

Abstract
Endothelial cells (EC) can present antigen to either CD8+ T lymphocytes through constitutively expressed major histocompatibility complex class I (MHC-I) or CD4+ T lymphocytes through gamma interferon (IFN-γ)-induced MHC-II. Kaposi's sarcoma-associated herpesvirus (KSHV) is the etiological agent of Kaposi's sarcoma (KS), an EC neoplasm characterized by dysregulated angiogenesis and a substantial inflammatory infiltrate. KSHV is understood to have evolved strategies to inhibit MHC-I expression on EC and MHC-II expression on primary effusion lymphoma cells, but its effects on EC MHC-II expression are unknown. Here, we report that the KSHV infection of human primary EC inhibits IFN-γ-induced expression of the MHC-II molecule HLA-DR at the transcriptional level. The effect is functionally significant, since recognition by an HLA-DR-restricted CD4+ T-cell clone in response to cognate antigen presented by KSHV-infected EC was attenuated. Inhibition of HLA-DR expression was also achieved by exposing EC to supernatant from KSHV-inoculated EC before IFN-γ treatment, revealing a role for soluble mediators. IFN-γ-induced phosphorylation of STAT-1 and transcription of CIITA were suppressed in KSHV-inoculated EC via a mechanism involving SOCS3 (suppressor of cytokine signaling 3). Thus, KSHV infection resulted in transcriptional upregulation of SOCS3, and treatment with RNA interference against SOCS3 relieved virus-induced inhibition of IFN-γ-induced STAT-1 phosphorylation. Since cell surface MHC-II molecules present peptide antigens to CD4+ T lymphocytes that can function either as direct cytolytic effectors or to initiate and regulate adaptive immune responses, inhibition of this antigen-presenting pathway would provide a survival advantage to the virus.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV) (11) is the etiologic agent of the endothelial tumor Kaposi's sarcoma (KS). It also causes primary effusion lymphoma (PEL) and is associated with multicentric Castleman's disease (7). Immunocompromised patients, such as those either infected with human immunodeficiency virus or individuals undergoing organ transplantation, are at risk of developing KS if infected with KSHV (14). KS lesions are histologically complex; there is aberrant angiogenesis through proliferation of KSHV-transformed spindle-shaped endothelial cells (EC) and a pronounced inflammatory cell infiltrate, including CD4+ and CD8+ T lymphocytes (18, 27, 45).
KSHV has evolved numerous immune evasion strategies against both innate and adaptive immune responses (4, 34). Cell surface major histocompatibility complex class I (MHC-I) molecules present antigenic peptides from endogenously expressed proteins to CD8+ T lymphocytes. KSHV has evolved to negatively regulate this process by causing lysosomal degradation of cell surface MHC-I through the viral E3 ubiquitin ligases K5 and K3 (16, 20, 41, 44). Cell surface MHC-II molecules present peptide antigens to CD4+ T lymphocytes that then initiate and regulate adaptive immune responses. Resting EC do not express MHC-II, but it is induced by gamma interferon (IFN-γ) (25), a cytokine present in KS lesions (27) and capable of reactivating latent KSHV (6, 12). This induction requires de novo synthesis of the “master regulator” of the expression of MHC-II genes: class II transactivator (CIITA) (39, 40). This protein is recruited to the MHC-II promoter by RFX5, where it functions as a scaffold to coordinate transcription factor complex assembly (50). Transactivation of CIITA occurs predominantly from its promoter IV (PIV) following IFN-γ engagement with its receptor that triggers the tyrosine phosphorylation of signaling components, including the receptor-associated tyrosine kinases, Jak1 and Jak2, and the transcription factor STAT-1 (29). Both STAT-1 and IRF-1 are required for IFN-γ-induced expression of CIITA from PIV (28), as is remodeling of its promoter region into a complex chromatin structure (30; see reference 33).
In addition to their role in initiating and regulating adaptive immune responses, it is now clear that CD4+ T lymphocytes specific for several herpesviruses can directly recognize and kill MHC-II-positive cells expressing their cognate antigen (5, 13, 24). Indeed, appreciation of the in vivo relevance of cytotoxic CD4+ T lymphocytes against virus-infected cells is increasing (8). Evasion of the CD4+ T-lymphocyte response should therefore provide a survival advantage to herpesviruses. Although less well studied than interference of the MHC-I pathway, some examples and mechanisms of viral interference of the MHC-II pathway have been identified for cytomegalovirus (CMV), Epstein-Barr virus (EBV), and herpes simplex virus 1 (HSV-1) (47).
In the present study the effects of KSHV infection on IFN-γ-induced expression and function of the MHC-II molecule HLA-DR in EC were examined. KSHV inhibited IFN-γ-induced STAT-1 phosphorylation, concomitant transcription of CIITA and HLA-DR, and EC expression of HLA-DR protein. The inhibition of STAT-1 phosphorylation was mediated by suppressor of cytokine signaling 3 (SOCS3), a negative regulator of the IFN-γ signaling pathway that was induced upon KSHV infection. Importantly, KSHV-mediated suppression of IFN-γ-induced HLA-DR expression attenuated an MHC-matched CD4+ T-lymphocyte response to cognate antigen.
MATERIALS AND METHODS
KSHV production.
The KSHV used for the present studies was the recombinant strain rKSHV.219 expressing red fluorescent protein (RFP) from the KSHV lytic PAN promoter, green fluorescent protein (GFP) from the EF-1α promoter, and a puromycin resistance gene as a selectable marker (46). The method of production of this virus was that of the originators, Vieira and O'Hearn (46), and was as described previously (9). Thus, latently infected VK219 cells were maintained in minimal essential Eagle medium, 2.2 g of NaHCO3/liter, 10% fetal calf serum, and puromycin at 5 μg/ml (all from Sigma-Aldrich) and penicillin and streptomycin (10 μg/ml; Invitrogen). To induce KSHV lytic reactivation, the cells were infected with BacK50 and treated with 1.25 mM sodium butyrate (Sigma-Aldrich). BacK50 is a recombinant baculovirus engineered to express the KSHV lytic switch replication and transcription activator (RTA) protein, encoded by ORF50 (46). BacK50 was produced in insect SF9 cells grown in Grace's insect medium (Invitrogen) supplied with 10% fetal bovine serum at 28°C in a 5% CO2 incubator. At 3 days after BacK50 infection the baculovirus-containing supernatant was separated from SF9 cells by centrifugation at 450 × g for 10 min. The supernatants were passed through a 0.45-μm-pore-size filter.
After reactivation of rKSHV.219 in VK219 cells for 48 h, the supernatant was removed, centrifuged at 500 × g for 15 min, and then ultracentrifuged at 65,000 × g for 4 h. The resultant pellet was resuspended overnight in EBM2 medium (Lonzo; Clonetics). The titer was determined by infecting HEK293 cells with serial dilutions of rKSHV.219 and enumerating the GFP-positive cells at 48 h postinfection by fluorescence microscopy. One GFP-positive cell was assumed to result from infection by one infectious unit of virus; indeed, immunofluorescence assay (IFA) staining confirmed that the majority of GFP-positive cells contained a single LANA dot (data not shown). In some experiments, rKSHV.219 was inactivated by UV irradiation prior to EC infection.
