

Abstract
Osteoclasts are multinuclear bone-resorbing cells formed by the fusion of monocyte/macrophage-lineage precursor cells. Activation of the transcription factor NFATc1 (nuclear factor of activated T cells c1) by the receptor activator of NF-κB ligand (RANKL) is critical for osteoclast differentiation. In our previous report (Y. Kuroda, C. Hisatsune, T. Nakamura, K. Matsuo, and K. Mikoshiba. Proc. Natl. Acad. Sci. U. S. A. 105:8643, 2008), we demonstrated that osteoblasts induce osteoclast differentiation via Ca2+ oscillation/calcineurin-dependent and -independent NFATc1 activation pathways; however, the mechanism underlying the latter remained unclear. Here we show that Cot, a serine/threonine kinase also known as tumor progression locus 2 (Tpl-2), directly phosphorylates all Ca2+/calcineurin-regulated NFAT family members (NFATc1 through NFATc4) and increases their protein levels. Moreover, Cot activity in osteoclasts was enhanced via cell-cell interaction with osteoblasts, and Cot promoted Ca2+ oscillation/calcineurin-independent osteoclastogenesis by increasing NFATc1 stability through phosphorylation. We propose that NFAT activation in vivo occurs via phosphorylation-induced protein stabilization, even in the absence of Ca2+ oscillation and calcineurin activity.
INTRODUCTION
Maintenance of bone homeostasis is achieved by a continuous bone remodeling that involves two opposing processes, bone resorption by osteoclasts and bone formation by osteoblasts. Interaction between these two cell types is important for proper bone remodeling (23). Accumulating data indicate that the bone destruction caused by osteoclast abnormalities occurs under various pathological conditions, such as postmenopausal osteoporosis, osteoarthritis, and bone metastasis of cancer (37), but the precise molecular mechanisms by which excessive osteoclast differentiation and activation are induced under these conditions remain unknown.
Mature osteoclasts are multinucleated cells derived from the monocyte/macrophage lineage that exhibit bone resorption activity. Macrophage colony-stimulating factor (M-CSF) and the receptor activator of NF-κB ligand (RANKL) are two indispensable cytokines that induce osteoclast differentiation from bone marrow-derived monocyte/macrophage precursor cells (BMMs). RANKL in particular contributes to osteoclast differentiation by activating the transcription factor NFATc1, a master regulator of osteoclast terminal differentiation (22, 35). NFATc1 upregulates genes important for osteoclast differentiation and function, such as those encoding the dendritic cell-specific transmembrane protein (DC-STAMP) and the vacuolar proton pump subunit Atp6v0d2, both of which are important for cell-cell fusion (15, 17, 41); the protease cathepsin K, which is secreted into resorption lacunae (21); and β3 integrin, which regulates sealing ring formation (6). NFATc1 activation is essential for osteoclastogenesis, as evidenced by the fact that NFATc1-deficient embryonic stem cells fail to differentiate into osteoclasts (35).
The canonical mechanism of NFATc1 activation is through dephosphorylation by calcineurin, a Ca2+/calmodulin-dependent phosphatase, and subsequent nuclear translocation. The NFAT family consists of five members: NFATc1 through NFATc4, which are regulated by Ca2+/calcineurin-signaling, and NFAT5, which is activated by osmotic stress. All four Ca2+/calcineurin-regulated isoforms exhibit two conserved domains: the NFAT homology region (NHR) in the N-terminal half and the Rel homology region (RHR), including the DNA binding domain, in the C-terminal half (20, 12). The NHR contains the transactivation domain and a regulatory domain exhibiting numerous serine residues. In general, NFAT activation is initiated by calcineurin-mediated dephosphorylation of the regulatory domain. However, we previously reported that NFATc1 expressed in osteoclast precursors is activated even in the presence of the calcineurin inhibitor FK506, when cells are cocultured with osteoblasts, promoting differentiation of precursors into multinuclear osteoclasts. We also showed that osteoblasts induce differentiation of inositol 1,4,5-trisphosphate receptor type 2 and type 3 double knockout (IP3R2/3KO) BMMs into osteoclasts without detectable RANKL-induced Ca2+ oscillation (16). These findings strongly suggest the existence of a Ca2+ oscillation/calcineurin-independent NFATc1 activation pathway for osteoclastogenesis.
Cot (cancer Osaka thyroid), a serine/threonine kinase gene also known as tumor progression locus 2 (Tpl-2), was initially identified in a screen for transforming genes expressed by a human thyroid carcinoma cell line (24). The endogenous Cot gene encodes a protein of the mitogen-activated protein kinase kinase kinase (MAPKKK) family. Overexpressed Cot activates the MAPK extracellular signal-regulated kinase (ERK), JNK, and p38 (5, 28, 32). Cot is also critical for upregulation of an inflammatory cytokine, tumor necrosis factor alpha (TNF-α), in macrophages in response to lipopolysaccharide (LPS) stimulation (8). Macrophages derived from Cot-deficient mice show defects in LPS-induced ERK phosphorylation but not in JNK, p38, or NF-κB activation, demonstrating that Cot physiologically functions as a MAPKKK selective for ERK in these cells. Exogenously overexpressed Cot in cultured cell lines also reportedly stimulates activity of transcription factors such as NFAT, NF-κB, and AP-1 (1, 2, 5, 18, 39). Cot can also induce calcineurin-independent NFAT transactivation via the NFAT N terminus (7). Thus, Cot potentially stimulates the Ca2+ oscillation/calcineurin-independent NFAT activation pathway during osteoclast differentiation.
In this study, we demonstrate that Cot stimulated via osteoclast-osteoblast interaction promotes Ca2+ oscillation/calcineurin-independent osteoclastogenesis. We also show that Cot increases NFATc1 protein stability through phosphorylation of residues distinct from those required for cytoplasmic-nuclear shuttling, thereby enhancing NFATc1 activation in a Ca2+ oscillation/calcineurin-independent manner. Collectively, our data provide novel mechanistic insight into how osteoblasts promote osteoclastogenesis through Ca2+ oscillation/calcineurin-independent NFATc1 activation.
MATERIALS AND METHODS
Mice and bone analysis.
Generation of IP3R2KO and CotKO mice has been described (9, 13). IP3R2/CotKO mice were generated by crossing IP3R2KO and CotKO mice in a 129/SvJ and C57BL/6 hybrid background. Mice showed no abnormality in growth rate or body weight. Histological experiments and histomorphometric analysis (27) were performed by Kureha Special Laboratory (Tokyo, Japan). The left tibia was fixed in 70% ethanol, and the undecalcified bone was embedded in glycol methacrylate. Sections 3 μm thick were cut longitudinally in the proximal region of the tibia and stained with toluidine blue and tartrate-resistant acid phosphatase (TRAP). Histomorphometry was performed with a semiautomatic image analyzing system (Osteoplan II; Carl Zeiss, Thornwood, NY) linked to a light microscope. Histomorphometric measurements were made at ×400 using a minimum of 21 optical fields in the secondary spongiosa. All animals were ethically treated according to guidelines of the Animal Experiments Committee of RIKEN Brain Science Institute.
In vitro osteoclastogenesis.
The method used for in vitro osteoclastogenesis was previously described (16). Briefly, nonadherent bone marrow cells were cultured with 10 ng/ml M-CSF (R&D Systems) for 3 days. Adherent cells served as BMMs and were further cultured with 100 ng/ml soluble RANKL (sRANKL) (PeproTech) and 10 ng/ml M-CSF. For immunocytochemistry, nonadherent bone marrow cells were cultured on cover glasses (Matsunami) coated with vitronectin (Promega). To analyze the effect of FK506 (Calbiochem and Fujisawa Pharmaceutical), inhibitor was added simultaneously with sRANKL. For coculture studies, bone marrow cells were cultured together with ST2 cells in the presence of 10−8 M 1,25-dihydroxyvitamin D3 (Calbiochem) and 10−7 M dexamethasone (Sigma) for 4 to 5 days. The medium was refreshed 3 days after RANKL stimulation in the sRANKL/M-CSF-induced osteoclast differentiation system and every 2 days in the coculture system.
