

Abstract
The secondary metabolite mediating the GacS-dependent growth-inhibitory effect exerted by the rice rhizosphere isolate Pseudomonas putida RW10S2 on phytopathogenic Xanthomonas species was identified as white-line-inducing principle (WLIP), a member of the viscosin group of cyclic lipononadepsipeptides. WLIP producers are commonly referred to by the taxonomically invalid name “Pseudomonas reactans,” based on their capacity to reveal the presence of a nearby colony of Pseudomonas tolaasii by inducing the formation of a visible precipitate (“white line”) in agar medium between both colonies. This phenomenon is attributed to the interaction of WLIP with a cyclic lipopeptide of a distinct structural group, the fungitoxic tolaasin, and has found application as a diagnostic tool to identify tolaasin-producing bacteria pathogenic to mushrooms. The genes encoding the WLIP nonribosomal peptide synthetases WlpA, WlpB, and WlpC were identified in two separate genomic clusters (wlpR-wlpA and wlpBC) with an operon organization similar to that of the viscosin, massetolide, and entolysin biosynthetic systems. Expression of wlpR is dependent on gacS, and the encoded regulator of the LuxR family (WlpR) activates transcription of the biosynthetic genes and the linked export genes, which is not controlled by the RW10S2 quorum-sensing system PmrR/PmrI. In addition to linking the known phenotypes of white line production and hemolytic activity of a WLIP producer with WLIP biosynthesis, additional properties of ecological relevance conferred by WLIP production were identified, namely, antagonism against Xanthomonas and involvement in swarming and biofilm formation.
INTRODUCTION
Plant root-associated fluorescent pseudomonads produce very diverse antagonistic secondary metabolites targeting competing microorganisms, including phytopathogens (18, 20). Synthesis of such molecules with antimicrobial properties is often triggered by the Gac/Rsm two-component signaling pathway, a global regulatory system in gammaproteobacteria (24). Lipopeptides (LPs) constitute a major group of antimicrobial molecules in the arsenal of Pseudomonas used for this biological warfare (33, 36, 37, 43). Several strains accommodate large gene clusters in their genomes to encode the nonribosomal peptide synthetases (NRPSs) that generate such compounds. These multimodular megaenzymes sequentially recruit, activate, and stereospecifically condense amino acids to generate linear or cyclic LPs. A typical NRPS module is composed of three domains for consecutive adenylation, thiolation, and condensation of a building block, whereas a separate thioesterase (TE) domain is required for product release, often with concurrent cyclization (18). Many of the LPs from nonpathogenic plant-associated Pseudomonas isolates exhibit antifungal activity and have adverse effects on oomycetes (37). As such, rhizosphere pseudomonads appear to be major players in suppressing plant diseases caused by these eukaryotic microorganisms (7, 29).
The inhibitory potential of Pseudomonas LPs toward bacteria has not been explored to the same extent, but a number of studies indicate that some Gram-positive bacteria, such as Bacillus, Staphylococcus, and Mycobacterium, are sensitive to some of these compounds (37). This was confirmed in a recent comparative study of five representative LPs (40). None of the Gram-negative strains tested (like Pseudomonas, all belonging to the gammaproteobacteria) was significantly inhibited by massetolide A, orfamide A, anthrofactin, entolysin B, syringomycin E, or the atypical LP SB-253514 that is tailored with a rhamnosyl and cyclocarbamate moiety (40).
Apart from bacteriocins, few compounds mediating antagonism between Pseudomonas and related bacteria have been identified. Promysalin, produced by Pseudomonas putida RW10S1, specifically targets other Pseudomonas organisms (26). Its amphipathic nature is reminiscent of LPs, but biosynthesis proceeds via a dedicated pathway, using salicylic acid in addition to an amino acid and a fatty acid as building blocks (26). Focusing on Xanthomonas, a major genus of phylogenetically related phytopathogens (45), as a target, we identified anti-Xanthomonas activity in P. putida RW10S2 isolated from rice rhizosphere (51). This strain was previously identified as a potential LP producer by NRPS-directed PCR screening (41). Here, we show that the antibacterial activity of strain RW10S2 is mediated by white-line-inducing principle (WLIP), an LP of the viscosin group.
WLIP, the white-line-inducing principle, is produced by so-called “Pseudomonas reactans” strains. When P. reactans is confronted with Pseudomonas tolaasii on solid medium, a white precipitate is produced between the colonies (54). This diagnostic test for the mushroom pathogen P. tolaasii, named “white line in agar,” relies on the interaction of WLIP with the fungitoxic lipopeptide tolaasin generating the precipitation line. In this study, we identified the NRPS system for biosynthesis of WLIP by P. putida RW10S2 and confirmed that it is required for the white-line reaction. A role for WLIP in biofilm formation and swarming, in addition to its antagonistic activity against Xanthomonas, is reported.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Bacterial strains, plasmids, and primers used for this study are listed in Tables S1 and S2 in the supplemental material. All Pseudomonas and Xanthomonas strains were grown in Trypticase soy broth (TSB) or agar (TSA) medium (BD Biosciences) at 30°C. Strains of Pseudomonas aeruginosa, Klebsiella, and Staphylococcus were cultured at 37°C in the same medium. Luria-Bertani (LB) medium was used to grow Escherichia, Bordetella, and Salmonella strains at 37°C and Aeromonas, Burkholderia, Mycobacterium, Rhodococcus, Serratia, Sphingomonas, Variovorax, and Yersinia strains at 30°C. Nutrient broth (NB) was used to culture Bacillus megaterium at 30°C and Citrobacter, Enterobacter, Proteus, and Shigella strains at 37°C. Bacillus subtilis was grown in TY medium (5 g/liter peptone, 3 g/liter yeast extract) at 30°C. Saccharomyces and Candida were grown in YPD (10 g of BactoYeast, 20 g of BactoPeptone, and 100 ml of 20% dextrose per liter) at 30°C. If needed, antibiotics were added at the following concentrations: kanamycin, 50 μg/ml; ampicillin, 50 μg/ml; and tetracycline, 20 μg/ml. Media were solidified with 1.5% agar. 5-Bromo-4-chloro-3-indolyl-beta-d-galactopyranoside (X-Gal) and isopropyl beta-d-thiogalactoside (IPTG) (40 mg/liter; Duchefa Biochemie) were added to detect the presence of insert DNA cloned in vectors in E. coli.
Mutant library construction, screening, and plasposon rescue.
Competent RW10S2 cells were prepared for electrotransformation in MEB buffer as described by Fredslund (16). All procedures for electroporation, random pTnMod-OKm′ mutagenesis, and plasposon rescue to clone mutated regions were performed as described by Parret et al. (34). Plasmid DNA was isolated using the Qiaprep Spin Miniprep kit (Qiagen). Routine DNA sequencing was performed at Macrogen Inc. (Seoul, South Korea). For the screening of mutants affected in antagonistic activity, mutants from the library were arrayed in a 96 (8 by 12)-spot pattern on square plates (12 cm by 12 cm) and overlaid with indicator Xanthomonas citri pv. malvacearum LMG 761. Mutants devoid of inhibitory activity were picked for further characterization.
Draft genome sequencing.
Genomic DNA was obtained using the Gentra Puregene Yeast/Bact kit (Qiagen Benelux B.V., Venlo, The Netherlands). Pair-end sequencing was performed using an Illumina Genome Analyzer (GA) II with 50 cycles (carried out at Baseclear, Leiden, The Netherlands). Velvet was used for de novo assembly of paired-end reads (55). A set of hash sizes were used to run Velvet multiple times, after which the quality of the assembly was judged by the N50 criterion. Contigs from the assembly showing the highest N50 value were automatically annotated using the RAST server (3). The annotations were verified manually for the regions of interest.
Southern blot analysis.
