

Abstract
Human coronaviruses are associated with upper respiratory tract infections that occasionally spread to the lungs and other organs. Although airway epithelial cells represent an important target for infection, the respiratory epithelium is also composed of an elaborate network of dendritic cells (DCs) that are essential sentinels of the immune system, sensing pathogens and presenting foreign antigens to T lymphocytes. In this report, we show that in vitro infection by human coronavirus 229E (HCoV-229E) induces massive cytopathic effects in DCs, including the formation of large syncytia and cell death within only few hours. In contrast, monocytes are much more resistant to infection and cytopathic effects despite similar expression levels of CD13, the membrane receptor for HCoV-229E. While the differentiation of monocytes into DCs in the presence of granulocyte-macrophage colony-stimulating factor and interleukin-4 requires 5 days, only 24 h are sufficient for these cytokines to sensitize monocytes to cell death and cytopathic effects when infected by HCoV-229E. Cell death induced by HCoV-229E is independent of TRAIL, FasL, tumor necrosis factor alpha, and caspase activity, indicating that viral replication is directly responsible for the observed cytopathic effects. The consequence of DC death at the early stage of HCoV-229E infection may have an impact on the early control of viral dissemination and on the establishment of long-lasting immune memory, since people can be reinfected multiple times by HCoV-229E.
INTRODUCTION
Coronaviruses (CoVs) are enveloped positive-strand RNA viruses from the Coronaviridae family. Five members have been reported to infect humans, including 229E, OC43, the newly discovered NL63 and HKU1, and the emerging SARS-CoV. Human CoVs (HCoVs) 229E and NL63 are closely related and belong to the alphacoronavirus genus, whereas OC43, HKU1, and SARS-CoV belong to betacoronavirus genus. HCoVs infect airways and are responsible for different respiratory diseases (19, 44). Although the SARS-CoV was associated with a severe acute respiratory disease during the 2002–2003 pandemic, most HCoVs cause only a mild respiratory infection (49). Epidemiological studies suggest that HCoVs account for 15 to 30% of common colds, with only occasional spreading to the lower respiratory tract. Airway epithelial cells represent the primary target of infection (19, 44). Nevertheless, in vitro experiments demonstrate that other cell types can be infected. For example, HCoV-229E was reported to infect and replicate in neural cells, hepatocytes, monocytes, and macrophages (3, 11, 12). The neurotropism of HCoV-229E and OC43 has also been documented in vivo, and a possible association with multiple sclerosis has been suggested (4). Because peripheral blood cells from the myeloid lineage can be infected by HCoVs, these cells have been proposed to serve as a vector for viral spread to neural tissues (13).
In addition to epithelial cells, the human airway epithelium possesses an elaborate network of dendritic cells (DCs). DCs serve as sentinels in the respiratory tract, where they detect inhaled pathogens through the recognition of pathogen-associated molecular patterns (PAMPs), e.g., bacterial lipopolysaccharides or viral nucleic acids. In order to mediate this function, DCs express pattern recognition receptors (PRRs), e.g., Toll-like receptors, NOD-like receptors, RIG-like receptors, and C-type lectin receptors. Upon engagement, these receptors induce the migration of DCs to the draining lymph nodes and their maturation into antigen-presenting cells. This maturation process determines the ability of DCs to stimulate an adaptive immune response against antigens that have been captured in the lungs. Therefore, these conventional DCs are at the nexus of innate and adaptive immunity in the lungs.
Several viruses that spread through the airways infect DCs. For example, infection of DCs by influenza virus induces their maturation and migration to the lymph nodes to efficiently prime an adaptive immune response (1, 29, 38). In contrast, measles virus (MV) uses DCs as a Trojan horse to spread from the lungs to other tissues (14, 15), while still inducing long-term immune responses. Although MV-infected DCs migrate properly to lymph nodes, their phenotypic maturation is perturbed, and infected DCs fail to stimulate interacting lymphocytes (42), indicating that cross-priming by uninfected DCs might be the route for eliciting an MV adaptive immune response (43). In addition, activation signals provided by lymphocytes dramatically enhance MV replication within infected DCs, and this likely contributes to the establishment of viremia (42). Thus, DC infection has different effects in disease pathogenesis, depending on the host-pathogen interactions. Indeed, DC infection supports priming of T cells, and yet infection of a migrating cell also contributes to the spread of the virus to distal tissues.
CoVs associated with the common cold, such as HCoV-229E, are likely to interact with lung DCs in vivo. However, the susceptibility of DCs to infection by these viruses is as yet unknown. We describe here the in vitro infection of monocyte-derived DCs (Mo-DCs) with human HCoV-229E. Infection resulted in dramatic cytopathic effects, with the formation of large syncytia, and cell death occurred within 24 h. In contrast, infected monocytes from the same donors were preserved from cytopathic effects and acquired sensitivity to cell death only after a short stimulation with granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin-4 (IL-4). Different hypotheses were tested to explain this observation.
MATERIALS AND METHODS
Production of HCoV-229E virus stocks and in vitro infection.
Virus stocks were established on MRC5 cells using HCoV-229E virus strain from ATCC (VR-740). After washing, 80 to 90% confluent cell cultures were infected in a minimal volume of serum-free medium for 2 h. Dulbecco modified Eagle medium (DMEM) containing 10% fetal calf serum (FCS) and antibiotics was added, and infected cultures were incubated for 4 to 5 days at 37°C and 5% CO2. The cytopathic effect was monitored by optical microscopy. Cell supernatants were harvested, centrifuged for 5 min at 4,000 rpm, and aliquoted into cryotubes for storage at −80°C. Virus titers were determined as 50% tissue culture infective doses (TCID50). MRC5 cells were seeded in 96-well plates and inoculated with serial dilutions of virus stock ranging from 10−1 to 10−8. Plates were incubated for 12 h at 37°C before adding DMEM supplemented with 10% FCS. The plates were incubated for another 6 days and then fixed with 4% paraformaldehyde before being stained with crystal violet. Infected wells were numbered for each virus dilution, allowing us to calculate a TCID50 (26, 45). To perform in vitro infections, cell suspensions of monocytes, Mo-DCs, or CD34-DCs were incubated for 2 h at 37°C with an appropriate volume of virus stock to match the indicated multiplicity of infection (MOI). Mock infections were performed using supernatant from uninfected MRC5 cell cultures. Finally, cells were dispensed at 106 cells/ml and harvested at the indicated time points.
Detection of HCoV-229E replication.
Viral replication was assessed in culture supernatants of infected cells by quantitative reverse transcription-PCR (qRT-PCR). Viral RNA was extracted from medium using an automated QiaSymphony system (Qiagen). HCoV-229E-specific primers and probe previously designed and targeting the HCoV-229E N gene were used (17). The viral quantification was calculated by using an external standard curve constituted by serial 10-fold dilutions of viral RNA transcripts (108 to 102 copies). These transcripts were in vitro transcribed with T7 polymerase from pCR-XL-TOPO plasmids containing the M and N genes of HCoV-229E previously cloned from HCoV-229E (strain ATCC-VR-740) MRC5 cell culture supernatants. The RNA transcripts were quantified in a UV spectrophotometer.
Production of SARS-CoV stocks and in vitro infection.