Endothelial cell culture.
Human umbilical vein endothelial cells (HUVEC) were isolated from umbilical cords as described previously (15) and maintained in medium 199 (M199; Invitrogen) containing 20% fetal calf serum, 28 μg of gentamicin/ml, 2.5 μg of amphotericin B/ml, 1 ng of epidermal growth factor/ml, and 1 μg of hydrocortisone/ml (all from Sigma-Aldrich) until confluent. Either the first or the second passage EC from a different donor were used in each experiment. In the T-lymphocyte recognition experiments, only the microvascular cell line HMEC-1 was used, which was also maintained in M199 supplemented as described above. Cultures of either HUVEC or HMEC-1 were dissociated with trypsin-EDTA (Sigma) and passaged into either tissue culture multiwell plates (Falcon; Becton Dickinson) as described previously (10) or prefabricated channel slides (μ-Slide VI; Ibidi GmbH). The seeding density yielded confluent monolayers for infection within 24 h.
Ethical approval.
Umbilical cords were collected after written informed consent, with the assistance of the Birmingham Women's Health Care National Health Service Trust and the approval of the South Birmingham Research Ethics Committee.
Infection of endothelial cells with KSHV and treatment with cytokines.
rKSHV.219 was diluted in EBM2 to give appropriate concentrations prior to inoculating either HUVEC at a multiplicity of infection (MOI) of 10 or HMEC-1 at an MOI of 5. Virus-inoculated cells were centrifuged at 450 × g for 30 min (noninoculated samples were treated in an identical manner, but without rKSHV.219). EC were incubated (37°C 5% CO2) for a further 90 min before the KSHV-containing medium was replaced with M199 and supplements. The KSHV-inoculated EC cultures are referred to as “KSHV-EC.” Within the KSHV-inoculated cultures, infected cells could be identified as those that were positive for GFP expression. The recombinant virus rKSHV219 expresses GFP from the EF-1α promoter and RFP from the KSHV lytic PAN promoter; RFP could occasionally be detected in a minority of EC at 24 h (<5%), demonstrating minimal lytic replication. Where necessary, supernatants from noninoculated or KSHV-inoculated EC were harvested at 24 h postinfection and stored at −80°C until required. In some experiments, EC were treated with recombinant human IFN-γ (PeproTech, Inc.) for 4 h before rinsing and replacement with fresh medium and culture for a further 72 h prior to assay.
Treatment of endothelial cells with siRNA against SOCS3.
EC were plated in six-well plates in M199 (supplemented as described above but without antibiotics) to reach ca. 70% confluence the following day. The cells were transfected either with a pool of three target-specific commercial prevalidated SOCS-3 small interfering RNAs (siRNAs) or an appropriate scrambled control (all from Santa Cruz Biotechnology), with Lipofectamine RNAiMAX reagent (Invitrogen) according to the manufacturer's instructions. Approximately 24 h later EC were inoculated with KSHV, as described above. Transfection did not affect the subsequent level of GFP-positive EC. Efficient knockdown of SOCS3 protein (a reduction in expression by more than 80%), at 24 h postinfection, was verified by Western blotting, as we previously described (9).
Flow cytometry and antibodies.
EC were either fixed and permeabilized for intracellular staining according to the manufacturer's instructions (Fix & Perm kit; Caltag) or surface stained on ice. The antibodies used were as follows: MHC-II (Alexa Fluor 647-conjugated mouse anti-human HLA-DR [Biolegend] and HLA-DP [BD Biosciences]); STAT-1 (Alexa Fluor 647-conjugated mouse anti-human STAT-1 [BD Biosciences]); and IFNγRα (unconjugated mouse anti-human IFNγRα [Santa Cruz Biotechnology]), followed by specific secondary conjugated antibody (Alexa Fluor 633-conjugated anti-mouse IgG [H&L; Invitrogen] or fluorescein isothiocyanate [FITC]-conjugated anti-mouse IgG [Dako]). Expression was analyzed by flow cytometry (Dako CyAn). KSHV-infected cells were positive for GFP expression. The data were analyzed using either Summit (Becton Dickinson) or FloJo (TreeStar, Inc.) software.
Evaluation of gene expression by qRT-PCR.
RNA was extracted from EC using TRIzol (Invitrogen), and quantitative reverse transcriptase PCR (qRT-PCR) was conducted either in one step with a QuantiTect probe PCR kit (Qiagen) (SOCS3 and CIITA) or in two steps with conversion to cDNA, followed by qPCR, using TaqMan reagents (Applied Biosystems). Assay-on-Demand primers and probes were used for SOCS3, CIITA, and HLA-DRA (Applied Biosystems) and 18S as a control gene (Eurogentec or Applied Biosystems). All products were used according to the manufacturer's instructions. Samples were amplified using the 7900 HT real-time PCR machine and analyzed using the software package SDS 2.2 (Applied Biosystems).
Immunocytochemistry and antibodies.
HUVEC were formaldehyde fixed, methanol permeabilized, and blocked with phosphate-buffered saline and 5% goat serum. Primary antibody against phospho-STAT-1 (unconjugated rabbit anti-pSTAT-1 [Tyr701]; Cell Signaling Technology) was added, followed by incubation overnight at 4°C, and then conjugated secondary antibody (Alexa Fluor 647-conjugated anti-rabbit IgG [H&L; Invitrogen]) was added for 1 to 2 h. Bisbenzimide (Sigma) served as a nuclear stain, and channels were coated with Prolong Gold antifade mountant (Invitrogen) prior to visualization. Images were captured by confocal microscopy (Zeiss LSM510) and analyzed with ImageJ software.
CD4+ T-lymphocyte antigen recognition assay.
HMEC-1 cells (1.5 × 105) were seeded in prefabricated channel slides (μ-Slide VI) prior to KSHV inoculation and treatment with IFN-γ, as described above. After IFN-γ treatment, some channels were exposed to PRS peptide (amino acid residues 276 to 295 of EBV nuclear antigen 2 [EBNA2]; 50 μg/ml) for 1 h and then washed. After washing, 1.5 × 104 of the CD4+ T-lymphocyte clone PRS c93 were added to each well, and the coculture was incubated at 37°C for 18 h. The PRS c93 clone is specific for the EBNA2 epitope aa276-295 presented by HLA-DRB3*02 and was generated by standard procedures as previously described (23). Extracellular IFN-γ released from T lymphocytes into the coculture supernatant was quantified by sandwich enzyme-linked immunosorbent assay (ELISA; Endogen) according to the manufacturer's protocol.
Statistical analyses.
Effects of multiple treatments were tested using analysis of variance (ANOVA) followed by the Bonferroni post hoc test. The effects of single treatments were tested by using the Student paired t test.
RESULTS
KSHV suppresses IFN-γ-induced surface expression of HLA-DR by EC.