Genomic DNAs of wild-type P. putida RW10S2 and representative mutants were digested by EcoRI. Restriction fragments were separated by agarose gel electrophoresis (1%, 120 V, 2.5 h). Primer pair PGPRB-7325/PGPRB-7326 was used to synthesize a probe specific for the kanamycin (Km) resistance cassette from plasposon TnMod-OKm′ using the digoxigenin (DIG) labeling mix from Roche Applied Science (Germany). Blotting, hybridization, and chemiluminescent detection were performed as recommended by Roche Applied Science. After hybridization at 45°C, the membrane was washed twice in low-stringency solution (2× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate], 0.1% SDS) (5 min, room temperature) and twice in high-stringency solution (0.5× SSC, 0.1% SDS) (15 min, 65°C). Chemiluminescent detection was performed with ready-to-use disodium 3-{4-methoxyspiro[1,2-dioxetane-3,2′-(5′-chloro)tricyclo(3.3.1.13,7)decan]-4-yl}phenyl phosphate (CSPD) as recommended by Roche.
Bioinformatic analyses.
NRPS domain analysis was performed with the PKS/NRPS Analysis web-based tool (2). Using NRPSpredictor2, the amino acid specificity of each A domain was analyzed (44). Homology searches were performed using the BLAST programs on http://www.ncbi.nih.nlm.gov/blast.cgi and http://www.pseudomonas.com. DNA or protein sequence alignments, phylogenetic analyses, and local BLAST searches were performed with Geneious Pro (version 5.5.3) (12).
Complementation of regulatory mutants.
The RW10S2 wlpR gene was PCR amplified using Platinum Pfx DNA polymerase (Invitrogen) and primer pair PGPRB-6551/PGPRB-6552 and cloned into the pJB3Tc20 vector (pCMPG6125) using the XbaI and EcoRI sites. Similarly, the RW10S2 gacS gene was amplified using primer pair PGPRB-7317/PGPRB-7318 and cloned into the pJB3Tc20 vector (pCMPG6203) using HindIII and XbaI sites. Triparental conjugation was used to mobilize pCMPG6125 and pCMPG6203 from Escherichia coli DH5α to P. putida RW10S2 mutants CMPG2173 and CMPG2134, respectively, with helper strain HB101(pRK2013). Thirty microliters of each culture (optical density at 600 nm [OD600] of between 0.5 and 0.8) of E. coli DH5α containing the construct, Pseudomonas, and helper strain was mixed and spotted on a TSA plate, followed by overnight incubation at 30°C. The mixture was resuspended in 1 ml of sterile TSB, and aliquots of different dilutions were spread on TSA supplemented with ampicillin (50 μg/ml, to counterselect E. coli) and tetracycline (20 μg/ml). The same procedure was used to transfer the broad-host-range expression vector pHERD26T carrying the P. putida BW11M1 xtlR gene (pCMPG6116) to mutant strain CMPG2172. Likewise, pCMPG6113, a pJB3Tc20 construct carrying RW10S1 gacS (26), was introduced in mutant strain CMPG2134. The transconjugants were selected for further phenotypic analyses. Strains carrying a pHERD26T derivative were grown in the presence of l-arabinose (0.1%) to induce expression (35).
Extraction and purification of WLIP.
A crude extract of WLIP produced by P. putida RW10S2 and the reference WLIP producer Pseudomonas sp. strain LMG 2338 was obtained using the procedure reported by Mortishire-Smith et al. (31). Larger amounts of semipurified WLIP were obtained by solid-phase extraction (SPE) as previously described for viscosin (46). Five hundred milliliters of King's B medium in 1-liter Erlenmeyer flasks was inoculated with 2.5 ml overnight bacterial culture. The cells were grown at 25°C for 4 days with shaking (200 rpm). To separate cells, cultures were centrifuged at 9,000 × g for 20 min (Beckman J2-M1). The supernatant was filtered through Stericup membranes (0.22-μm pore size; Millipore), followed by acidification to pH 5 with HCl (1 M). The supernatant was kept at 4°C overnight to precipitate surface-active compounds. The SPE cartridge (C18 end capped, 25 ml/50 mg; Biotage), was conditioned and equilibrated with 15 ml methanol and Milli-Q water, respectively, using vacuum-accelerated elution (Vacmaster-20; Biotage). The cartridge was subsequently washed with 40%, 60%, and 80% (vol/vol) acetonitrile (ACN) in water to remove impurities. To elute WLIP, 100% ACN was used. A white precipitate was obtained after drying the collected eluate in a vacuum concentrator. The SPE-extracted material was dissolved in methanol containing 0.7% formic acid.
HPLC and mass spectrometry.
For high-pressure liquid chromatography (HPLC) experiments, a capillary chromatography system (CapLC; Waters, Milford, MA) was directly connected to the mass spectrometer. The column dimensions were 0.32 by 150 mm (XTerra C18 5 μm; Waters, Milford, MA), the injection volume was 0.1 μl, the flow rate was set to 5 μl,/min and the samples were eluted with a water-ACN gradient. Spectra were taken every second in the m/z range of 400 to 1500.
Mass spectra were acquired in positive ionization mode on a quadrupole orthogonal acceleration time-of-flight mass spectrometer (Q-Tof-2; Micromass, Manchester, United Kingdom) equipped with a standard electrospray probe (Z-Spray; Micromass, Manchester, United Kingdom) and controlled by the MassLynx 3.4 software. The capillary voltage and cone voltage were set to 3 kV and 30 V, respectively. Fragment ion spectra were obtained with argon as the collision gas and with a collision energy of 40 eV. Samples were dissolved in ACN-water (1:1, vol/vol) and infused at 5 μl/min.
NMR spectroscopy.
All nuclear magnetic resonance (NMR) measurements were performed either on a Bruker DRX spectrometer operating at 1H and 13C frequencies of 500.13 MHz and 125.76 MHz, respectively, or on a Bruker Avance II spectrometer operating at 1H and 13C frequencies of 700.13 MHz and 176.05 MHz, respectively. In both cases, a 1H,13C,15N TXI-Z probe was used. The sample temperature was set to 25°C throughout. High-quality NE-HP5-7 (New Era Enterprises Inc.) NMR tubes were used. Two-dimensional (2D) spectra measured for structure elucidation included a gradient-selected 1H-1H correlation spectrum (gCOSY), a 1H-1H total-correlation spectrum (TOCSY) with a 90-ms MLEV-17 spin lock, an off-resonance rotating-frame nuclear Overhauser effect spectrum (ROESY) with a 400-ms mixing time, and a sensitivity-improved gradient-selected heteronuclear single-quantum correlation 1H-13C (gHSQC) spectrum applying adiabatic 180° pulses. Standard pulse sequences as present in the Bruker library were used throughout. All experiments sampled 4,096 data points in the direct dimension and 512 data points in the indirect dimension, except for the COSY, which was run with 256 points in the indirect dimension. The spectral width in the 1H dimension was set to 14 ppm in the TOCSY and the gHSQC spectrum, 16 ppm in the COSY, and 20 ppm in the off-resonance ROESY, while the spectral width in the 13C dimension was set to 90 ppm. For 2D processing, the spectra were zero filled to a 2,048-by-2,048 real data matrix. Prior to Fourier transformation, all spectra were multiplied with a squared cosine bell function in both dimensions, except for the COSY, which was multiplied with a squared sine bell function. The chemical shift axes of all spectra were calibrated to the DMF-d7 methyl peak positioned at the highest chemical shift (1H 1.92 ppm; 13C 34.89 ppm).
Antimicrobial activity assays.
