

Abstract
Soft rot (type II) fungi belonging to the family Xylariaceae are known to substantially degrade hardwood by means of their poorly understood lignocellulolytic system, which comprises various hydrolases, including feruloyl esterases and laccase. In the present study, several members of the Xylariaceae were found to exhibit high feruloyl esterase activity during growth on lignocellulosic materials such as wheat straw (up to 1,675 mU g−1) or beech wood (up to 80 mU g−1). Following the ester-cleaving activity toward methyl ferulate, a hydrolase of Xylaria polymorpha was produced in solid-state culture on wheat straw and purified by different steps of anion-exchange and size-exclusion chromatography to apparent homogeneity (specific activity, 2.2 U mg−1). The peptide sequence of the purified protein deduced from the gene sequence and verified by de novo peptide sequencing shows high similarity to putative α-l-rhamnosidase sequences belonging to the glycoside hydrolase family 78 (GH78; classified under EC 3.2.1.40). The purified enzyme (98 kDa by SDS-PAGE, 103 kDa by size-exclusion chromatography; pI 3.7) converted diverse glycosides (e.g., α-l-rhamnopyranoside and α-l-arabinofuranoside) but also natural and synthetic esters (e.g., chlorogenic acid, hydroxycinnamic acid glycoside esters, veratric acid esters, or p-nitrophenyl acetate) and released free hydroxycinnamic acids (ferulic and coumaric acid) from arabinoxylan and milled wheat straw. These catalytic properties strongly suggest that X. polymorpha GH78 is a multifunctional enzyme. It is the first fungal enzyme that combines glycosyl hydrolase with esterase activities and may help this soft rot fungus to degrade lignocelluloses.
INTRODUCTION
Lignocellulose results from plant synthesis of complex cell wall polymers, which provide rigidity and mechanical stability and are thought to protect the plant from microbial attack (32, 35). The particular properties of lignocelluloses are based on the structure of their major components. In the secondary cell wall of wood, cellulose (40 to 50%) forms the fiber backbone, hemicelluloses (mainly xylan, 20 to 40%) cover and link the cellulose fibers, and lignin (20 to 35%) acts as molecular glue between the cell wall polysaccharides (35).
To get access to the fermentable polysaccharides and sugars, it is necessary to remove, at least in part, the persistent lignin polymer. There are different microbial strategies to accomplish this, but the most efficient biocatalytic systems were developed by filamentous fungi, which belong to the basidiomycetes and ascomycetes. Ecophysiologically, they can be divided into three different groups, white rot, brown rot, and soft rot fungi, all of which colonize compact wood (e.g., trunks, branches, and stumps of trees). Whereas a lot of scientific work on white and brown rot, exclusively caused by basidiomycetes, has been done over the last 20 years, soft rot caused by specialized ascomycetes has been less well studied. On the other hand, soft rot fungi (e.g., fungi of the family Xylariaceae) are found in almost all broad-leaved forests, indicating their substantial involvement in the recycling of woody lignocelluloses and hence in the carbon cycle (22, 30, 44). Wood-dwelling ascomycetes are seemingly lacking ligninolytic peroxidases, the key enzymes of lignin degradation, but possess an alternative enzyme system, consisting of a set of versatile hydrolases and laccases, which allows them to efficiently degrade the lignocellulosic complex (30, 32). Among the hydrolases, esterases may be of particular importance as they can cleave ester bonds between plant cell wall polysaccharides and the hydroxycinnamic acid units (e.g., ferulic or p-coumaric acid) of lignin, thereby releasing large lignin fragments (30). Enzymes involved in the hydrolysis of these bonds are classified as feruloyl esterase (ferulic acid esterases [FAEs]; EC 3.1.1.73) belonging to the sub-subclass of carboxylic ester hydrolases (EC 3.1.1.x) and release phenolic acids and their dimers from lignocellulosic materials. FAE acts on the side chains of cell wall polysaccharide structures (e.g., arabinofuranosyl units of arabinoxylan), cleaves the cross-linkages between xylan chains and between xylan and lignin parts, and thereby plays a role in the initial degradation of lignocelluloses (45, 46). Just a few microbial esterases related to lignocellulose degradation have been characterized, and among them are mostly those of bacteria and microscopic ascomycetes (e.g., Clostridium spp., Pseudomonas spp., Aspergillus spp., Penicillium spp., Fusarium spp., and Aureobasidium pullulans) (6, 46). Isolation and characterization of extracellular esterases from wood- and litter-decomposing basidiomycetes have been reported for Coprinopsis cinerea, Schizophyllum commune, Volvariella volvacea, and Termitomyces clypeatus (6, 20, 25, 31).
During the last few years, an increasing number of glycoside hydrolases (GH)-encoding genes were identified in fungal genome sequencing projects. The diversity of these carbohydrate-degrading enzymes has been summarized and is being continuously evaluated in the Carbohydrate-Active EnZymes (CAZy) database (http://www.cazy.org) (10). Within one sequence-based GH family, different enzymatic activities and diverse side activities can sometimes be found, which makes it difficult to deduce the actual function of a GH protein and its correct EC number. Most of the listed GH sequences refer to putative enzymes whose particular catalytic properties are unknown. To fill this gap of knowledge and to improve enzyme classification, more experimentally based data from proteins of wild-type organisms are required to reveal their enzymatic specificities and possible physiological functions.
We describe here the production, purification, as well as catalytic and molecular characterization of the first GH78 protein from a wood-dwelling xylariaceous fungus which cleaves both glycosidic and ester bonds and partly hydrolyzes native lignocellulose (milled wheat straw).
MATERIALS AND METHODS
Fungal organisms.
Fungal strains used in this study were preselected by an agar plate screening test using ethyl ferulate (EFA) as the indicator substance (6). They belong to 11 different litter- and wood-decomposing basidio- and ascomycetous species (Agrocybe aegerita DSMZ22459, Coprinellus radians DSMZ888, Mycetinis [Marasmius] alliaceus Zi06, Pleurotus ostreatus K5, Pycnoporus cinnabarinus ATCC 200478, Daldinia concentrica A20, D. vernicosa A31, Kretzschmaria deusta A29, Morchella elata A30, Xylaria hypoxylon A38, X. polymorpha A34, A35, A36) and are deposited in the fungal culture collection of the International Graduate School of Zittau.
Media and culture conditions.
For the FAE activity screening under conditions close to those in nature, solid-state cultures in 100-ml Erlenmeyer flasks were prepared (30). They contained sterilized (30 min at 121°C) beech wood shavings or chopped wheat straw (3 g each), and the shavings and wheat straw were moistened with 10 and 20 ml distilled water, respectively. Each flask was inoculated with three agar plugs (diameter, 1 cm) from a fully overgrown malt extract agar plate (1 to 2 weeks old). The flasks were incubated at 23°C for 3 weeks. In 5-day intervals, three flasks of each fungus were harvested and extracted with distilled water by shaking (4 h on a rotary shaker at 150 rpm). Aliquots (1 ml) were centrifuged and used for the measurement of FAE activity and pH; enzyme activities (mU) were calculated per gram of dry mass of wood or straw material.
Enzymatic activity.
FAE activity was determined by following the hydrolysis of methyl ferulate [MFA; 2-propenoic acid 3-(4-hydroxy-3-methoxyphenyl)-methyl ester, 1 mM] to ferulic acid at 37°C in 3-(N-morpholino)-propane sulfonic acid buffer (MOPS; 100 mM) at pH 6.0. The reaction was initiated by the addition of enzyme solution and terminated after 5 to 30 min by an equal volume of the stop solution (11.3 vol % acetic acid-methanol) (15), which led to the complete inhibition of the enzymatic reaction (proved by a control experiment).
