568 or Norway3487 at a multiplicity of infection of 0.001. At different times postinfection, virus titers in the supernatants of infected cells were determined by use of plaque assays in MDCK cells. The mean from triplicate independent cultures ± standard deviation is shown. The mean titers of Norway3487 virus were significantly higher than those of Norway3568 virus (*, P < 0.001; **, P < 0.01).
Next, we examined the replicative ability of Norway3487 and Norway3568 viruses in ferrets, a widely used animal model in influenza virus research because the disease manifestations resemble those of infected humans. Ferrets were intranasally inoculated with 106 PFU of virus (in 500 μl), and on days 3 and 6 postinfection, three ferrets per group were euthanized for virus titration. The virus titers in organs were determined by plaque assays in MDCK cells. On day 3 postinfection, both viruses replicated well in the nasal turbinates, trachea, and lung tissues of the infected ferrets (Fig. 2). On day 6 postinfection, however, only Norway3487 virus was recovered from the lungs and tracheas of infected animals, whereas no virus was detected in those organs of Norway3568-infected animals (Fig. 2).
Fig 2.
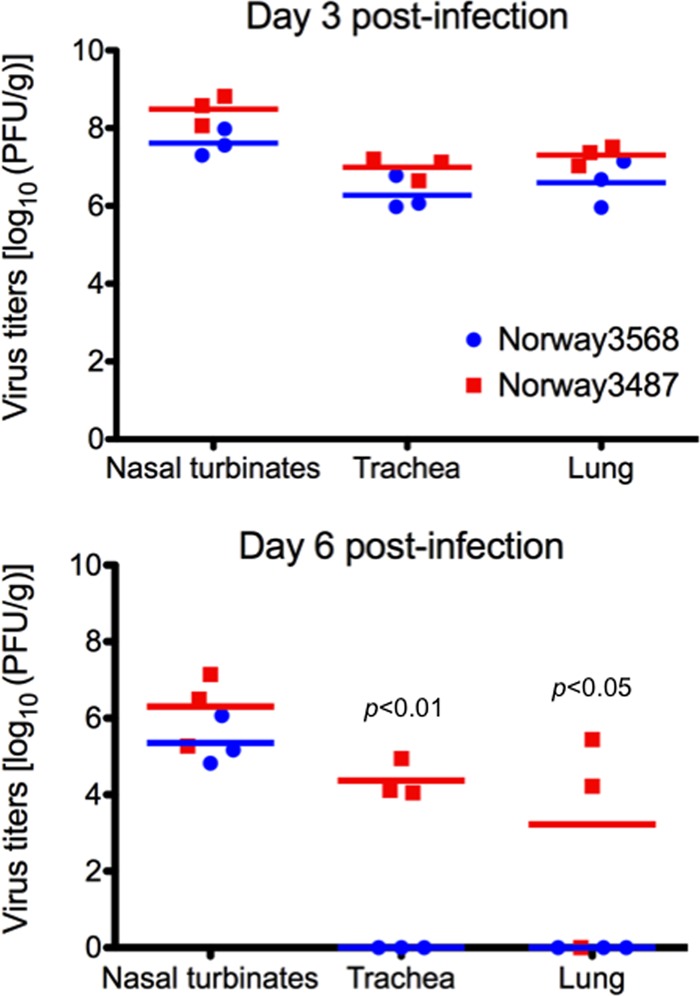
Virus replication in respiratory organs of ferrets. Ferrets were intranasally infected with 106 PFU/500 μl of virus. Three animals from each group were euthanized on days 3 and 6 postinfection for virus titration. Red and blue horizontal bars show the mean titers for Norway3487 and Norway3568 viruses, respectively. No statistically significant differences between virus titers in respiratory tissues of ferrets infected with Norway3487 virus and those infected with Norway3568 virus were found at day 3 postinfection. The titers of Norway3487 virus in trachea and lungs were significantly higher than those of Norway3568 virus at day 6 postinfection (P < 0.01 in trachea; P < 0.05 in lungs).