Conventional deferred-antagonism assays were performed as previously described (34). This agar diffusion method relies on detection of halo formation in an overlay of indicator cells (∼107 CFU/ml) due to growth inhibition by chloroform-killed producer cells (a 2-μl spot of ∼107 CFU/ml). To analyze the activity of purified WLIP, the method of Lo Cantore et al. was applied (28). From a stock solution of 2.5 mg/ml (2.22 mM) in dimethyl sulfoxide (DMSO), 2-fold serial dilutions (in DMSO) were prepared starting from 1 mM, and 10-μl samples were spotted on an agar plate and overlaid with indicator cells. DMSO spots were used as negative controls. The radius of the inhibition halo was recorded. For each sample, the antagonistic activity assay was done in three repeats.
White-line-in-agar assay.
The white-line-in-agar test was performed in triplicate on King's B medium (54). Bacterial strains were streaked next to P. tolaasii, CH36 and white precipitate formation in the agar was evaluated after 48 h of growth at 25°C.
Hemolytic activity assay.
Hemolytic activity was analyzed by spotting the bacterial culture (2 μl) or extracted compound solution (5 μl) on horse blood TSA (containing 5% defibrinated horse blood; Oxoid) and incubation at 30°C for 16 h. Three repeats were performed for each sample.
Swarming motility assay.
To test swarming motility, an overnight culture was spotted on 1%, 0.9%, 0.8%, and 0.7% TSA plates. Plates were incubated at 30°C, and swarming was evaluated after incubation for 16 h (three repeats).
Biofilm formation assay.
Biofilm analysis was done essentially as described by De Keersmaecker et al. (11). A platform of 96 polystyrene pegs (Nunc no. 445497) fitting into a microtiter plate (Nunc no. 269789) was used. Overnight bacterial culture was diluted using TSB to an OD600 of 0.5. This cell suspension was diluted further 100 times, and 200 μl of cells was loaded into a Nunc microtiter plate (eight repeats for each sample). The pegged lid was inserted into the plate, sealed with Parafilm, and incubated for 48 h at 30°C without shaking. To wash the cells, the lid was transferred to a new plate with 200 μl phosphate-buffered saline in each well. Pegs were then stained with 200 μl of 0.1% crystal violet in an isopropanol–methanol–phosphate-buffered saline solution (1:1:18) for 30 min. Subsequently, excess stain was rinsed off by placing the pegs into a 96-well plate filled with 200 μl distilled water per well. After the pegs were air dried (30 min), the dye bound to the adherent cells was extracted with 30% glacial acetic acid (200 μl) for 10 min, and then 135 μl of each extract was used to measure the absorbance at 570 nm (SynergyMX Multimode reader; BioTek).
RNA extraction and reverse transcription-PCR (RT-PCR).
Total RNA was isolated from RW10S2 and selected mutant cells after overnight culture in TSB, using the TRIzol Plus RNA purification system protocol (Invitrogen). In order to remove contaminating genomic DNA, RNA extracts were treated using a Turbo DNA-free kit (Ambion). Total RNA was quantified using the NanoDrop ND-1000 system (Thermo Scientific) and quality checked by running on a 1% agarose gel. A 1.5-μg amount of RNA was used to synthesize first-strand cDNA using Random Decamers (50 μM; Ambion), Moloney murine leukemia virus (M-MuLV) reverse transcriptase (Westburg), and a deoxynucleoside triphosphate (dNTP) set (Westburg). cDNA was subjected to PCR with specific primers listed in Table S2 in the supplemental material. The 16S rRNA gene was used as an internal control. The primer pairs PGPRB-6231/PGPRB-6232, PGPRB-7319/PGPRB-7320, PGPRB-6233/PGPRB-6234, PGPRB-7321/PGPRB-7322, PGPRB-7323/PGPRB-7324, and PGPRB-6353/PGPRB-6354 were used to amplify cDNAs derived from transcripts corresponding to wlpA (849 bp), covering the adjacent gene pairs wlpR-wlpF (670 bp), wlpB-wlpC (801 bp), wlpC-wlpD (606 bp), and wlpD-wlpE (568 bp), and specific for the 16S rRNA gene (563 bp), respectively.
Nucleotide sequence accession numbers.
The gene sequences have been submitted to GenBank under accession numbers JN982331 (gacS), JN982332 (wlpR and wlpA), JN982333 (wlpBC), and JN982334 (16S rRNA).
RESULTS
Genetic characterization of P. putida RW10S2 mutants lacking Xanthomonas antagonism.
Using a panel of phytopathogenic Xanthomonas strains, P. putida RW10S2, an isolate from rice rhizosphere in Sri Lanka (51), showed significant antibacterial activity against several of these strains (Fig. 1). Differences in sizes of the growth inhibition halos indicated that sensitivities vary among Xanthomonas strains. To identify RW10S2 genes involved in production of anti-Xanthomonas activity, a plasposon mutant library (∼15,000 mutants) was constructed using pTnMod-OKm′. This library was screened for mutants lacking inhibitory activity against X. citri pv. malvacearum LMG 761, one of the most sensitive strains. By plasposon rescue, the flanking regions of the respective inserts were cloned and sequenced for selected null mutants. Taking advantage of a draft genome sequence of RW10S2 (unpublished data), the genes affected by plasposon insertion were identified (Fig. 2A and 3A).
Fig 1.

Growth inhibition of Xanthomonas species by P. putida RW10S2. Spotted RW10S2 cells (2 μl; 107 CFU/ml) were overlaid with Xanthomonas indicator cells. For each indicator, the growth inhibition halo (radius in mm; mean ± standard deviations [SD] of three repeats) is indicated in parentheses: (A) X. citri pv. malvacearum LMG 761 (8.3 ± 0.6); (B) X. axonopodis pv. manihotis LMG 784 (7.6 ± 0.3); (C) X. translucens pv. cerealis LMG 679 (4.6 ± 0.6); (D) X. albilineans LMG 494 (4.5 ± 0.5); (E) X. hortorum pv. hederae LMG 7411 (4.3 ± 0.3); (F) Xanthomonas sp. pv. zinnia LMG 8692 (2.6 ± 0.6); (G) X. translucens pv. graminis LMG 726 (2.8 ± 0.3); (H) X. campestris pv. pelargonii LMG 10342 (3.1 ± 0.6); (I) X. alfalfae pv. alfalfae LMG 497 (2.6 ± 0.3).
Fig 2.

Organization of the wlp gene cluster in P. putida RW10S2 (A) and its transcriptional analysis (B). (A) The labeled circles show predicted NRPS domains encoded by wlpA (NRPS1), wlpB (NRPS2), and wlpC (NRPS3): C, condensation; A, adenylation; T, thiolation; TE, thioesterase. The condensation domains with putative dual condensation/epimerization function are underlined. The condensation domain with predicted lipo-initiation activity, attaching a fatty acid (FA), is labeled Ci. For the nine modules, amino acid specificity based on in silico analysis of the respective A domains is indicated. The vertical bold lines mark the plasposon insertion sites leading to WLIP production deficiency. The mutants in bold were verified by Southern blot analysis (see Fig. S1 in the supplemental material) and phenotypically characterized. (B) The composite panel shows the amplicons obtained by RT-PCR transcript analysis using primer couples for the specified pairs of adjacent convergent genes and for 16S rRNA (internal control). RNA was extracted from the wild type (WT) and from mutants CMPG2173 (wlpR) and CMPG2134 (gacS).
Fig 3.