Construction of retroviral vectors and infection of primary osteoclasts.
Mouse Cot cDNA in pBluescript was obtained from a FANTOM3 clones set (3). With Cot cDNA as the template, PCR was performed to synthesize the truncated form of Cot (trCot). The primers used were as follows: forward, CGC TCG AGA TGG AGT ACA TGA GCA, and reverse, GGG AAT TCT CAG TTA ACT CGT GGC TGG TCC TCT CT. The PCR fragment was then subcloned into the XhoI and EcoRI sites of the pMSCV-GFP retroviral expression vector. pMSCV-HA-trCot-IRES-GFP was generated by inserting a hemagglutinin (HA) tag into the 5′ end of trCot. Recombinant retroviruses were produced by cotransfecting pMSCV-GFP, pMSCV-trCot-IRES-GFP, or pMSCV-HA-trCot-IRES-GFP with pVSV-G (Clontech) into the GP2-293 packaging cell line (Clontech) using Lipofectamine 2000 (Invitrogen). The medium was replaced 8 h later, and viral supernatants were harvested 2 days posttransfection and stored at −80°C. For viral infections, 5 × 105 cells were plated in each well of a 24-well plate and cultured with 10 ng/ml M-CSF. After 2 days, medium was replaced with 350 μl of viral supernatant containing 8.0 μg/ml Polybrene (Sigma) and 10 ng/ml M-CSF. Twenty-four hours later, supernatants were removed, and cells were further cultured with 10 ng/ml M-CSF and 100 ng/ml RANKL for 3 to 4 days.
Bone resorption assay.
Retrovirally infected cells were cultured on bone slices for 8 days in the presence of RANKL/M-CSF. Resorption pits on bone slices were visualized by wheat germ agglutinin (WGA) staining as described previously (33). Total resorption area per bone slice was quantified using ImageJ software (NIH).
DNA constructs and mutagenesis.
To construct the mouse trCot expression vector (pME18S-trCot), the trCot fragment was excised from pMSCV-trCot-IRES-GFP and inserted into the pME18S vector (36), and an HA tag was inserted at the 5′ end. A Cot kinase-deficient mutant (KMCot) was generated by introducing the K167M mutation (18) using a QuikChange II site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. Full-length NFATc1 (amino acids [aa] 1 to 716) was amplified by PCR using pSH160c as the template, and the product was subcloned into pME18S. N-terminal (aa 1 to 418) and C-terminal (aa 419 to 716) fragments of the wild type and an active form of NFATc1 were amplified by PCR using pSH160c or pMSCV-caNFATc1-IRES-GFP as the template, and products were subcloned into the pGEX-4T-1 vector (Amersham). Human NFATc2 cDNA was a kind gift from Tim Hoey (11). Mouse NFATc3 and NFATc4 cDNAs were obtained from a FANTOM3 clone set (3). Full-length NFATc2, NFATc3, and NFATc4 were PCR amplified and subcloned into pcDNA3.1/myc-His (Invitrogen). Full-length NFATc3 C-terminally tagged with the Myc epitope and 6His was further subcloned into pME18S. The N-terminal fragment of NFATc2 (aa 1 to 407), NFATc3 (aa 1 to 431), or NFATc4 (aa 1 to 416) was PCR amplified using pcDNA-myc-His-NFATc2, NFATc3, or NFATc4 as the template, and products were subcloned into pGEX-4T-1 (Amersham). Nucleotide sequences of all constructs were confirmed by DNA sequencing.
Transfection and immunoprecipitation.
HeLa, COS-7, or RAW264.7 cells were transfected with various plasmids using TransIT-LT1 (Mirus) or FuGENE (Roche), according to the manufacturer's recommendations. HeLa cells were lysed with TNE buffer (10 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1.0% Nonidet P-40, 1 mM EDTA, 25 mM NaF), and COS-7 and RAW264.7 cells were lysed with TNE buffer containing 0.1% sodium dodecyl sulfate (SDS) for 30 min at 4°C. Lysates were centrifuged at 20,000 × g for 10 min. Then, 0.5 μg of anti-HA antibody 3F10 (Roche) and 20 μl of protein G-Sepharose (GE Healthcare) were added to the supernatants. After incubation for 3 h at 4°C, immunocomplexes were washed five times with the same buffer. For immunoprecipitation of RAW264.7 cells, cells were divided into three groups 24 h after transfection and further cultured for 24 h with medium containing 0.5% serum under control, sRANKL treatment, or ST2 cell coculture conditions.
Immunoblotting and immunocytochemistry.
Methods for immunoblotting and immunocytochemistry were previously described (16). For immunocytochemistry of transfected HeLa cells, normal growth medium was replaced with serum-free medium 16 h after transfection, and cells were incubated another 4 h with or without 1 μM FK506. The primary antibodies used were anti-NFATc1 MAb 7A6 (Santa Cruz Biotechnology), anti-Cot antibody M-20 (Santa Cruz Biotechnology), anti-phospho-Tpl2 Ser400 (Cell Signaling), anti-β-tubulin clone TUB 2.1 (Sigma), anti-β-actin MAb AC-15 (Sigma), anti-HA antibody 3F10 (Roche), and anti-PKCα antibody (Sigma). Membranes were scanned with an LAS-4000 luminescent image analyzer (Fujifilm) or by using the Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE). Immunoreacted coverslips were examined with a confocal laser scanning microscope (FV10i; Olympus). The mean signal intensity and integral intensity in the entire cell or nucleus were measured by freehand region of interest (ROI) surrounding areas of interest using ImageJ software (NIH) to quantify NFATc1 expression levels in HeLa cells (32 to 37 cells).
Luciferase reporter assay.
The calcitonin receptor promoter-luciferase reporter plasmid (pCTR-luc) (35) was transfected into RAW264.7 cells. RANKL was added 2 h after transfection, and luciferase activity was measured 24 h after RANKL treatment using a dual-luciferase reporter assay system (Promega).
Kinase assay.
COS-7 cells transfected with a vector encoding HA-tagged trCot or KMCot were lysed with TNE buffer containing 0.1% SDS. Recombinant proteins purified from Escherichia coli expressing GST-fused full-length NFATc1 (aa 1 to 716), GST–N-terminal NFATc1 (aa 1 to 418), GST–C-terminal NFATc1 (aa 419 to 716), GST-caNFATc1 (aa 1 to 418), GST–N-terminal NFATc2 (aa 1 to 407), GST–N-terminal NFATc3 (aa 1 to 431), or GST–N-terminal NFATc4 (aa 1 to 416) were added to immunoprecipitated Cot in kinase buffer (40 mM HEPES [pH 7.4], 10 mM MgCl2, 3 mM MnCl2, 2 mM dithiothreitol [DTT], and 5 mM NaF) containing 200 mM [γ-32P]ATP (Amersham Biosciences) for 20 min at 30°C. Phosphorylated NFATc1 fusion proteins were separated by SDS-polyacrylamide gel electrophoresis (PAGE), and radioactivity was quantified using the BAS 5000 system (Fujifilm).
Subcellular fractionation.
At 16 h after transfection of cells, growth medium was changed to serum-depleted medium for 4 h, and subcellular fractionation was performed as described previously (31). Briefly, HeLa cells resuspended in buffer (10 mM Tris-Cl [pH 7.5], 10 mM NaCl, 3 mM MgCl2, 0.5 mM DTT, 0.1 mM EGTA, 0.05% Nonidet P-40, and a protease inhibitor cocktail [Roche]) were centrifuged at 650 × g to pellet nuclei.
Alkaline phosphatase treatment.
Lysates of transfected HeLa cells were prepared in buffer containing 20 mM Tris-HCl [pH 7.5], 2 mM EDTA, 0.5% Triton X-100, 0.1% SDS, 25 mM NaF, and a protease inhibitor cocktail (Roche). Lysates were incubated with calf intestine alkaline phosphatase (TaKaRa) (10 U/50 μl) in the presence of 5 mM MgCl2 for 30 min at 37°C.
