

Abstract
Escherichia coli contains multiple 3′ to 5′ RNases, of which two, RNase PH and polynucleotide phosphorylase (PNPase), use inorganic phosphate as a nucleophile to catalyze RNA cleavage. It is known that an absence of these two enzymes causes growth defects, but the basis for these defects has remained undefined. To further an understanding of the function of these enzymes, the degradation pattern of different cellular RNAs was analyzed. It was observed that an absence of both enzymes results in the appearance of novel mRNA degradation fragments. Such fragments were also observed in strains containing mutations in RNase R and PNPase, enzymes whose collective absence is known to cause an accumulation of structured RNA fragments. Additional experiments indicated that the growth defects of strains containing RNase R and PNPase mutations were exacerbated upon RNase PH removal. Taken together, these observations suggested that RNase PH could play a role in structured RNA degradation. Biochemical experiments with RNase PH demonstrated that this enzyme digests through RNA duplexes of moderate stability. In addition, mapping and sequence analysis of an mRNA degradation fragment that accumulates in the absence of the phosphorolytic enzymes revealed the presence of an extended stem-loop motif at the 3′ end. Overall, these results indicate that RNase PH plays a novel role in the degradation of structured RNAs and provides a potential explanation for the growth defects caused by an absence of the phosphorolytic RNases.
INTRODUCTION
Like most other organisms, Escherichia coli contains a number of exo-RNases that participate in many aspects of RNA metabolism. Among the main cellular functions of these factors are the degradation of fragments generated by the endonucleolytic cleavage of unstable RNAs and 3′-end processing of stable RNAs, such as tRNA and rRNA (3, 14). Of the eight known E. coli exo-RNases, the enzymes RNase II, RNase R, polynucleotide phosphorylase (PNPase), and Oligoribonuclease (Orn) are the main effectors of RNA fragment digestion. The known properties of these enzymes are consistent with some degree of specialization of function. RNase II is an abundant enzyme that digests unstructured fragments but is unable to digest through secondary structures. RNase R contains a helicase-like domain that makes it especially effective at digesting through relatively strong secondary structures. PNPase functions in digesting both unstructured RNA fragments and those that have a moderate degree of secondary structure. Orn is capable of acting on very short RNA fragments that are not effectively bound by the other exo-RNases. With respect to their role in RNA processing, since mature tRNAs and rRNAs contain base-paired residues near their 3′ ends, the removal of precursor sequences requires the action of exo-RNases that can digest close to structured regions. RNase T and RNase PH are the most effective in this regard, and these enzymes are primarily involved in the 3′ end maturation of the stable RNAs (21, 23, 24).
Among the E. coli exo-RNases, PNPase and RNase PH differ from the others in that they utilize inorganic phosphate, rather than water, as a nucleophile to catalyze RNA degradation. It has been an open question whether the mechanistic differences in RNA digestion by these enzymes have any functional consequences for cellular RNA metabolism. Interestingly, an absence of either factor has a minimal effect, but an absence of both causes a significant impairment of cell growth (19, 39). Such observations suggest that the phosphorolytic RNases could collectively be involved in some important aspects of RNA metabolism. However, the specific joint functions of these enzymes are not known. In particular, much less is understood about RNase PH compared to PNPase, which has been studied for more than 50 years.
To address the common functions of the phosphorolytic enzymes, different RNAs were analyzed in strains lacking PNPase and RNase PH using Northern blot analysis. Interestingly, in strains lacking both enzymes, an accumulation of mRNA fragments was observed. Similar fragments were also observed in strains containing mutations in PNPase and RNase R, a combination of enzymes previously shown to be involved in the degradation of structured RNAs (9). The evidence presented here suggests that RNase PH has a role in the degradation of a subset of RNA fragments that contain significant amounts of secondary structure, thereby revealing a novel function for this enzyme in RNA metabolism.
MATERIALS AND METHODS
Strains and plasmids.