These results suggest that infection with Norway3487 virus, which was isolated from a severe case, resulted in more efficient replication in vitro and a delay in viral clearance from ferret organs compared with the efficiency of replication and time of clearance of Norway3568 virus, which was isolated from a mild case.
Pathogenicity of Norwegian viruses in nonhuman primates.
Nonhuman primates are increasingly being used as a model to assess highly pathogenic influenza virus infections because of their close genetic relationship to humans (1, 17, 27). To evaluate the pathogenic potential of the cluster II Norway3487 and Norway3568 viruses in a nonhuman primate model, Chinese-origin cynomolgus macaques (Macaca fascicularis) were inoculated with a suspension containing a total of 6.7 × 107 PFU of Norway3487 or Norway3568 virus via multiple routes, as described in Materials and Methods.
One animal infected with Norway3487 (animal 374) died during the BAL procedure on day 3 postinfection and had extensive respiratory tract lesions. In the upper respiratory tract, higher levels of Norway3568 virus than Norway3487 virus were detected in nasal wash samples at day 1 postinfection (P < 0.01; Fig. 3 and Table 3). In BAL fluid samples, the titers of Norway3487 virus were significantly higher than those of Norway3568 virus on day 5 postinfection (P < 0.001; Fig. 3). On day 7 postinfection, three animals per group were euthanized for virologic and pathological analyses. Some animals used in this study had Plasmodium spp. in their erythrocytes (Table 3); however, their erythrocyte counts were normal. For animals euthanized on day 7 postinfection, no virus was detected in any of the organs tested, with the exception of the trachea obtained from an animal infected with Norway3487 virus (data not shown). Pathological examination revealed bronchointerstitial pneumonia in all infected monkeys. We observed severe lung edema with proteinaceous fluid and severe inflammatory changes in large areas of the examined tissue of the animals infected with Norway3487 virus (Fig. 4A). More severe damage was found in the right cranial lung lobes of the animals infected with Norway3487 virus than those infected with Norway3568 virus (Student t test, P = 0.0096; Fig. 4B). On day 14 postinfection, no virus was detected in any of the organs tested (data not shown). These findings demonstrate that, to some extent, infection with Norway3487 virus causes more severe disease than infection with Norway3568 virus.
Fig 3.
Virus titers in respiratory washes from infected cynomolgus macaques. Cynomolgus macaques were infected with 6.7 × 107 PFU of viruses through multiple routes. Nasal wash, tracheal brush, and BAL fluid samples were collected every other day for virus titration. Red and blue horizontal bars show the mean titers for Norway3487 and Norway3568 viruses, respectively. The titers of Norway3487 virus were significantly higher than those of Norway3568 virus in BAL fluid samples on day 5 postinfection (P < 0.001), whereas those of Nowary3487 in nasal wash samples were lower than those of Norway3568 virus at day 1 postinfection (P < 0.01).
Table 3.