Phenotypic characterization of P. putida RW10S2 and representative mutants lacking WLIP production. (A) Antagonism against X. citri pv. malvacearum LMG 761. (B) White-line formation obtained by confronting P. putida RW10S2 with P. tolaasii CH36 (lower bacterial streak) (C) Hemolysis on a horse blood TSA plate. (D) Swarming on 0.8% TSA. (E) Biofilm formation on polystyrene pegs visualized by staining of adherent cells. WT, RW10S2 wild type; wlpA through wlpR, mutants CMPG2170, CMPG2169, CMPG2120, and CMPG2173, respectively; wlpR+, mutant CMPG2173 with pCMPG6125 containing wlpR from P. putida RW10S2; wlpR*, mutant CMPG2173 with pCMPG6116 containing xtlR from P. putida BW11M1; gacS, mutant CPMG2134; gacS+, mutant CMPG2134 with pCMPG6203 containing gacS from P. putida RW10S2; gacS*, mutant CMPG2134 with pCMPG6113 containing gacS from P. putida RW10S1. The phenotypes shown for the selected wlpA, wlpB, wlpC, and gacS mutants are representative for the other wlp NRPS and gacS mutants (Fig. 2; see Table S1 in the supplemental material). The corresponding quantitative data are shown in Fig. 4 (biofilm formation) and in Table S3 in the supplemental material (antagonism, hemolysis, and swarming).
Large numbers of independent mutants (three, five, and eight, respectively) with mutations in three NRPS genes were identified: CMPG2170 to CMPG2172 (NRPS1), CMPG2165 to CMPG2169 (NRPS2), and CMPG2120 to CMPG2127 (NRPS3) (Fig. 2A). Southern blot analysis confirmed the single-copy insertion of the plasposon for selected NRPS1 (CMPG2170), NRPS2 (CMPG2169), and NRPS3 (CMPG2120) mutants (see Fig. S1 in the supplemental material). In one plasposon insertion mutant (CMPG2173), a LuxR family gene was disrupted (Fig. 2A), equally causing loss of anti-Xanthomonas activity (Fig. 3A). This putative regulatory gene was located upstream of the first NRPS gene, positioned divergently to it. The RW10S2 luxR-like gene product showed about 70% identity with the NRPS-linked LuxR-type regulators PsoR of P. putida PCL1445 (13) and EtlR of P. entomophila (50).
The gene organization visualized in Fig. 2 is quite similar to those of some three-membered NRPS systems identified in Pseudomonas strains: a first NRPS gene is linked with a cognate luxR-like gene but not linked with the two other NRPS genes that occur as a tandem in a separate operon. This analogy in genomic organization with the genes involved in biosynthesis of a viscosin-like LP (9), massetolide A (8), and entolysin (50) suggests that the three RW10S2 NRPSs equally operate consecutively to synthesize a nonribosomal peptide.
In addition to NRPS-related mutations, seven independent insertions in the RW10S2 gacS gene (see Table S1 in the supplemental material) also knocked out inhibitory activity against Xanthomonas (Fig. 3A). GacS is the sensor kinase member of the Gac/Rsm two-component system, a global regulatory system that controls the expression of many secondary metabolites in gammaproteobacteria (24). The RW10S2 GacS protein is most similar (about 89% amino acid identity) to the P. putida GB-1 (PputGB1_1252) and P. entomophila (PSEEN1359) GacS proteins.
In silico analysis of P. putida RW10S2 NRPS genes.
Analysis by the NRPS-PKS tool indicated that the first, second, and third encoded NRPSs consist of two, four, and three modules, respectively (Fig. 2A). Each module contains a condensation (C) domain, an adenylation (A) domain, and a thiolation (T) domain. The carboxy-terminal region of the third NRPS carries two tandem thioesterase (TE) domains. Alignments of the RW10S2 NRPS amino acid sequences with those of the similarly organized triple-NRPS systems for massetolide and viscosin biosyntheses (8, 9) revealed moderate homologies (60 to 65% amino acid identity) with MassA/ViscA (60.9%/60.6%), MassB/ViscB (64.2%/65.3%), and MassC/ViscC (61.8%/61.9%). The NRPSpredictor2 tool was used to analyze the presumed specificity of the nine A domains. Assuming colinear biosynthesis, starting with NRPS1, continued by NRPS2, and ending with NRPS3, the putative peptide produced would have the sequence Leu-Glu/Asp-Thr-Val-Leu-Ser-Leu-Ser-Ile. Phylogenetic analysis of the A domains (see Fig. S2 in the supplemental material) supported this prediction. It pointed to Glu specificity of the second module and probable allo-Thr preference for the third module.
This amino acid sequence is very similar to those of members of the viscosin group, in particular to those of WLIP and viscosin (18). Both LPs contain a nonapeptide moiety but differ only by the configuration (d or l, respectively) of a leucine residue at the fifth position. According to Balibar et al. (4), the d configuration of amino acids in the Pseudomonas LP arthrofactin is generated by a specific condensation domain, with additional epimerization activity, acting on the amino acid loaded on the previous module. Such C/E domains constitute a distinct cluster separated from the clusters with regular condensation domains and the lipo-initiation domain cluster in phylogenetic analysis of their sequences (8, 41, 50). Application of this sequence comparison to the RW10S2 C domains (see Fig. S3 in the supplemental material) identified a putative lipo-initiation domain in the first module of NRPS1 (Fig. 2A). Among the other C domains, only the one in the eighth module (second module of NRPS3) clustered with the conventional (nonepimerizing) domains, suggesting incorporation of an l-Leu residue at position 7 (conserved in members of the viscosin group) and a d configuration at positions 1 through 6 and 8. The predicted d-Leu5 would match the equivalent residue in WLIP but not that in viscosin (l-Leu5). The invariant l-Leu1 of viscosin members would be in conflict with the current prediction of a d-Leu1. However, this particular exception to the “Balibar rule” has been noted also for another viscosin group member, massetolide A (8), and for the cyclic decadepsipeptides orfamides A to D (19). Overall, this analysis strongly suggests that the RW10S2 NRPS system mediates biosynthesis of a member of the viscosin group.
WLIP-linked phenotypes of P. putida RW10S2.
Domain analysis of the RW10S2 NRPSs strongly points to a system synthesizing a lipopeptide closely related to WLIP or, possibly, viscosin. When a WLIP producer is grown close to P. tolaasii, a white-line precipitate will be formed, attributed to the interaction between WLIP and the LP tolaasin produced by P. tolaasii (54). Such a precipitate is not observed with a viscosin producer confronting P. tolaasii, indicating that the minor structural difference in viscosin compared to WLIP (l-Leu instead of d-Leu) affects the outcome of this diagnostic test (41). Strain RW10S2 generated a heavy white line when confronting P. tolaasii, while all mutants with a disrupted NRPS gene displayed a lack of white precipitate formation (Fig. 3B). The same phenotypic change was observed for the mutant with an inactivated NRPS-linked regulatory gene and the gacS mutants.
In a phenotypic variant of P. reactans LMG 5329, loss of white-line production coincided with disappearance of the capacity to lyse erythrocytes as visualized in blood agar (32). Using purified LPs, Lo Cantore et al. (28) demonstrated that purified WLIP from P. reactans NCPPB1311 causes red blood cell lysis through transmembrane pore formation, being a more potent hemolytic agent than tolaasin I (38). Strong hemolysis was also observed for P. putida RW10S2 cells grown on horse blood TSA (Fig. 3C; see Table S3 in the supplemental material). Notably, all mutants impaired in anti-Xanthomonas antagonism and the white-line phenotype were also deficient in hemolytic activity (Fig. 3A to C; see Table S3 in the supplemental material).
Given the consistent correlation between gene activity and WLIP-related phenotypes in P. putida RW10S2, the NRPS genes (encoding NRPS1, NRPS2, and NRPS3) and the NRPS1-linked LuxR family gene were designated wlpA, wlpB, wlpC, and wlpR, respectively. These genes are coexpressed as shown by RT-PCR analysis (Fig. 2B). Cotranscription was demonstrated for the wlpB-wlpC genes (with overlapping stop-start codons) along with the downstream wlpD-wlpE (macA-macB homologues, also with overlapping stop-start codons). Likewise, cotranscription was observed for wlpF (oprM homologue) located downstream of wlpR (Fig. 2B).