Virus stocks were established on VeroE6 cells using the FFM-1 strain of SARS-CoV (kindly provided by H. W. Doerr, Institute of Medical Virology, Frankfurt University Medical School, Frankfurt, Germany), as previously described (7). All viral stocks were stored at −80°C in single-use aliquots and titrated in a standard limiting dilution assay on FRhK-4 cell monolayers. Infectious titers were determined as the TCID50 with optical microscopic reading of the cytopathic effect as described above. All work involving infectious SARS-CoV was performed in an enhanced biosafety level 3 containment laboratory with rigorous safety procedures according to World Health Organization (WHO) guidelines. In vitro infections of monocytes, Mo-DCs, or CD34-DCs with SARS-CoV were performed as described above for HCoV-229E infections. Mock infections were performed using supernatant from uninfected VeroE6 cell cultures.
Preparation of monocytes and Mo-DCs.
Blood cells were obtained from leukapheresis samples freshly collected from normal donors after informed consent was obtained and according to institutional guidelines. Mononuclear cells were separated on a Ficoll-Hypaque gradient (density, 1.077 /ml). CD14+ cells corresponding to monocytes were purified by positive selection using immunomagnetic beads coated with anti-CD14 MAbs (Miltenyi Biotech, Cologne, Germany). Purity was assessed by anti-CD14 immunostaining and flow cytometry analysis (Pharmingen, San Diego, CA). DC-SIGN+ and BDCA-1+ populations were also purified by positive selection using immunomagnetic beads. CD14+ cells were plated at 2 × 106 cells/ml in RPMI 1640 medium containing 10% FCS in the presence of IL-4 (20 ng/ml; Gentaur Molecule Products, Belgium), and GM-CSF (100 ng/ml; Gentaur Molecule Products, Belgium). Monocyte differentiation into Mo-DCs was obtained in 5 days as previously described (10). In some experiments, DC maturation was induced using trimeric CD40-L (250 ng/ml; R&D Systems) or tumor necrosis factor alpha (TNF-α; 5 ng/ml; R&D Systems) plus prostaglandin E2 (PGE2; 1 μg/ml), or polyinosinic-poly(C) sodium salt (poly-IC; 1 μg/ml; Sigma).
Preparation of CD34-DCs.
Peripheral blood CD34+ cells were purified from leukapheresis products collected from patients after stem cell mobilization with granulocyte colony-stimulating factor (G-CSF) and cyclophosphamide. Permission to obtain leukapheresis samples was obtained after obtaining informed consent from the patients and approval by the institutional review board. CD34+ cells were purified using immunomagnetic beads as described elsewhere (32). Differentiation of CD34+ cells into CD34-DCs was adapted from Movassagh et al. (32). Briefly, CD34+ cells were cultured for 10 days. The medium was changed at days 3, 5, and 8, and cells were seeded at 2 × 106 cells/ml in RPMI containing 10% FCS, 1% glutamine, 2% antibiotics, 100 ng of GM-CSF/ml, 50 ng of SCF/ml, 50 ng of IL-4/ml, 5 ng of TNF-α/ml, and 300 ng of Flt3-L/ml. At day 10, the CD34+ cells had achieved their differentiation into CD34-DCs.
MAbs and flow cytometry analysis.
Fluorescein isothiocyanate (FITC)-conjugated anti-CD14, phycoerythrin (PE)-conjugated anti-CCR7, allophycocyanin (APC)-conjugated anti-CD83, FITC-conjugated anti-CD14, APC-conjugated anti-CD209 (DC-SIGN), PE-conjugated anti-TRAIL, FITC-conjugated anti-active caspase-3, PE-conjugated anti-caspase-3, and APC-conjugated anti-CD13 monoclonal antibodies (MAbs) were from Pharmingen (San Diego, CA). HCoV-229E spike glycoprotein was detected using MAb 5-11H.6 (2, 19). The SARS-CoV anti-M serum recognizes the C-terminal domain of SARS-CoV membrane (M) protein (Proscience, Poway, CA). TRITC (tetramethyl rhodamine isothiocyanate)-conjugated goat anti-mouse secondary antibody was from Zymed Laboratories (San Francisco, CA), and Alexa Fluor 488 goat anti-rabbit was from Invitrogen Molecular Probes (Eugene, OR). Negative controls were appropriate irrelevant isotype-matched MAbs.
For immunostaining, 2 × 105 cells were washed twice with phosphate-buffered saline (PBS) and 2% FCS and then fixed in PBS containing 4% paraformaldehyde (PFA). Cells were permeabilized or not with PermWash buffer (BD Bioscience), incubated with the appropriate MAb at 4°C for 20 min, and washed in PBS before analysis by flow cytometry using a FACSCalibur (Becton Dickinson). At least 20,000 gated events were collected and analyzed with FlowJo software (Tree Star). The results are expressed as a percentage of positive cells and/or as the mean fluorescence intensity compared to negative controls. Dead cells were stained according to the manufacturer's instructions with the Dead/Live cell kit (Invitrogen Corp., Carlsbad, CA).
Cell culture imaging.
Monocytes or Mo-DCs were infected with HCoV-229E at an MOI of 0.05 and with SARS-CoV at an MOI of 1 and cultured in the culture chambers of microscopy slides (Ibidi, Munich, Germany). At 24 h postinfection, the cells were fixed for 15 min with 4% PFA for HCoV-229E and 1 h for SARS-CoV, for safety reasons. Free aldehyde groups were quenched in 50 mM NH4Cl, and the cells were permeabilized for 5 min at 4°C in PBS containing 0.1% Triton X-100. The cells were incubated for 1 h in PBS supplemented with 5% goat serum and incubated again in the same solution containing the respective viral antibody: anti-spike glycoprotein MAb 5-11H.6 for HCoV-229E-infected cells and anti-M antibody (Proscience) for SARS-CoV experiments. HCoV-229E- and SARS-CoV-infected cells were washed twice with PBS and incubated for 1 h with a respective secondary antibody. Finally, the cells were washed with PBS and stained for 5 min with a PBS solution containing DAPI (4′,6′-diamidino-2-phenylindole). After a wash with PBS, 100 μl of Fluoromount-G was added to the wells (Southern Biotech, Birmingham, AL). The slides were analyzed, and image acquisition was performed using fluorescence microscope using a ×40 oil immersion objective lens.
Human primary Mo-DCs were infected with HCoV-229E at an MOI of 0.05 and incubated for 11 h at 37°C in the culture chamber of a microscopy slide (Ibidi). The microscopy slide was then placed under an optical microscope for live cell imaging. Snapshots were captured every 30 s over a period of 9 h using a ×10 objective lens and a ×1.5 magnifying lens. Finally, images were compiled into a movie and played back at 24 frames per s.
Cytokine detection and neutralization.
Cytokines produced from monocytes and Mo-DCs were measured from cell culture supernatants by using a multiplex bead-based Luminex assay (Biosource; Invitrogen Corp.) according to the manufacturer's instructions. IFN-α/β production was determined by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions (PBL Interferon Source, Piscataway, NJ). For the neutralization of type I IFNs, monocytes were treated with sheep polyclonal antibody against human IFN-α (2,000 IU/ml; PBL Biomedical Laboratories, Piscataway, NJ) and with sheep polyclonal antibody against human IFN-β (500 IU/ml; PBL Biomedical Laboratories). For neutralization of human IL-6 bioactivity, monocyte cultures were treated with anti-human IL-6 antibody (10 μg/ml; R&D Systems).