Virus titers in respiratory washes from infected cynomolgus macaquesa
| Specimen and day | Virus titer (log10 PFU/ml) in the indicated animals infected with: |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Norway3568 virus |
Norway3487 virus |
|||||||||
| 367c | 368e | 369 | 370c | 371c,e | 372e | 373c,e | 374d | 375c | 376c | |
| Nasal wash | ||||||||||
| 1 | 3.6 | 5.1 | 3.9 | —b | 3.1 | — | 2.6 | — | — | — |
| 3 | 1.8 | — | 1.9 | 1.8 | — | 1.9 | 1.6 | — | — | — |
| 5 | 1.3 | 1. 8 | 2.5 | — | — | 1.6 | — | NAf | — | — |
| 7 | — | — | — | — | — | — | — | NA | — | — |
| 10 | NA | — | — | NA | NA | — | NA | NA | NA | NA |
| 14 | NA | — | — | NA | NA | — | NA | NA | NA | NA |
| Tracheal brush | ||||||||||
| 1 | 1. 8 | 4.2 | 5.3 | — | 3.0 | 2. 8 | 4.8 | 1.6 | 2.2 | 2.2 |
| 3 | 1.3 | 3.6 | 1.3 | 4.0 | — | — | 3.4 | — | — | — |
| 5 | 3.9 | 5.0 | — | 2.6 | — | — | 3.6 | NA | 2.2 | 2.7 |
| 7 | — | 2.2 | 3.9 | — | — | 3.1 | 3.6 | NA | — | — |
| 10 | NA | — | — | NA | NA | — | NA | NA | NA | NA |
| 14 | NA | — | — | NA | NA | — | NA | NA | NA | NA |
| BALg fluid | ||||||||||
| 1 | 4.4 | 4.1 | 6.5 | 4.1 | 4.5 | 4.6 | 6.7 | 6.3 | 6.6 | 5.3 |
| 3 | 3.2 | 3.6 | 2.5 | 3.9 | 2.7 | 4.7 | 4.1 | 4.7 | 4.3 | 4.6 |
| 5 | 1.6 | 2.6 | 1.3 | 3.6 | 2.2 | 5.8 | 4.3 | NA | 4.5 | 4.1 |
| 7 | — | — | — | — | — | — | — | NA | — | — |
| 10 | NA | — | — | NA | NA | — | NA | NA | NA | NA |
| 14 | NA | — | — | NA | NA | — | NA | NA | NA | NA |
Cynomolgus macaques were infected with 6.7 × 107 PFU through multiple routes. Nasal wash and tracheal brush samples were collected every other day for virus titration.
—, virus not detected (detection limit, 1.3 log10 PFU/ml.
These animals were euthanized on day 7 postinfection for virologic and pathological analyses.
This animal died during the bronchoalveolar lavage procedure on day 3 postinfection.
These animals had Plasmodium spp. in their erythrocytes; however, their erythrocyte counts were normal.
NA, not applicable.
BAL, bronchoalveolar lavage.
Fig 4.

Pathological analyses of the lungs of infected macaques. (A) Infection with Norway3568 virus (panel 1) resulted in moderate to severe bronchointerstitial pneumonia (panel 4) with some minimally affected to unaffected regions (panel 7). Infection with Norway3487 virus (panel 2) resulted in moderate to severe bronchointerstitial pneumonia with severe lung edema and inflammatory changes (panel 5), even in areas with less severe gross changes (panel 8). Lungs derived from a noninfected control animal did not have any gross or histological changes (panels 3 and 6). Arrowheads, gross lesions. Boxes drawn with dotted lines depict the areas shown in the microscopic images. Bars, 200 μm. (B) Pathological severity scores in infected animals on day 7 postinfection. To represent comprehensive histological changes, respiratory tissue slides were evaluated by scoring the pathological changes. The pathological scores were determined for each animal in each group (n = 4 and n = 3 for Norway3487 and Norway3568 viruses, respectively). Error bars denote standard deviations. The mean pathological severity score in the right cranial lung lobe was higher for Norway3487 virus than for Norway3568 virus (Student t test, P = 0.0096).
We also investigated proinflammatory responses in nonhuman primates infected with Norway3487 and Norway3568 viruses by measuring the levels of cytokines and chemokines in the BAL fluid of infected monkeys. The cytokine responses to infection occurred in two separate peaks (Fig. 5). The first peak occurred within 1 day of infection and involved the induction of proinflammatory cytokines and chemokines such as granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-1β (IL-1β), macrophage inflammatory protein 1α (MIP-1α), MIP-1β, IL-18, monocyte chemotactic protein-1 (MCP-1), tumor necrosis factor alpha (TNF-α), IL-6, and IL-12. The second peak was observed at day 5 postinfection and included the induction of MCP-1, TNF-α, IL-6, and IL-8. Increases in IL-12, granulocyte colony-stimulating factor (G-CSF), IL-1 receptor α (IL-1Rα), transforming growth factor α (TGF-α), and vascular endothelial growth factor (VEGF) were observed at day 7 postinfection, although the expression levels varied among the animals. The induction of IL-12, IL-1Rα, G-CSF, and VEGF appeared to be more remarkable for Norway3568 virus. There were no statistically significant differences between the cytokine/chemokine levels observed for Norway3487 virus and those observed for Norway3568 virus.