The reaction product (ferulic acid) was analyzed by high-performance liquid chromatography (HPLC) using a reversed-phase C18 column (4 μm, 4.6 mm by 125 mm; Phenomenex Synergi Fusion-RP 80A; Aschaffenburg, Germany) and an Agilent 1200 Infinity liquid chromatograph (Waldbronn, Germany) operating under isocratic conditions (40% [vol/vol] acetonitrile in 15 mM phosphoric acid; flow rate, 1.0 ml min−1). FAE activities were expressed in international units (1 international unit = 1,000 mU), defined as the amount of enzyme that forms 1 μmol of free ferulic acid per minute. To test for possible intracellular FAE activity, fungal mycelium was disrupted with dry ice in a mortar and suspended in MOPS buffer. After centrifugation, the FAE activity in the cell crude extract was measured as described above.
Production and purification of GH78 hydrolase from X. polymorpha (XpoGH78).
Approximately 2.0 kg wheat straw was presoaked with distilled water overnight and then placed in large autoclavable plastic bags (containing ∼500 g wet straw per bag) and sterilized twice at 121°C for 30 min. For inoculation, the content of two overgrown agar plates of X. polymorpha (strain A35) was homogenized in 160 ml of a sterile NaCl solution (0.9%), and the mycelial suspension obtained was added to each bag with straw. After 8 weeks of incubation, the bags with colonized wheat straw were harvested and extracted with distilled water. The enzyme-containing straw extract was removed from the mycelium and straw particles by centrifugation (12,000 rpm) and subsequent filtration (filter GF6; Whatman, Dassel, Germany) and then concentrated and dialyzed in a tangential flow ultrafiltration system at 11°C (10-kDa cutoff; Pall-Filtron, Dreieich, Germany).
The crude enzyme preparation, which exhibited FAE activity, was purified by three steps of fast protein liquid chromatography (FPLC) using an ÄKTA system (GE Healthcare, Freiburg, Germany). The first separation step on DEAE-Sepharose was carried out at pH 4.5, and the final purification step was carried out on a Mono Q column at pH 5.0 using sodium acetate buffer (10 mM) as the mobile phase. Elution of the target protein was performed with the same buffer and an increasing sodium chloride gradient (0 to 1.5 M for DEAE-Sepharose and 0 to 1.0 M for the Mono Q column, respectively). The second purification step was performed by size-exclusion chromatography (SEC; Superdex 75) using sodium acetate buffer (50 mM, pH 6.5) containing sodium chloride (100 mM) as the eluent. In all cases, elution of protein was monitored at 280 nm. FAE-containing fractions were collected, combined, concentrated, and dialyzed against 10 mM sodium acetate buffer (pH 6.0) and stored at −80°C.
Part of the XpoGH78 fraction obtained after separation on the Mono Q column was further purified (polished) by semipreparative HPLC-SEC (using an HPLC system). For this, an Agilent 1200 system fitted with a Biosep-SEC-S-2000 column (300 by 7.8 mm Phenomenex) was used under isocratic conditions (solvent, 100 mM sodium acetate, 100 mM sodium chloride; pH 6.5; flow rate, 1 ml min−1). A total of 400 μl was repeatedly injected into the system in the form of 10- to 30-μl samples, and the major peak of XpoGH78 was collected by a scale fraction collector (Agilent type).
Enzyme characterization.
The molecular mass of purified XpoGH78 was determined by SDS-PAGE (Novex Xcell SureLock minicell; Invitrogen, Karlsruhe, Germany) and native PAGE using precast gels (NuPAGE Novex 10% Bis-Tris gel; Invitrogen). Analytical isoelectric focusing (IEF) was performed with the same electrophoresis system but using IEF precast gels (Novex IEF gel; Invitrogen) under the conditions described previously (29). After SDS electrophoresis and isoelectric focusing, the protein bands were visualized in the gels with a colloidal blue staining kit (Invitrogen). Protein concentrations were determined by using a Roti-Nanoquant protein assay kit (Roth, Karlsruhe, Germany) with bovine serum albumin as the standard (29).
To verify the enzymatic activity of polished XpoGH78 after the HPLC-SEC step, it was applied to semipreparative IEF analysis under the conditions described above. After electrophoresis, the gel was cut into two pieces and the position of the native enzyme in one half of the gel was determined by active staining using p-nitrophenyl α-l-rhamnopyranoside (p-NPRP) and sodium carbonate (1.0 M). The appearance of a distinct yellow band (p-nitrophenol) in the gel marked the position of the target protein, and from the second half of the gel, the respective position was cut out to obtain an electrophoretically pure fraction of XpoGH78. The gel plug was transferred into sodium acetate buffer (100 mM, pH 6.0) and gently homogenized; after centrifugation and concentration, this protein preparation was used to detect different enzymatic activities.
Effects of temperature and pH on enzyme activity and stability.
The pH stability of purified XpoGH78 was tested in citrate-phosphate buffer (50 mM) at pH 3.0, 7.0, and 10.0 at room temperature (20°C). The effect of temperature on enzyme stability was studied at 25°C, 40°C, and 60°C in MOPS buffer (100 mM, pH 6.0). After incubation intervals of 2, 4, 6, and 8 h, aliquots of the sample were taken and the remaining FAE activity was measured as described above (hydrolysis of MFA). The pH optimum of purified XpoGH78 was determined in MOPS buffer (100 mM) at pH values ranging from pH 4 to 10 using MFA (0.1 mM), veratric acid methyl ester (0.5 mM), and 3,4-dimethoxybenzylic acetate (0.5 mM) as the substrates. The temperature optimum was determined by following the hydrolysis of MFA at different temperatures (10 to 80°C) in MOPS buffer (100 mM) at pH 6.0. All experiments were performed in triplicate.
Kinetic parameters and substrate specificity.
XpoGH78 activity (U mg−1) toward numerous p-nitrophenyl substrates was tested, e.g., for p-nitrophenyl acetate (p-NPA [30]), p-nitrophenyl α-l-arabinofuranoside (p-NPAF [40]), p-nitrophenyl β-d-glucopyranoside and p-nitrophenyl xylopyranoside (p-NPGP and p-NPXP, respectively [40]), p-nitrophenyl cellobioside (p-NPCB [40]), as well as naringin (4′,5,7-trihydroxyflavanon-7-rhamnoglucoside [17]) at pH 5.0 in sodium acetate buffer (50 mM) at 405 nm. The Michaelis-Menten constant (Km), catalytic constant (kcat), and substrate specificity (U mg−1) of pure XpoGH78 were determined for the eponymous p-nitrophenyl α-l-rhamnopyranoside as well as for different aryl-alkyl esters (methyl p-coumarate [MpCA], MFA, EFA, methyl sinapate [MSA], methyl caffeate [MCA], ethyl/methyl-3,4-dimethoxybenzoate [veratric acid ethyl/methyl ester]), two hydroxycinnamic acid glycoside esters (methyl-6-O-sinapoyl- and methyl-6-O-feruloyl-glucopyranoside [28]), and chlorogenic acid (CGA) under similar reaction conditions as described above (100 mM MOPS buffer, pH 6.0, 37°C). After terminating the enzymatic reactions (after 5 to 30 min) by addition of the stop solution (11.3 vol % acetic acid-methanol), the products formed (e.g., hydroxycinnamic acids and 3,4-dimethoxybenzoic acid) were detected and quantified by HPLC as described above either under isocratic conditions or by using an appropriate solvent gradient. Dibenzoyl tartrate, the specific substrate of certain esterases from bacteria and some basidiomycetous yeasts (e.g., Rhodotorula mucilaginosa [49]), was tested with XpoGH78 as well. Lineweaver-Burk plots were made from the initial rates obtained at various substrate concentrations.