Structural characterization of the P. putida RW10S2 LP.
The module composition of the RW10S2 NRPS system and phenotypic characteristics of NRPS mutants suggested the LP produced by this strain to be very similar, if not identical, to WLIP. To resolve this issue, the LP was purified for chemical characterization. The mass spectrum of the RW10S2 LP extract contained an abundant protonated pseudo-molecular ion (M + H)+ at m/z 1126 (exact mass, 1,126.7 Da), corresponding to the molecular weight (1,125) of WLIP (31). HPLC analysis confirmed that the corresponding peak, displaying Xanthomonas-antagonistic and hemolytic activities, was absent in extracts from a wlpC mutant and a gacS mutant (see Fig. S4 in the supplemental material). The mass fragmentation fingerprint of the RW10S2 compound was compared with its equivalent produced by strain LMG 2338 and found to be identical (see Fig. S5 in the supplemental material). Strain LMG 2338 (NCPPB 387) represents the original P. reactans strain used to elucidate the structure of WLIP (31). The inferred identity with WLIP was further examined by NMR analysis of a solution of the RW10S2 LP in DMF-d7 (see Fig. S6 in the supplemental material). The presence of the 3-hydroxydecanoic acid (HDA) moiety and all the amino acid spin systems of WLIP were confirmed by the 2D 1H-1H COSY and 1H-1H TOCSY, while the N→C primary sequence (HDA-Leu-Glu-Thr-Val-Leu-Ser-Leu-Ser-Ile) was confirmed by analysis of the 2D 1H-1H ROESY. The NMR spectra were found to be very similar to those of pseudodesmin A (48), whose primary sequence differs from WLIP by a Glu→Gln substitution but whose stereochemical configurations and conformational properties in solution are the same (47). To confirm the configurations of the amino acid stereocenters, the 1H and 13C chemical shifts of the RW10S2 LP were compared to those of pseudodesmin A, obtained as described before (48) but now in DMF-d7 (see Fig. S7 in the supplemental material). As the chemical shift is a very sensitive indicator of even subtle changes in conformation, one or more differences in stereochemistry between the two LPs should lead to significant differences in the chemical shifts as well. Except at the level of the Glu→Gln substitution, no significant chemical shift differences were found between the compounds, fully confirming the identity of the RW10S2 LP to be WLIP.
Antimicrobial spectrum of WLIP from P. putida RW10S2.
Using the method of Lo Cantore et al. (28), the antimicrobial activity of purified RW10S2 WLIP was tested against a panel of phylogenetically diverse bacteria (see Table S2 in the supplemental material). Among the proteobacteria tested, WLIP inhibited only Xanthomonas strains, and of the Gram-positive bacteria, it inhibited only Bacillus megaterium and Bacillus subtilis (Table 1). Among 17 strains of Xanthomonas, X. citri pv. malvacearum LMG 761, X. axonopodis pv. manihotis LMG 784, and X. albilineans LMG 494 were the most sensitive strains. For X. axonopodis pv. manihotis, the minimal inhibitory quantity (MIQ) was determined using a serial dilution series (see Fig. S8 in the supplemental material). The MIQ of 0.7 μg for WLIP is about twice the MIQ (0.32 μg) for Bacillus megaterium ITM100 reported by Lo Cantore et al. (28). Only a very small but clear halo was noted for five strains, and an intermediate halo size was observed for the other sensitive strains. No inhibition of X. oryzae pv. oryzae was observed. The activity spectrum of purified WLIP matched well the Xanthomonas-inhibitory profile of P. putida RW10S2. No growth-inhibitory activity of RW10S2 WLIP was observed for a Saccharomyces cerevisiae strain and a Candida albicans strain. This observation is consistent with the low antifungal activity and lack of yeast-antagonistic activity of WLIP obtained from P. reactans NCPPB1311 (28).
Table 1.
Antagonistic activity spectrum of purified WLIP
| Straina | Inhibition halo (mm)b |
|---|---|
| Xanthomonas sp. pv. zinnia LMG 8692 | 2.3 ± 0.6 |
| X. albilineans LMG 494 | 6.5 ± 0.5 |
| X. alfalfae pv. alfalfa LMG 497 | 2.6 ± 0.6 |
| X. axonopodis pv. manihotis LMG 784 | 6.6 ± 0.3 |
| X. campestris pv. campestris LMG 582 | 1.8 ± 0.3 |
| X. campestris pv. pelargonii 10342 | 3.6 ± 0.8 |
| X. citri pv. malvacearum LMG 761 | 7.3 ± 0.6 |
| X. hortorum pv. hederae LMG 7411 | 3.5 ± 0.9 |
| X. sacchari LMG 471 | 2.6 ± 0.6 |
| X. translucens LMG 12921 | 3.3 ± 0.6 |
| X. translucens pv. cerealis LMG 679 | 4.5 ± 0.5 |
| X. translucens pv. graminis LMG 726 | 3.3 ± 0.6 |
| X. translucens pv. hordei LMG 737 | 1.5 ± 0.9 |
| X. vasicola pv. holcicola LMG 736 | 1.3 ± 0.6 |
| Bacillus megaterium ATCC 13632 | 15.0 ± 1.0 |
| B. subtilis LMG 7135 | 2.5 ± 0.5 |
All other bacteria listed in Table S2 in the supplemental material were not inhibited by strain RW10S2 and purified WLIP.
Spots of purified WLIP (22.5 μg) were overlaid with bacterial cells. Each value (radius of halo) is the mean ± standard deviations of three repeats.
Role of WLIP in swarming and biofilm formation by P. putida RW10S2.
The involvement of LPs in swarming and biofilm formation has been reported for several Pseudomonas strains (5, 23, 36, 37, 42). Therefore, swarming and biofilm formation of P. putida RW10S2 and its WLIP-deficient mutants were investigated. Strain RW10S2 showed swarming on TSA at agar concentrations below 0.9%. In all mutants deficient in WLIP production (wlpA, wlpB, wlpC, wlpR, and gacS mutants), swarming was abolished (Fig. 3D; see Table S3 in the supplemental material). Similarly, biofilm formation was reduced about 10-fold in all these mutants (Fig. 3E and 4).
Fig 4.
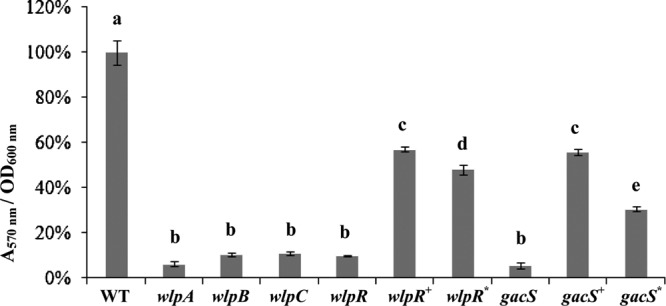
Biofilm formation by P. putida RW10S2 and selected mutants affected in WLIP production (abbreviations are as in Fig. 3). Error bars indicate standard deviations. Analysis of variance (ANOVA) was used to evaluate significant differences (P < 0.001) between the wild type (set to 100%), mutants, and complemented mutants (indicated with different letters above the bars).
Regulatory genes of WLIP production.