Fig 5.

Cytokine/chemokine production in infected cynomolgus macaques. The concentrations of various cytokines and chemokines in the BAL fluid of infected cynomolgus macaques were measured on days 1, 3, 5, and 7 postinfection by use of protein array analysis with a Milliplex MAP nonhuman primate cytokine/chemokine panel (premixed 23-plex; Millipore, Bedford, MA) with a Bio-Plex 200 system (Bio-Rad Laboratories, Hercules, CA). No statistically significant differences between cytokine/chemokine levels for Norway3487 virus and those for Norway3568 virus were found.
In vitro characterization of Norwegian viruses: receptor specificity and polymerase activity.
As described above, there are 10 amino acid differences between Norway3487 and Norway3568 viruses (Table 2). Amino acid residues that determine receptor-binding specificity have been identified in the HA receptor-binding pocket. The Gly at position 222 of HA has been shown to confer binding to avian-type receptors, whereas the Asp at position 222 of HA confers binding to human-type receptors (20). To examine the receptor-binding specificity of Norway3487 and Norway3568 viruses, we conducted glycan microarrays as described elsewhere (35). As expected, Norway3568 virus, which possesses HA222-Asp (5), efficiently bound to α2,6Gal-sialylated glycans (human-type receptor), whereas Norway3487 virus, which possesses HA222-Gly, bound to multiple α2,3Gal-sialylated glycans (avian-type receptor) as well as to α2,6Gal-sialylated glycans (Fig. 6). In agreement with previous studies (5, 19, 35, 38), these results suggest that an Asp-to-Gly amino acid change at position 222 of the HA of Norway3487 virus confers the ability to recognize both avian-type and human-type receptors.
Fig 6.
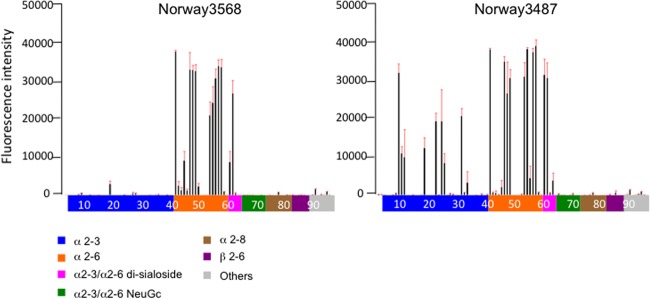
Receptor specificity of Norwegian viruses. Sialylated glycan binding by Norway3568 and Norway3487 viruses; purified whole virions were analyzed by glycan microarray. The microarrays displayed 86 sialylated and 9 asialo glycans printed on coated glass slides. Different types of glycans on the array (x axis) are highlighted in different colors; the identity of each numbered glycan is provided in Table S1 in the supplemental material. Black bars denote the mean fluorescent binding signal intensity (y axis) of 4 spots; the standard error is shown as a red extension.