The wild-type strain used in the present study is MG1655*, a derivative of the sequenced MG1655 strain that has a defective rph gene that was corrected by recombineering (8). Derivatives of these strains containing deletion alleles have been described earlier or were made by transduction of the deletion alleles marked with a kanamycin-resistant (Kanr) cassette from the Keio strain collection into MG1655* (6, 19). For the Δrph, Δrnr, Δhfq, and ΔpcnB alleles, the Kanr cassette was further excised by FLP-mediated recombination. Strains containing a ΔssrA mutation were made by transduction of a ΔssrA::cat allele (37). Strains containing a pnp(Ts) mutation were constructed by P1 transduction of this allele using a chloramphenicol-resistant marker closely linked to pnp (38).
Cell growth measurements.
For each set of growth rate measurements, three colonies for each strain were picked and grown to saturation. The saturated cultures were diluted into prewarmed Luria-Bertani (LB) medium at 37°C, and the optical density at 600 nm was measured four to five times during the exponential growth phase. The doubling time and standard deviation for each strain was determined by linear regression.
Northern blot analysis.
Total RNA was prepared as described previously (2). RNA samples were fractionated on a 1.2% agarose–2.2% formaldehyde gel and transferred to positively charged Nytran membrane. For probing mRNAs, digoxigenin-labeled antisense probes were prepared by in vitro transcription according to the manufacturer's (Roche) instructions. In some instances, oligonucleotide probes were used after labeling with 32P at the 5′ end using polynucleotide kinase. The sequences of the oligonucleotide probes were as follows: probe 1 (5′-CACGGTTAAATCCTTCACCGGGGGATCTGCTC-3′), probe 2 (5′-CAAAGCCAACATACGGGTTAACCTGGTAACCACC-3′), probe 3 (5′-CCAGACGGGTAGCGATTTCAGGAGTGATCGC-3′), probe 4 (5′-CCCAGAACAACTACGGAACCGTCTTTCGGATCC-3′), and probe 5 (5′-GCCTGCGGCTGAGTTACAACGTCTTTGATA-3′).
RNA digestion assays.
Duplex substrates were prepared by annealing a 5′-32P-labeled 34-mer RNA (Fig. 3) with a 2-fold excess of complementary oligonucleotide. A total of 200 to 400 cpm of duplex substrates was left untreated or treated with 0.25 μg of each purified RNase in exonuclease buffer (50 mM morpholinepropanesulfonic acid [pH 7.0], 3 mM MgCl2, 5 mM sodium phosphate, 1 mM dithiothreitol) at 37°C for 1 h, followed by electrophoresis of the resulting products on a 20% polyacrylamide–8 M urea gel. The gels were dried, and the extent of substrate digestion was quantified by phosphorimaging.
Fig 3.

Digestion of duplex RNA by exonucleases. (A to C) Three partial duplex substrates—duplex I, duplex II, and duplex III—were prepared by annealing a 5′-labeled 34-mer RNA with 17-, 14-, and 12-nt complementary oligonucleotides. Each substrate was untreated or treated with the indicated RNase, and the extent of substrate digestion was assessed following gel electrophoresis. The sequence of each duplex is shown on the left of each panel. The migration of the undigested labeled RNA and a product derived by the removal of the 3′ single-stranded residues from the labeled RNA are indicated by dotted lines. (A) Digestion of duplex I. A 17-nt control RNA that corresponds to the part of the labeled RNA that is base-paired in duplex I was also run on the gel (lane 1). (B) Digestion of duplex II. (C) Digestion of duplex III. (D) For each substrate, phosphorimaging of dried gels was performed to quantify the extent of digestion through the duplex region with each enzyme. The mean extent of digestion and standard errors by each enzyme for the different duplexes, derived from five experiments, is indicated. Blue circles, digestion by RNase II; green squares, digestion by RNase T; red diamonds, digestion by RNase PH.
Mapping the boundaries of the OmpA mRNA degradation fragment.
Portions (2 μg) of the total RNA derived from MG1655* or MG1655*Δrph Δpnp ΔpcnB were ligated with 0.2 U of T4 RNA ligase (New England Biolabs) for 3 h at room temperature alongside parallel reactions in which no ligase was added to the RNA. Each set of ligated and mock-ligated RNA samples was annealed with 25 fmol of the oligonucleotide OmpA-Rev (5′-CAGACTTCAGAGTGAAGTGC-3′) by slow cooling from 65 to 35°C and reverse transcribed as described previously (15). The resulting cDNAs were amplified by PCR using the oligonucleotides OmpA-Fwd (5′-GGATCCGAAAGACGGTTCC-3′) and OmpA-Rev. The PCR-derived products were separated on a 2% agarose gel, and the major ∼300-bp fragment derived from ligation of RNA from MG1655*Δrph Δpnp ΔpcnB was sequenced at a commercial facility (GeneWiz).