No transcription of the WLIP biosynthetic genes was detectable in the wlpR and gacS mutants (Fig. 2B). Phenotypes associated with WLIP production were restored partially or to near-wild-type levels in the RW10S2 wlpR and gacS mutants by introduction of the cloned RW10S2 wlpR and gacS, respectively (Fig. 3; see Table S3 in the supplemental material). Swarming capacity was regained by the wlpR-complemented mutant, but some difference in pattern (irregular swarm edge) was evident (Fig. 3D). This may be due to unbalanced plasmid-driven expression of a chromosomal regulatory gene participating in control of a complex phenotype that involves several other determinants. Interestingly, partial complementation was also achieved by introduction of the cloned NRPS-linked LuxR family gene xtlR of the LP producer P. putida BW11M1 (sharing 66% amino acid identity [25, 41]). Biofilm formation by this heterologously complemented mutant attained about ∼50% of the wild-type capacity, slightly lower than the partially restored biofilm phenotype of the homologously wlpR-complemented and gacS-complemented mutants (Fig. 4). The RW10S2 gacS mutant could also be complemented to some extent by gacS from P. putida RW10S1 (sharing 86% amino acid identity [26]). Introducing this heterologous gacS gene resulted in restored swarming capacity but with a modified dendritic pattern (Fig. 3D). Interestingly, RW10S2 pmrR and pmrI mutants with a deficient N-acylhomoserine lactone-dependent quorum-sensing system (49) displayed wild-type WLIP-related phenotypes (data not shown), indicating quorum-sensing-independent regulation of WLIP production.
DISCUSSION
In this study, the antagonistic activity of P. putida RW10S2 against pathogenic Xanthomonas species was investigated, which resulted in the isolation and biological characterization of the cyclic LP WLIP and identification of its biosynthetic and regulatory genes.
Genetic backbone of WLIP biosynthesis.
Although the structure of WLIP has been known for more than 2 decades (31), genetic analysis of its biosynthesis was not yet reported. In contrast to the single-operon organization of several Pseudomonas multiple-NRPS systems (arthrofactin, orfamide, syringafactin, syringopeptin, and putisolvin [43]), we show here that three NRPS genes located in two regions of the RW10S2 genome are responsible for WLIP biosynthesis. The wlpA gene, encoding an NRPS with the first two modules, is separated from the wlpBC operon, encoding 4- and 3-modular NRPSs, respectively. This type of spread gene organization has also been observed for synthesis of entolysin from P. entomophila L48 (50), massetolide A from P. fluorescens SS101 (8), and viscosin from P. fluorescens SBW25 (9). Massetolides and viscosin belong to the same structural group of lipononadepsipeptides, but entolysin is built from 14 amino acids. In the latter case, the isolated NRPS gene (etlA) also encodes the first two modules. The unlinked etlBC operon provides the remaining modules (8 and 4, respectively, compared to a 4-3 modular distribution for the WLIP, viscosin, and massetolide syntheses). The in silico analysis of A-domain specificity, using both the NRPSpredictor2 tool (44) and a phylogenetic comparison with A domains of known Pseudomonas LPs, combined with Pseudomonas genomic synteny of the three NRPS genes and application of the colinearity rule in modular biosynthesis proved to be valuable for reliable prediction of the amino acid sequence and fatty acid modification of the secondary metabolite synthesized. In line with some previously noted exceptions to the “Balibar rule” (4, 8, 19), the prediction of the d/l configurations in WLIP by phylogenetic analysis of C domains in its NRPSs was confirmed except for one residue.
The genomic sequence context of the mutated NRPS genes indicates the presence of three genes encoding a candidate export system for the secondary metabolite synthesized. Immediately downstream of wlpR, a coexpressed gene (wlpF) encoding a putative OprM-like outer membrane efflux protein (Pfam PF02321) is located (Fig. 2). Similarly, a coexpressed gene tandem downstream of wlpBC (wlpDE) encodes proteins related to MacA (Pfam PF12700) and MacB (Pfam PF12704) (Fig. 2). The Escherichia coli MacA-MacB-TolC system is an ATP-dependent tripartite macrolide efflux transporter (30). Since OprM represents the functional and structural Pseudomonas equivalent of TolC (1) and wlpF is cotranscribed with the RW10S2 wlp genes, the three NRPS-linked genes likely encode an LP export system, WlpDEF. In several known Pseudomonas LP gene clusters, the oprM-like gene and macAB-like genes flank the initiating and terminating NRPS genes, respectively (8, 13, 50). Likewise, polycistronic transcription of the arthrofactin biosynthetic genes arfA, arfB, and arfC with the downstream exporter genes arfD (macA homologue) and arfE (macB homologue) was demonstrated in Pseudomonas sp. strain MIS38 (27). Mutational analysis of the Mac-like Pseudomonas genes downstream of the putisolvin (13) and arthrofactin (27) biosynthetic operons revealed reduced LP production, indicating a primary but not exclusive role in export of the corresponding product. This residual production level could explain why in our screening for mutants devoid of antagonistic activity, no insertions in these flanking genes were indentified. The involvement of an OprM homologue in LP production by Pseudomonas has not yet been experimentally verified.
Regulation of WLIP production.
As reported earlier for syringomycin (21), tolaasin (17), amphisin (22), and syringopeptin (52), WLIP production by P. putida RW10S2 is dependent on a functional GacS, the sensor of the global regulatory system Gac/Rsm (24). It is not triggered by the RW10S2 N-acylhomoserine lactone-mediated quorum-sensing system PmrR-PmrI (49). The LuxR family protein WlpR, encoded by a gacS-dependent wlpA-linked gene, apparently represents the cognate regulator of WLIP biosynthesis. Inactivation of wlpR abolished the expression of the other wlp genes, WLIP production, and WLIP-linked phenotypes. LuxR-type regulators are found in the flanking regions of most Pseudomonas LP-synthesizing NRPS genes (10). Among these homologues of wlpR, also located upstream of the respective initiating NRPS genes, several were also shown to be essential for production of syringafactin (syfR) (5), putisolvin (psoR) (13), viscosin (viscAR) (10), arthrofactin (arfF) (53), and entolysin (etlR) (50). A downstream-positioned luxR-type regulator gene (viscBCR) is also involved in viscosin production, but inactivation of its counterpart (pspto2833) did not affect production of syringafactin (5). No additional luxR-type gene similar to viscBCR is present within 2 kb downstream of the wlpBC operon.
The LuxR-type regulator gene located upstream of the initiating NRPS genes for massetolide production can restore viscosin biosynthesis in a mutant with the equivalent viscosin-regulatory gene inactivated (10). Such functional exchangeability was demonstrated here as well for the corresponding regulator of P. putida RW10S2, whose function can be taken over, at least partially, by its counterpart from P. putida BW11M1, a lipopeptide-producing banana rhizosphere isolate from Sri Lanka (25, 41).
Clearly, the involvement of a LuxR-type regulator(s), lacking the autoinducer domain of quorum-sensing-associated LuxR regulators and encoded by regulatory genes clustered with the biosynthetic ones, constitutes a recurrent theme in LP production by Pseudomonas (37), reiterated in WLIP biosynthesis. In only few Pseudomonas strains, N-acylhomoserine lactone-dependent regulation has been reported (6, 14).
Physiological role of WLIP.