The viral polymerase complex has been shown to affect viral replicative ability and pathogenicity (8, 11, 18, 33). To assess the viral polymerase activity of Norway3487 and Norway3568 viruses, we conducted a luciferase activity-based minireplicon assay as described previously (25). Since the amino acid sequences of PA and NP are identical for these two viruses, we used the same plasmids for the expression of Norway3487 and Norway3568 virus PA and NP proteins, respectively. As shown in Fig. 7, the polymerase activity of the Norway3487 polymerase complex was about 1.5-fold higher than that of the Norway3568 polymerase complex at 37°C (P < 0.001). Moreover, a polymerase complex possessing Norway3487 PB2 and Norway3568 PB1 possessed stronger polymerase activity than a complex possessing PB2 and PB1 from Norway3568 virus (P < 0.001), whereas one possessing Norway3568 PB2 and Norway3487 PB1 did not (Fig. 7). These findings suggest that Norway3487 PB2 contributes to enhanced polymerase activity, presumably resulting in the efficient replication of this virus in vitro and in vivo.
Fig 7.
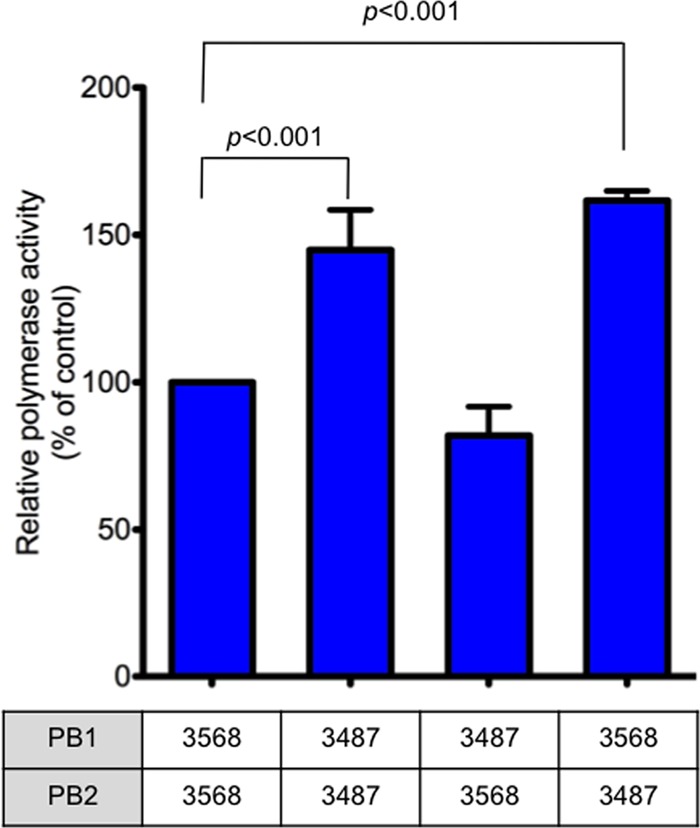
Polymerase activity of Norwegian viruses measured in a minigenome replicon assay. Four protein expression plasmids for PB2, PB1, PA, and NP derived from Norway3487 or Norway3568, pPolI-WNA-Flu expressing the NA gene encoding the firefly luciferase gene, and the pGL4.74[hRuc/TK] control plasmid were transfected into 293T cells and assayed for luciferase activity after a 24-h incubation at 37°C. Since Norway3487 and Norway3568 viruses do not differ in their PA and NP sequences, only combinations of PB1 and PB2 proteins were tested. 3487 and 3568, Norway3487 and Norway3568 viruses, respectively. The values shown are means ± standard deviations for the results of three independent experiments and are standardized to the activities of the expression plasmids for the Norway3568 RNP complex proteins (100%). Polymerase activities of replication complexes possessing the PB2 protein from Norway3487 virus were higher than those possessing PB2 from Norway3568 virus (P < 0.001).