Enzymatic hydrolysis of esterified biopolymers.
Insoluble wheat arabinoxylan (Megazymes, Wicklow, Ireland) and milled wheat straw were used as the substrates to investigate the ability of XpoGH78 to attack ester bond-containing cell wall polymers via the release of hydroxycinnamic acids. Arabinoxylan was suspended in distilled water (1%, wt/vol) according to the manufacturer's protocol; the wheat straw was ground to a fine powder (particle size, ∼40 by 40 μm, determined microscopically) by a planetary ball mill (Fritsch, Oberstein, Germany) and then suspended in distilled water (1%, wt/vol). Enzymatic hydrolysis was initiated by addition of Mono Q column-purified XpoGH78 (0.05 μM) to the reaction mixture containing either arabinoxylan or wheat straw (final concentration, 0.5%) in MOPS buffer (100 mM, pH 6.0). Controls contained heat-inactivated enzyme (treated at 95°C for 30 min). Reaction solutions were incubated for 2, 8, 12, and 24 h at 37°C. The amount of released hydroxycinnamic acids was quantified by HPLC (isocratic elution with 10 to 15% acetonitrile; flow rate, 1 ml min−1) and expressed in mg per g wheat straw or arabinoxylan used (5 mg ml−1). In a control experiment, a wheat straw sample was saponified under mild alkaline conditions (1.0 M sodium hydroxide for 2 h) in order to determine the total amount of ester-linked hydroxycinnamic acids in the straw (7).
Protein sequencing and peptide analysis.
For peptide sequencing and N-terminal analysis, the protein was electrophoretically separated and electroblotted as described previously (29). Preparation and analysis of peptide fragments were performed by Protagen AG (Dortmund, Germany).
Tryptic peptides of the purified XpoGH78 preparation exhibiting both α-l-rhamnosidase and FAE activity were analyzed using a Proxeon (now Thermo Scientific, Waltham, MA) nano-HPLC system coupled with a nanoelectrospray ion source (PicoTip emitter; New Objective, Woburn, MA) and an LTQ Orbitrap Velos mass spectrometer (Thermo Scientific, Waltham, MA; also compare the information in the supplemental material).
Molecular work.
To identify the XpoGH78-encoding gene, mycelium of X. polymorpha (strain A35) grown on wheat straw was harvested every third day beginning at culture day 10. Fungal biomass was used to isolate total RNA (TRIzol Plus RNA purification system; Life Technologies, Carlsbad, CA), which was then reverse transcribed into cDNA (RevertAid H Minus first-strand cDNA synthesis kit; Fermentas, St. Leon-Rot, Germany) using a poly(dT)-anchor primer (see Table S1 in the supplemental material), thereby adding the anchor sequence to the 3′ end. Furthermore, by adding 1 μl primer TS-Short (10 μM) to the reaction mix, an anchor sequence was added to the 5′ end of the cDNA using a protocol described previously (33). PCR amplifications were performed in a MasterCycler EP Gradient S gradient cycler (Eppendorf, Hamburg, Germany). All primers used were obtained from biomers.net (Ulm, Germany) and applied as a 10 μM stock solution in the case of specific primers or a 100 μM stock solution in the case of degenerated primers (see Table S1 in the supplemental material). The PCR mixtures (25 μl) contained 1 μl of cDNA (0.25 μg), 10 μl PCR master mix (2.5-fold concentrated; 5Prime, Hamburg, Germany), 1 μl MgCl2 (25 mM), and 1 μl of each primer.
PCRs were started with an initial denaturation at 94°C for 2 min, followed by 40 cycles of denaturation at 94°C for 30 s, annealing for 30 s at a temperature gradient with touchdown in case of degenerated primers (15 cycles, in which the temperature gradient decreases by 1°C per cycle beginning with 52 to 72°C; 25 cycles, in which the temperature gradient decreases by 1°C per cycle in the range of 37 to 57°C) or temperatures according to the 4 + 2 rule (13) in case of specific primers, and elongation for 2 to 3 min at 70°C. Final elongation was performed over 10 min at 70°C. Positive PCR products were purified (QIAquick PCR purification kit; Qiagen, Hilden, Germany) and either directly sequenced by Eurofins MWG Operon (Ebersberg, Germany) or sequenced after cloning into Escherichia coli using a TOPO TA cloning kit (Life Technologies). BioEdit software (version 7.0.9) was used for sequences analysis. To predict specific parameters of the deduced protein sequence, the programs iPSort (4), NetNGlyc (version 1.0) and NetOGlyc (version 3.1) (21), ProtParam tool (18), and SignalP (37) were used. Phylogenetic analysis of the deduced protein sequence and reference sequences obtained from GenBank (http://www.ncbi.nlm.nih.gov/GenBank) was performed using the MEGA5 program (41).
Sequencing strategy for the XpoGH78-encoding gene.
By using the sequences of four internal de novo peptide fragments of the purified XpoGH78 protein, degenerated primers were designed (see Table S1 in the supplemental material; forward primers FP_R317, FP_R6_A, and FP_R176_B and reverse primer RP_R176_B). Furthermore, three primers (RP_R406_A, RP_R406_B, RP_R755_A) were designed on the basis of protein alignments of putative fungal α-l-rhamnosidases belonging to glycoside hydrolase (GH) family 78.
These primers were used with cDNA to amplify fragments of the XpoGH78 gene (several specific PCR products in the range of 800 to 1,200 bp in size). A 3′ rapid amplification of cDNA end (RACE) experiment was performed using the specific primer FP_SRH_002 or FP_SRH_006 and the AP3 anchor, resulting in products of approximately 1,560 and 800 bp in size, respectively, which were directly sequenced. For completion of the sequence at the cDNA level, a 5′ RACE (33) was performed using specific primer RP_SRH_002 and heel-carrier anchor primers. The 1:100-diluted PCR product was then used in a nested PCR with heel-specific primer and degenerated primer RP_R176_B. The resulting product with a size of approximately 600 bp was excised from the gel, purified, and cloned. Finally, after completion of the cDNA sequence, the specific primers Xpo-FAE-START-For and Xpo-FAE-STOP-Rev were used in PCR to amplify the complete coding DNA sequence (CDS) of the XpoGH78-encoding gene from genomic DNA (2,921 bp). The PCR product was purified and cloned, and finally, five independent clones were fully sequenced.
Nucleotide sequence accession numbers.
The nucleotide sequences of XpoGH78 identified have been deposited in the GenBank nucleotide database under the following accession numbers: JN815084 (X. polymorpha gh78-1 gene) and JN815083 (X. polymorpha gh78-1 mRNA).
RESULTS
FAE activity of ascomycetous and basidiomycetous fungi during solid-state cultivation.