RESULTS
Accumulation of mRNA degradation fragments in strains lacking PNPase and RNase PH.
In an effort to define common functions of the phosphorolytic RNases, Northern blot analysis was performed on RNA derived from strains lacking either or both of the phosphorolytic RNases. A first set of studies evaluated four relatively well-characterized mRNAs: rpsO, trxA, lpp, and ompA (4, 5, 18, 28). As shown in Fig. 1A, single or double deletion of either pnp or rph had a moderate effect on the rpsO, trxA, or lpp mRNAs. However, with ompA mRNA, an absence of both enzymes resulted in a substantial accumulation of discrete mRNA fragments. Lower levels of these fragments were observed with strains lacking either enzyme, suggesting that a collective absence of both enzymes is responsible for high-level ompA mRNA fragment accumulation. Such observations are reminiscent of other robust processes in which significant RNA metabolism defects are observed only when two or more exonucleases are inactivated (16, 22).
Fig 1.
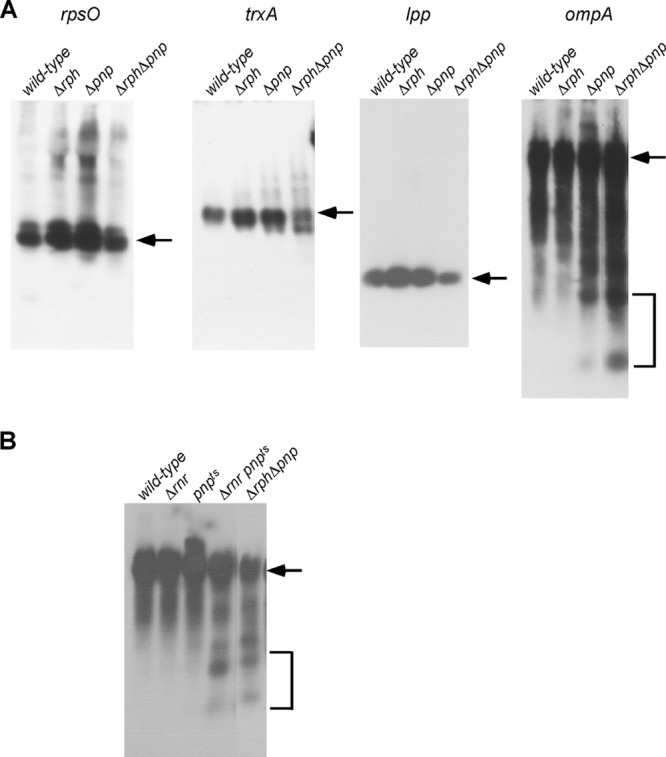
Accumulation of mRNA degradation fragments in strains lacking phosphorolytic exonucleases. (A) Total RNA was isolated from MG1655* (wild type), MG1655*Δrph, MG1655*Δpnp, or MG1655*Δrph Δpnp strains following growth in LB medium at 37°C and was analyzed by Northern blotting using probes to rpsO, trxA, lpp, or ompA mRNA. The positions of the full-length RNAs and the degradation products that accumulate in the absence of the phosphorolytic RNases are indicated by arrows and brackets, respectively. (B) Strains MG1655* or derivatives that contain Δrnr, pnp(Ts), or Δrnr pnp(Ts) mutations were grown to saturation in LB medium at 30°C, diluted 1,000-fold into fresh medium, transferred to 44°C, and harvested for RNA preparation upon reaching mid-log phase. RNA samples were analyzed by Northern blotting with an ompA mRNA probe. RNA derived from MG1655*Δrph Δpnp was loaded as a control.