Apoptosis and infection inhibition.
To block apoptosis, Mo-DCs were incubated prior to infection with caspase inhibitor Z-VAD-FMK (BD Biosciences) for 30 min at room temperature and then maintained in cell culture medium. Similar experiments were performed to block apoptosis using hIL-6 (100 ng/ml; Miltenyi Biotech) and hIFN-β (PBL Biomedical Laboratories). To inhibit death ligand signaling, Mo-DCs were treated during and after infection with either an anti-hTRAIL MAb (100 ng/ml; R&D Systems), a recombinant human Fas-Fc chimera (1 μg/ml; R&D Systems), or a recombinant human TNFR-Fc chimera (1 μg/ml; R&D Systems). To block HCoV-229E infection, Mo-DCs were treated 1 h at room temperature with 15 μg of anti-CD13 MAb (Biolegend, San Diego, CA) prior to infection as described elsewhere (39). Anti-CD13 was maintained in cell culture medium.
RESULTS
HCoV-229E induces massive cytopathic effects in Mo-DC cultures but not in monocytes.
Although CoVs associated with the common cold, such as HCoV-229E, are likely to interact with DCs in the upper respiratory tract, the in vitro susceptibility of human DCs to infection by these viruses is not yet known. To address this question, we infected human primary monocytes and monocyte-derived DCs (Mo-DCs) with HCoV-229E. We first assessed the expression on the surface of monocytes and Mo-DCs of aminopeptidase N (CD13), a membrane-bound metalloprotease previously identified as a receptor for human HCoV-229E (52). CD13 is expressed on epithelial cells from the lungs and the intestine but also on monocytes, granulocytes, and neuronal cells. Both monocytes and Mo-DCs expressed high levels of CD13 (Fig. 1D). Human primary monocytes and Mo-DCs were then infected with HCoV-229E at an MOI of 0.05 and cultivated for 24 h. Bright-field observation of cells at 24 h postinfection revealed a massive cytopathic effect in Mo-DCs, whereas monocyte cultures remained largely unaffected (Fig. 1A). The cytopathic effect in Mo-DCs was assessed by the presence of cellular aggregates, large syncytia, and numerous cellular debris. Infectious virus was required for killing cells since no cytopathic effect was observed when HCoV-229E virus was UV inactivated (Fig. 1A, right panel). To evaluate the kinetics of cell death after infection, time-lapse microscopy was recorded on infected Mo-DC cultures (see Movie S1 in the supplemental material). This recording started 11 h after infection, and snapshots were captured every 30 s for 9 h. Small syncytia connected to each other by branched projections were visible as early as 11 h postinfection. These structures progressively fused with neighboring cells to form large syncytial structures. Finally, a rapid and near complete lysis of both syncytia and isolated cells was observed at 20 h postinfection.
Fig 1.

Cytopathic effects of HCoV-229E on Mo-DCs. (A) Bright-field microscopy of monocytes and Mo-DC cultures either mock treated or infected with HCoV-229E (MOI = 0.05) and then cultured for 24 h. (B) Monocytes (●) and Mo-DCs (▲) were infected with HCoV-229E at MOIs of 0.05, 0.1, and 0.5. Viable cells were enumerated at the indicated time points by trypan blue exclusion (mean of three donors). (C) Detection of the active form of caspase-3 in HCoV-229E-infected Mo-DCs and monocytes by cytometry (the results of one representative experiment out of five are shown). (D) CD13 expression on monocytes and Mo-DCs. Cells were stained with an anti-CD13 MAb (open line) or an isotypic control (closed line) and analyzed by flow cytometry. The data shown are representative of three different donors. (E) Mo-DCs infected with SARS CoV (MOI = 1 for 24 h). The upper part shows a bright-field image of Mo-DCs either mock treated or infected with SARS-CoV. The lower part shows the immunostaining of viral spike glycoprotein and membrane envelope protein (M) (red, anti-spike; green, anti-M; blue, DAPI). The data are representative of three independent experiments.
To quantify cell death induced by HCoV-229E in Mo-DC cultures, we determined the number of living cells in infected cultures by trypan blue exclusion at different time points and different MOIs, including 0.05, 0.1, and 0.5 (Fig. 1B). Cell death was massive in Mo-DC cultures, even at the lowest MOI of 0.05, with virtually no viable cells detected at 24 h postinfection. In contrast, monocytes were resistant to cell death, with limited loss of viable cells at 48 h after inoculation, even with the highest MOI of 0.5. In the same experiment, we also quantified the status of caspase-3 activation since this protease plays a central role in the execution phase of programmed cell death. In agreement with our observation showing that only Mo-DCs were killed by HCoV-229E infection, caspase-3 was activated in Mo-DCs but not in monocytes (Fig. 1C). Thus, the death of Mo-DCs induced after HCoV-229E infection is associated with caspase-3 activation. We then compared the effects of SARS-CoV, which is much more pathogenic in humans than HCoV-229E and spreads to lower respiratory tract, possibly through dendritic cell transfer (51). Although viral infection was detected by anti-spike and anti-M immunostaining, no cytopathic effect was induced by SARS-CoV infection in Mo-DCs (Fig. 1E), in agreement with previous reports (28, 46). The same result was obtained when a 20-fold-higher MOI of SARS-CoV was used (data not shown). Altogether, these observations show that HCoV-229E (but not SARS-CoV) induces massive cytopathic effects in Mo-DC cultures, whereas monocytes do not exhibit this unusual susceptibility.
Mo-DCs, but not monocytes, are highly susceptible to HCoV-229E infection.
To determine whether cytopathic effects of HCoV-229E correlate with a higher susceptibility to infection, Mo-DCs and monocytes were infected, cultured, and then immunostained for the expression of viral spike glycoprotein. As expected, the large syncytia of Mo-DCs were positive for HCoV-229E spike protein expression, thus demonstrating viral infection (Fig. 2A). In contrast, only a small fraction of monocytes expressed the viral spike glycoprotein after exposure to a similar inoculum of HCoV-229E, and no cell fusion occurred. Immunostaining and flow cytometry analysis of infected cells revealed that a large fraction of Mo-DCs (>85%) expressed the spike glycoprotein of HCoV-229E at 10 h postinfection, while only a few monocytes were positively stained regardless of the MOI used (Fig. 2B). These results suggest that Mo-DCs are much more susceptible to infection by HCoV-229E than are monocytes. This was confirmed when viral RNA load was measured by qRT-PCR in cell cultures supernatants (Fig. 2C). Viral RNA load increased during the first 8 h postinfection in both Mo-DCs and monocytes, thus demonstrating viral replication, but was much higher in Mo-DC cultures. Interestingly, the viral RNA load detected in monocyte cultures increased when cells were infected with a higher MOI, thus demonstrating that monocytes are permissive to HCoV-229E infection, but Mo-DCs are clearly much more susceptible. Furthermore, HCoV-229E never induced massive cytopathic effects in monocyte cultures, even when performing infections with higher MOIs (Fig. 1B, right panel). In conclusion, Mo-DCs are not only more susceptible to viral replication but also to HCoV-229E-induced cytopathic effects.