DISCUSSION
There are several known virulence markers for influenza viruses (6, 8, 11, 14, 16, 18, 24, 29, 33), and the polymerase complex and HA are known to be key players in host range determination and pathogenicity (8, 11, 16, 18, 21, 33). Despite the lack of known virulence markers in A(H1N1)pdm09 viruses, many severe cases of infection in people with no recognized risk factors were reported (2). Here, we found that two Norwegian isolates (Norway3487 and Norway3568, which belong to cluster II) not only differed in their clinical outcomes, despite their genetic similarity (i.e., only 10 amino acid differences), but also differed in their biological phenotypes in vitro and in vivo (i.e., replication ability in cultured cells and animals, viral clearance in ferrets, and pathogenicity in nonhuman primates). These distinct phenotypes for the Norwegian isolates may result from differences in their biological properties, such as the receptor-binding specificity of their HA and their viral polymerase activity. The more pathogenic Norway3487 virus has the Asp-to-Gly change at position 222 of its HA, conferring avian-type receptor-binding specificity. Thus, this change may result in more efficient infection of human alveolar type II pneumocytes, which express avian-type receptors, reducing the availability of progenitor cells for essential lung functions and thus leading to severe pulmonary impairment, diffuse alveolar damage, and respiratory distress, culminating in the increased pathogenicity in nonhuman primates described previously (35, 37). Moreover, we found that PB2 derived from Norway3487 virus contributed to higher polymerase activity, possibly leading to more efficient viral replication in vitro and in vivo. Since higher viral replication may cause increased pathogenicity in animals, it may be that PB2 also contributes to the pathogenicity of Norway3487 virus.
We showed that the cytokine/chemokine responses to infection occurred as two peaks in the infected nonhuman primates (Fig. 5). The magnitude of induction of the first peak of cytokines correlated in most animals with high viral titers in the BAL fluid at day 1 postinfection (Fig. 3 and 5 and Table 3). One of the animals infected with Norway3568 virus (animal 369) and three animals infected with Norway3487 had high viral titers in the BAL fluid and showed strong induction of the first peak of cytokines/chemokines (i.e., GM-CSF, IL-18, TNF-α, MIP-1α, MIP-1β, IL-1β, and IL-6) relative to that in animals with lower viral titers. Thus, higher levels of viral replication were associated with strong proinflammatory responses, which potentially augmented lung damage. The increased levels of IL-1R and VEGF in Norway3568 virus-infected animals after day 7 postinfection might signify tissue repair and remodeling of the alveolar architecture following viral clearance. This mechanism may be delayed in Norway3487 virus-infected animals (22, 26), possibly as a result of the increased tropism of the HA-222G for type II pneumocytes (35). Based on these data, we speculate that infection with Norway3487 virus leads to increased viral titers in the lungs, which trigger stronger inflammatory responses, augmenting tissue damage and delaying healing in the lungs of infected animals.
In summary, here we characterized two A(H1N1)pdm09 viruses that belong to the currently circulating cluster II viruses and caused distinct clinical symptoms, despite their genetic similarity. Our results suggest the potential involvement of the receptor-binding specificity of HA and of the polymerase activity in both the replicative ability in vitro and in vivo and the pathogenicity of (H1N1)pdm09 viruses in a nonhuman primate model. Continued surveillance and more detailed investigations will further our understanding of the mechanism of pathogenicity of the currently circulating A(H1N1)pdm09 viruses.
Supplementary Material
ACKNOWLEDGMENTS
We thank Li-Mei Chen, Pierre Rivailler, and Ruben Donis (Centers for Disease Control and Prevention, Atlanta, GA) for receptor-binding analyses. We also thank Ben Wolter, Carissa Boettcher, Nichole Goecks, Jennifer Post, Martha McGregor, Kelly Moore, Ashley Luka, Yuko Sakai-Tagawa, and Mutsumi Ito for technical support. We thank Susan Watson for editing the manuscript.
The glycan microarray was produced for CDC by using a glycan library generously provided by the Consortium for Functional Glycomics, funded by National Institute of General Medical Sciences grant GM62116. This work was supported by National Institute of Allergy and Infectious Diseases (NIAID) Public Health Service research grants; by the NIAID-funded Center for Research on Influenza Pathogenesis (CRIP; HHSN266200700010C); by a Grant-in-Aid for Specially Promoted Research; by the Japan Initiative for Global Research Network on Infectious Diseases from the Ministry of Education, Culture, Sports, Science, and Technology; by grants-in-aid from the Ministry of Health, Labor and Welfare of Japan; and by ERATO (the Japan Science and Technology Agency). This work was also supported by NIH National Center for Research Resources grant P51 RR000167 to the Wisconsin National Primate Research Center.