To establish the effects of KSHV on EC HLA-DR expression, surface levels were quantified on either IFN-γ-treated or untreated cells and either with or without prior KSHV inoculation. Early-passage HUVEC were inoculated with recombinant rKSHV.219 expressing GFP from the EF-1α promoter to identify latently infected cells (46). The EC monolayer remained intact and viable when inoculated with KSHV at an MOI of 10. As we previously reported, spindle-shaped morphology was apparent at 24 h postinoculation and was always accompanied by GFP expression (9). In this system, GFP was a reliable surrogate marker of KSHV infection since GFP-expressing cells also showed typical punctate immunofluorescence staining for the latency-associated nuclear antigen (LANA; data not shown). Approximately 30% of KSHV-inoculated EC expressed GFP within 24 h.
Without either virus inoculation or cytokine treatment, EC were negative for HLA-DR (Fig. 1Ai and B), and expression was not induced by KSHV inoculation per se (Fig. 1Aii and B). IFN-γ treatment alone induced a marked increase in expression of HLA-DR (Fig. 1Aiii and B), whereas the levels induced on KSHV-inoculated EC were significantly lower (Fig. 1Aiv and B). Importantly, after IFN-γ treatment, both the GFP-positive and the GFP-negative EC within the KSHV-inoculated cell population expressed lower levels of HLA-DR than EC not inoculated with virus (Fig. 1B), suggesting the involvement of a soluble factor. Inoculation of EC with KSHV inactivated by UV irradiation did not inhibit HLA-DR expression in response to IFN-γ treatment, indicating that viral gene expression was required for the effect (data not shown). The intracellular levels of HLA-DR were also reduced: the protein levels for IFN-γ-treated KSHV-EC were reduced by 77.2% ± 1.73%, compared to IFN-γ-treated EC (i.e., as determined from the mean fluorescence intensity [MFI] values from three experiments). These data are commensurate with the extracellular levels shown in Fig. 1B. In order to determine whether this inhibition was specific to HLA-DR, we measured the levels of another MHC-II antigen HLA-DP on EC and KSHV-EC treated with IFN-γ. Indeed, we observed that the expression of HLA-DP in response to IFN-γ was also inhibited by KSHV inoculation (Fig. 1C).
Fig 1.
Effect of KSHV on IFN-γ-induced EC MHC-II expression. EC were inoculated with KSHV at an MOI of 10 (KSHV-EC) and cultured for 24 h prior to treatment with IFN-γ (400 U/ml, 4 h). After a further 72 h, the cells were analyzed by flow cytometry for MHC-II (HLA-DR) expression. (A) Representative scatter plots of HLA-DR expression on noninoculated EC (EC) and KSHV-EC with or without IFN-γ treatment. (B) MFI values for HLA-DR expression on IFN-γ-treated EC and the GFP-positive and -negative EC within the KSHV-inoculated (KSHV-EC) population. Data are means ± the SEM from six independent experiments. **, P < 0.01 (EC versus KSHV-EC as determined by paired Student t test). (C) MFI values for HLA-DP expression on IFN-γ-treated EC and KSHV-inoculated EC (KSHV-EC). Data are means ± the SEM from three independent experiments. *, P < 0.05 (EC versus KSHV-EC as determined by paired Student t test).
The KSHV-encoded proteins K3 and K5 can downregulate the expression of the IFN-γ receptor (IFNγR) (21). The possibility was therefore considered that such downregulation could explain the reduced expression of HLA-DR on KSHV-inoculated EC compared to noninoculated cells. KSHV-infected (GFP-positive) EC within KSHV-inoculated cultures did not express IFNγR to significantly different levels than EC that had not been inoculated with virus (Fig. 2). KSHV-uninfected (GFP-negative) EC within KSHV-inoculated cultures expressed IFNγR to levels slightly higher than those of noninoculated cells, but this difference also was not statistically significant (Fig. 2). Thus, modification of IFNγR expression levels was not responsible for the reduced HLA-DR expression seen on both GFP-positive and -negative cells within KSHV-inoculated EC cultures.
Fig 2.
Effect of KSHV on EC IFNγR expression. EC were inoculated with KSHV at an MOI of 10 (KSHV-EC) and cultured for 24 h prior to flow cytometry analysis of IFNγR expression. MFI values are shown as means ± the SEM from three independent experiments for noninoculated EC (EC) and KSHV-EC. US, unstained cells. IgG, cells stained with an isotype control antibody for the anti-IFNγR antibody.
Supernatant from KSHV-inoculated EC cultures inhibits IFN-γ-induced expression of HLA-DR.
Since suppression of MHC-II expression in response to IFN-γ occurred on both GFP-positive and -negative EC within the KSHV-inoculated EC population, we tested whether KSHV-infected EC released soluble factor(s) that modulated HLA-DR expression. Treatment with supernatants for 24 h from either KSHV-inoculated or noninoculated EC did not induce HLA-DR expression on otherwise-untreated EC (Fig. 3Ai, Aii, and B). However, when the supernatant from KSHV-inoculated EC was used to condition EC prior to their IFN-γ treatment, HLA-DR expression was significantly reduced by more than 50% compared to the levels for EC treated with supernatants from uninfected EC (Fig. 3Aiii, Aiv, and B).
Fig 3.
Effect of pretreating EC with conditioned supernatant from KSHV-inoculated EC on IFN-γ-induced MHC-II expression. EC were treated with conditioned supernatants from either noninoculated EC (EC supernatant) or KSHV-inoculated EC (KSHV-EC supernatant) for 24 h. They were then treated with IFN-γ (+IFNγ) or not (No IFNγ). After a further 72 h, the cells were analyzed by flow cytometry for MHC-II (HLA-DR) expression. (A) Representative scatter plots of HLA-DR expression on EC conditioned with either EC supernatant or KSHV-EC supernatant and then either treated with IFN-γ or not. (B) MFI values for HLA-DR expression. Data are means ± the SEM from five independent experiments. *, P < 0.05 (EC supernatant- versus KSHV-EC supernatant-conditioned EC as determined by paired Student t test).
KSHV inhibits IFN-γ-induced expression of HLA-DRA and CIITA by EC.
To establish whether the inhibition of HLA-DR expression by KSHV occurred at the transcriptional level, we measured IFN-γ-induced HLA-DRA mRNA in KSHV-inoculated and noninoculated EC. As expected, the HLA-DRA mRNA levels were upregulated in response to IFN-γ (Fig. 4A). However, this upregulation was significantly inhibited in KSHV-inoculated EC compared to noninoculated EC (Fig. 4A). MHC-II gene expression is dependent upon the transactivator CIITA (40). We therefore determined whether its expression was modified by KSHV. As expected, CIITA mRNA levels were upregulated in response to IFN-γ, but this upregulation was significantly inhibited in KSHV-inoculated EC (Fig. 4B). These data indicated that KSHV-mediated inhibition of HLA-DR expression occurs upstream of CIITA transcription.
Fig 4.