Reverse transcriptase PCR.
Total RNAs were isolated using TRIzol reagent (Invitrogen). First-strand cDNA was produced from total RNA using SuperScript II (Invitrogen) reverse transcriptase and oligo(dT) primers. Primer sequences were as follows: NFATc1 forward, CTG AAT TCA TGC CAA GCA CCA GCT TTC C; NFATc1 reverse, ATC TCG AGT TAG CCC TCC TCG GGG TCC GTG G; glyceraldehyde-3-phosphate dehydrogenase (Gapdh) forward, ATG GTG AAG GTC GGT GTG AAC G; and Gapdh reverse, AAA CAT GGG GGC ATC GGC AGA A. A total of 20 or 25 cycles were used to amplify NFATc1, and 25 cycles were used to amplify Gapdh.
Cycloheximide (CHX) chase assay.
Transfected HeLa cells were incubated in serum-depleted medium for 4 h and treated with 50 μg/ml CHX (Wako). Cocultured osteoclasts were pretreated with 1 μM FK506 for 2 h and then with 10 μg/ml CHX in the presence of FK506. Total proteins were collected at different time points and immunoblotted. Densitometric analysis was performed using ImageJ software (NIH) to quantify NFATc1 expression. NFATc1 band intensity was normalized to β-actin and then normalized to that at time zero.
Statistical analysis.
Statistical analysis was performed using Student's t test.
RESULTS
Cot induces Ca2+ oscillation/calcineurin-independent osteoclastogenesis and NFATc1 activation.
We previously demonstrated that osteoblasts induce Ca2+ oscillation/calcineurin-independent NFATc1 activation in osteoclast precursors via osteoclast-osteoblast interaction (16). To examine the molecular mechanisms underlying this activity, we focused on the serine/threonine protein kinase Cot, since it reportedly enhances NFAT activity via a calcineurin-independent pathway in other cell types (1, 7, 40). We first ectopically expressed a truncated, constitutively active form of Cot (trCot) (4) in IP3R2/3KO BMMs, in which Ca2+ oscillation-triggered NFATc1 activation is defective and sRANKL/M-CSF-induced osteoclastogenesis is impaired. Remarkably, IP3R2/3KO BMMs overexpressing trCot differentiated into tartrate-resistant acid phosphatase (TRAP)-positive multinuclear osteoclasts upon sRANKL/M-CSF treatment, while control cells expressing GFP alone did not (Fig. 1A). trCot-expressing IP3R2/3KO osteoclasts exhibited a number of nuclei similar to that seen in GFP-overexpressing wild-type (WT) osteoclasts (see Fig. S1 in the supplemental material) and resorbed bone to an extent similar to that of control WT osteoclasts (Fig. 1B). trCot overexpression in WT BMMs also enhanced sRANKL/M-CSF-induced osteoclastogenesis (Fig. 1A, graph), similar to the effect seen following expression of constitutively active NFATc1 (caNFATc1) (16). Treatment of cells with the calcineurin inhibitor FK506 did not inhibit trCot-induced osteoclastogenesis of WT or IP3R2/3KO BMMs, further demonstrating that osteoclastogenesis induced by trCot is independent of Ca2+ oscillation/calcineurin (Fig. 1A). We next examined whether NFATc1 transcriptional activity was enhanced by Cot kinase in RAW264.7 monocytes/macrophages, which differentiate into osteoclasts in response to sRANKL. Luciferase assays with the calcitonin receptor promoter, a direct NFATc1 target during osteoclast differentiation (32), showed that NFATc1-dependent transcriptional activity was enhanced by trCot but not by kinase-deficient mutant KMCot (18). This effect was not inhibited by FK506 treatment (Fig. 1C), suggesting that Cot induces NFATc1 transcriptional activity in a calcineurin-independent manner. NFATc1 is reportedly subject to positive autoamplification during osteoclastogenesis (12, 35). Thus, if trCot induces NFATc1 activation, NFATc1 upregulation should be observed in osteoclasts. Indeed, trCot expression significantly increased NFATc1 protein levels in primary osteoclasts (Fig. 1D, left). This effect was also seen in cells treated with FK506 (Fig. 1D, right), consistent with the finding that trCot induces BMM osteoclastogenesis, even in the presence of FK506 (Fig. 1A). Notably, FK506 treatment inhibited calcineurin activity, as judged by a single, slow-migrating band of phosphorylated NFATc1 seen on SDS-PAGE (Fig. 1D, right), as opposed to multiple faster-migrating NFATc1 bands indicative of dephosphorylation by calcineurin (Fig. 1D, left). Canonical NFATc1 activation occurs through dephosphorylation by calcineurin and subsequent nuclear translocation. Since a dephosphorylated form of NFATc1 was not evident in trCot-expressing cells treated with FK506 (Fig. 1C), we asked whether NFATc1 was translocated into nuclei in these cells. In WT cells infected with control or trCot virus, NFATc1 nuclear translocation was observed in differentiated multinuclear osteoclasts (Fig. 1E). In IP3R2/3KO or FK506-treated cells infected with control virus, NFATc1 expression levels were low and NFATc1 was cytoplasmic in mononuclear cells (Fig. 1E). In contrast, infection with trCot virus promoted NFATc1 nuclear translocation in IP3R2/3KO and FK506-treated cells (Fig. 1E). Since dephosphorylated forms of NFATc1 were not seen in cells treated with FK506, trCot likely activates NFATc1 via a noncanonical mechanism. Misexpressed trCot protein was cytoplasmic, and levels of cytoplasmic NFATc1 increased in trCot-expressing cells compared with control cells (Fig. 1E), suggesting that Cot kinase interacts with NFATc1 in the cytoplasm.
Fig 1.

Cot induces Ca2+ oscillation/calcineurin-independent osteoclastogenesis. (A) (Left) TRAP staining of wild-type (WT) and IP3R2/3KO (2/3KO) BMMs infected with retroviruses encoding either an active form of Cot plus GFP (trCot) or GFP alone (−) in the absence or presence of FK506. Bar, 50 μm. (Right) Percentage of TRAP-positive multinuclear osteoclasts (MNCs). Values are means ± standard deviations (SD); n = 4 for each group. (B) Bone resorption assay. Arrows show bone resorption pits. Bars, 200 μm. (C) NFATc1 transcriptional activity based on luciferase reporter assays in RAW264.7 cells following overexpression of an active form of Cot (trCot), a kinase-deficient mutant of Cot (KMCot) or empty vector (vec.) in the absence or presence of FK506. Values are means ± SD; n = 3 for each group. **, P < 0.01. (D) The expression levels of NFATc1 protein in BMMs infected with retroviruses encoding either an active form of Cot plus GFP (tr) or GFP alone (−) in the presence or absence of FK506. β-Tubulin signal served as an internal control. (E) Expression and localization of NFATc1 (red) in WT and 2/3KO osteoclasts infected with retroviruses encoding HA-tagged trCot plus GFP (HA-trCot, dark blue) or GFP alone (−) in the absence or presence of FK506. Nuclei are stained with DAPI (light blue). Bar, 50 μm.
Cot knockout BMMs show impaired Ca2+ oscillation/calcineurin-independent osteoclastogenesis.