Fig 2.

Susceptibility of monocytes and Mo-DCs to HCoV-229E. (A) Viral spike (S) glycoprotein expression in monocytes and Mo-DC cultures mock treated or infected with HCoV-229E (MOI = 0.05) and then cultured for 10 h (red, anti-spike immunostaining; blue, DAPI staining). (B) Cell surface expression of HCoV-229E spike glycoprotein on mock-treated and HCoV-229E-infected cells (the results of one representative experiment out of three are shown). (C) Kinetics of viral RNA produced in medium from monocytes and Mo-DCs infected with HCoV-229E at MOIs of 0.05, 0.1, and 0.5 (mean of two donors).
Blocking cell entry or replication of HCoV-229E into Mo-DCs prevents cytopathic effects.
Once inside Mo-DCs, HCoV-229E replicates actively, as demonstrated by cell surface expression of the viral spike glycoprotein and detection of the viral RNA load (Fig. 2). Interestingly, UV-inactivated virus was ineffective at inducing cytopathic effect in Mo-DCs, suggesting that viral replication in infected cells is linked to the cytopathic effect (Fig. 1A, right panel). To demonstrate that HCoV-229E-induced cytopathic effects and cell death are dependent on viral entry, Mo-DC infection was performed in the presence of anti-CD13 MAb to block virus binding to its receptor. Pretreating Mo-DC cultures with anti-CD13 MAb protected cells from virus-induced cytopathic effects (Fig. 3A), confirming that virus entry into Mo-DCs is essential. In addition, we tested whether type I IFN (IFN-β) could rescue Mo-DCs from HCoV-229E-induced cytopathic effects. Indeed, IFN-α/β are essential antiviral cytokines that can control viral replication by inducing a large cluster of immune factors. Mo-DCs were pretreated with increasing doses of IFN-β for 24 h before HCoV-229E infection. As shown in Fig. 3B, even a low dose of IFN-β (100 IU/ml) was sufficient to prevent HCoV-229E replication, cytopathic effects, and massive cell death in Mo-DC cultures. Altogether, these results demonstrate that HCoV-229E-induced cytopathic effects require viral entry in Mo-DCs and sustained viral replication.
Fig 3.

Role HCoV-229E cell entry and replication on Mo-DC death. (A) Mo-DCs were incubated or not with anti-CD13 MAb prior to infection with HCoV-229E. Cell death was determined by the observation of cytopathic effects and the formation of syncytia by bright-field microscopy (the results of one representative experiment out of three are shown). (B) Mo-DCs were treated or not with increasing amounts of IFN-β prior to infection with HCoV-229E, and the percentages of cells expressing spike viral protein or activated caspase-3 were determined by flow cytometry (the results of one representative experiment out of three are shown).
Different populations of conventional DCs show a similar susceptibility to HCoV-229E-induced cytopathic effects.
Several subsets of conventional DCs have been described (20, 30, 35, 36). To determine whether other DC populations also show a high susceptibility to HCoV-229E, we tested DCs derived from CD34+ precursor cells (9, 40). Mo-DCs and CD34-derived DCs (CD34+-DCs) show similar morphologies and antigen uptake/presentation capacities (18, 24). We first demonstrated that CD34+-DCs express significant amounts of CD13 on their surface (Fig. 4B). Then, we investigated whether this DC population was as susceptible as Mo-DCs to HCoV-229E infection. We observed massive cell death at 24 h postinfection in both DC populations, as determined by trypan blue exclusion (Fig. 4A). Furthermore, infection of CD34+-DCs with HCoV-229E was assessed by viral spike glycoprotein expression on their surface (Fig. 4C), thus demonstrating viral infection. Caspase-3 activation was also observed in HCoV-229E-infected CD34+-DCs (Fig. 4D). In addition, we also tested peripheral blood DCs purified on the basis of BDCA-1 or DC-SIGN expression (BDCA-1+-DCs or DC-SIGN+-DCs). Both BDCA-1+-DCs or DC-SIGN+-DCs rapidly died upon HCoV-229E infection similarly as for Mo-DCs (Fig. 4A). Altogether, these results show that all tested populations of conventional DCs are highly susceptible to HCoV-229E-induced cell death.
Fig 4.

Killing of conventional DCs by HCoV-229E. (A) CD34+-DCs, BDCA-1+-DCs, and DC-SIGN+-DCs were either mock treated (open symbols) or infected with HCoV-229E (closed symbols) at different MOIs (0.05, 0.1, and 0.5). Viable cells were enumerated at the indicated time points by trypan blue exclusion (mean of two donors). (B, C, and D) CD34+-DCs were infected with HCoV-229E, cultured for 24 h, and then immunostained for CD13, spike glycoprotein, or active caspase-3 expression. Cells were analyzed by flow cytometry. (E) Mo-DCs were treated with poly-IC, CD40L, or PGE2+TNF-α to induce maturation and then infected with HCoV-229E. The CD80 and HLA-DR expression levels were determined by flow cytometry. (F) Cell death was determined by using Dead/Live staining.
Together, GM-CSF and IL-4 induce monocyte differentiation into immature DCs. However, DCs' ability to stimulate T cells still requires their maturation into professional antigen-presenting cells. This maturation is induced by PAMPs or by cellular factors such as CD40L, TNF-α, or PGE2. To determine whether Mo-DCs' susceptibility to HCoV-229E infection depends on their maturation state, we preincubated cells in the presence of poly-IC, CD40L, or PGE2+TNF-α for 16 h before infection with HCoV-229E, and the expression of CD80 and HLA-DR maturation markers was analyzed. Mo-DCs cultured 16 h in the presence of poly-IC, CD40L, or PGE2+TNF-α upregulated the expression of CD80 and HLA-DR (Fig. 4E). However, regardless of the maturation agent used, Mo-DC maturation did not confer resistance to infection and did not protect cells from HCoV-229E-induced cell death (Fig. 4F).
Cell death induced by HCoV-229E infection is independent of FasL, TRAIL, and TNF-α.
Death receptors are transmembrane proteins that transmit apoptotic signals initiated by specific ligands such as FasL, TNF-α, and TRAIL. They recruit and activate cysteine proteases of the caspase family that are essential mediators of programmed cell death or apoptosis and were found to be activated in HCoV-229E-infected Mo-DCs (Fig. 1C). To evaluate the role of FasL, TNF-α, and TRAIL receptors on the death induced by HCoV-229E infection of Mo-DCs, we neutralized the binding of these ligands during infection by using different inhibitors: an anti-TRAIL MAb, a recombinant human TNFR-Fc chimera, and a recombinant human Fas-Fc chimera (Fig. 5A). We observed that blocking Fas, TNF-α, or TRAIL pathways did not rescue Mo-DCs from HCoV-229E-induced death. We controlled in Jurkat cells that anti-TRAIL, TNF-R/Fc, and FAS/Fc were efficient to block apoptosis induced by their cognate ligands, i.e., TRAIL, TNF-α, and FasL, respectively (data not shown). Caspases can be activated through pathways that are independent of membrane death receptors. To determine whether cell death induced by HCoV-229E relies on caspases, we pretreated cells with Z-VAD-FMK, a general caspase inhibitor that irreversibly blocks their catalytic site (48). As shown in Fig. 5B, Z-VAD-FMK did not prevent the death of HCoV-229E-infected Mo-DCs. We controlled in Jurkat cells that Z-VAD-FMK efficiently blocked the apoptosis induced by TNF-α (data not shown). Altogether, these observations suggest that even if HCoV-229E infection activates caspases in Mo-DC (Fig. 1C), cell death does not require caspase activation but rather results from cytolysis and/or necrosis of multinucleated syncytia induced by replicative infection.