Effect of KSHV on IFN-γ-induced EC HLA-DRA and CIITA gene expression. EC were either inoculated with KSHV at an MOI of 10 (KSHV-inoculated) or not (KSHV-noninoculated) and cultured for 24 h. They were then treated with 40 0U of IFN-γ/ml for up to 2 h before being harvested at the time points indicated on the x axis. Samples were harvested in TRIzol for mRNA extraction and qRT-PCR analysis. (A) HLA-DR mRNA levels. The data are means ± the SEM from three independent experiments. ANOVA showed a significant effect of KSHV-infection. **, P < 0.001 (as determined by the Bonferroni post hoc test [KSHV-EC versus EC]). (B) CIITA mRNA levels. The data are means ± the SEM from three independent experiments. ANOVA showed a significant effect of time (P < 0.0005) and KSHV infection (P < 0.0001). *, P < 0.05 (as determined by the Bonferroni post hoc test [KSHV-EC versus EC]).
KSHV inhibits IFN-γ-induced phosphorylation of STAT-1.
Upon engagement of the IFN-γ receptor, STAT-1 monomers are phosphorylated and form homodimers that translocate to the nucleus and transactivate IFN-γ-responsive genes (17), including CIITA (28). To establish whether the inhibition of CIITA transcription was a consequence of inhibition of STAT-1 phosphorylation, the levels of STAT-1 phosphorylated at Tyr701 (phospho-STAT-1) were compared in KSHV-inoculated (KSHV-EC) and noninoculated EC (EC) with or without IFN-γ treatment. First, we established that the basal (nonphosphorylated) STAT-1 levels were not modified by KSHV inoculation as determined by flow cytometry (KSHV-EC, 9.8 ± 1.1, and EC, 9.3 ± 1.2 [mean MFI ± the SEM for three experiments]). However, in the absence of IFN-γ treatment, the phospho-STAT-1 levels were higher in KSHV-inoculated EC compared to noninoculated EC (Fig. 5). These data indicated that de novo KSHV infection induces STAT-1 phosphorylation.
Fig 5.
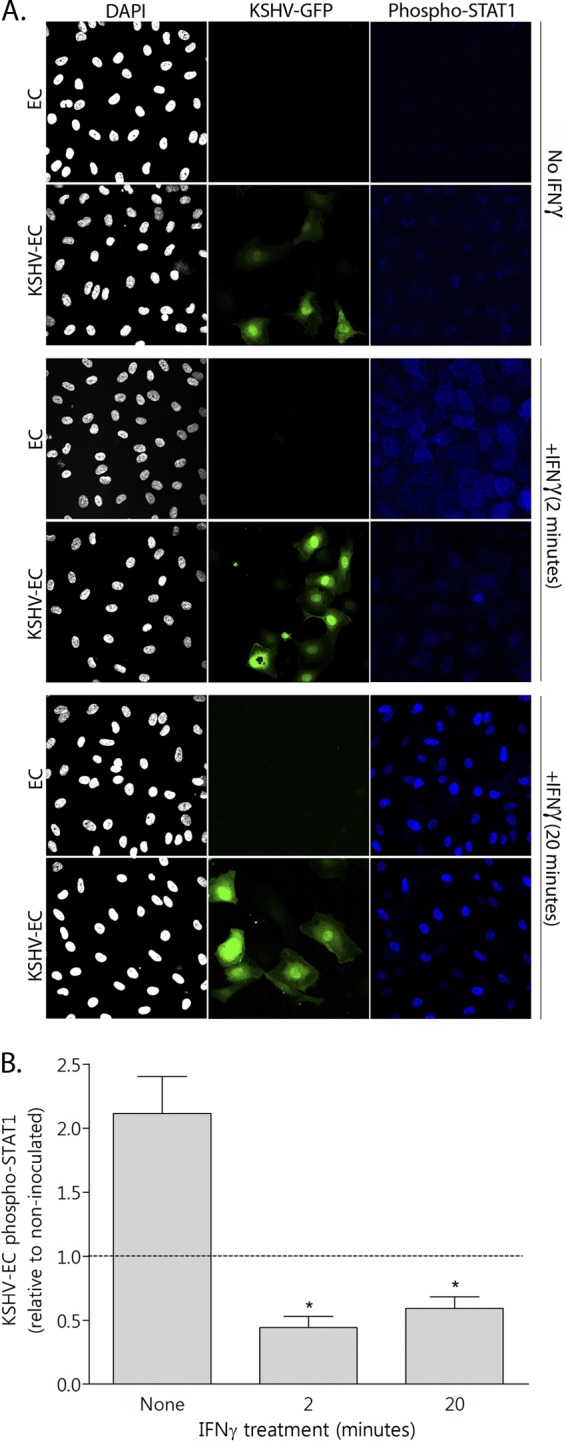
Effect of KSHV on IFN-γ-induced EC STAT-1 phosphorylation. EC were either inoculated with KSHV at an MOI of 10 (KSHV-EC) or not (EC) and cultured for 24 h. They were then treated with 400 U of IFN-γ/ml for 0 (none), 2, or 20 min before formaldehyde fixation and staining for phosphorylated STAT-1 (phospho-STAT1). (A) Confocal microscopy images representative of three independent experiments. KSHV infection is revealed by GFP expression (KSHV-GFP) and nuclear staining (DAPI [4′,6′-diamidino-2-phenylindole]) revealed the total cell population. (B) The levels of STAT-1 phosphorylation (phospho-STAT1) were quantified from confocal images of KSHV-EC, relative to non-KSHV-inoculated EC, using ImageJ software to determine fluorescence intensity, and these values were normalized to the cell number. The data are means ± the SEM from three independent experiments. *, P < 0.05 (for KSHV-EC versus EC as determined by paired Student t test).
As expected, STAT-1 was rapidly phosphorylated in response to IFN-γ treatment, since the fluorescence intensity of labeled phospho-STAT-1 increased by 85.3% ± 3.3% after 2 min of IFN-γ treatment (means ± the SEM for three experiments). However, in EC that were inoculated with KSHV, the phospho-STAT-1 levels were significantly lower compared to noninoculated EC after IFN-γ treatment for either 2 or 20 min (Fig. 5B). Importantly, the reduction in phospho-STAT-1 levels in KSHV-inoculated EC could be seen in both the GFP-positive and -negative cells (Fig. 5A), a finding consistent with our observations on the inhibition of HLA-DR expression on the surface of cells from both of these populations (Fig. 1).
Inhibition of phosphorylation of STAT-1 is mediated by KSHV-induced upregulation of SOCS3.
Since IFN-γ-induced STAT-1 phosphorylation was inhibited in KSHV-inoculated EC, the possible contribution of SOCS1 or SOCS3, negative regulators of the JAK/STAT signaling pathway (see reference 49), was evaluated. Significant upregulation of SOCS3 mRNA was observed by qRT-PCR in KSHV-inoculated EC compared to noninoculated EC (Fig. 6A, left panel), a finding consistent with our previous observations (9). In parallel assays, SOCS1 mRNA levels were not upregulated by KSHV infection (Fig. 6A, left panel), showing that modification of IFN-γ signaling could not be attributed to induction of SOCS1 expression. Furthermore, significant upregulation of SOCS3 was also observed in EC that had been treated with supernatant from KSHV-inoculated EC compared to those cells treated with supernatant from noninoculated EC (Fig. 6A, right panel).