Footnotes
Published ahead of print 20 June 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Baskin CR, et al. 2009. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc. Natl. Acad. Sci. U. S. A. 106:3455–3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bautista E, et al. 2010. Clinical aspects of pandemic 2009 influenza A (H1N1) virus infection. N. Engl. J. Med. 362:1708–1719 [DOI] [PubMed] [Google Scholar]
- 3. Blixt O, et al. 2004. Printed covalent glycan array for ligand profiling of diverse glycan binding proteins. Proc. Natl. Acad. Sci. U. S. A. 101:17033–17038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Christman MC, Kedwaii A, Xu J, Donis RO, Lu G. 2011. Pandemic (H1N1) 2009 virus revisited: an evolutionary retrospective. Infect. Genet. Evol. 11:803–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chutinimitkul S, et al. 2010. Virulence-associated substitution D222G in the hemagglutinin of 2009 pandemic influenza A(H1N1) virus affects receptor binding. J. Virol. 84:11802–11813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Conenello GM, Zamarin D, Perrone LA, Tumpey T, Palese P. 2007. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 3:1414–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fereidouni SR, Beer M, Vahlenkamp T, Starick E. 2009. Differentiation of two distinct clusters among currently circulating influenza A(H1N1)v viruses, March-September 2009. Euro Surveill. 14(46):pii=19409 [PubMed] [Google Scholar]
- 8. Gabriel G, Herwig A, Klenk HD. 2008. Interaction of polymerase subunit PB2 and NP with importin alpha1 is a determinant of host range of influenza A virus. PLoS Pathog. 4:e11 doi:10.1371/journal.ppat.0040011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Garten RJ, et al. 2009. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 325:197–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gibbs AJ, Armstrong JS, Downie JC. 2009. From where did the 2009 ‘swine-origin’ influenza A virus (H1N1) emerge? Virol. J. 6:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hatta M, Gao P, Halfmann P, Kawaoka Y. 2001. Molecular basis for high virulence of Hong Kong H5N1 influenza A viruses. Science 293:1840–1842 [DOI] [PubMed] [Google Scholar]
- 12. Herfst S, et al. 2010. Pandemic 2009 H1N1 influenza virus causes diffuse alveolar damage in cynomolgus macaques. Vet. Pathol. 47:1040–1047 [DOI] [PubMed] [Google Scholar]
- 13. Itoh Y, et al. 2009. In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature 460:1021–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jackson D, Hossain MJ, Hickman D, Perez DR, Lamb RA. 2008. A new influenza virus virulence determinant: the NS1 protein four C-terminal residues modulate pathogenicity. Proc. Natl. Acad. Sci. U. S. A. 105:4381–4386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Katz JM, Wang M, Webster RG. 1990. Direct sequencing of the HA gene of influenza (H3N2) virus in original clinical samples reveals sequence identity with mammalian cell-grown virus. J. Virol. 64:1808–1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kawaoka Y, Webster RG. 1988. Sequence requirements for cleavage activation of influenza virus hemagglutinin expressed in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 85:324–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kobasa D, et al. 2007. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 445:319–323 [DOI] [PubMed] [Google Scholar]