Fig 5.
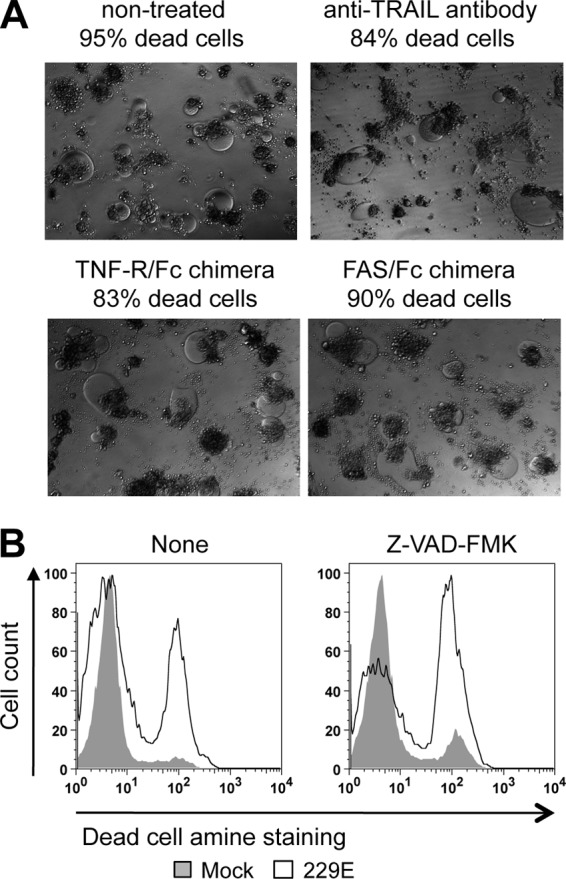
Death of HCoV-229E-infected Mo-DCs is independent of TRAIL, TNF-α, FasL, and caspase activity. (A) Mo-DCs were infected with HCoV-229E and incubated for 24 h in the presence of anti-TRAIL blocking antibodies, TNFR-Fc chimera, or FAS-Fc chimera. Cell death was determined by trypan blue exclusion (the results of one representative experiment out of three are shown). (B) Inhibition of caspase activity does not prevent cell death in Mo-DC cultures. Mo-DCs were preincubated for 24 h in the presence of Z-VAD-FMK, a potent broad-spectrum caspase inhibitor, and then infected by HCoV-229E in the presence of Z-VAD-FMK. After 24 h of culture, cell death was determined by using Dead/Live staining (the results if one representative experiment out of three are shown).
Cytokines produced by monocytes do not account for their resistance to HCoV-229E-induced cell death and cytopathic effects.
We searched for cellular soluble factors that could account for monocyte resistance to HCoV-229E-induced cell death and cytopathic effects. Specific cytokines such as IFN-α/β and IL-6 could provide resistance to viral infection. We performed multiplex bead-based Luminex assays and ELISA to determine the cytokine/chemokine expression profiles in culture supernatants of monocytes and Mo-DCs collected 6, 12, and 24 h after infection with HCoV-229E. The results obtained at 24 h are presented in Table 1. Tested cytokines and chemokines were all induced in monocytes as soon as 6 h after infection with HCoV-229E, reaching prominent inductions of TNF-α (×27), IFN-α (×19), MCP1 (×16), IL-6 (×11), and IFN-γ (×9) at 24 h. In contrast, for Mo-DCs, no increased induction was observed at early time points (not shown), and a more restricted induction profile limited to IFN-α (×24), IL-8 (×16), and TNF-α (×9) at 24 h after infection. Some of these cytokines that were differentially induced, such as IL-6, could account for the resistance of monocytes to HCoV-229E infection, cytopathic effects, and cell death. To test this hypothesis, Mo-DCs were pretreated for 24 h with UV-inactivated supernatants from infected monocyte cultures. Then, Mo-DCs susceptibility to infection by HCoV-229E was determined. As shown in Fig. 6A, monocyte culture supernatant did not confer resistance to HCoV-229E-induced cell death and cytopathic effects. Thus, resistance of monocytes to HCoV-229E-induced cell death is probably not mediated by a soluble factor but rather relies on an intrinsic property of this cell type that is lost during differentiation into Mo-DCs. It should be noted that monocyte culture supernatants contained 113 pg of IFN-α/ml and 31 pg of IFN-β/ml (Table 1), which correspond to 17 and 5 IU/ml, respectively. These levels are much lower than the 100- to 1,000-IU/ml doses of IFN-β used to inhibit HCoV-229E replication in Mo-DCs in Fig. 3B.
Table 1.
Cytokine and chemokine levels detected in culture supernatants from mock- and HCoV-229E-infected monocytes and Mo-DCs at 24 h postinfection
| Cytokine | Mean cytokine concna (pg/ml) ± SD in: |
|||||
|---|---|---|---|---|---|---|
| Monocytes |
Mo-DCs |
|||||
| Mock | 229E | Fold increase | Mock | 229E | Fold increase | |
| IL-6 | 87 ± 6 | 937 ± 508 | 11 | 7 ± 5 | 7 ± 2 | 1 |
| IL-8 | 15,295 ± 9,444 | 31,297 ± 2,656 | 2 | 122 ± 5 | 1,891 ± 1,107 | 16 |
| IL-12 | 20 ± 14 | 87 ± 4 | 4 | 22 ± 11 | 37 ± 19 | 2 |
| IL-15 | 33 ± 11 | 187 ± 18 | 6 | 72 | 64 ± 8 | 0.9 |
| TNF-α | 2 ± 1 | 54 ± 17 | 27 | 4 ± 1 | 34 ± 29 | 9 |
| IFN-γ | 7 ± 1 | 64 ± 2 | 9 | 0 | 4 ± 1 | 4 |
| IFN-α | 6 ± 4 | 113 ± 6 | 19 | 6 ± 4 | 141 ± 47 | 24 |
| IFN-β | 9 ± 2 | 31 ± 15 | 3 | 9 ± 2 | 27 ± 15 | 3 |
| MIP-1α | 40 ± 2 | 340 ± 73 | 9 | 19 ± 2 | 37 ± 16 | 2 |
| MIP-1β | 893 ± 393 | 4,363 ± 192 | 5 | 119 ± 112 | 223 ± 108 | 2 |
| RANTES | 39 ± 1 | 173 ± 8 | 4 | 37 ± 4 | 18 ± 8 | 0.5 |
| MCP-1 | 564 ± 234 | 8,821 ± 4,929 | 16 | 128 ± 7 | 240 ± 58 | 2 |
All cytokines and chemokines were measured by a multiplex bead-based Luminex assay (mean of three donors), except for IFN-α and IFN-β, which were measured by ELISA (mean of six donors). The fold increase for mock-infected versus HCoV-229E-infected cells is also indicated.