The presence of the ompA mRNA fragments suggested that this transcript could contain some features that are different from the other mRNAs. One possibility was that the ompA mRNA fragments could contain higher levels of secondary structure, requiring a different set of enzymes for digestion. Currently, the exonucleases implicated in the degradation of structured RNAs in vivo are RNase R and PNPase. RNases R has a unique ability to digest RNA duplexes, possibly due to the presence of a helicase-like domain that opens up RNA duplexes to promote digestion (9, 35). PNPase does not efficiently digest RNA duplexes in vitro, but this enzyme associates strongly with RhlB RNA helicase in vivo, and RNA unwinding by RhlB allows PNPase to digest through moderately stable secondary structures (12, 25, 26, 30). RNase R and PNPase appear to share overlapping roles in structured RNA digestion since high levels of fragment accumulation are only observed when both enzymes are rendered defective (10).
If the ompA mRNA fragments that accumulates in strains lacking PNPase and RNase PH contain high levels of secondary structure, one prediction would be that similar fragments would also be observed in strains containing defects in RNase R and PNPase. Although a removal of both enzymes is not feasible due to the synthetic lethality of rnr and pnp deletions (10), this hypothesis was tested using a strain containing a combination of a deletion mutation in rnr and a temperature-sensitive mutation in pnp. No significant ompA fragment accumulation was observed in a wild-type strain or in strains containing single mutations in either enzyme (Fig. 1B). However, in the strain containing mutations in both rnr and pnp, ompA mRNA fragments similar to those observed in strains lacking RNase PH and PNPase were found to accumulate at a nonpermissive temperature. Because mutations in rnr and pnp are known to be associated with the accumulation of structured RNA fragments, it can be inferred that the ompA mRNA fragments observed in the Δrph Δpnp double-mutant strain also contained high levels of secondary structure, suggesting that RNase PH, like PNPase and RNase R, could be involved in the degradation of such structured RNAs inside the cell.
RNase PH, PNPase, and RNase R function in a common pathway.
If RNase PH functions in a degradation pathway common to RNase R and PNPase to digest structured RNAs, one prediction would be that the introduction of a Δrph mutation into a strain containing RNase R and PNPase mutations would result in an exacerbated growth defect. Noting that a combination of rnr deletion and a pnp(Ts) mutation renders cells sensitive to temperature, a specific prediction was that the incorporation of a Δrph mutation into a strain carrying these mutations would increase the temperature sensitivity of the resulting strain. To determine whether that is the case, a doubly mutated Δrnr pnp(Ts) strain and a triply mutated Δrph Δrnr pnp(Ts) strain were streaked on agar plates at different temperatures. As controls, the wild-type strain MG1655* and MG1655*Δrph were also streaked. At 30°C each strain grew comparably well, indicating that the temperature-sensitive PNPase enzyme retains considerable activity at this temperature (Fig. 2A). In contrast, when streaked at 42°C, both MG1655*Δrnr pnp(Ts) and MG1655*Δrph Δrnr pnp(Ts) failed to form colonies, indicating a nearly complete inactivation of PNPase at this temperature. At an intermediate temperature (37°C), however, the growth of the triply mutated strain was noticeably worse than the double mutant, indicating that the introduction of a Δrph mutation into a Δrnr pnp(Ts) strain background has a negative effect on growth. These observations were corroborated by growth rate measurements on the four strains at 37°C (Fig. 2B). Both MG1655* and MG1655*Δrph grew with a doubling time of 25 to 30 min, indicating that the Δrph mutation, by itself, does not cause any significant defects, whereas doubling time of MG1655*Δrnr pnp(Ts) was moderately increased to 38 min. In contrast, the triply mutated MG1655*Δrph Δrnr pnp(Ts) strain grew with a doubling time of 87 min, considerably slower than the other strains. Thus, the Δrph mutation confers a growth defect only when the other two enzymes involved in structured RNA turnover have been partially inactivated. These observations reinforce the notion that RNase PH functions in a pathway that involves both PNPase and RNase R.
Fig 2.

RNase PH shares overlapping functions with RNase R and PNPase. (A) MG1655*, MG1655* Δrph, MG1655* Δrnr pnp(Ts), and MG1655* Δrph Δrnr pnp(Ts) strains were streaked on LB plates and incubated overnight at 30, 42, or 37°C as indicated. (B) Growth rates. Strains analyzed in panel A were grown in LB medium at 37°C, and the doubling times were determined by monitoring the optical density at different times during the exponential phase. The measured doubling times and standard deviations are indicated.