An agar plate screening using EFA as the indicator substrate showed that three out of six tested ascomycetous species (K. deusta, three strains of X. polymorpha, and X. hypoxylon, all members of the Xylariaceae) and four of five basidiomycetes were able to hydrolyze the substrate, which was proved by the formation of transparent zones around the fungal mycelium (see Table S2 in the supplemental material). This result was confirmed by MFA-hydrolyzing activities (later attributed to an FAE activity) detected during growth on wheat straw (13 to 1,675 mU g−1) and beech wood (40 to 80 mU g−1). It is noteworthy that the three ascomycetous species causing soft rot type II produced considerably higher levels of esterase than the tested basidiomycetes (see Table S2 in the supplemental material). X. polymorpha strain A35, which showed the highest FAE activity, was selected for more detailed investigations concerning the nature of the ester-cleaving enzyme.
Five days after inoculation of solid-state cultures with X. polymorpha, FAE activity (100 and 114 mU g−1 in straw and beech wood, respectively) was already detectable in both lignocellulosic substrates; at the same time, the pH slightly decreased from 7.0 to 6.5 in wheat straw and from 6.0 to 5.3 in beech wood. Afterwards, in the wheat straw cultures, the FAE level steadily increased up to 1,675 mU g−1 on day 25 of cultivation (Fig. 1A), while in beech wood, the highest activity (164 mU g−1) was observed on day 10, but this then decreased to below 100 mU g−1 on day 25. Intracellular esterase activities were not detectable in crude cell crude extracts of X. polymorpha.
Fig 1.

(A) Time course of FAE activity in solid-state culture using wheat straw (circles) and beech wood (squares) as growth substrates. Data points represent mean values of three culture flasks with a standard deviation of <5%. (B) FPLC elution profile of the 3rd purification step of XpoGH78 performed on a Mono Q column: absorbance at 280 nm (solid line), FAE activity (black circles), and NaCl gradient (dashed line). (C) Elution profile of XpoGH78 before (dashed line) and after (solid line) semipreparative HPLC-SEC; the inset shows the UV-visible spectrum of the protein peak exhibiting rhamnosidase and FAE activities. (D) SDS-PAGE (left), native PAGE (middle), and native isoelectric focusing (right) of XpoGH78 (lane 2, 4, and 5) after HPLC-SEC; lanes 1, 3, and 6, protein markers.
On the basis of these findings, wheat straw was chosen as the growth substrate to produce larger amounts of the ester-hydrolyzing enzyme for subsequent purification studies (on the basis of molecular findings, the enzyme was later designated XpoGH78, for Xylaria polymorpha glycoside hydrolase 78; compare Fig. 4 and the information in the supplemental material).
Fig 4.

Phylogenetic analysis of eukaryotic and prokaryotic GH78 α-l-rhamnosidases defining two major groups (I and II). The analysis was performed using the neighbor-joining method with Poisson-corrected protein distances and 500 bootstrap replicates. A total of 371 protein sequences with a total of 3,939 positions were included. The protein alignment comprises two conserved stretches involved in substrate recognition of fungal and bacterial representatives of both main groups as well as the novel characterized X. polymorpha enzyme (XpoGH78). Arrows indicate structural and functional important positions deduced from the three-dimensional structure of Bacillus sp. strain G1 (PDB entry 2OKX; GenBank accession number BAB62315 [11]). A ter, A. terreus; B sp, Bacillus sp.; L aci, Lactobacillus acidophilus; L pla, Lactobacillus plantarum; T bac, Thermomicrobia bacterium; A acu, Aspergillus aculeatus; A kaw, A. kawachii; A nid, A. nidulans.
Purification and physical characterization of XpoGH78.
As shown in Table 1, XpoGH78 was purified 43-fold starting from the wheat straw extract and resulting in specific activities of 2.6 and 8.0 U mg−1 for MFA and p-NPRP, respectively, after a three-step purification procedure with a final step performed on a Mono Q column (Fig. 1B). A portion of the XpoGH78 protein obtained that way could be further polished by HPLC-SEC, whereby some protein impurities (∼0.2 mg) were removed, leading to a homogeneous protein preparation that gave a single protein peak with an apparent molecular mass of 103 kDa in the SEC elution profile (Fig. 1C). The protein purity was confirmed by comprehensive mass spectral analyses and a label-free protein quantitation method by averaging the mass spectrometry signal response of the three most intense peptides, since this directly correlates with the input amount of the protein (19).
Table 1.
Purification of GH78 protein from X. polymorpha (measured by demethylation of methyl ferulate) produced during solid-state fermentation of wheat strawa
| Purification step | Total activity (U) | Total protein (mg) | Sp act (U/mg) | Yield (%) | Purification (fold) |
|---|---|---|---|---|---|
| Wheat straw extraction | 57.4 | 910.8 | 0.06 | 100 | 1.0 |
| Ultrafiltration | 54.5 | 620.5 | 0.09 | 94.9 | 1.5 |
| DEAE-Sepharose separation | 43.9 | 68.2 | 0.64 | 76.5 | 10.7 |
| Superdex Sepharose SEC | 20.6 | 11.5 | 1.79 | 35.9 | 29.8 |
| Mono Q column separation | 4.9 | 1.9 | 2.58 | 8.5 | 43.0 |
| Biosep-SEC-S-2000 column separation | 3.8 | 1.7 | 2.24 | 6.6 | 37.3 |
Cultures were harvested after 25 days and extracted with water.
Along with major bands at 93 kDa in the native polyacrylamide gel and at 98 kDa in the SDS-polyacrylamide gel, two additional slight bands appeared in the latter gel (at 39 and 62 kDa). These probably corresponded to XpoGH78 fragments or subunits (of a heterodimer) which may have originated from the rather drastic conditions of denaturing electrophoresis (Fig. 1D). Interestingly, the sum of both bands gives 101 kDa, which is in the range of the molecular size determined under native (SEC) and denaturing (SDS-PAGE) conditions. This observation was proved by the peptide analyses of the 39- and 62-kDa fragments that clearly show their affiliation to the XpoGH78 sequence. In fact, peptide mapping data turned out that the lower-mass fragment obviously belongs to the first one-third of the protein and the 62-kDa fragment seems to match the remaining two-thirds of the identified amino acid sequence (see Fig. S2 in the supplemental material).
The specific activities of the polished XpoGH78 preparation slightly decreased to 2.24 and 6.70 U mg−1 for MFA and p-NPRP, respectively, probably due to the harsh conditions during the HPLC-SEC procedure.
XpoGH78 showed an acidic pI of 3.7 and appeared as one distinct band in the IEF gel both after colloidal blue (Fig. 1D) and activity (data not shown) staining. The isolation of the electrophoretically homogeneous XpoGH78 band from the IEF gel resulted in a protein preparation that showed both rhamnosidase and esterase activities. This finding strongly indicates that XpoGH78 is a fungal hydrolase with multiple enzymatic activities.
The temperature and pH optima of XpoGH78 were found to be 45°C and pH 6 to 8, respectively (see Fig. S1B in the supplemental material). XpoGH78 was found to be relatively stable at neutral and alkaline pH but lost about 90% of its activity within 2 h at pH 3 (see Fig. S1D in the supplemental material). A temperature of 60°C caused an activity loss of over 90% within 2 h, and at 40°C, 50% of the initial activity was still detectable after 8 h of incubation (see Fig. S1C in the supplemental material).
Specific activities and kinetic constants.
Purified XpoGH78 hydrolyzed p-NPRP with the highest specific activity (calculated as Vmax/protein amount) of 24.4 U mg−1 (Table 2), which is consistent with its sequence signature as GH78. The enzyme also converted the well-known rhamnosidase substrate naringin (1.2 U mg−1) into the corresponding α-l-rhamnose and monoglycosylated prunin (naringenin 7-glucoside), which was not further hydrolyzed into the aglycon naringenin (data not shown). Thus, the low specific β-glucosidase activity observed for p-NPGP (0.1 U mg−1; Table 2) could not be confirmed for prunin. Other side activities were found for p-NPAF (1.5 U mg−1) and p-NPCB (0.07 U mg−1), while p-NPXP was not hydrolyzed by XpoGH78.