Fig 6.

Analysis of monocyte resistance to cytopathic effects and cell death. (A) Mo-DCs were infected with HCoV-229E at an MOI of 0.05 after preincubation for 24 h with culture supernatants from mock-treated monocytes, HCoV-229E-infected monocytes, or UV-treated supernatants from HCoV-229E-infected monocytes. Viable cells were enumerated at the indicated time points by trypan blue exclusion. The results correspond to the mean of three donors. (B) Phenotype analysis of monocytes incubated for 24 h in the presence of GM-CSF and IL-4. Cells were stained with an anti-CD14 MAb or anti-CD11c (open lines) or an isotypic control (closed lines) and analyzed by flow cytometry. The data shown are representative of three different donors. (C) Monocytes become susceptible to HCoV-229E-induced cell death when induced to differentiate into Mo-DCs. Monocytes were treated for 24 h with GM-CSF, IL-4, or both combined and then infected with HCoV-229E. After 24 h, the cells were immunostained for spike glycoprotein and active caspase-3 expression.
HCoV-229E susceptibility is early acquired during monocyte differentiation into Mo-DCs.
The differentiation of monocytes into immature DCs is usually achieved after 5 days of culture in the presence of both GM-CSF and IL-4. To determine at which stage of differentiation monocytes acquire their susceptibility to infection, we cultivated purified CD14+ monocytes either in medium alone or in GM-CSF, IL-4, or both cytokines. At different time points, cells were harvested and tested for the induction of cytopathic effects by HCoV-229E infection (MOI of 0.05). After only 24 h of culture in the presence of both GM-CSF and IL-4, although cells retain a cell surface phenotype of monocytes, expressing CD14 but no CD11c (Fig. 6B), they became susceptible to HCoV-229E, as assessed by the presence of large syncytia, dead cells and cellular debris (Fig. 6C, upper panel). Again, this susceptibility paralleled with high levels of viral spike glycoprotein expression and activation of caspase 3 (Fig. 6C, lower panel). The strong increase of viral spike protein on cell membrane, as in Mo-DCs, argues that this protein, which is responsible for membrane fusion, is the cause of syncytia formation and cell death. Interestingly, monocytes treated with IL-4 alone also became susceptible to HCoV-229E infection, and massive cytopathic effects were observed. A similar trend was observed when treating monocytes with GM-CSF alone, but the cytopathic effects were much less pronounced. This suggests that signaling events predominantly induced by IL-4, but partially overlapping with GM-CSF, are responsible for monocytes sensitization.
DISCUSSION
Although infection with the emerging SARS-CoV was associated with a severe acute respiratory disease, most human CoVs are responsible for mild upper respiratory tract infections, such as common colds, with only occasional spreading to the lower respiratory tract. Most respiratory viruses interact with DCs in the upper respiratory tract, which results in initiating an antiviral immune response but may also result in the spreading of the virus as a result of DC migration to the draining lymph nodes. In the present study, we investigated the interaction between HCoV-229E and human DCs. We observed that HCoV-229E infection causes massive cytopathic effects, resulting in the rapid death of DCs. Cell death correlates with a high surface expression of the HCoV-229E spike protein, which is responsible for cell-cell fusion, and the formation of large syncytia that blow up in a very short time (10 h, see the Movie S1 in the supplemental material). In contrast, monocytes from the same donors are less susceptible to infection and resist cytopathic effects and cell death despite similar expression levels of CD13, the cell surface receptor for HCoV-229E. Monocytes rapidly acquire susceptibility to HCoV-299E infection upon a short differentiation in the presence of GM-CSF and/or IL-4. DC differentiation might downregulate a restriction factor present in monocytes or induce the expression of an unknown cellular factor increasing susceptibility to HCoV-229E infection such as a coreceptor on the surface of the DCs. The infection and killing of DCs is dependent upon viral entry and viral replication, since blocking virus entry with an anti-CD13 antibody or inactivating the virus with UV protected cells. Pretreating cells with blocking antibodies against TNF-α, FAS-L, TRAIL, and IFN-α/β or with the caspase inhibitor Z-VAD-FMK did not protect infected Mo-DCs from death. We therefore suggest that cell death induced by HCoV-229E is not an apoptotic process but rather a direct consequence of virus replication and viral spike protein expression on the cell surface. Consistent with this interpretation, the only situation in which we observed protection was when cells were pretreated with IFN-β, which prevented infection by HCoV-229E. However, when Mo-DCs were infected with HCoV-229E and only then treated with IFN-β, viral replication occurred, inducing cytopathic effects and massive cell death (data not shown). This suggests that, as previously described for other respiratory viruses (5, 6), HCoV-229E encodes a virulence factor that blocks IFN-α/β signaling and prevents the induction of antiviral IFN-stimulated genes in Mo-DCs. This might explain why endogenous type I IFNs induced after HCoV-229E infection are unable to block viral replication and cytopathic effects in Mo-DCs. Monocytes exposed to HCoV-229E also responded by producing type I IFNs, and yet the blockade of IFN-α/β binding did not alter susceptibility to infection. Altogether, this suggests that monocyte resistance to HCoV-229E-induced cytopathic effects is independent of endogenous type I IFN production but rather relies on the expression of an as-yet-unknown restriction factor or the absence of a cellular factor increasing susceptibility to viral infection such as a coreceptor.
Because DCs are major sensors to detect viral infection and prime adaptive immunity, viruses have evolved strategies to interfere with their development, maturation, function, or viability to suppress or escape immune response. In this regard, killing DCs can be an efficient viral strategy to delay or prevent the establishment of adaptive immune responses. Infections by measles virus, human immunodeficiency virus, or lymphocytic choriomeningitis virus deplete DC populations in infected hosts (12, 22, 37). In vitro experiments have also demonstrated that filoviruses, vaccinia virus, herpes simplex virus, H5N1 influenza virus, and measles virus induce DC apoptosis or cytolysis within a few days (16, 31, 33, 41, 47). Also, human echovirus is extremely cytopathic toward DCs and induces their death in less than 24 h (27). DC killing by HCoV-229E, if it happens in vivo in human infection, could delay the induction of an adaptive immune response, thus providing time to replicate in the infected host. Furthermore, this could affect the establishment of a long-term immunological memory to the virus, explaining why people can be reinfected multiple times by HCoV-229E (8).
Massive death of infected DCs may also act as a host defense mechanism to prevent virus spreading in the body. Because DCs are located at every possible entry site of the body, they are one of the first cell types encountered by incoming viruses. Upon stimulation by PAMPs, DCs migrate from peripheral tissues to the draining lymph nodes where they elicit antigen-specific T lymphocytes. Many viruses use DCs not only as a vehicle to penetrate draining lymphoid organs but also to interfere with their antigen-presenting cell functions so that the immune response is skewed toward inappropriate cytokine profiles. In the case of HCoV-229E, the extreme susceptibility of DCs to infection probably prevents this virus from using them as a “Trojan horse.” Although this hypothesis needs to be further supported by experimental data, it was recently shown that DCs infected by Legionella pneumophila undergo apoptosis to restrict bacterial replication and spreading (34). Interestingly, SARS-CoV, which spreads to the lower respiratory tract and is therefore associated with a much more severe respiratory disease than HCoV-229E, does not induce massive cell death and cytopathic effects in DCs, arguing in favor of this hypothesis.