Fig 6.

Effect of KSHV-induced SOCS3 expression on IFNγ-induced STAT-1 phosphorylation. (A) EC were either inoculated with KSHV at an MOI of 10 (KSHV-EC) or not (EC) (left panel) or treated with conditioned supernatants from either noninoculated (EC supernatant treatment) or KSHV-EC (KSHV-EC supernatant treatment) (right panel). The EC were then cultured for 24 h prior to harvesting for mRNA extraction and quantification of SOCS3 and SOCS1 gene expression by real-time qRT-PCR. (B) EC were either untransfected (UT) or transfected with siRNA against either SOCS3 (SOCS3) or a nonspecific siRNA control (NS). The EC were then either inoculated with KSHV at an MOI of 10 (KSHV-EC) or not (EC) and cultured for 24 h prior to treatment with IFN-γ (400 U/ml, 2 min). The EC were then fixed with formaldehyde and stained for phosphorylated STAT-1 (pSTAT1). The data are means ± the SEM from two to four independent experiments. *, P < 0.05; **, P < 0.01 (EC versus KSHV-EC or EC supernatant versus KSHV-EC supernatant conditioned EC as determined by paired Student t test).
To determine whether SOCS3 played a role in the KSHV-mediated inhibition of STAT-1 phosphorylation, its KSHV-induced expression was inhibited with siRNA prior to IFN-γ treatment. Western blotting confirmed that SOCS3 protein was upregulated by KSHV inoculation of EC and that >80% knockdown was achieved with SOCS3 siRNA, while there was no effect of the nonspecific control siRNA (data not shown; see also reference 9). In noninoculated EC, nonspecific siRNA (NS) or SOCS3 siRNA (SOCS3) did not significantly modify IFN-γ-induced phospho-STAT-1 levels compared to EC receiving no siRNA (UT) (Fig. 6B). Consistent with Fig. 5, KSHV-inoculated EC expressed lower levels of phospho-STAT-1 after IFN-γ treatment than noninoculated EC. This inhibition of phospho-STAT-1 levels was abolished when the EC were pretreated with SOCS3 siRNA (Fig. 6B) but was not affected by the nonspecific siRNA. These data correlate with a requirement for SOCS3 expression in KSHV-mediated inhibition of phosphorylation of STAT-1.
Effect of human IL-6 preconditioning on IFN-γ-induced EC HLA-DR expression.
SOCS3 expression can be induced by human interleukin-6 (hIL-6) (2), and KSHV infection of EC induces hIL-6 (9). We therefore sought to determine whether KSHV-induced hIL-6 played a role in the inhibition of HLA-DR expression in response to IFN-γ treatment. EC were preconditioned with hIL-6 for 24 h prior to IFN-γ treatment and subsequent measurement of the HLA-DR levels. hIL-6 preconditioning significantly inhibited HLA-DR expression but did not completely recapitulate the effect of the virus (Fig. 7). Indeed, the addition of hIL-6 neutralizing antibody to KSHV-EC did not rescue HLA-DR expression in response to IFN-γ (the HLA-DR levels for IFN-γ-treated EC were as follows: noninoculated EC, 604.8 ± 195.4; KSHV-inoculated EC, 109.4 ± 28.8; and KSHV-inoculated hIL-6 neutralizing antibody-treated EC, 75.7 ± 18.1 [mean MFI ± the SEM from four experiments]). These data suggest that hIL-6 alone is not responsible for the KSHV-mediated inhibition of HLA-DR expression.
Fig 7.
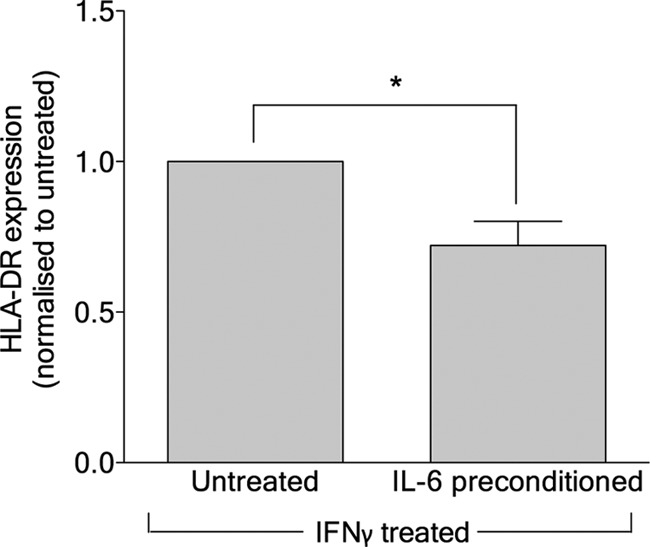
Effect of human IL-6 preconditioning on IFN-γ-induced EC HLA-DR expression. EC were treated either with (IL-6 preconditioned) or without (untreated) recombinant human IL-6 for 24 h prior to being treated with IFN-γ (400 U/ml, 4 h; IFN-γ treated). The HLA-DR expression levels were measured by flow cytometry 3 days later and normalized to the level obtained with IL-6-untreated cells. The data are means ± the SEM from seven independent experiments. *, P < 0.05 (hIL-6 preconditioning versus untreated as determined by paired Student t test).
KSHV-mediated reduction in IFN-γ-induced MHC-II expression inhibits antigen-specific CD4+ T-lymphocyte responses.
To assess the functional impact of KSHV inhibition of IFN-γ-induced MHC-II expression, the ability of KSHV-inoculated versus noninoculated EC to present antigen to MHC-matched antigen-specific human CD4+ T lymphocytes was examined. Since T-lymphocyte clones specific for KSHV antigens were not available, a CD4+ T-lymphocyte clone specific for a defined epitope (PRS aa275-296) within EBNA2 was utilized. The target EC were the MHC-II-matched human microvascular cell line (HMEC-1). Since HMEC-1 were more susceptible to infection by KSHV than HUVEC, these cells were infected with KSHV at an MOI of 5 to give levels of infection comparable to those seen on HUVEC in previous experiments. The level of CD4+ T-lymphocyte stimulation after coculture with target cells was measured by quantifying IFN-γ released by the responding cells.
HMEC-1 cells were negative for HLA-DR expression without either IFN-γ treatment or KSHV inoculation (Fig. 8A). IFN-γ treatment induced HLA-DR expression but, as for the HUVEC (Fig. 1), the levels were lower on HMEC-1 that had been inoculated with KSHV (Fig. 8A). UV irradiation of KSHV did not alter the level of IFN-γ-induced MHC-II (Fig. 8A), demonstrating that viral gene transcription is required.
Fig 8.