Table 2.
Catalytic constants and substrate specificities of purified X. polymorpha GH78
| Substrate | Sp act (U/mg) | Km (μM) | kcat (s−1) | kcat/Km (s−1 M−1) |
|---|---|---|---|---|
| p-Nitrophenyl α-l-rhamnopyranoside | 24.4a | 971 | 32.9 | 3.4 × 104 |
| p-Nitrophenyl α-l-arabinofuranoside | 1.5 | NDb | ND | ND |
| Naringine | 1.2 | ND | ND | ND |
| p-Nitrophenyl β-d-glucopyranoside | 0.1 | ND | ND | ND |
| p-Nitrophenyl β-d-cellobioside | 0.07 | ND | ND | ND |
| Methyl ferulate | 7.3a | 80 | 9.3 | 1.2 × 105 |
| Methyl p-coumarate | 4.1a | 21 | 6.2 | 2.9 × 105 |
| Methyl sinapate | 2.8a | 103 | 3.7 | 3.6 × 104 |
| Ethyl ferulate | 2.1a | 94 | 2.6 | 2.7 × 104 |
| Methyl caffeate | 0.92a | 151 | 1.4 | 9.3 × 103 |
| Veratric acid ethyl ester | 0.01a | 78 | 0.1 | 1.3 × 103 |
| Veratric acid methyl ester | 0.01a | 62 | 0.1 | 1.6 × 103 |
| Methyl-6-O-sinapoyl glucopyranoside | 1.2 | ND | ND | ND |
| Methyl-6-O-feruloyl glucopyranoside | 2.2 | ND | ND | ND |
| Chlorogenic acid | 0.3 | ND | ND | ND |
| p-Nitrophenyl acetate | 0.09 | ND | ND | ND |
| Dibenzoyl tartrate | 0 | ND | ND | ND |
Calculated as Vmax/protein amount.
ND, not determined.
The broadness of the substrate spectrum of XpoGH78 became further evident by the hydrolysis of a number of aryl-alkyl esters (e.g., MpCA), hydroxycinnamic acid glycoside esters (e.g., methyl-6-O-feruloyl-glucopyranoside), and quinic acid esters (e.g., CGA) as well as acetylated substrates (p-NPA) (Table 2). Michaelis-Menten constants (Km), turnover numbers (kcat) and catalytic efficiencies (kcat/Km) were determined for typical FAE substrates, and the lowest Km of 21 μM (i.e., the highest affinity) and the highest kcat/Km (2.9 × 105 s−1 M−1) were observed for MpCA followed by MFA (1.2 × 105 s−1 M−1); the catalytic efficiencies for veratric acid esters were about 2 orders of magnitude lower. The picture is similar when comparing the specific activities of XpoGH78: the highest values of 7.3 and 4.1 U mg−1 were found for MFA and MpCA, respectively, while the activities for all other ester substrates were lower. Relatively high activities were also observed for model compounds representing glycoside ester structures within the network of cell wall polysaccharides (28). Thus, XpoGH78 exhibited specific activities of 2.2 and 1.2 U mg−1 for a feruloyl glucoside ester and a sinapoyl glucoside ester, respectively. Minor activities were detected for CGA (0.3 U mg−1), an ester of cyclitol quinic acid, and polyphenolic caffeic acid (always present in fruits, vegetables, or cereals) and for the artificial ester p-NPA (∼0.1 U mg−1). On the other hand, dibenzoyl tartrate was not a substrate of XpoGH78. All these findings indicate that the Xylaria GH78 protein is, from the catalytic point of view, an α-l-rhamnosidase (EC 3.2.1.40) with strong esterase and other moderate side activities.
Hydrolysis of wheat arabinoxylan and wheat straw.
XpoGH78 partly hydrolyzed native cell wall polymers, which was proven by the release of hydroxycinnamic acids (p-coumaric and ferulic acid) from insoluble arabinoxylan or milled wheat straw (Fig. 2A and B). The treatment of arabinoxylan with XpoGH78 resulted in the release of 0.37 mg g−1 ferulic acid within 2 h, which further increased to 0.45 mg g−1and finally 0.54 mg g−1 over another 6 and 22 h of incubation (Fig. 3). Remarkably, XpoGH78 also acted on native wheat straw. The major metabolites, ferulic and p-coumaric acid, were already detectable after 2 h of incubation, and their concentrations of 0.24 mg g−1 and 0.14 mg g−1, respectively, corresponded to 14.5% and 10.1% of the total amount of these acids alkaline extractable from wheat straw.
Fig 2.

Partial hydrolysis of arabinoxylan (A) and milled wheat straw (B) by XpoGH78. HPLC elution profiles were obtained under slightly different, isocratic separation conditions (15% and 10% acetonitrile) and monitored at 323 nm after 12 h of incubation of the suspended polymeric materials with active XpoGH78 (solid lines) or boiled enzyme (dashed lines). Peak 1, ferulic acid; peak 2, p-coumaric acid. The insets show the UV-visible spectra of hydroxycinnamic acids.
Fig 3.
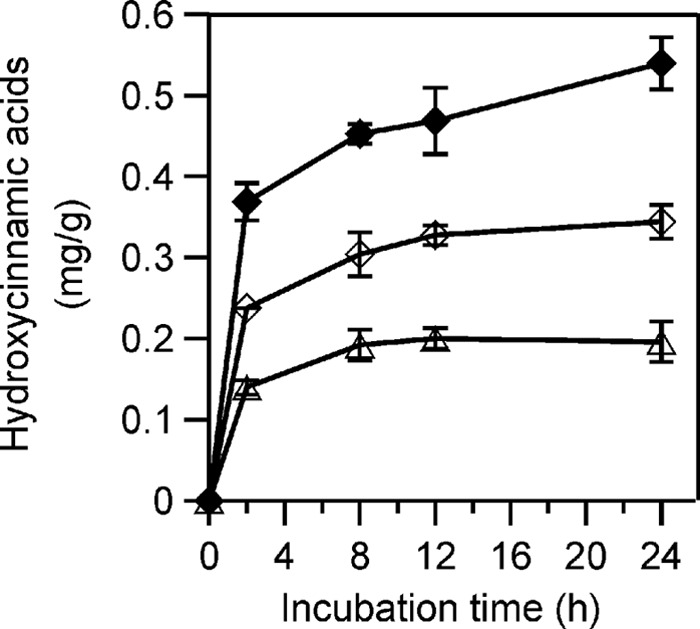
Release of hydroxycinnamic acids from xylan and straw by Mono Q column-purified XpoGH78: ferulic acid from suspended arabinoxylan (closed diamonds), ferulic acid (open diamonds), and p-coumaric acid (open triangles) from milled wheat straw. The amount of released acids was calculated in mg per g of polymeric substrate.
Gene sequence and predicted features of XpoGH78.