We next used Cot knockout (CotKO) mice (8, 13) to determine whether Cot loss of function alters osteoclast differentiation. Lack of expression of Cot in these cells was confirmed by Western blotting (Fig. 2A). We hypothesized that if Cot was responsible for the Ca2+ oscillation/calcineurin-independent pathway, treatment of CotKO BMMs with FK506 should strongly inhibit osteoblast-induced osteoclastogenesis. When we employed an in vitro osteoclastogenesis protocol using sRANKL and M-CSF, we observed that CotKO BMMs differentiated normally into TRAP-positive multinuclear osteoclasts (Fig. 2B). FK506 treatment of CotKO BMMs blocked differentiation, as it did in WT BMMs (Fig. 2B), indicating that sRANKL-induced differentiation occurs through a Ca2+ oscillation/calcineurin-dependent pathway. When osteoclastogenesis was induced by coculture with mouse stromal ST2 cells, CotKO BMMs differentiated into osteoclasts to the same extent as did WT BMMs (Fig. 2C). Remarkably, in the presence of FK506, osteoclast differentiation of CotKO BMMs was much more strongly inhibited than that seen in WT BMMs (Fig. 2C). Conditional knockout of the regulatory subunit of calcineurin (CnB) in osteoblasts reportedly impairs osteoclastogenesis due to decreased RANKL expression and increased expression of osteoprotegerin (OPG), a decoy receptor for RANKL in osteoblasts (42). Thus, our findings might be a consequence of the effect of FK506 on osteoblasts. To exclude this possibility, we examined the effect of FK506 treatment on RANKL and OPG expression in ST2 cells by quantitative PCR. RANKL expression, which was induced in ST2 cells by treatment with 1,25-dihydroxyvitamin D3 and dexamethasone (Dex), was not significantly reduced by treatment with FK506 at concentrations up to 5 μM (see Fig. S2A in the supplemental material). OPG expression was slightly increased by FK506 treatment (see Fig. S2B); however, RANKL expression remained much higher than OPG expression (see Fig. S2), indicating that strong inhibition of osteoclastogenesis seen in CotKO BMMs following FK506 treatment was not due to decreased RANKL expression in ST2 cells.
Fig 2.
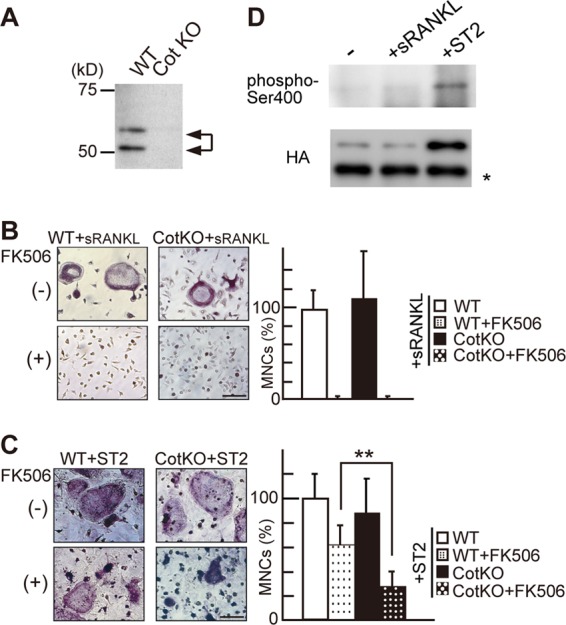
Cot mediates osteoblast-induced Ca2+ oscillation/calcineurin-independent osteoclastogenesis. (A) Cot expression in wild-type (WT) and Cot knockout (CotKO) primary osteoclasts, as detected by Western blotting. (B and C) Effect of FK506 on sRANKL/M-CSF-induced (B) and ST2 cell-induced (C) osteoclastogenesis of WT and CotKO BMMs. Note that the number of TRAP-positive MNCs in FK506-treated CotKO BMMs was significantly decreased compared with that seen in FK506-treated WT BMMs (C, right). Values are means ± SD; n = 6 for each group. **, P < 0.01. Bar, 50 μm. (D) Phosphorylation of Ser400 and expression levels of HA-Cot in RAW264.7 cells under control (−), sRANKL-treated (+sRANKL), and ST2 cell coculture (+ST2) conditions. *, antibody IgG band.
Next, we asked whether osteoblasts stimulate Cot kinase activity in osteoclasts via cell-cell interaction. Since ST2 cells also express Cot (data not shown), we expressed HA-tagged wild-type Cot (wtCot) in RAW264.7 cells and assessed kinase activity and protein levels of Cot purified from control, sRANKL-treated, and ST2-cocultured cells. Cot is activated by upstream kinases, and phosphorylation on Ser400 is required for Cot kinase activity in macrophages (30). If Cot is activated by interaction with ST2 cells, levels of Ser400-phosphorylated Cot should increase. We found that levels of Cot kinase protein were dramatically increased after coculture with ST2 cells but not following treatment with sRANKL (Fig. 2D, bottom) and that increased Cot protein was phosphorylated on Ser400 (Fig. 2D, top). These findings suggest that activated Cot is increased in osteoclast precursors via interaction with osteoblasts and functions in Ca2+ oscillation/calcineurin-independent NFATc1 activation during osteoclastogenesis.
Cot stimulated by osteoclast-osteoblast interaction phosphorylates NFATc1 and enhances its levels.
To gain mechanistic insight into how Cot activates NFATc1, we asked whether Cot phosphorylates NFATc1. First, we coexpressed NFATc1 and either HA-tagged wtCot, trCot, or KMCot in HeLa cells and examined potential interactions between Cot and NFATc1 by coimmunoprecipitation. We found that all three forms of Cot interacted with NFATc1, indicating that the Cot kinase activity is not required for interaction (Fig. 3A). Interestingly, we observed that coexpression of trCot, but not wtCot or KMCot, significantly increased NFATc1 protein levels and reduced NFATc1 mobility on SDS-PAGE (Fig. 3B), suggesting that activated Cot alters both NFATc1 protein levels and posttranslational modification. When HeLa cell lysates were treated with alkaline phosphatase (AP), the band mobility of NFATc1 coexpressed with trCot shifted downward to that seen in KMCot-coexpressing cells (Fig. 3C). These results strongly suggest that Cot forms a complex with NFATc1 and that its kinase activity induces NFATc1 phosphorylation. To determine whether Cot directly phosphorylates NFATc1, we next performed an in vitro kinase assay using immunopurified Cot and glutathione S-transferase (GST) fusions of full-length NFATc1 or the N-terminal or C-terminal half as the substrates (Fig. 3D). trCot directly phosphorylated full-length and N-terminal NFATc1 protein but not the C-terminal half (Fig. 3E, top), modifications that were not induced by KMCot. Interestingly, when assayed with a mutant form of the NFATc1 N terminus in which 21 Ser residues are replaced with Ala residues, mimicking a dephosphorylated active state (25) (see Fig. S3 in the supplemental material), Cot phosphorylated mutant NFATc1 to the same extent as it did wild-type NFATc1 (Fig. 3F). These findings suggest that Cot phosphorylates amino acid residues different from those required for cytoplasmic-nuclear shuttling. The N-terminal halves of NFATc2, NFATc3, and NFATc4 (termed the NFAT homology region [NHR]) are homologous to that of NFATc1 (Fig. 3D). Thus, we asked whether Cot phosphorylates these other Ca2+/calcineurin-regulated NFAT family proteins. Similarly to NFATc1, the N termini of other NFATs were also directly phosphorylated by trCot, as revealed by an in vitro kinase assay (Fig. 3G). Levels of each NFAT were drastically increased when proteins were coexpressed with trCot in HeLa cells (Fig. 3H), indicating that NFAT family proteins employ a common regulatory mechanism involving Cot-induced phosphorylation to enhance protein levels.
Fig 3.