Digestion of RNA duplexes by RNase PH in vitro.
The experiments described above suggested that RNase PH could be contributing to the digestion of structured RNA in vivo. To determine whether RNase PH can digest duplex RNA in vitro, three related RNA duplexes were prepared and treated with purified RNases. Each duplex contained a common 5′ labeled 34-nucleotide (nt) RNA that was annealed to a complementary oligonucleotide of different lengths (Fig. 3). These three substrates form 17, 14, and 12-bp duplexes and are denoted as duplexes I, II, and III, respectively. In each case, a common 17-nt single-stranded poly(A) tail on the labeled RNA constituted a loading platform for RNases to initiate digestion. In addition to RNase PH, three other RNases were used as controls. RNase R, well known for its ability to digest through duplex RNA, was used as a positive control. RNase II, which is known to exhibit low activity on RNA duplexes and RNase T were used as additional controls (11).
The addition of each enzyme to duplex I resulted in some degree of RNA digestion in each case (Fig. 3A). With RNase R, all of the labeled RNA, including the base-paired region, was digested to limit products (Fig. 3A, lane 6), indicating that a 17-bp RNA duplex is not inhibitory to its action, as has been shown previously for this substrate (9). With RNase II and RNase T, digestion of the single-stranded segment of the labeled RNA was initiated efficiently, but each enzyme stalled near the interface of the single-stranded and double-stranded regions. With RNase II, the digested substrates retained three to nine single-stranded residues (Fig. 3A, lane 5), a finding consistent with its inability to digest close to a duplex (27, 33). With RNase T, digestion proceeded further and the major product was one that had all of the single-stranded residues removed (Fig. 3A, lane 3). A quantification of the extent of digestion indicated that both RNase II and RNase T digested ca. 25% of the substrate through the duplex region, whereas the remainder of the substrate had only the aforementioned single-stranded residues removed (Fig. 3D). With RNase PH, the extent of single-strand digestion was similar to that observed with RNase T (Fig. 3A, lane 4). A quantification of the data, however, revealed that a significantly higher percentage of the substrate, ca. 60%, was digested to limit products, indicating an increased propensity to digest through duplexes compared to either RNase II or RNase T.
Qualitatively similar results were obtained using Duplex II, which contained three fewer base pairs compared to duplex I. In each case, both the pattern and the extent of digestion of this substrate by each of the four enzymes was comparable to that observed with duplex I (Fig. 3B and D). Finally, digestions were performed with duplex III, which contained two fewer base-pairs compared to duplex II (Fig. 3C). In this case, an increased degree of digestion through the duplex region was observed with each enzyme (Fig. 3D), possibly due to increased breathing of the shortened duplex. Remarkably, compared to RNase II and RNase T, the efficiency of RNase PH digestion increased sharply, resulting in nearly complete digestion through the duplex, similar to what was observed with RNase R. Thus, RNase PH appears to have a high propensity to digest through short regions of duplex RNA. Overall, these studies show that RNase PH can digest through structured RNA, and the efficiency of digestion is dramatically increased on substrates that contain short duplexes.
Characterization of an ompA mRNA degradation fragment.
Given the ability of RNase PH to digest secondary structures, it was of interest to further characterize the ompA mRNA degradation fragments observed in strains lacking RNase PH and PNPase (Fig. 1). One prediction was that these enzymes are needed for digestion because the fragments contain secondary structures near the 3′ end that impede digestion by single-strand specific RNases.