The diagnostic tool introduced by Wong and Preece in 1979 to identify mushroom-pathogenic bacteria producing the toxin tolaasin has found wide application (54). It relies on the formation of a sharp precipitation line attributed to interaction of tolaasin with another LP, WLIP, secreted by juxtaposed colonies of P. tolaasii and a WLIP-producing P. reactans. The latter name is a phenotypic designation, referring to the white-line reaction, for an ill-defined group of Pseudomonas strains. In this study, the P. putida strain RW10S2 was identified as a WLIP producer, and characterization of defined biosynthetic and regulatory mutants lends support to the LP interaction hypothesis. Previously, Rainey and colleagues reported the lack of white-line production by P. tolaasii mutants with inactivated tolaasin biosynthetic enzymes (39). The ecological significance of this interaction remains unclear. Sequestering an opponent's toxic LP may have a protective effect. In contrast to a previous report (38), an inhibitory effect on P. reactans growth was reported for purified tolaasin (28). Such an effect was not observed for purified WLIP on P. tolaasii (28), which was confirmed in our study. WLIP's moderate antifungal activity and inhibition of Gram-positive bacteria were previously documented (28), with both properties being shared with many other Pseudomonas LPs (37, 40). The general recalcitrance of Gram-negative bacteria, including an X. campestris pv. phaseoli indicator (strain ICMP3035), to LP inhibition was also observed by Lo Cantore et al. (28). We noticed, however, that the growth of several other Xanthomonas strains is inhibited by WLIP, but the severity appears to be species/strain dependent. Etchegaray et al. (15) demonstrated disintegration of Xanthomonas cells caused by a concentrated Bacillus subtilis LP extract containing iturin and surfactin. Xanthomonas campestris pv. campestris was significantly more sensitive than X. axonopodis pv. citri. The major rice pathogen X. oryzae pv. oryzae is not sensitive to WLIP produced by strain RW10S2, which itself originates from the rhizosphere of rice plants. In line with the properties of several Pseudomonas LPs (37), our study further revealed that WLIP promotes solid-surface translocation and biofilm formation by its producer. These features may reflect an important role for WLIP in the colonization of structured environments.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge assistance of J. Desair (CMPG, KU Leuven) with HPLC purification, of M. Ghequire (CMPG, KU Leuven) with the antimicrobial spectrum tests, and of D. De Coster (CMPG, KU Leuven) with Southern blot analysis. We thank M. Höfte and J. Xu (Laboratory of Phytopathology, Ghent University, Belgium) for assistance in testing the WLIP sensitivities of Xanthomonas oryzae pv. oryzae strains.
D.S. acknowledges Ghent University for a postdoctoral research grant through the Bijzonder Onderzoeksfonds (BOF). The 700-MHz NMR apparatus is part of the Interuniversitary Facility funded by the Flemish Government (FFEU-ZWAP) and jointly operated by UGent, VUB, and UA. This work was supported by a KU Leuven-Zhejiang University SBA fellowship to W.L. and grant GOA/011/2008 (KU Leuven Research Council).
Footnotes
Published ahead of print 27 April 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Akama H, et al. 2004. Crystal structure of the drug discharge outer membrane protein, OprM, of Pseudomonas aeruginosa: dual modes of membrane anchoring and occluded cavity end. J. Biol. Chem. 279: 52816–52819 [DOI] [PubMed] [Google Scholar]
- 2.Ansari MZ, Yadav G, Gokhale RS, Mohanty D. 2004. NRPS-PKS: a knowledge-based resource for analysis of NRPS/PKS megasynthases. Nucleic Acids Res. 32: W405–W413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aziz RK, et al. 2008. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balibar CJ, Vaillancourt FH, Walsh CT. 2005. Generation of d amino acid residues in assembly of arthrofactin by dual condensation/epimerization domains. Chem. Biol. 12: 1189–1200 [DOI] [PubMed] [Google Scholar]
- 5.Berti AD, Greve NJ, Christensen QH, Thomas MG. 2007. Identification of a biosynthetic gene cluster and the six associated lipopeptides involved in swarming motility of Pseudomonas syringae pv. tomato DC3000. J. Bacteriol. 189: 6312–6323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cui X, Harling R. 2005. N-Acyl-homoserine lactone-mediated quorum sensing blockage, a novel strategy for attenuating pathogenicity of Gram-negative bacterial plant pathogens. Eur. J. Plant Pathol. 111: 327–339 [Google Scholar]
- 7.D'aes J, De Maeyer K, Pauwelyn E, Höfte M. 2010. Biosurfactants in plant-Pseudomonas interactions and their importance to biocontrol. Environ. Microbiol. Rep. 2: 359–372 [DOI] [PubMed] [Google Scholar]
- 8.de Bruijn I, de Kock MJ, de Waard P, van Beek TA, Raaijmakers JM. 2008. Massetolide A biosynthesis in Pseudomonas fluorescens. J. Bacteriol. 190: 2777–2789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Bruijn I, et al. 2007. Genome-based discovery, structure prediction and functional analysis of cyclic lipopeptide antibiotics in Pseudomonas species. Mol. Microbiol. 63: 417–428 [DOI] [PubMed] [Google Scholar]
- 10.de Bruijn I, Raaijmakers JM. 2009. Diversity and functional analysis of LuxR-type transcriptional regulators of cyclic lipopeptide biosynthesis in Pseudomonas fluorescens. Appl. Environ. Microbiol. 75: 4753–4761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Keersmaecker SC, et al. 2005. Chemical synthesis of (S)-4,5-dihydroxy-2,3-pentanedione, a bacterial signal molecule precursor, and validation of its activity in Salmonella typhimurium. J. Biol. Chem. 280: 19563–19568. [DOI] [PubMed] [Google Scholar]
- 12.Drummond A, et al. 2011. Geneious, version 5.5.3. Geneious, Auckland, New Zealand [Google Scholar]
- 13.Dubern JF, Coppoolse ER, Stiekema WJ, Bloemberg GV. 2008. Genetic and functional characterization of the gene cluster directing the biosynthesis of putisolvin I and II in Pseudomonas putida strain PCL1445. Microbiology 154: 2070–2083 [DOI] [PubMed] [Google Scholar]
- 14.Dubern JF, Lugtenberg BJ, Bloemberg GV. 2006. The ppuI-rsaL-ppuR quorum-sensing system regulates biofilm formation of Pseudomonas putida PCL1445 by controlling biosynthesis of the cyclic lipopeptides putisolvins I and II. J. Bacteriol. 188: 2898–2906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Etchegaray A, et al. 2008. Effect of a highly concentrated lipopeptide extract of Bacillus subtilis on fungal and bacterial cells. Arch. Microbiol. 190: 611–622 [DOI] [PubMed] [Google Scholar]
- 16.Fredslund L. 2008. Microbial degradation of xenobiotics in soil-spatial variation, bacterial motility and chemotaxis. Ph.D. ; thesis. University of Copenhagen, Copenhagen, Denmark [Google Scholar]
- 17.Grewal SI, Han B, Johnstone K. 1995. Identification and characterization of a locus which regulates multiple functions in Pseudomonas tolaasii, the cause of brown blotch disease of Agaricus bisporus. J. Bacteriol. 177: 4658–4668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gross H, Loper JE. 2009. Genomics of secondary metabolite production by Pseudomonas spp. Nat. Prod. Rep. 26: 1408–1446 [DOI] [PubMed] [Google Scholar]
- 19.Gross H, et al. 2007. The genomisotopic approach: a systematic method to isolate products of orphan biosynthetic gene clusters. Chem. Biol. 14: 53–63 [DOI] [PubMed] [Google Scholar]
- 20.Haas D, Défago G. 2005. Biological control of soil-borne pathogens by fluorescent pseudomonads. Nat. Rev. Microbiol. 3: 307–319 [DOI] [PubMed] [Google Scholar]