- 18. Li Z, et al. 2005. Molecular basis of replication of duck H5N1 influenza viruses in a mammalian mouse model. J. Virol. 79:12058–12064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu Y, et al. 2010. Altered receptor specificity and cell tropism of D222G haemagglutinin mutants from fatal cases of pandemic A(H1N1) 2009 influenza. J. Virol. 84:12069–12074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Matrosovich M, et al. 2000. Early alterations of the receptor-binding properties of H1, H2, and H3 avian influenza virus hemagglutinins after their introduction into mammals. J. Virol. 74:8502–8512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matrosovich MN, Gambaryan AS, Klenk H-D. 2008. Receptor specificity of influenza viruses and its alteration during interspecies transmission, p 134–155 In Klenk H-D, Matrosovich MN, Stech J. (ed), Avian influenza, vol 27 Karger, Basel, Switzerland [Google Scholar]
- 22. Mura M, dos Santos CC, Stewart D, Liu M. 2004. Vascular endothelial growth factor and related molecules in acute lung injury. J. Appl. Physiol. 97:1605–1617 [DOI] [PubMed] [Google Scholar]
- 23. Neumann G, et al. 1999. Generation of influenza A viruses entirely from cloned cDNAs. Proc. Natl. Acad. Sci. U. S. A. 96:9345–9350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Obenauer JC, et al. 2006. Large-scale sequence analysis of avian influenza isolates. Science 311:1576–1580 [DOI] [PubMed] [Google Scholar]
- 25. Ozawa M, et al. 2007. Contributions of two nuclear localization signals of influenza A virus nucleoprotein to viral replication. J. Virol. 81:30–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Park JW, et al. 2004. Interleukin-1 receptor antagonist attenuates airway hyperresponsiveness following exposure to ozone. Am. J. Respir. Cell Mol. Biol. 30:830–836 [DOI] [PubMed] [Google Scholar]
- 27. Rimmelzwaan GF, et al. 2001. Pathogenesis of influenza A (H5N1) virus infection in a primate model. J. Virol. 75:6687–6691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Safronetz D, et al. 2011. Pandemic swine-origin H1N1 influenza A virus isolates show heterogeneous virulence in macaques. J. Virol. 85:1214–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Seo SH, Hoffmann E, Webster RG. 2002. Lethal H5N1 influenza viruses escape host anti-viral cytokine responses. Nat. Med. 8:950–954 [DOI] [PubMed] [Google Scholar]
- 30. Shiino T, et al. 2010. Molecular evolutionary analysis of the influenza A(H1N1)pdm, May-September, 2009: temporal and spatial spreading profile of the viruses in Japan. PLoS One 5:e11057 doi:10.1371/journal.pone.0011057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shinya K, et al. 2006. Avian flu: influenza virus receptors in the human airway. Nature 440:435–436 [DOI] [PubMed] [Google Scholar]
- 32. Smith GJ, et al. 2009. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 459:1122–1125 [DOI] [PubMed] [Google Scholar]
- 33. Subbarao EK, London W, Murphy BR. 1993. A single amino acid in the PB2 gene of influenza A virus is a determinant of host range. J. Virol. 67:1761–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Trifonov V, Khiabanian H, Rabadan R. 2009. Geographic dependence, surveillance, and origins of the 2009 influenza A (H1N1) virus. N. Engl. J. Med. 361:115–119 [DOI] [PubMed] [Google Scholar]
- 35. Watanabe T, et al. 2011. Avian-type receptor-binding ability can increase influenza virus pathogenicity in macaques. J. Virol. 85:13195–13203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Weinfurter JT, et al. 2011. Cross-reactive T cells are involved in rapid clearance of 2009 pandemic H1N1 influenza virus in nonhuman primates. PLoS Pathog. 7:e1002381 doi:10.1371/journal.ppat.1002381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu L, et al. 2010. A single-amino-acid substitution in the HA protein changes the replication and pathogenicity of the 2009 pandemic A (H1N1) influenza viruses in vitro and in vivo. Virol. J. 7:325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang H, Carney P, Stevens J. 2010. Structure and receptor binding properties of a pandemic H1N1 virus hemagglutinin. PLoS Curr. 2:RRN1152 doi:10.1371/currents.RRN1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.