Cot binds to and directly phosphorylates NFATc1. (A) Interaction of wild-type (wt), active (tr), and kinase-deficient (KM) forms of HA-tagged Cot with NFATc1 (c1) in HeLa cells by immunoprecipitation (IP) assay. In, input; IB, immunoblotting. (B) Increased expression and slower migration of NFATc1 in HeLa cells cotransfected with trCot, as revealed by comparison with the input lanes in panel A (detected by short exposure time). (C) NFATc1 is hyperphosphorylated in HeLa cells coexpressing trCot but not in cells expressing KMCot. Alkaline phosphatase (AP) treatment increased mobility of NFATc1, presumably through removal of Cot-induced phosphate groups. (D) Schematic representation of the structure of various GST fusion fragments of NFATc1. SRD, serine-rich domain. SP1, SP2, and SP3 are highly conserved Ser-Pro (SP) repeat motifs reportedly responsible for nuclear localization. NHR, NFAT homology region; RHR, Rel homology region; ca, constitutively active form. (E) Cot directly phosphorylates NFATc1. (Top) Cot phosphorylates the full-length (left, arrowhead) and the N-terminal region (right, arrowhead), but not the C-terminal region of NFATc1, as detected by autoradiography. (Bottom) Coomassie brilliant blue (CBB) staining. Closed arrowhead, full-length (left) or N-terminal region (right) of NFATc1. Open arrowhead, C-terminal region of NFATc1. (F) The N-terminal region of a constitutively active form (ca) of NFATc1 is also phosphorylated by Cot. Arrow, autophosphorylated trCot. (G) Cot directly phosphorylates the N-terminal region of Ca2+/calcineurin-regulated NFAT family proteins. (Top) trCot phosphorylates the N-terminal regions of NFATc2, NFATc3, and NFATc4 as detected by autoradiography. (Bottom) Coomassie brilliant blue (CBB) staining. (H) Increased protein levels of indicated Ca2+/calcineurin-regulated NFAT family proteins in HeLa cells cotransfected with trCot.
Cot enhances NFATc1 protein stability through phosphorylation.
Our findings suggest that Cot-induced phosphorylation positively regulates NFATc1 activity. To determine if this is the case, we first examined the effect of Cot on NFATc1 subcellular localization in a heterologous expression system using HeLa cells. These studies confirmed that the canonical NFATc1 activation pathway functions in HeLa cells: treatment of HeLa cells overexpressing NFATc1 with ionomycin induced NFATc1 dephosphorylation presumably via calcineurin, as judged by a downward mobility shift of NFATc1 and increased levels of NFATc1 in the nuclear fraction (Fig. 4A, compare lanes 1 and 4). Coexpression of NFATc1 with trCot drastically increased NFATc1 protein levels, and additive phosphorylation of NFATc1 was observed, as evidenced by an upward mobility shift (Fig. 4A, lane 2). In trCot coexpressing cells, NFATc1 protein was mostly cytoplasmic; however, nuclear NFATc1 protein levels were also increased, irrespective of ionomycin treatment (Fig. 4A, right, lanes 2 and 5). KMCot coexpression did not alter NFATc1 protein level or localization (Fig. 4A, lanes 1 and 3 and lanes 4 and 6), suggesting that the Cot kinase activity is required for enhanced levels of NFATc1 in the whole-cell and nuclear fractions.
Fig 4.

Cot increases NFATc1 expression level through protein stabilization. (A) Subcellular fractionation of NFATc1 in HeLa cells overexpressing NFATc1 alone (lanes 1 and 4), NFATc1 and trCot (lanes 2 and 5), or NFATc1 and KMCot (lanes 3 and 6). HeLa cells were treated without (lanes 1 to 3) or with (lanes 4 to 6) 2.5 μM ionomycin (iono) for 10 min. Note that the amount loaded of the nuclear fraction is 16-fold that of the cytosol fraction. (B) NFATc1 localization in HeLa cells coexpressing trCot or KMCot in the absence (left) or presence (right) of FK506. Bar, 50 μm. (C) NFATc1 signal intensity in the entire cell (left) and in the nucleus (middle) and the ratio between the nucleus and the entire cell (right). Shown are means ± standard errors of the means derived from analysis of 32 to 37 cells. *, P < 0.05; **, P < 0.01. N.S., difference was not significant. (D) NFATc1 and GAPDH mRNA levels in HeLa cells, as detected by RT-PCR. (E) Increased expression levels and hyperphosphorylation of NFATc1 (arrowhead) in HeLa cells cotransfected with trCot, detected with the Odyssey system. These samples were used for the degradation assay whose results are shown in panel F. (F) NFATc1 protein degradation dynamics in HeLa cells treated with cycloheximide (CHX). (Left) NFATc1 levels in cells after CHX treatment. β-Actin was used for protein normalization. (Right) Relative amounts of NFATc1 protein. NFATc1 coexpressed with trCot showed a decreased degradation rate compared to protein coexpressed with KMCot or alone. n = 4 for each group. Values are means ± SD. *, P < 0.05. (G) NFATc1 protein degradation dynamics in primary WT and CotKO osteoclasts cocultured with ST2 cells and treated with CHX. The experimental procedure is depicted in the left upper panel. Note that the CHX chase assay was performed in the presence of FK506. (Bottom left) NFATc1 protein levels in osteoclasts after CHX treatment. (Right) Relative amounts of NFATc1. NFATc1 in CotKO osteoclasts showed a decreased degradation rate compared to protein in WT osteoclasts. n = 3 for each group. Values are means ± SD. *, P < 0.05.
Immunofluorescence analysis of HeLa cells further revealed that NFATc1 protein levels were clearly increased by coexpression of NFATc1 with trCot but not KMCot and that overexpressed NFATc1 localized primarily to the cytoplasm (Fig. 4B, left, and C), suggesting that Cot contributes to enhanced protein levels rather than to nuclear translocation. In trCot-coexpressing RAW 264.7 cells, overexpressed NFATc1 was also primarily cytoplasmic and RANKL stimulation partly enhanced NFATc1 translocation to the nucleus (see Fig. S4A in the supplemental material). In contrast, less abundant endogenous NFATc1 was predominantly nuclear in multinuclear osteoclasts in the presence of trCot (Fig. 1E). When NFATc1 and trCot were coexpressed in HeLa cells, the nuclear intensity of NFATc1 staining increased; however, the ratio of nuclear to whole-cell NFATc1 intensity did not (Fig. 4C). These data suggest that trCot contributes primarily to enhanced NFATc1 protein levels in the cytoplasm, leading to a subsequent increase in nuclear NFATc1.
The NFATc1 expression vector lacked both the authentic promoter and the 3′ untranslated region, which potentially affects mRNA stability, making it unlikely that Cot increased NFATc1 mRNA expression. Indeed, we observed that NFATc1 mRNA levels were comparable among cells expressing NFATc1 alone, NFATc1 plus trCot, and NFATc1 plus KMCot (Fig. 4D). Overall, these observations suggest that Cot regulates NFATc1 protein stability. Therefore, we examined NFATc1 protein stability by quantitative Western blotting using the Odyssey system. We again observed that NFATc1 levels were increased by trCot coexpression, and we also detected hyperphosphorylated NFATc1, particularly in trCot-coexpressing cells (Fig. 4E, arrowhead). To quantify stability changes, we treated HeLa cells with cycloheximide (CHX) and examined the NFATc1 degradation rate. At 2 to 3 h after CHX treatment, NFATc1 protein stability in trCot coexpressing cells increased about 2.5-fold compared to that in cells expressing NFATc1 alone or NFATc1 together with KMCot (Fig. 4F). These results indicate that observed increases in NFATc1 protein levels are due to enhanced protein stability mediated by Cot, which likely leads to nuclear localization and activation of NFATc1.
We next performed a CHX chase assay using primary osteoclasts to determine whether endogenous Cot stabilized endogenous NFATc1 protein (Fig. 4G), as exogenous trCot did in HeLa cells (Fig. 4F). As noted, we had observed significant differences in efficiency of osteoclastogenesis between CotKO and WT BMMs cocultured with ST2 cells in the presence of FK506 (Fig. 2C). Based on these observations, we first induced osteoclastogenesis in the absence of FK506 to accumulate equivalent levels of NFATc1 in both WT and CotKO cells. We then treated cells with FK506 prior to CHX treatment (Fig. 4G, top left). NFATc1 protein levels were comparable between WT and CotKO osteoclasts before CHX treatment; however, NFATc1 was more rapidly degraded in CotKO than WT osteoclasts by 1 h after CHX treatment (Fig. 4G). These results suggest that Cot activated in osteoclasts via interaction with osteoblasts functions in part by stabilizing NFATc1 protein.