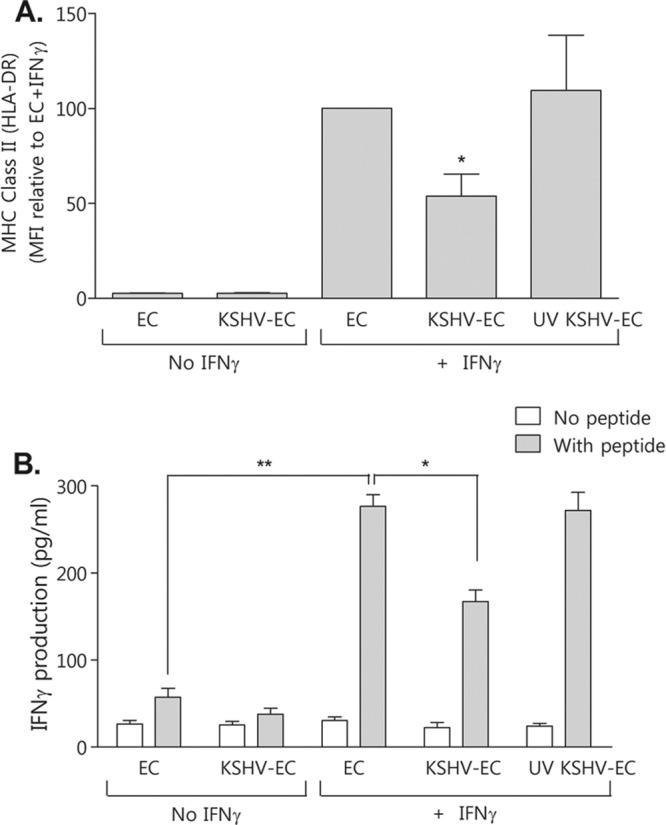
Effect of KSHV attenuation of IFNγ-induced HMEC-1 MHC-II expression on CD4+ T-lymphocyte antigen-specific activation. Human microvascular endothelial cells (HMEC-1) were either inoculated with KSHV at an MOI of 5 (KSHV-EC) or not (EC) or inoculated with UV-irradiated KSHV (UV KSHV-EC). They were then cultured for 24 h prior to being treated with IFN-γ (400 U/ml, 4 h) (+ IFN-γ) or not (No IFN-γ). After a further 72 h, cells were either analyzed by flow cytometry for MHC-II (HLA-DR) expression (A) or pulsed with peptide (PRS aa275-296 of EBV EBNA2) for 1 h before the addition of antigen-specific MHC-II-matched CD4+ T-lymphocyte clone cells for a further 18 h (B). The supernatants were then collected and assayed for IFN-γ by sandwich ELISA. The data are means ± the SEM from three independent experiments. *, P < 0.05; **, P < 0.01 (as determined by paired Student t test).
Upon the addition of CD4+ T lymphocytes to either KSHV-inoculated or noninoculated HMEC-1 cells, there was a low background level of IFN-γ produced that did not increase with the addition of cognate peptide (Fig. 8B). These data demonstrate that the unstimulated, MHC-II-negative HMEC-1 could not present peptide to the CD4+ T lymphocytes. However, pretreatment of non-KSHV-inoculated HMEC-1 with IFN-γ prior to peptide exposure led to the induction of >5-fold-greater IFN-γ secretion from the CD4+ T lymphocytes (Fig. 8B), corresponding to the induction of MHC-II expression. Consistent with the KSHV-mediated inhibition of HLA-DR expression, the CD4+ T-lymphocyte response was significantly inhibited by KSHV inoculation of HMEC-1 (Fig. 8B).
DISCUSSION
KSHV is the etiological agent of KS, an EC neoplasm characterized by dysregulated angiogenesis and a substantial inflammatory infiltrate. In the present study we have shown that KSHV inhibits IFN-γ-induced MHC-II expression on the surfaces of EC, as determined by quantifying extracellular and intracellular HLA-DR levels. This inhibition was apparent on the surfaces of both infected and uninfected EC within the KSHV-inoculated culture, indicating a role for one or more soluble mediators released from infected EC. Indeed, when uninfected (noninoculated) EC were conditioned with supernatant from KSHV-inoculated EC, prior to IFN-γ stimulation, the inhibition of HLA-DR expression seen on KSHV-inoculated EC was recapitulated. Upregulation of HLA-DP in response to IFN-γ treatment was also inhibited by KSHV infection, indicating the effect was not restricted to HLA-DR and was most likely a consequence of modification of upstream IFN-γ signaling processes. Indeed, IFN-γ-induced transactivation of both HLA-DR and CIITA genes was inhibited in KSHV-inoculated EC, as was the phosphorylation of STAT-1 at tyrosine residue 701. Using RNAi, we showed that this effect was attributable to KSHV-induced upregulation of SOCS3, a cytokine-inducible gene product that is a negative regulator of the JAK/STAT signaling pathway. Thus, we propose that KSHV infection of EC induces the expression of SOCS3, as we and others have previously described (9, 43), which inhibits early components of the IFN-γ signaling pathway and the subsequent upregulation of MHC-II gene expression.
Importantly, EC presentation of epitope peptide to an MHC-II-matched antigen-specific human CD4+ T-lymphocyte clone was impaired by KSHV, demonstrating a functional consequence of the inhibition of EC MHC-II expression. To our knowledge, this is the first report of the ability of KSHV to inhibit IFN-γ induction of EC MHC-II, identifying a new viral immune evasion strategy. In addition, our findings reveal a potentially more general role for SOCS3 in the regulation of antigen presentation in inflammation through its ability to modulate expression of MHC-II.
Previous studies have shown that EC can present antigen to both CD8+ and CD4+ T lymphocytes in vitro and in vivo (19, 35). Given the importance of CD4+ T lymphocytes to the immune system for the initiation and regulation of adaptive immunity and as direct cytotoxic effectors, the expression of MHC-II on EC, the predominant cell type in KS lesions (32), should be an important component of the host anti-KSHV response. Resting EC do not constitutively express MHC-II. However, in agreement with others (25), we observed an increase in expression 72 h after treatment with IFN-γ, a cytokine that is present in KS lesions (27). This expression was attenuated when EC were inoculated with KSHV 24 h prior to IFN-γ treatment. Modulation of MHC-II expression by KSHV would provide a selective advantage to the virus, by repressing CD4+ T-lymphocyte function in terms of both direct cytolytic activity and the regulation of adaptive responses.
Schmidt et al. (36) recently reported that the KSHV protein vIRF-3 could inhibit the constitutive expression of MHC-II on PEL cells, through inhibition of CIITA. However, in the present study the inhibition of MHC-II expression was similar on both GFP-positive and GFP-negative EC within the KSHV-inoculated cell population and could be attributed to the action of a soluble mediator. Thus, it is unlikely that vIRF-3 or any other viral protein was directly and exclusively responsible for the entire HLA-DR downregulation observed in the present study.
The KSHV proteins K3 and K5 can induce IFNγR ubiquitination, endocytosis, and degradation, thus depleting its cell surface expression (21). In the present study, IFNγR expression was not downregulated in the entire KSHV-infected culture but IFN-γ signaling was inhibited in both KSHV-infected and uninfected EC. These data are inconsistent with K3 and K5 being responsible for the downregulation of MHC-II from the surfaces of KSHV-infected EC cultures.