- 21.Hrabak EM, Willis DK. 1992. The lemA gene required for pathogenicity of Pseudomonas syringae pv. syringae on bean is a member of a family of two-component regulators. J. Bacteriol. 174: 3011–3020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koch B, et al. 2002. Lipopeptide production in Pseudomonas sp. strain DSS73 is regulated by components of sugar beet seed exudate via the Gac two-component regulatory system. Appl. Environ. Microbiol. 68: 4509–4516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuiper I, et al. 2004. Characterization of two Pseudomonas putida lipopeptide biosurfactants, putisolvin I and II, which inhibit biofilm formation and break down existing biofilms. Mol. Microbiol. 51: 97–113 [DOI] [PubMed] [Google Scholar]
- 24.Lapouge K, Schubert M, Allain FH, Haas D. 2008. Gac/Rsm signal transduction pathway of γ-proteobacteria: from RNA recognition to regulation of social behaviour. Mol. Microbiol. 67: 241–253 [DOI] [PubMed] [Google Scholar]
- 25.Li W. 2012. Molecular study of Pseudomonas putida antagonism against pathogenic Pseudomonas and Xanthomonas. Ph.D. thesis. KU Leuven, Leuven, Belgium [Google Scholar]
- 26.Li W, et al. 2011. Promysalin, a salicylate-containing Pseudomonas putida antibiotic, promotes surface colonization and selectively targets other Pseudomonas. Chem. Biol. 18: 1320–1330 [DOI] [PubMed] [Google Scholar]
- 27.Lim SP, Roongsawang N, Washio K, Morikawa M. 2009. Flexible exportation mechanisms of arthrofactin in Pseudomonas sp. MIS38. J. Appl. Microbiol. 107: 157–166 [DOI] [PubMed] [Google Scholar]
- 28.Lo Cantore P, et al. 2006. Biological characterization of white line-inducing principle (WLIP) produced by Pseudomonas reactans NCPPB1311. Mol. Plant Microbe Interact. 19: 1113–1120 [DOI] [PubMed] [Google Scholar]
- 29.Mendes R, et al. 2011. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 332: 1097–1100 [DOI] [PubMed] [Google Scholar]
- 30.Modali SD, Zgurskaya HI. 2011. The periplasmic membrane proximal domain of MacA acts as a switch in stimulation of ATP hydrolysis by MacB transporter. Mol. Microbiol. 81: 937–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mortishire-Smith RJ, et al. 1991. Determination of the structure of an extracellular peptide produced by the mushroom saprotroph Pseudomonas reactans. Tetrahedron 47: 3645–3654 [Google Scholar]
- 32.Munsch P, Alatossava T. 2002. Several pseudomonads, associated with the cultivated mushrooms Agaricus bisporus or Pleurotus sp., are hemolytic. Microbiol. Res. 157: 311–315 [DOI] [PubMed] [Google Scholar]
- 33.Nybroe O, Sørensen J. 2004. Production of cyclic lipopeptides by fluorescent Pseudomonads, p 147–172 In Ramos J-L. (ed), Pseudomonas, vol 3 Kluwer Academic/Plenum Publishers, New York, NY [Google Scholar]
- 34.Parret AHA, Schoofs G, Proost P, De Mot R. 2003. Plant lectin-like bacteriocin from a rhizosphere-colonizing Pseudomonas isolate. J. Bacteriol. 185: 897–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiu D, Damron FH, Mima T, Schweizer HP, Yu HD. 2008. PBAD-based shuttle vectors for functional analysis of toxic and highly regulated genes in Pseudomonas and Burkholderia spp. and other bacteria. Appl. Environ. Microbiol. 74: 7422–7426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raaijmakers JM, de Bruijn I, de Kock MJ. 2006. Cyclic lipopeptide production by plant-associated Pseudomonas spp.: diversity, activity, biosynthesis, and regulation. Mol. Plant Microbe Interact. 19: 699–710 [DOI] [PubMed] [Google Scholar]
- 37.Raaijmakers JM, de Bruijn I, Nybroe O, Ongena M. 2010. Natural functions of lipopeptides from Bacillus and Pseudomonas: more than surfactants and antibiotics. FEMS Microbiol. Rev. 34: 1037–1062 [DOI] [PubMed] [Google Scholar]
- 38.Rainey PB, Brodey CL, Johnstone K. 1991. Biological properties and spectrum of activity of tolaasin, a lipodepsipeptide toxin produced by the mushroom pathogen Pseudomonas tolaasii. Physiol. Mol. Plant Pathol. 39: 57–70 [Google Scholar]
- 39.Rainey PB, Brodey CL, Johnstone K. 1993. Identification of a gene cluster encoding three high-molecular-weight proteins, which is required for synthesis of tolaasin by the mushroom pathogen Pseudomonas tolaasii. Mol. Microbiol. 8: 643–652 [DOI] [PubMed] [Google Scholar]
- 40.Reder-Christ K, et al. 2012. Model membrane studies for characterization of different antibiotic activities of lipopeptides from Pseudomonas. Biochim. Biophys. Acta 1818: 566–573 [DOI] [PubMed] [Google Scholar]
- 41.Rokni-Zadeh H, Mangas-Losada A, De Mot R. 2011. PCR detection of novel non-ribosomal peptide synthetase genes in lipopeptide-producing Pseudomonas. Microb. Ecol. 62: 941–947 [DOI] [PubMed] [Google Scholar]
- 42.Roongsawang N, et al. 2003. Cloning and characterization of the gene cluster encoding arthrofactin synthetase from Pseudomonas sp. MIS38. Chem. Biol. 10: 869–880 [DOI] [PubMed] [Google Scholar]
- 43.Roongsawang N, Washio K, Morikawa M. 2010. Diversity of nonribosomal peptide synthetases involved in the biosynthesis of lipopeptide biosurfactants. Int. J. Mol. Sci. 12: 141–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rottig M, et al. 2011. NRPSpredictor2—a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 39: W362–W367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ryan RP, et al. 2011. Pathogenomics of Xanthomonas: understanding bacterium-plant interactions. Nat. Rev. Microbiol. 9: 344–355 [DOI] [PubMed] [Google Scholar]
- 46.Saini HS, et al. 2008. Efficient purification of the biosurfactant viscosin from Pseudomonas libanensis strain M9-3 and its physicochemical and biological properties. J. Nat. Prod. 71: 1011–1015 [DOI] [PubMed] [Google Scholar]
- 47.Sinnaeve D, et al. 2009. The solution structure and self-association properties of the cyclic lipodepsipeptide pseudodesmin A support its pore-forming potential. Chemistry 15: 12653–12662 [DOI] [PubMed] [Google Scholar]
- 48.Sinnaeve D, et al. 2009. Structure and X-ray conformation of pseudodesmins A and B, two new cyclic lipodepsipeptides from Pseudomonas bacteria. Tetrahedron 65: 4173–4181 [Google Scholar]
- 49.Steindler L, Bertani I, De Sordi L, Bigirimana J, Venturi V. 2008. The presence, type and role of N-acyl homoserine lactone quorum sensing in fluorescent Pseudomonas originally isolated from rice rhizospheres are unpredictable. FEMS Microbiol. Lett. 288: 102–111 [DOI] [PubMed] [Google Scholar]
- 50.Vallet-Gely I, et al. 2010. Association of hemolytic activity of Pseudomonas entomophila, a versatile soil bacterium, with cyclic lipopeptide production. Appl. Environ. Microbiol. 76: 910–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vlassak K, Holm L, Duchateau L, Vanderleyden J, De Mot R. 1992. Isolation and characterization of fluorescent Pseudomonas associated with the roots of rice and banana grown in Sri Lanka. Plant Soil 145: 51–63 [Google Scholar]
- 52.Wang N, Lu SE, Wang J, Chen ZJ, Gross DC. 2006. The expression of genes encoding lipodepsipeptide phytotoxins by Pseudomonas syringae pv. syringae is coordinated in response to plant signal molecules. Mol. Plant Microbe Interact. 19: 257–269 [DOI] [PubMed] [Google Scholar]
- 53.Washio K, Lim SP, Roongsawang N, Morikawa M. 2010. Identification and characterization of the genes responsible for the production of the cyclic lipopeptide arthrofactin by Pseudomonas sp. MIS38. Biosci. Biotechnol. Biochem. 74: 992–999 [DOI] [PubMed] [Google Scholar]
- 54.Wong WC, Preece TF. 1979. Identification of Pseudomonas tolaasii: the white line in agar and mushroom tissue block rapid pitting tests. Appl. Bacteriol. 47: 401–407 [Google Scholar]
- 55.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18: 821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.