Cot-mediated NFATc1 activation contributes to osteoclastogenesis in vivo.
To determine the physiological significance of the Ca2+ oscillation/calcineurin-independent osteoclast differentiation pathway, we generated mice lacking both IP3R2 and Cot (IP3R2/CotKO mice). As shown in Fig. 5A, IP3R2/CotKO BMMs, in which both the Ca2+/calcineurin-dependent and -independent osteoclast differentiation pathways are altered, showed significantly decreased numbers of TRAP-positive multinuclear osteoclasts when cocultured with ST2 cells, compared with the BMMs from WT or IP3R2KO mice (Fig. 5A). Although IP3R2KO BMMs also showed decreased TRAP-positive cell numbers in the coculture system compared with WT BMMs, IP3R2KO mice did not exhibit differences in osteoclast parameters and bone volume compared to WT mice (see Fig. S5 in the supplemental material) (16), suggesting that the Ca2+ oscillation/calcineurin-independent pathway compensates for the loss of the Ca2+ oscillation/calcineurin-dependent one in vivo. The number of osteoclasts, osteoclast surface area, and eroded surface area were significantly decreased in the tibias of IP3R2/CotKO mice compared with values seen in IP3R2KO mice (Fig. 5B and C), suggesting aberrant osteoclast differentiation in IP3R2/CotKO mice. Despite these changes, the IP3R2/CotKO mice did not exhibit increased bone volume (Fig. 5C). We next examined in vitro osteoblastogenesis of WT, IP3R2KO, and IP3R2/CotKO calvarial osteoblasts and observed no significant difference in osteoblastogenesis among these groups (see Fig. S6A in the supplemental material). We also examined in vivo osteoblastic parameters, namely, osteoblast surface, osteoid surface, and bone formation rate, in WT, IP3R2KO, and IP3R2/CotKO mice. Osteoblast and osteoid surfaces did not differ significantly between groups, but the bone formation rate was significantly decreased only in IP3R2/CotKO mice (see Fig. S6B and C in the supplemental material). In IP3R2/CotKO mice, both bone resorption and bone formation rates were reduced, thereby maintaining bone volume.
Fig 5.
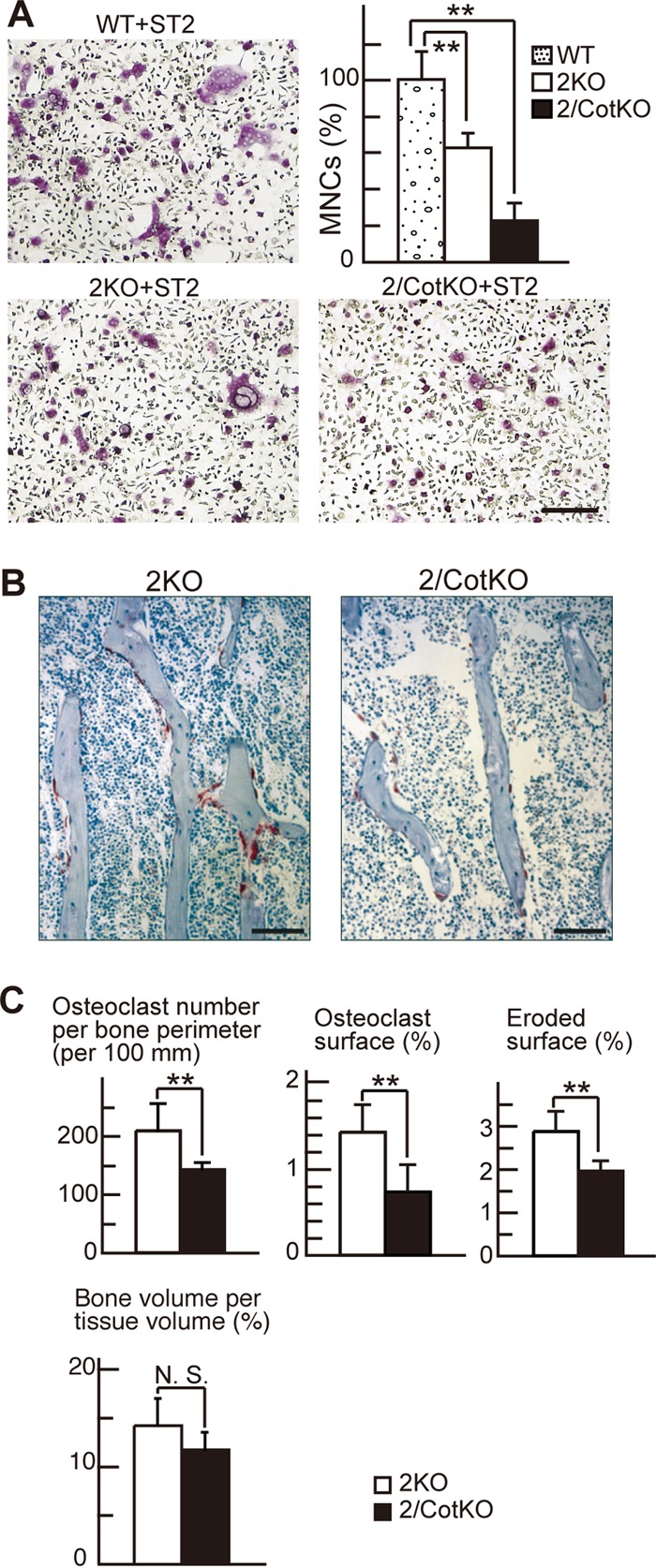
Cot contributes to osteoclastogenesis through Ca2+ oscillation/calcineurin-independent osteoclast differentiation in vivo. (A) The number of TRAP-positive MNCs lacking both IP3R2 and Cot kinase (IP3R2/CotKO) was significantly decreased compared to those in WT and IP3R2KO mice in a coculture system. n = 3 for each group. Values are means ± SD. **, P < 0.01. Bar, 100 μm. (B) Histology (TRAP and toluidine blue staining) of the tibias of IP3R2KO and IP3R2/CotKO mice at 10 weeks of age. Bars, 100 μm. (C) Histomorphometric parameters of osteoclastic bone resorption and bone volume in the tibias of IP3R2KO and IP3R2/CotKO mice at 10 weeks of age. Values are means ± SD. **, P < 0.01. N.S., difference was not significant. n = 4 for each group.
DISCUSSION
In this study, we show that Cot promotes Ca2+ oscillation/calcineurin-independent osteoclastogenesis by increasing NFATc1 protein stability through phosphorylation and that osteoclastogenesis is impaired in IP3R2/CotKO mice. We also demonstrate that Cot directly phosphorylates NFATc1 in vitro and that Cot activation in osteoclasts is regulated by cell-cell interaction with osteoblasts. These results reveal the molecular mechanism of Ca2+ oscillation/calcineurin-independent NFATc1 activation by Cot and a physiological role for Cot in osteoclastogenesis.
Induction of Ca2+ oscillation/calcineurin-independent NFATc1 activation by Cot.