The SOCS family consists of eight structurally related proteins that attenuate signaling by blocking JAK tyrosine kinase activity or STAT activation and are typically induced by different cytokines and other stimuli (49). SOCS3 was recognized as one of the most highly and consistently upregulated EC genes following KSHV infection (43). In the present study and our previous work (9), SOCS3 was expressed at very low levels in untreated EC and significantly upregulated upon either KSHV inoculation or treatment with supernatant from KSHV-inoculated EC. Our data indicate KSHV infection per se induces STAT-1 phosphorylation. A previous report showed such effects can be mediated by KSHV vFLIP (3). However, the entire increase in STAT1 phosphorylation in our system could not be attributed to vFLIP alone, since increased levels were seen in both GFP-positive and -negative cells. Here, our data are commensurate with KSHV-induced SOCS3 either inhibiting or delaying STAT-1 phosphorylation after IFN-γ stimulation. Furthermore, they are consistent with previous studies reporting that stable overexpression of SOCS3 inhibited tyrosine phosphorylation of STAT-1 in response to IFN-γ in HeLa and MCF-7 cell lines (38) and that transfection of SOCS3 into macrophages inhibited the transcriptional response to IFN-γ by reducing JAK1 activation (42). Our studies reveal that hIL-6 can contribute to HLA-DR downregulation, since preconditioning EC with this cytokine reduced IFN-γ-induced EC expression of this antigen. The mechanism is presumably through the induction of SOCS3, as we showed previously (9). However, the function-neutralizing antibody to hIL-6 did not rescue HLA-DR expression in IFN-γ-treated KSHV-EC. Therefore, other factors acting in concert with this cytokine may downregulate HLA-DR. In addition, very high local intercellular concentrations of hIL-6 produced by infected cells may contribute to this activity.
SOCS3 upregulation is also a consequence of infection with viruses, including other members of the Herpesviridae, such as HSV-1 (48) and EBV (22), as well as influenza virus A (31). These findings suggest a more widespread role for SOCS3, and possibly other SOCS proteins, in viral pathogenesis. Our study is the first to link virus-induced SOCS3 expression to the inhibition of signaling elements necessary for the induction of MHC-II expression and the potential consequences for immune surveillance and viral clearance. This mechanism may explain the inhibition in IFN-γ-induced MHC-II expression described following infection of EC by other herpesviruses, such as CMV (26, 37) and varicella-zoster virus (1).
We highlight here the role of paracrine signaling in viral immune evasion mechanisms. This signaling process is likely to play a key role in KSHV immune modulation in KS lesions. Since cell surface MHC-II molecules present antigenic peptides that can be recognized by CD4+ T lymphocytes, inhibition of MHC-II expression in the KS lesion would provide a survival advantage to both the virus and the transformed EC in which it resides. In summary, we demonstrated here not only a previously unrecognized immunomodulatory strategy of KSHV in EC but also suggest a role for SOCS3 in the regulation of antigen presentation.
ACKNOWLEDGMENTS
This study was supported by Cancer Research UK grant C7934 to G.B.N., L.M.B., and D.J.B. and by Medical Research Council grants G0400408 and G0800154 to D.J.B.
We acknowledge Jeff Vieira for the generous provision of rKSHV219-infected Vero cells and BacK50 baculovirus and Martin Rowe for critical reading of the manuscript.
Footnotes
Published ahead of print 24 April 2012
REFERENCES
- 1. Abendroth A, et al. 2000. Modulation of major histocompatibility class II protein expression by varicella-zoster virus. J. Virol. 74:1900–1907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander WS, Hilton DJ. 2004. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu. Rev. Immunol. 22:503–529 [DOI] [PubMed] [Google Scholar]
- 3. Alkharsah KR, et al. 2011. Deletion of Kaposi's sarcoma-associated herpesvirus FLICE inhibitory protein, vFLIP, from the viral genome compromises the activation of STAT1-responsive cellular genes and spindle cell formation in endothelial cells. J. Virol. 85:10375–10388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aresté C, Blackbourn DJ. 2009. Modulation of the immune system by Kaposi's sarcoma-associated herpesvirus. Trends Microbiol. 17:119–129 [DOI] [PubMed] [Google Scholar]
- 5. Arvin AM, et al. 1991. Equivalent recognition of a varicella-zoster virus immediate-early protein (IE62) and glycoprotein I by cytotoxic T lymphocytes of either CD4+ or CD8+ phenotype. J. Immunol. 146:257–264 [PubMed] [Google Scholar]
- 6. Blackbourn DJ, Fujimura S, Kutzkey T, Levy JA. 2000. Induction of HHV-8 gene expression by recombinant interferon-gamma. AIDS 14:12–13 [DOI] [PubMed] [Google Scholar]
- 7. Bouvard V, et al. 2009. A review of human carcinogens. B. Biological agents. Lancet Oncol. 10:321–322 [DOI] [PubMed] [Google Scholar]
- 8. Brown DM. 2010. Cytolytic CD4 cells: direct mediators in infectious disease and malignancy. Cell. Immunol. 262:89–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Butler LM, et al. 2011. Kaposi's sarcoma-associated herpesvirus infection of endothelial cells inhibits neutrophil recruitment through an IL-6-dependent mechanism: a new paradigm for viral immune evasion. J. Virol. 85:7321–7332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Butler LM, Rainger GE, Rahman M, Nash GB. 2005. Prolonged culture of endothelial cells and deposition of basement membrane modify the recruitment of neutrophils. Exp. Cell Res. 310:22–32 [DOI] [PubMed] [Google Scholar]
- 11. Chang Y, et al. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869 [DOI] [PubMed] [Google Scholar]
- 12. Cheng F, et al. 2009. KSHV reactivation from latency requires Pim-1 and Pim-3 kinases to inactivate the latency-associated nuclear antigen LANA. PLoS Pathog. 5:e1000324 doi:10.1371/journal.ppat.1000324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Christensen JP, Cardin RD, Branum KC, Doherty PC. 1999. CD4+ T cell-mediated control of a gamma-herpesvirus in B cell-deficient mice is mediated by IFN-γ. Proc. Natl. Acad. Sci. U. S. A. 96:5135–5140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Colman R, Blackbourn DJ. 2008. Risk factors in the development of Kaposi's sarcoma. AIDS 22:1629–1632 [DOI] [PubMed] [Google Scholar]
- 15. Cooke BM, Usami S, Perry I, Nash GB. 1993. A simplified method for culture of endothelial cells and analysis of adhesion of blood cells under conditions of flow. Microvasc. Res. 45:33–45 [DOI] [PubMed] [Google Scholar]
- 16. Coscoy L, Ganem D. 2000. Kaposi's sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc. Natl. Acad. Sci. U. S. A. 97:8051–8056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Darnell JE, Jr, Kerr IM, Stark GR. 1994. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264:1415–1421 [DOI] [PubMed] [Google Scholar]