Our most striking observation is that, compared to DCs, monocytes from the same donors are resistant to HCoV-229E infection. However, when stimulated for only 24 h with IL-4 alone, and to some extent with GM-CSF alone, monocyte cultures became susceptible to infection. This suggests that signaling events induced by IL-4, and also somewhat induced by GM-CSF, are responsible for monocyte sensitization to HCoV-229E infection. For example, both IL-4 and GM-CSF lead to the activation of phosphatidylinositol 3-kinase and GRB2/mitogen-activated protein kinase pathways, which seem to be critical in the biological functions of DCs (23, 25). In a previously published work, Collins et al. found that monocytes/macrophages undergo apoptosis when infected with HCoV-229E (11). However, these authors did not describe the massive cell death and cytopathic effects that we observed in Mo-DCs. This difference could be due to different cell isolation and purification methods. Indeed, monocytes obtained by adhering peripheral blood mononuclear cells quickly differentiate into macrophages, as acknowledged by the authors themselves. This could account for their susceptibility to HCoV-229E infection, whereas monocytes positively selected by magnetic beads are resistant, as shown in the present report.
What are the mechanisms allowing monocytes, but not DCs, to prevent cell death and massive cytopathic effects upon HCoV-229E infection? We observed that monocytes produced 16 times more IL-6 than did Mo-DCs upon infection. This could explain the resistance of monocytes since IL-6 is an IFN-like cytokine with antiviral properties. However, neutralizing IL-6 in infected monocyte cultures did not confer susceptibility to HCoV-229E, and adding recombinant IL-6 into infected DC cultures did not confer resistance to the virus (data not shown). As well, supernatants collected from infected monocytes did not protect Mo-DCs. Thus, a soluble cofactor does not account for monocyte resistance to HCoV-229E infection. Interestingly, it has been shown that bovine viral diarrhea virus, a positive-strand RNA virus that belongs to the Flaviviridae family, has opposite effects on these two cell types, DCs being resistant, whereas monocytes are rapidly killed by the infection (21). HIV is another example demonstrating that cells from the macrophage lineage have different levels of susceptibility to infection. Although macrophages and monocytes both express HIV-1 entry receptors, monocytes freshly purified from peripheral blood are resistant to HIV-1 infection. In contrast, monocyte-derived macrophages are highly susceptible to infection. As an explanation for this, Wang et al. demonstrated that freshly isolated monocytes express higher levels of anti-HIV-1 microRNAs than monocyte-derived macrophages (50). This suggests that the expression of specific antiviral miRNA is a possible mechanism underlying monocyte resistance to HCoV-229E. The precise mechanism that triggers the susceptibility of monocytes to infection after a short stimulation with IL-4 or GM-CSF remains to be elucidated.
In conclusion, we demonstrate here that HCoV-229E, which is commonly spread among human population, infects and destroys human Mo-DCs rapidly, whereas monocytes are resistant. The next step of this work is to identify not only the molecular basis of the DC susceptibility and monocyte resistance to HCoV-229E infection but also the mechanisms that regulate this phenotype and how this affects the severity of the disease.
Supplementary Material
ACKNOWLEDGMENTS
We thank Chung Cheung (Department of Microbiology, Li Ka Shing Faculty of Medicine, University of Hong Kong) for providing HCoV-229E virus and MRC5 cells. We thank François M. Lemoine (UPMC, Pitié-Salpêtrière, Paris, France) for providing CD40-L, TNF-α, and PGE2. We thank Pierre Talbot (Institut Armand-Frappier, Laval, Quebec, Canada) for providing the MAb 5-11H.6 for the HCoV-229E spike glycoprotein detection.
Footnotes
Published ahead of print 2 May 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Abt M, Gassert E, Schneider-Schaulies S. 2009. Measles virus modulates chemokine release and chemotactic responses of dendritic cells. J. Gen. Virol. 90:909–914 [DOI] [PubMed] [Google Scholar]
- 2. Arbour N, et al. 1999. Acute and persistent infection of human neural cell lines by human coronavirus OC43. J. Virol. 73:3338–3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arbour N, Day R, Newcombe J, Talbot PJ. 2000. Neuroinvasion by human respiratory coronaviruses. J. Virol. 74:8913–8921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bonavia A, Arbour N, Yong VW, Talbot PJ. 1997. Infection of primary cultures of human neural cells by human coronaviruses 229E and OC43. J. Virol. 71:800–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Caignard G, Bourai M, Jacob Y, Tangy F, Vidalain PO. 2009. Inhibition of IFN-alpha/beta signaling by two discrete peptides within measles virus V protein that specifically bind STAT1 and STAT2. Virology 383:112–120 [DOI] [PubMed] [Google Scholar]
- 6. Caignard G, et al. 2007. Measles virus V protein blocks Jak1-mediated phosphorylation of STAT1 to escape IFN-alpha/beta signaling. Virology 368:351–362 [DOI] [PubMed] [Google Scholar]
- 7. Callendret B, et al. 2007. Heterologous viral RNA export elements improve expression of severe acute respiratory syndrome (SARS) coronavirus spike protein and protective efficacy of DNA vaccines against SARS. Virology 363:288–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Callow KA, Parry HF, Sergeant M, Tyrrell DA. 1990. The time course of the immune response to experimental coronavirus infection of man. Epidemiol. Infect. 105:435–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Caux C, et al. 1996. CD34+ hematopoietic progenitors from human cord blood differentiate along two independent dendritic cell pathways in response to GM-CSF+TNF alpha. J. Exp. Med. 184:695–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chapuis F, et al. 1997. Differentiation of human dendritic cells from monocytes in vitro. Eur. J. Immunol. 27:431–441 [DOI] [PubMed] [Google Scholar]
- 11. Collins AR. 2002. In vitro detection of apoptosis in monocytes/macrophages infected with human coronavirus. Clin. Diagn. Lab. Immunol. 9:1392–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Desforges M, Miletti T, Gagnon M, Talbot PJ. 2006. HCoV-229E infects and activates monocytes. Adv. Exp. Med. Biol. 581:511–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Desforges M, Miletti TC, Gagnon M, Talbot PJ. 2007. Activation of human monocytes after infection by human coronavirus 229E. Virus Res. 130:228–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Swart RL, et al. 2007. Predominant infection of CD150+ lymphocytes and dendritic cells during measles virus infection of macaques. PLoS Pathog. 3:e178 doi:10.1371/journal.ppat.0030178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Witte L, et al. 2008. DC-SIGN and CD150 have distinct roles in transmission of measles virus from dendritic cells to T lymphocytes. PLoS Pathog. 4:e1000049 doi:10.1371/journal.ppat.1000049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Engelmayer J, et al. 1999. Vaccinia virus inhibits the maturation of human dendritic cells: a novel mechanism of immune evasion. J. Immunol. 163:6762–6768 [PubMed] [Google Scholar]