Transcriptional activity of Ca2+-dependent isoforms of NFATc1 to -c4 is canonically regulated by calcineurin via dephosphorylation of their N-terminal regulatory domains in the NHR. In addition to calcineurin, several Ser/Thr kinases, such as the MAPKs p38 and JNK, GSK3β, PKA, and CK1, regulate NFAT transcriptional activity by promoting nuclear export or inhibiting nuclear import (12). While in most cases phosphorylation is postulated to inhibit NFAT activation, increasing evidence indicates that phosphorylation can also positively regulate NFAT activation. PMA (phorbol 12-myristate 13-acetate)/ionomycin stimulation-induced phosphorylation of the N-terminal transactivation domain has been observed in NFATc2, and mutation of modified Ser residues reportedly impairs transcriptional activity (26). Pim-1, a Ser/Thr kinase, potentially activates NFATc1 (29). Although it is not known how Pim-1 induces NFAT activity, Pim-1 also directly phosphorylates the NFATc1 N-terminal half in vitro and enhances its transcriptional activity without altering its subcellular localization (29). We demonstrate here that Cot increases NFATc1 protein stability through phosphorylation, resulting in Ca2+ oscillation/calcineurin-independent NFATc1 activation. We found that a hyperphosphorylated form of NFATc1 seen in the presence of coexpressed trCot shows enhanced resistance to degradation (Fig. 4E and F) and that Cot induces increases in NFATc1 protein levels (Fig. 1D and 4A). Nuclear translocation of Cot-phosphorylated NFATc1 is more easily observed in osteoclasts than in HeLa cells (Fig. 1E and 4B) and is efficiently induced by RANKL stimulation in RAW264.7 cells (see Fig. S4A in the supplemental material). These data suggest that osteoclasts harbor a mechanism for inducing nuclear NFATc1 by increasing its total levels, as well as through inducing calcineurin-dependent nuclear translocation. Both mechanisms are likely activated during osteoclastogenesis. An in vitro kinase assay showed that Cot directly phosphorylates sites other than the 21 Ser residues previously identified as phosphorylation sites critical for cytoplasmic retention (Fig. 3F), implying that direct phosphorylation by Cot may regulate NFATc1 stability via a mechanism distinct from that previously identified to modulate its subcellular distribution, DNA binding affinity, or transcriptional activity. Furthermore, this Cot-induced positive regulation was common to all Ca2+/calcineurin-dependent NFAT family proteins (Fig. 3G and H), strongly suggesting that Cot-induced NFAT activation contributes to activities besides osteoclastogenesis.
Recently, it was reported that Pim-1 enhances osteoclastogenesis in a phosphorylation-dependent manner (14). At present, osteoblast-derived molecules that activate Cot kinase in osteoclast precursors are not known. Our preliminary data showing that sonicated ST2 cell membrane fragments can induce osteoclastogenesis of IP3R2KO BMMs strongly suggest that a membrane-bound molecule(s) is a plausible candidate as a Cot kinase activator (our unpublished data). Overall, our findings indicate that NFATc1 phosphorylation by protein kinases, such as Cot and Pim-1, contributes to NFATc1 activation in cooperation with calcineurin-induced dephosphorylation during osteoclast differentiation.
Physiological role of Cot in osteoclastogenesis.
In this study, we demonstrated that CotKO BMMs exhibit severely impaired osteoclastogenesis when cocultured with ST2 cells in the presence of FK506 (Fig. 2B and C) and that activated Cot is increased in osteoclast precursors interacting with ST2 cells (Fig. 2D). These findings are consistent with the idea that Ca2+ oscillation/calcineurin-independent NFATc1 activation requires osteoblasts (16). Hirata et al. also reported a function for Cot in osteoclast differentiation (10). However, in contrast to our findings, they used a Cot inhibitor to show that Cot acts downstream of sRANKL signaling (10). The discrepancy is likely due to differences in experimental strategy, such as the use of genetic versus pharmacological inhibition. The higher concentrations of the inhibitor required to block osteoclastogenesis than to block ERK activity in BMMs could potentially affect signaling other than the Cot-ERK pathway. We also cannot rule out the possibility that expression patterns of additional factors are altered in Cot knockout mice.
Although osteoclast formation by IP3R2/CotKO BMMs was substantially impaired (Fig. 5A and C), IP3R2/CotKO mice exhibited normal bone density (Fig. 5C). The most likely explanation for the discrepancy between in vitro and in vivo phenotypes is that bone formation is also impaired in IP3R2/CotKO mice (see Fig. S5C in the supplemental material). This may not be an osteoblast cell-autonomous effect because differentiation of IP3R2/CotKO calvarial osteoblasts in vitro did not differ from that of WT or IP3R2KO cells (see Fig. 5A in the supplemental material), even though Cot is also expressed in osteoblasts (13). This view is further supported by the normal osteoblast and osteoid surfaces seen in IP3R2/CotKO mice in vivo. Thus, it is tempting to speculate that reduced bone formation is due to impaired osteoclast function in coupling of bone resorption and formation (23). Analysis of osteoclast-specific Cot knockout mice is necessary to further analyze the role of Cot in coupling.
In addition to Cot kinase, another unidentified molecule(s) may function in Ca2+ oscillation/calcineurin-independent osteoclastogenesis. Indeed, some IP3R2/CotKO BMMs were still able to differentiate into multinuclear osteoclasts (Fig. 5A). We are currently searching for such molecules that function in this pathway. Cot-mediated signaling might be activated under pathological situations associated with excess osteoclastic bone resorption. For example, osteoclast abnormalities induce bone destruction in an inflammation locus (37). In the case of periodontitis, LPS reportedly induces excessive osteoclast formation and bone resorption through intercellular communication between T cells or osteoblasts and osteoclasts (13, 38). Since Toll-like receptor 4 (TLR4), the LPS receptor, is expressed in osteoclasts (34), Cot is activated by LPS stimulation (8), and LPS promotes osteoclastogenesis of RANKL-treated osteoclast precursors (19, 34), osteoclast abnormalities in periodontitis could be partially explained by our finding that Cot mediates NFATc1 activation. Our results encourage further investigation of the molecular mechanism for osteoclastogenesis in various situations, including normal developmental and pathological conditions, in order to understand bone homeostasis in vivo and develop new therapies for skeletal disease.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. Miyoshi for Cot kinase knockout mice, N. A. Clipstone for the pMSCV-GFP and pMSCV-caNFATc1-IRES-GFP vector plasmids, H. Takayanagi for the pCTR-Luc vector plasmid, G. Crabtree for the pSH160c vector plasmid, and Tim Hoey for the pREP-NFATp plasmid. We are grateful to N. Hatano for excellent technical support in the determination of phosphorylation sites and the Support Unit for Bio-material Analysis, RIKEN BSI Research Resources Center, for help with FANTOM3 clone distribution and DNA sequencing analysis. We also thank all members of our laboratories, especially K. Kawaai for fruitful discussions and E. Ebisui, A. Oide, and T. Takada for technical assistance, and E. Lamar for critical reading of the manuscript.
This study was supported by Grants-in-Aid from the JST to K. Mikoshiba and by Grants-in-Aid from JSPS for Young Scientists (B) to Y. Kuroda (23790265) and for Scientific Research (B) to K. Matsuo (21390425).