The gh78-1 gene (2,921 bp) contains four introns in the middle of the sequence and encodes a protein of 866 amino acids with a calculated molecular mass of 93.4 kDa and a theoretical pI of 4.69. Peptide fragments of XpoGH78 obtained by peptide mapping matched perfectly with the deduced amino acid sequence according to the identified gh78-1 gene and cover 68.82% of the overall protein. Furthermore, several conserved amino acids (e.g., catalytic residues), GH78-specific domains (e.g., pfam08531), calcium binding positions, as well as six potential N-glycosylation sites were found in the translated protein sequence (see Fig. S2 in the supplemental material). Surprisingly, no signal peptide sequence could be predicted with the programs SignalP and iPSort.
DISCUSSION
Three ascomycetous soft rot fungi belonging to the family Xylariaceae were found to produce high levels of a methyl ferulate-hydrolyzing activity during growth on lignocellulosic materials. One responsible enzyme with strong FAE activity was purified from X. polymorpha. Since the amino acid sequence of the isolated hydrolase indicates its phylogenetic affiliation to the glycoside hydrolases (EC 3.2.1.40; GH family 78, group I of the α-l-rhamnosidases), it was designated XpoGH78. The isolated enzyme exhibits a broad substrate specificity and was found to hydrolyze both glycosidic and ester bonds.
The highest level of ester-cleaving activity of X. polymorpha (1,675 mU g−1, ∼250 U liter−1 in relation to the water content of straw [30]) was reached when the fungus was grown on wheat straw in solid-state culture. Numerous microbial FAEs were found to be produced in solid-state culture when complex carbon sources rich in esterified hydroxycinnamic acids, such as agricultural waste materials (e.g., wheat or maize bran, sugar beet pulp), were used (6, 46). Generally, the maximum FAE levels reported vary considerably (2 to 180 U liter−1), with the variation depending on the particular microorganism but also on the ester substrates used for enzyme detection (e.g., methyl ester, p-NPA, or 4-nitrophenyl-5-O-feruloyl-α-l-arabinofuranoside) (5, 9).
Interestingly, the amino acid sequence of the isolated ester-cleaving protein does not show any similarity to known fungal esterase sequences deposited in the GenBank or CAZy database. Using BLAST searches, strong similarities of the identified peptide sequence with fungal and bacterial α-l-rhamnosidases of GH family 78 (EC 3.2.1.40) were found, which led to the designation XpoGH78 for the enzyme. All of the approximately 400 homologous protein sequences of this family contain highly conserved regions (Fig. 4) and amino acid residues crucial for substrate catalysis of both glycosides (e.g., Asp474 and Glu467) and esters (e.g., Asp741, Asp474, Glu467, Ser457, His476, and Cys462). Further, the motif AASVA, which corresponds to Sm-X-Nu-X-Sm (where X is any amino acid and Sm is a small amino acid), a characteristic sequence of the catalytic triad, was found at position 631.
A phylogenetic analysis revealed a division of the GH78 family into two large groups, I and II (Fig. 4; see Fig. S3 and S4 in the supplemental material). XpoGH78 exhibits the highest sequence similarity to group I, comprising 47 ascomycetous, 1 oomycetous, as well as 200 bacterial sequences. To our best knowledge, the vast majority of these sequences refer to putative proteins with so far unknown catalytic properties, and all fungal representatives are lacking N-terminal signal peptides. The only exception is a characterized bacterial α-l-rhamnosidase from a Bacillus sp. with 38% identity and 56% similarity to XpoGH78 (BAB62314 [11]). Within group I, the XpoGH78 sequence shows the highest similarities to putative sequences of phylogenetically related ascomycetes (e.g., Aspergillus terreus, 58% identity/72% similarity; Magnaporthe oryzae, 54% identity/69% similarity). In contrast, the XpoGH78 sequence did not match well (<20% similarity) with members of group II of the GH78 family, which includes 77 fungal and 42 bacterial sequences. Like group I, most members of group II are putative protein sequences (only a few characterized fungal proteins have been reported [47]), but contrary to group I, many of the fungal sequences contain predicted N-terminal signal peptides (see Fig. S4 in the supplemental material).
XpoGH78 is a glycosylated protein (10%; data not shown) with molecular masses of 103 and 93 kDa determined under native conditions (SEC and native PAGE, respectively) and 98 kDa estimated by denaturing SDS-PAGE (Fig. 1D). The native protein may be a heterodimer that consists of two subunits (39 and 62 kDa), or it is rather labile and may partially split under denaturing conditions. Similar observations were made for different recombinant bacterial rhamnosidases which can occur as monomers and dimers depending on native or denaturing conditions (3, 8, 50).
All in all, the physicochemical and catalytic properties of XpoGH78 are comparable to those of the characterized group II α-l-rhamnosidases of Aspergillus kawachii and A. terreus (90 kDa [47]), and it efficiently hydrolyzes p-NPRP but also naringin, the canonical substrates of GH family 78 (11). The specific activity of XpoGH78 for p-NPRP (24.4 U mg−1) is within the activity range reported for fungal and bacterial α-l-rhamnosidases, e.g., of A. terreus (84 U mg−1 [17]), Aspergillus nidulans (9.3 U mg−1 [47]), and Clostridium stercorarium (82 U mg−1 [50]).
Interestingly, the enzyme was found to act on carboxylate ester bonds, a catalytic property that is associated with hydrolases of the carboxyl esterase (CE) family (EC 3.1.1.x). Thus, XpoGH78 hydrolyzed diverse aryl esters, and it turned out that an increasing number of substituents on the aromatic ring of the substrate affected its affinity and the catalytic efficiency (from MpCA to MSA). Thus, the additional hydroxyl group at the C-3 position of MCA led to a decrease in the catalytic efficiency by a factor of 30 compared to that for MpCA. An even more drastic activity loss was observed for an FAE from Aspergillus niger, which hydrolyzed MpCA but not MCA (15). The activity toward veratric acid esters increased with the decreasing number of C atoms of the alcohol moiety (i.e., from ethyl to methyl), probably due to steric hindrance.
The production of bifunctional and multifunctional glycosidases has been reported for several filamentous fungi, mainly ascomycetes (e.g., arabinofuranosidase of Penicillium chrysogenum and Aspergillus awamori; arabinofuranosidase/β-xylosidase of Fusarium graminearum; arabinofuranosidase/xylobiohydrolase of Penicillium purpurogenum [40]) and some bacteria (e.g., bifunctional xylanase-deacetylase [24] and bifunctional xylanase-ferulic acid esterase [14, 34]). Ester hydrolysis by β-glucosidases (EC 3.2.1.21) has been reported for a few bacterial proteins (e.g., from Flavobacterium johnsonae and Clavibacter michiganense), which were found to cleave β-glycosyl ester linkages present in phytohormone conjugates (23, 38). Overlapping substrate specificities were also observed for FAE-B of A. niger, an enzyme that converted different esters, including feruloylated oligosaccharides, wheat arabinoxylan, and alky-aryl esters; interestingly, this enzyme showed significant sequence similarity to an Aspergillus tannase (EC 3.1.1.20) but without possessing the respective enzymatic activity (6). A chlorogenic acid hydrolase (EC 3.1.1.42) with broad substrate specificity was described for another A. niger strain and suggested to be a new esterase/protein type, since it did not show any homology to known cinnamoyl esterases (2). Eventually, Koseki et al. (2010) described an FAE from A. kawachii that contains the carbohydrate-binding module from GH family 54 (α-l-arabinofuranosidase, EC 3.2.1.55) (27).