- 17. Esposito S, et al. 2006. Impact of human coronavirus infections in otherwise healthy children who attended an emergency department. J. Med. Virol. 78:1609–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ferlazzo G, Wesa A, Wei WZ, Galy A. 1999. Dendritic cells generated either from CD34+ progenitor cells or from monocytes differ in their ability to activate antigen-specific CD8+ T cells. J. Immunol. 163:3597–3604 [PubMed] [Google Scholar]
- 19. Gagneur A, et al. 2002. Coronavirus-related nosocomial viral respiratory infections in a neonatal and paediatric intensive care unit: a prospective study. J. Hosp. Infect. 51:59–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Geijtenbeek TB, et al. 2000. Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell 100:575–585 [DOI] [PubMed] [Google Scholar]
- 21. Glew EJ, et al. 2003. Differential effects of bovine viral diarrhoea virus on monocytes and dendritic cells. J. Gen. Virol. 84:1771–1780 [DOI] [PubMed] [Google Scholar]
- 22. Hahm B, Trifilo MJ, Zuniga EI, Oldstone MB. 2005. Viruses evade the immune system through type I interferon-mediated STAT2-dependent, but STAT1-independent, signaling. Immunity 22:247–257 [DOI] [PubMed] [Google Scholar]
- 23. Hamdorf M, Berger A, Schule S, Reinhardt J, Flory E. 2010. PKCδ-induced PU.1: phosphorylation promotes hematopoietic stem cell differentiation to dendritic cells. Stem Cells 29:297–306 [DOI] [PubMed] [Google Scholar]
- 24. Herbst B, et al. 1997. CD34+ peripheral blood progenitor cell and monocyte derived dendritic cells: a comparative analysis. Br. J. Haematol. 99:490–499 [DOI] [PubMed] [Google Scholar]
- 25. Hoarau C, et al. 2008. Supernatant from bifidobacterium differentially modulates transduction signaling pathways for biological functions of human dendritic cells. PLoS One 3:e2753 doi:10.1371/journal.pone.0002753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kärber G. 1931. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Arch. Exp. Pathol. Pharmakol. 162:480–483 [Google Scholar]
- 27. Kramer M, et al. 2007. Echovirus infection causes rapid loss-of-function and cell death in human dendritic cells. Cell Microbiol. 9:1507–1518 [DOI] [PubMed] [Google Scholar]
- 28. Law HK, et al. 2005. Chemokine upregulation in SARS-coronavirus-infected, monocyte-derived human dendritic cells. Blood 106:2366–2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Legge KL, Braciale TJ. 2003. Accelerated migration of respiratory dendritic cells to the regional lymph nodes is limited to the early phase of pulmonary infection. Immunity 18:265–277 [DOI] [PubMed] [Google Scholar]
- 30. MacDonald KP, et al. 2002. Characterization of human blood dendritic cell subsets. Blood 100:4512–4520 [DOI] [PubMed] [Google Scholar]
- 31. Mahanty S, et al. 2003. Cutting edge: impairment of dendritic cells and adaptive immunity by Ebola and Lassa viruses. J. Immunol. 170:2797–2801 [DOI] [PubMed] [Google Scholar]
- 32. Movassagh M, et al. 1999. High level of retrovirus-mediated gene transfer into dendritic cells derived from cord blood and mobilized peripheral blood CD34+ cells. Hum. Gene Ther. 10:175–187 [DOI] [PubMed] [Google Scholar]
- 33. Muller DB, Raftery MJ, Kather A, Giese T, Schonrich G. 2004. Frontline: induction of apoptosis and modulation of c-FLIPL and p53 in immature dendritic cells infected with herpes simplex virus. Eur. J. Immunol. 34:941–951 [DOI] [PubMed] [Google Scholar]
- 34. Nogueira CV, et al. 2009. Rapid pathogen-induced apoptosis: a mechanism used by dendritic cells to limit intracellular replication of Legionella pneumophila. PLoS Pathog. 5:e1000478 doi:10.1371/journal.ppat.1000478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. O'Doherty U, et al. 1994. Human blood contains two subsets of dendritic cells, one immunologically mature and the other immature. Immunology 82:487–493 [PMC free article] [PubMed] [Google Scholar]
- 36. Olweus J, et al. 1997. Dendritic cell ontogeny: a human dendritic cell lineage of myeloid origin. Proc. Natl. Acad. Sci. U. S. A. 94:12551–12556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pacanowski J, et al. 2001. Reduced blood CD123+ (lymphoid) and CD11c+ (myeloid) dendritic cell numbers in primary HIV-1 infection. Blood 98:3016–3021 [DOI] [PubMed] [Google Scholar]
- 38. Piqueras B, Connolly J, Freitas H, Palucka AK, Banchereau J. 2006. Upon viral exposure, myeloid and plasmacytoid dendritic cells produce 3 waves of distinct chemokines to recruit immune effectors. Blood 107:2613–2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rosenzwajg M, Tailleux L, Gluckman JC. 2000. CD13/N-aminopeptidase is involved in the development of dendritic cells and macrophages from cord blood CD34+ cells. Blood 95:453–460 [PubMed] [Google Scholar]
- 40. Sallusto F, Lanzavecchia A. 1994. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 179:1109–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Servet-Delprat C, et al. 2000. Consequences of Fas-mediated human dendritic cell apoptosis induced by measles virus. J. Virol. 74:4387–4393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Servet-Delprat C, et al. 2000. Measles virus induces abnormal differentiation of CD40 ligand-activated human dendritic cells. J. Immunol. 164:1753–1760 [DOI] [PubMed] [Google Scholar]
- 43. Servet-Delprat C, Vidalain PO, Valentin H, Rabourdin-Combe C. 2003. Measles virus and dendritic cell functions: how specific response cohabits with immunosuppression. Curr. Top. Microbiol. Immunol. 276:103–123 [DOI] [PubMed] [Google Scholar]
- 44. Sizun J, Soupre D, Giroux JD, Legrand MC. 1995. Nosocomial respiratory infection due to coronavirus in neonatal intensive care units: prospective evaluation. Arch. Pediatr. 2:1020–1021 (In French.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Spearman C. 1908. The method of right and wrong cases (constant stimuli) with Gauss formulae. Br. J. Psychol. 2:227–242 [Google Scholar]
- 46. Spiegel M, Schneider K, Weber F, Weidmann M, Hufert FT. 2006. Interaction of severe acute respiratory syndrome-associated coronavirus with dendritic cells. J. Gen. Virol. 87:1953–1960 [DOI] [PubMed] [Google Scholar]
- 47. Thitithanyanont A, et al. 2007. High susceptibility of human dendritic cells to avian influenza H5N1 virus infection and protection by IFN-alpha and TLR ligands. J. Immunol. 179:5220–5227 [DOI] [PubMed] [Google Scholar]
- 48. Thornberry NA. 1998. Caspases: key mediators of apoptosis. Chem. Biol. 5:R97–R103 [DOI] [PubMed] [Google Scholar]
- 49. Vabret A, Dina J, Brison E, Brouard J, Freymuth F. 2009. Human coronaviruses. Pathol. Biol. (Paris) 57:149–160 (In French.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang X, et al. 2009. Cellular microRNA expression correlates with susceptibility of monocytes/macrophages to HIV-1 infection. Blood 113:671–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang ZY, et al. 2004. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J. Virol. 78:5642–5650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yeager CL, et al. 1992. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 357:420–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.