Footnotes
Published ahead of print 21 May 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Ballester A, Velasco A, Tobena R, Alemany S. 1998. Cot kinase activates tumor necrosis factor-α gene expression in a cyclosporin A-resistant manner. J. Biol. Chem. 273:14099–14106 [DOI] [PubMed] [Google Scholar]
- 2. Belich MP, Salmeron A, Johnston LH, Ley SC. 1999. TPL-2 kinase regulates the proteolysis of the NF-κB-inhibitory protein NF-κB1 p105. Nature 397:363–368 [DOI] [PubMed] [Google Scholar]
- 3. Carninci P, et al. 2005. The transcriptional landscape of the mammalian genome. Science 309:1559–1563 [DOI] [PubMed] [Google Scholar]
- 4. Ceci JD, et al. 1997. Tpl-2 is an oncogenic kinase that is activated by carboxy-terminal truncation. Genes Dev. 11:688–700 [DOI] [PubMed] [Google Scholar]
- 5. Chiariello M, Marinissen MJ, Gutkind JS. 2000. Multiple mitogen-activated protein kinase signaling pathways connect the cot oncoprotein to the c-jun promoter and to cellular transformation. Mol. Cell. Biol. 20:1747–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Crotti TN, et al. 2006. NFATc1 regulation of the human β3 integrin promoter in osteoclast differentiation. Gene 372:92–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de Gregorio R, Iniguez MA, Fresno M, Alemany S. 2001. Cot kinase induces cyclooxygenase-2 expression in T cells through activation of the nuclear factor of activated T cells. J. Biol. Chem. 276:27003–27009 [DOI] [PubMed] [Google Scholar]
- 8. Dumitru CD, et al. 2000. TNF-α induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell 103:1071–1083 [DOI] [PubMed] [Google Scholar]
- 9. Futatsugi A, et al. 2005. IP3 receptor types 2 and 3 mediate exocrine secretion underlying energy metabolism. Science 309:2232–2234 [DOI] [PubMed] [Google Scholar]
- 10. Hirata K, et al. 2010. Inhibition of tumor progression locus 2 protein kinase suppresses receptor activator of nuclear factor-κB ligand-induced osteoclastogenesis through down-regulation of the c-Fos and nuclear factor of activated T cells c1 genes. Biol. Pharm. Bull. 33:133–137 [DOI] [PubMed] [Google Scholar]
- 11. Hoey T, Sun YL, Williamson K, Xu X. 1995. Isolation of two new members of the NF-AT gene family and functional characterization of the NF-AT proteins. Immunity 2:461–472 [DOI] [PubMed] [Google Scholar]
- 12. Hogan PG, Chen L, Nardone J, Rao A. 2003. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 17:2205–2232 [DOI] [PubMed] [Google Scholar]
- 13. Kikuchi T, et al. 2003. Cot/Tpl2 is essential for RANKL induction by lipid A in osteoblasts. J. Dent. Res. 82:546–550 [DOI] [PubMed] [Google Scholar]
- 14. Kim K, Kim JH, Youn BU, Jin HM, Kim N. 2010. Pim-1 regulates RANKL-induced osteoclastogenesis via NF-κB activation and NFATc1 induction. J. Immunol. 185:7460–7466 [DOI] [PubMed] [Google Scholar]
- 15. Kim K, Lee SH, Ha Kim J, Choi Y, Kim N. 2008. NFATc1 induces osteoclast fusion via up-regulation of Atp6v0d2 and the dendritic cell-specific transmembrane protein (DC-STAMP). Mol. Endocrinol. 22:176–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kuroda Y, Hisatsune C, Nakamura T, Matsuo K, Mikoshiba K. 2008. Osteoblasts induce Ca2+ oscillation-independent NFATc1 activation during osteoclastogenesis. Proc. Natl. Acad. Sci. U. S. A. 105:8643–8648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee SH, et al. 2006. v-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat. Med. 12:1403–1409 [DOI] [PubMed] [Google Scholar]
- 18. Lin X, Cunningham ET, Jr, Mu Y, Geleziunas R, Greene WC. 1999. The proto-oncogene Cot kinase participates in CD3/CD28 induction of NF-κB acting through the NF-κB-inducing kinase and IκB kinases. Immunity 10:271–280 [DOI] [PubMed] [Google Scholar]
- 19. Liu J, et al. 2009. Molecular mechanism of the bifunctional role of lipopolysaccharide in osteoclastogenesis. J. Biol. Chem. 284:12512–12523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mancini M, Toker A. 2009. NFAT proteins: emerging roles in cancer progression. Nat. Rev. Cancer 9:810–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matsumoto M, et al. 2004. Essential role of p38 mitogen-activated protein kinase in cathepsin K gene expression during osteoclastogenesis through association of NFATc1 and PU. 1. J. Biol. Chem. 279:45969–45979 [DOI] [PubMed] [Google Scholar]
- 22. Matsuo K, et al. 2004. Nuclear factor of activated T-cells (NFAT) rescues osteoclastogenesis in precursors lacking c-Fos. J. Biol. Chem. 279:26475–26480 [DOI] [PubMed] [Google Scholar]
- 23. Matsuo K, Irie N. 2008. Osteoclast-osteoblast communication. Arch. Biochem. Biophys. 473:201–209 [DOI] [PubMed] [Google Scholar]
- 24. Miyoshi J, Higashi T, Mukai H, Ohuchi T, Kakunaga T. 1991. Structure and transforming potential of the human cot oncogene encoding a putative protein kinase. Mol. Cell. Biol. 11:4088–4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Neal JW, Clipstone NA. 2001. Glycogen synthase kinase-3 inhibits the DNA binding activity of NFATc. J. Biol. Chem. 276:3666–3673 [DOI] [PubMed] [Google Scholar]
- 26. Okamura H, et al. 2000. Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Mol. Cell 6:539–550 [DOI] [PubMed] [Google Scholar]
- 27. Parfitt AM, et al. 1987. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 2:595–610 [DOI] [PubMed] [Google Scholar]
- 28. Patriotis C, Makris A, Chernoff J, Tsichlis PN. 1994. Tpl-2 acts in concert with Ras and Raf-1 to activate mitogen-activated protein kinase. Proc. Natl. Acad. Sci. U. S. A. 91:9755–9759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rainio EM, Sandholm J, Koskinen PJ. 2002. Cutting edge: transcriptional activity of NFATc1 is enhanced by the Pim-1 kinase. J. Immunol. 168:1524–1527 [DOI] [PubMed] [Google Scholar]
- 30. Robinson MJ, Beinke S, Kouroumalis A, Tsichlis PN, Ley SC. 2007. Phosphorylation of TPL-2 on serine 400 is essential for lipopolysaccharide activation of extracellular signal-regulated kinase in macrophages. Mol. Cell. Biol. 27:7355–7364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ruff VA, Leach KL. 1995. Direct demonstration of NFATp dephosphorylation and nuclear localization in activated HT-2 cells using a specific NFATp polyclonal antibody. J. Biol. Chem. 270:22602–22607 [DOI] [PubMed] [Google Scholar]
- 32. Salmeron A, et al. 1996. Activation of MEK-1 and SEK-1 by Tpl-2 proto-oncoprotein, a novel MAP kinase kinase kinase. EMBO J. 15:817–826 [PMC free article] [PubMed] [Google Scholar]
- 33. Selander K, Lehenkari P, Vaananen HK. 1994. The effects of bisphosphonates on the resorption cycle of isolated osteoclasts. Calcif. Tissue Int. 55:368–375 [DOI] [PubMed] [Google Scholar]
- 34. Takami M, Kim N, Rho J, Choi Y. 2002. Stimulation by toll-like receptors inhibits osteoclast differentiation. J. Immunol. 169:1516–1523 [DOI] [PubMed] [Google Scholar]
- 35. Takayanagi H, et al. 2002. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 3:889–901 [DOI] [PubMed] [Google Scholar]
- 36. Takebe Y, et al. 1988. SRα promoter: an efficient and versatile mammalian cDNA expression system composed of the simian virus 40 early promoter and the R-U5 segment of human T-cell leukemia virus type 1 long terminal repeat. Mol. Cell. Biol. 8:466–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tanaka S, Nakamura K, Takahasi N, Suda T. 2005. Role of RANKL in physiological and pathological bone resorption and therapeutics targeting the RANKL-RANK signaling system. Immunol. Rev. 208:30–49 [DOI] [PubMed] [Google Scholar]
- 38. Taubman MA, Kawai T. 2001. Involvement of T-lymphocytes in periodontal disease and in direct and indirect induction of bone resorption. Crit. Rev. Oral Biol. Med. 12:125–135 [DOI] [PubMed] [Google Scholar]
- 39. Tsatsanis C, Patriotis C, Bear SE, Tsichlis PN. 1998. The Tpl-2 protooncoprotein activates the nuclear factor of activated T cells and induces interleukin 2 expression in T cell lines. Proc. Natl. Acad. Sci. U. S. A. 95:3827–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tsatsanis C, Patriotis C, Tsichlis PN. 1998. Tpl-2 induces IL-2 expression in T-cell lines by triggering multiple signaling pathways that activate NFAT and NF-κB. Oncogene 17:2609–2618 [DOI] [PubMed] [Google Scholar]
- 41. Yagi M, et al. 2005. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J. Exp. Med. 202:345–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yeo H, et al. 2007. Conditional disruption of calcineurin B1 in osteoblasts increases bone formation and reduces bone resorption. J. Biol. Chem. 282:35318–35327 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.