The hydrolysis of complex esters indicates that XpoGH78 may be part of the lignocellulolytic system of xylariaceous soft rot fungi. This assumption was supported by the hydrolysis of hydroxycinnamic acid glycoside esters naturally occurring in different plant tissues (28) or of quinic acid esters present in bark but also by the release of coumaric and/or ferulic acids (the main ester-linked hydroxycinnamic acids in graminaceous plants [6]) from polymeric wheat arabinoxylan and milled wheat straw. Most degradation studies demonstrating the release of hydroxycinnamic acids by fungal esterases were carried out either in the presence of auxiliary enzymes (e.g., xylanases and arabinofuranosidases [12, 26, 48]) or after pretreatment of the complex lignocellulosic substrate, e.g., by steam explosion (6, 7).
As indicated by an internal de novo peptide located just one amino acid downstream of the translational start methionine as well as by the SignalP and iPSort prediction, XpoGH78 does not contain an N-terminal signal peptide that can be employed for standard protein translocation and secretion. Such findings regarding obviously secreted enzymes without a signal peptide are rare but have been reported for a number of extracellular bacterial proteins (e.g., the lipase of Serratia marcescens [1] and the hemolysin of Escherichia coli [16]) and fungal enzymes, such as the β-xylosidase of the phytopathogenic ascomycete Cochliobolus carbonum (43). XpoGH78 is a glycosylated (10% carbohydrates; data not shown) as well as a pH- and thermostable protein that is not detectable intracellularly, as determined with cell lysates (data not shown). Due to these facts, we assume that XpoGH78 may be secreted either by the classic secretory pathway through the endoplasmic reticulum (ER)-Golgi pathway (36, 43) or by an untypical protein-releasing mechanism independent of the ER-Golgi pathway. Similar findings have been proposed for an increasing number of proteins lacking N-terminal signal sequences (39). On the other hand, this unusual structural property of XpoGH78 fits well into the picture of α-l-rhamnosidase group I, all putative fungal members of which and the majority of the bacterial members of which are lacking N-terminal signal peptides.
In spite of the increasing number of glycoside hydrolase-encoding genes which have been identified in fungal genomes over the last few years, the catalytic activities and physical properties of the corresponding proteins are often unknown. Here, we followed the classic approach starting from esterase activities detected in lignocellulosic materials and ended up with a glycoside hydrolase 78 protein (according to its sequence) whose esterase activity would otherwise have been overlooked. Thus, XpoGH78 is the first fungal protein of GH family 78 that exhibits aryl-alkyl esterase activity on numerous natural and synthetic esters and enables the soft rot fungus to partially hydrolyze the lignocellulosic complex. Such a catalytic versatility combined in one protein with activities toward both glycosides and esters may be advantageous for the efficient degradation of the plant cell wall complex that contains both diverse sugar residues and esterified structures (42). On the basis of these results, it will be worthwhile to test other members of the family GH78 (as well as, beyond those, other glucosidases) for possible ester-cleaving activities, and vice versa, possible glucoside-hydrolyzing activities of known esterases (in particular, of FAEs) might be checked as well.
Supplementary Material
ACKNOWLEDGMENTS
Financial support in the form of a doctorate scholarship by the German Academic Exchange Service (DAAD; A/08/90704), the DBU project Fungal Secretoms (13211-32), the integrated EU project BIORENEW (NMP2-CT-2006-026456), the BMBF-founded DLR project (609220), and the administration of the International Graduate School Zittau is gratefully acknowledged.
Furthermore, we thank the colleagues of our lab for useful comments and discussions as well as Ulrike Schneider and Monika Brandt for their technical assistance.
Footnotes
Published ahead of print 27 April 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Akatsuka H, et al. 1994. The lipA gene of Serratia marcescens which encodes an extracellular lipase having no N-terminal signal peptide. J. Bacteriol. 176:1949–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asther M, et al. 2005. Purification and characterization of a chlorogenic acid hydrolase from Aspergillus niger catalysing the hydrolysis of chlorogenic acid. J. Biotechnol. 115:47–56 [DOI] [PubMed] [Google Scholar]
- 3.Avila M, et al. 2009. Physiological and biochemical characterization of the two alpha-l-rhamnosidases of Lactobacillus plantarum NCC245. Microbiology 155:2739–2749 [DOI] [PubMed] [Google Scholar]
- 4.Bannai H, Tamada Y, Maruyama O, Nakai K, Miyano S. 2002. Extensive feature detection of N-terminal protein sorting signals. Bioinformatics 18:298–305 [DOI] [PubMed] [Google Scholar]
- 5.Bartolome B, et al. 2003. Growth and release of hydroxycinnamic acids from brewer's spent grain by Streptomyces avermitilis CECT 3339. Enzyme Microb. Technol. 32:140–144 [Google Scholar]
- 6.Benoit I, Danchin EG, Bleichrodt RJ, de Vries RP. 2008. Biotechnological applications and potential of fungal feruloyl esterases based on prevalence, classification and biochemical diversity. Biotechnol. Lett. 30:387–396 [DOI] [PubMed] [Google Scholar]
- 7.Benoit I, et al. 2006. Feruloyl esterases as a tool for the release of phenolic compounds from agro-industrial by-products. Carbohydr. Res. 341:1820–1827 [DOI] [PubMed] [Google Scholar]
- 8.Birgisson H, et al. 2004. Two new thermostable α-l-arabinofuranosidases from a novel thermophilic bacterium. Enzyme Microb. Technol. 34:561–571 [Google Scholar]
- 9.Christov LP, Prior BA. 1993. Esterases of xylan-degrading microorganisms: production, properties, and significance. Enzyme Microb. Technol. 15:460–475 [DOI] [PubMed] [Google Scholar]
- 10.Coutinho PM, Henrissat B. 1999. Carbohydrate-active enzymes: an integrated database approach, p 3–12. In Gilbert HJ, Davies G, Henrissat B, Svensson B. (ed), Recent advances in carbohydrate bioengineering. The Royal Society of Chemistry, Cambridge, United Kingdom [Google Scholar]
- 11.Cui Z, Maruyama Y, Mikami B, Hashimoto W, Murata K. 2007. Crystal structure of glycoside hydrolase family 78 alpha-l-rhamnosidase from Bacillus sp. GL1. J. Mol. Biol. 374:384–398 [DOI] [PubMed] [Google Scholar]
- 12.de Vries RP, Kester HCM, Poulsen CH, Benen JAE, Visser J. 2000. Synergy between enzymes from Aspergillus involved in the degradation of plant cell wall polysaccharides. Carbohydr. Res. 327:401–410 [DOI] [PubMed] [Google Scholar]
- 13.Dieffenbach CW, Lowe TM, Dveksler GS. 1993. General concepts for PCR primer design. PCR Methods Applic. 3:30–37 [DOI] [PubMed] [Google Scholar]