- 18. Ensoli B, et al. 2001. Biology of Kaposi's sarcoma. Eur. J. Cancer 37:1251–1269 [DOI] [PubMed] [Google Scholar]
- 19. Epperson DE, Pober JS. 1994. Antigen-presenting function of human endothelial cells. Direct activation of resting CD8 T cells. J. Immunol. 153:5402–5412 [PubMed] [Google Scholar]
- 20. Ishido S, Wang C, Lee BS, Cohen GB, Jung JU. 2000. Downregulation of major histocompatibility complex class I molecules by Kaposi's sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol. 74:5300–5309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li Q, Means R, Lang S, Jung JU. 2007. Downregulation of gamma interferon receptor 1 by Kaposi's sarcoma-associated herpesvirus K3 and K5. J. Virol. 81:2117–2127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lo AK, et al. 2006. Epstein-Barr virus infection alters cellular signal cascades in human nasopharyngeal epithelial cells. Neoplasia 8:173–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Long HM, et al. 2005. CD4+ T-cell responses to Epstein-Barr virus (EBV) latent-cycle antigens and the recognition of EBV-transformed lymphoblastoid cell lines. J. Virol. 79:4896–4907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Long HM, et al. 2011. Cytotoxic CD4+ T cell responses to EBV contrast with CD8 responses in breadth of lytic cycle antigen choice and in lytic cycle recognition. J. Immunol. 187:92–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McDouall RM, Batten P, McCormack A, Yacoub MH, Rose ML. 1997. MHC class II expression on human heart microvascular endothelial cells: exquisite sensitivity to interferon-gamma and natural killer cells. Transplantation 64:1175–1180 [DOI] [PubMed] [Google Scholar]
- 26. Miller DM, et al. 1998. Human cytomegalovirus inhibits major histocompatibility complex class II expression by disruption of the Jak/Stat pathway. J. Exp. Med. 187:675–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Milligan S, Robinson M, O'Donnell E, Blackbourn DJ. 2004. Inflammatory cytokines inhibit Kaposi's sarcoma-associated herpesvirus lytic gene transcription in in vitro-infected endothelial cells. J. Virol. 78:2591–2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morris AC, Beresford GW, Mooney MR, Boss JM. 2002. Kinetics of a gamma interferon response: expression and assembly of CIITA promoter IV and inhibition by methylation. Mol. Cell. Biol. 22:4781–4791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Muhlethaler-Mottet A, Di Berardino W, Otten LA, Mach B. 1998. Activation of the MHC class II transactivator CIITA by interferon-gamma requires cooperative interaction between Stat1 and USF-1. Immunity 8:157–166 [DOI] [PubMed] [Google Scholar]
- 30. Ni Z, Abou El Hassan M, Xu Z, Yu T, Bremner R. 2008. The chromatin-remodeling enzyme BRG1 coordinates CIITA induction through many interdependent distal enhancers. Nat. Immunol. 9:785–793 [DOI] [PubMed] [Google Scholar]
- 31. Pauli EK, et al. 2008. Influenza A virus inhibits type I IFN signaling via NF-κB-dependent induction of SOCS-3 expression. PLoS Pathog. 4:e1000196 doi:10.1371/journal.ppat.1000196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Regezi JA, et al. 1993. Human immunodeficiency virus-associated oral Kaposi's sarcoma. A heterogeneous cell population dominated by spindle-shaped endothelial cells. Am. J. Pathol. 143:240–249 [PMC free article] [PubMed] [Google Scholar]
- 33. Reith W, Boss JM. 2008. New dimensions of CIITA. Nat. Immunol. 9:713–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rezaee SA, Cunningham C, Davison AJ, Blackbourn DJ. 2006. Kaposi's sarcoma-associated herpesvirus immune modulation: an overview. J. Gen. Virol. 87:1781–1804 [DOI] [PubMed] [Google Scholar]
- 35. Rothermel AL, et al. 2004. Endothelial cells present antigens in vivo. BMC Immunol. 5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schmidt K, Wies E, Neipel F. 2011. Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor 3 inhibits IFN-γ and MHC II expression. J. Virol. 85:4530–4537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sedmak DD, et al. 1994. Cytomegalovirus inhibits major histocompatibility class II expression on infected endothelial cells. Am. J. Pathol. 144:683–692 [PMC free article] [PubMed] [Google Scholar]
- 38. Song MM, Shuai K. 1998. The suppressor of cytokine signaling (SOCS) 1 and SOCS3 but not SOCS2 proteins inhibit interferon-mediated antiviral and antiproliferative activities. J. Biol. Chem. 273:35056–35062 [DOI] [PubMed] [Google Scholar]
- 39. Steimle V, Otten LA, Zufferey M, Mach B. 1993. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome). Cell 75:135–146 [PubMed] [Google Scholar]
- 40. Steimle V, Siegrist CA, Mottet A, Lisowska-Grospierre B, Mach B. 1994. Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science 265:106–109 [DOI] [PubMed] [Google Scholar]
- 41. Stevenson PG, Efstathiou S, Doherty PC, Lehner PJ. 2000. Inhibition of MHC class I-restricted antigen presentation by gamma 2- herpesviruses. Proc. Natl. Acad. Sci. U. S. A. 97:8455–8460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stoiber D, et al. 1999. Lipopolysaccharide induces in macrophages the synthesis of the suppressor of cytokine signaling 3 and suppresses signal transduction in response to the activating factor IFN-γ. J. Immunol. 163:2640–2647 [PubMed] [Google Scholar]
- 43. Sturzl M, et al. 2009. The contribution of systems biology and reverse genetics to the understanding of Kaposi's sarcoma-associated herpesvirus pathogenesis in endothelial cells. Thromb. Haemost. 102:1117–1134 [DOI] [PubMed] [Google Scholar]
- 44. Tomescu C, Law WK, Kedes DH. 2003. Surface downregulation of major histocompatibility complex class I, PE-CAM, and ICAM-1 following de novo infection of endothelial cells with Kaposi's sarcoma-associated herpesvirus. J. Virol. 77:9669–9684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Valcuende-Cavero F, Febrer-Bosch MI, Castells-Rodellas A. 1994. Langerhans' cells and lymphocytic infiltrate in AIDS-associated Kaposi's sarcoma: an immunohistochemical study. Acta Derm. Venereol. 74:183–187 [DOI] [PubMed] [Google Scholar]
- 46. Vieira J, O'Hearn PM. 2004. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325:225–240 [DOI] [PubMed] [Google Scholar]
- 47. Wiertz EJ, Devlin R, Collins HL, Ressing ME. 2007. Herpesvirus interference with major histocompatibility complex class II-restricted T-cell activation. J. Virol. 81:4389–4396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yokota S, et al. 2004. Induction of suppressor of cytokine signaling-3 by herpes simplex virus type 1 contributes to inhibition of the interferon signaling pathway. J. Virol. 78:6282–6286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yoshimura A, Naka T, Kubo M. 2007. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 7:454–465 [DOI] [PubMed] [Google Scholar]
- 50. Zhu XS, et al. 2000. Transcriptional scaffold: CIITA interacts with NF-Y, RFX, and CREB to cause stereospecific regulation of the class II major histocompatibility complex promoter. Mol. Cell. Biol. 20:6051–6061 [DOI] [PMC free article] [PubMed] [Google Scholar]