- 14.Dodd D, et al. 2009. Biochemical analysis of a β-d-xylosidase and a bifunctional xylanase-ferulic acid esterase from a xylanolytic gene cluster in Prevotella ruminicola 23. J. Bacteriol. 191:3328–3338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faulds CB, Williamson G. 1994. Purification and characterisation of a ferulic acid esterase (FAE-III) from Aspergillus niger. Specificity for the phenolic moiety and binding to microcrystalline cellulose. Microbiology 140:779–787 [Google Scholar]
- 16.Felmlee T, Pellet S, Welch RA. 1985. Nucleotide sequence of an Escherichia coli chromosomal hemolysin. J. Bacteriol. 163:94–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gallego MV, Pinaga F, Ramon D, Valles S. 2001. Purification and characterization of an α-l-rhamnosidase from Aspergillus terreus of interest in winemaking. J. Food Sci. 66:204–209 [Google Scholar]
- 18.Gasteiger E, et al. 2005. Protein identification and analysis tools on the ExPASy server, p 571–607. In Walker JM. (ed), The proteomics protocols handbook. Humana Press, Totowa, NJ [Google Scholar]
- 19.Grossmann J, et al. 2010. Implementation and evaluation of relative and absolute quantification in shotgun proteomics with label-free methods. J. Proteomics 73:1740–1746 [DOI] [PubMed] [Google Scholar]
- 20.Hashimoto K, Kaneko S, Yoshida M. 2010. Extracellular carbohydrate esterase from the basidiomycete Coprinopsis cinerea released ferulic and acetic acids from xylan. Biosci. Biotechnol. Biochem. 74:1722–1724 [DOI] [PubMed] [Google Scholar]
- 21.Julenius K, Molgaard A, Gupta R, Brunak S. 2005. Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology 15:153–164 [DOI] [PubMed] [Google Scholar]
- 22.Kendrick B. 2000. The fifth kingdom, 3rd ed Focus Publishing, Newbury, MA [Google Scholar]
- 23.Ketudat Cairns JR, Esen A. 2010. β-Glucosidases. Cell. Mol. Life Sci. 67:3389–3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khandeparker R, Numan MT. 2008. Bifunctional xylanases and their potential use in biotechnology. J. Ind. Microbiol. Biotechnol. 35:635–644 [DOI] [PubMed] [Google Scholar]
- 25.Kontkanen H, et al. 2009. Novel Coprinopsis cinerea polyesterase that hydrolyzes cutin and suberin. Appl. Environ. Microbiol. 75:2148–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koseki T, Hori A, Seki S, Murayama T, Shiono Y. 2009. Characterization of two distinct feruloyl esterases, AoFaeB and AoFaeC, from Aspergillus oryzae. Appl. Microbiol. Biotechnol. 83:689–696 [DOI] [PubMed] [Google Scholar]
- 27.Koseki T, et al. 2010. Characterization of a chimeric enzyme comprising feruloyl esterase and family 42 carbohydrate-binding module. Appl. Microbiol. Biotechnol. 86:155–161 [DOI] [PubMed] [Google Scholar]
- 28.Kylli P, et al. 2008. Antioxidant potential of hydroxycinnamic acid glycoside esters. J. Agric. Food Chem. 56:4797–4805 [DOI] [PubMed] [Google Scholar]
- 29.Liers C, Ullrich R, Pecyna M, Schlosser D, Hofrichter M. 2007. Production, purification and partial enzymatic and molecular characterization of a laccase from the wood-rotting ascomycete Xylaria polymorpha. Enzyme Microb. Technol. 41:785–793 [Google Scholar]
- 30.Liers C, Ullrich R, Steffen KT, Hatakka A, Hofrichter M. 2006. Mineralization of 14C-labelled synthetic lignin and extracellular enzyme activities of the wood-colonizing ascomycetes Xylaria hypoxylon and Xylaria polymorpha. Appl. Microbiol. Biotechnol. 69:573–579 [DOI] [PubMed] [Google Scholar]
- 31.Liu X, Ding S. 2009. Molecular characterization of a new acetyl xylan esterase (AXEII) from edible straw mushroom Volvariella volvacea with both de-O-acetylation and de-N-acetylation activity. FEMS Microbiol. Lett. 295:50–56 [DOI] [PubMed] [Google Scholar]
- 32.Martínez AT, et al. 2005. Biodegradation of lignocellulosics: microbial, chemical, and enzymatic aspects of the fungal attack of lignin. Int. Microbiol. 8:195–204 [PubMed] [Google Scholar]
- 33.Matz M, et al. 1999. Amplification of cDNA ends based on template-switching effect and step-out PCR. Nucleic Acids Res. 27:1558–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montanier C, et al. 2009. The active site of a carbohydrate esterase displays divergent catalytic and noncatalytic binding functions. PLoS Biol. 7:e71 doi:10.1371/journal.pbio.1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Monties B, Fukushima Y. 2001. Occurrence, function and biosynthesis of lignins, p 1–64. In Hofrichter M, Steinbüchel A. (ed), Biopolymers, vol 1 Wiley-VCH, Weinheim, Germany [Google Scholar]
- 36.Muesch A, et al. 1990. A novel pathway for secretory proteins? Trends Biochem. Sci. 15:86–88 [DOI] [PubMed] [Google Scholar]
- 37.Nielsen H, Engelbrecht J, Brunak S, von Heijne G. 1997. A neural network method for identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Int. J. Neural Syst. 8:581–599 [DOI] [PubMed] [Google Scholar]
- 38.Okamoto K, Nakano H, Yatake T, Kiso T, Kitahata S. 2000. Purification and some properties of a beta-glucosidase from Flavobacterium johnsonae. Biosci. Biotechnol. Biochem. 64:333–340 [DOI] [PubMed] [Google Scholar]
- 39.Prudovsky I, et al. 2003. The non-classical export routes: FGF1 and IL-1α point the way. J. Cell Sci. 116:4871–4881 [DOI] [PubMed] [Google Scholar]
- 40.Ravanal MC, Callegari E, Eyzaguirre J. 2010. Novel bifunctional α-l-arabinofuranosidase/xylobiohydrolase (ABF3) from Penicillium purpurogenum. Appl. Environ. Microbiol. 76:5247–5253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596–1599 [DOI] [PubMed] [Google Scholar]
- 42.van den Brink J, De Vries RP. 2011. Fungal enzyme sets for plant polysaccharide degradation. Appl. Microbiol. Biotechnol. 91:1477–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wegener S, Ransom RF, Walton JD. 1999. A unique eukaryotic beta-xylosidase gene from the phytopathogenic fungus Cochliobolus carbonum. Microbiology 145:1089–1095 [DOI] [PubMed] [Google Scholar]
- 44.Whalley AJS. 1996. The xylariaceous way of life. Mycol. Res. 100:897–922 [Google Scholar]
- 45.Williamson G, Kroon PA, Faulds CB. 1998. Hairy plant polysaccharide: a close shave with microbial esterases. Microbiology 144:2011–2023 [DOI] [PubMed] [Google Scholar]
- 46.Wong DW. 2006. Feruloyl esterase: a key enzyme in biomass degradation. Appl. Biochem. Biotechnol. 133:87–112 [DOI] [PubMed] [Google Scholar]
- 47.Yadav V, Yadav PK, Yadav S, Yadav KDS. 2010. α-l-Rhamnosidase: a review. Process Biochem. 45:1226–1235. [Google Scholar]
- 48.Yu P, Maenz DD, McKinnon JJ, Racz VJ, Christensen DA. 2002. Release of ferulic acid from oat hulls by Aspergillus ferulic acid esterase and Trichoderma xylanase. J. Agric. Food Chem. 50:1625–1630 [DOI] [PubMed] [Google Scholar]
- 49.Zimmer C, Platz T, Cadez N, Giffhorn F, Kohring GW. 2006. A cold active (2R,3R)-(−)-di-O-benzoyl-tartrate hydrolyzing esterase from Rhodotorula mucilaginosa. Appl. Microbiol. Biotechnol. 73:132–140 [DOI] [PubMed] [Google Scholar]
- 50.Zverlov VV, et al. 2000. The thermostable alpha-l-rhamnosidase RamA of Clostridium stercorarium: biochemical characterization and primary structure of a bacterial alpha-l-rhamnoside hydrolase, a new type of inverting glycoside hydrolase. Mol. Microbiol. 35:173–179 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.