First, to understand the potential role of other trans-acting factors in the formation of the ompA mRNA degradation fragments, three different deletion mutants were introduced into strain MG1655*Δrph Δpnp. A first mutation that inactivates Hfq was introduced to determine whether the formation of the degradation fragments could be dependent upon small-regulatory RNAs (sRNAs). Hfq has been shown to be important for mediating the pairing of many sRNAs to mRNAs leading to mRNA destabilization in many instances (1, 32). In particular, Hfq promotes the association of the micA sRNA to ompA, resulting in the downregulation of the latter (34). It was therefore of interest to investigate whether the formation of the ompA fragments is dependent upon Hfq. Accordingly, RNA derived from MG1655*Δrph Δpnp and a derivative containing a Δhfq mutation were analyzed by Northern blotting (Fig. 4A). A significant reduction in the abundance of the ompA mRNA degradation fragment was observed in the Δhfq derivative, suggesting that these fragments are generated by the action of sRNAs. It should be noted that Hfq has been found to interact with RNase E, a major E. coli endonuclease, and therefore it is feasible that the tripartite interaction between ompA RNA, sRNAs, and Hfq promotes RNase E cleavage to generate the observed ompA mRNA degradation fragments. However, this point remains to be established. A second mutation tested (ΔssrA) deletes tmRNA, a key molecule involved in the release of stalled ribosomes from mRNAs containing translational stalling sequences, including those lacking a stop codon (20). This mutation was tested to ascertain whether the generation of ompA mRNA fragments could be related to translational stalling. However, only nominal differences were found in the levels of ompA mRNA fragments between strains lacking or containing the ΔssrA mutation, suggesting that tmRNA does not have a significant role in the generation of these fragments. Finally, a ΔpcnB mutation was tested to investigate the role of poly(A) polymerase (PAP), an enzyme that is encoded by pcnB. When RNA fragments contain secondary structures at the 3′ end, the incorporation of poly(A) residues by PAP to the RNA 3′ end, in many instances, provides a toehold for RNases to attach to these RNAs and promote their degradation (31). Since the ompA mRNA fragments being investigated were expected to contain secondary structures, it was feasible that polyadenylation of these fragments could be important for their turnover. This turned out to be the case, because when tested, a dramatic increase in the amount of the ompA mRNA degradation fragment was observed in the strain containing a ΔpcnB mutation. Thus, PAP appears to be critically important for promoting the degradation of these structured mRNA fragments.
Fig 4.

Analysis of an ompA mRNA degradation fragment. (A) Northern blot analysis of total RNA derived from strain MG1655*Δrph Δpnp (lane 1), as well as derivatives containing Δhfq (lane 2), ΔssrA (lane 3), or ΔpcnB (lane 4) mutations, using an ompA mRNA probe. The positions of the full-length RNAs and a major degradation product that accumulates in the absence of the phosphorolytic RNases are indicated by thick and thin arrows, respectively. (B) Schematic description of ompA mRNA. The mRNA is denoted by a horizontal line with the 5′ UTR, coding region, and the 3′ UTR separated, as indicated. The approximate location of the oligonucleotide probes used for mapping of the ompA degradation fragment is shown. (C) Northern blot analysis of ompA mRNA derived from MG1655*Δrph Δpnp using the ompA-specific oligonucleotide probes shown in panel B. The positions of the full-length ompA RNA and the degradation product are indicated by thick and thin arrows, respectively. (D) On the left is the strategy for identifying the boundaries of the ompA mRNA degradation fragment. Total RNA was first ligated, followed by reverse transcription using an ompA-specific primer (OmpA-Rev) that lies within the ompA degradation fragment. The resulting cDNA population was amplified using the divergently oriented ompA primers OmpA-Rev and OmpA-Fwd and was expected to yield a DNA product derived from the ompA degradation fragment whose 5′ and 3′ ends have become ligated. On the right, gel electrophoresis of the PCR products generated following RNA ligation and amplification was performed. Lane M, 100-bp marker; lanes 1 and 2, PCR products derived using unligated or ligated RNA from strain MG1655*; lanes 3 and 4, PCR products derived using unligated or ligated RNA from strain MG1655* Δrph Δpnp ΔpcnB. A product of the size expected for the ompA mRNA degradation fragment is indicated by an arrow. (E) Sequence of the 5′ and 3′ termini of the PCR product of the ompA mRNA degradation fragment. The PCR product derived from ligation of total RNA from strain MG1655*Δrph Δpnp ΔpcnB was sequenced using the primers OmpA-Rev (top panel) or OmpA-Fwd (bottom panel). The sequences and traces corresponding to the 5′ and 3′ extremities of the core ompA mRNA degradation fragment are shown. The trace of the top panel corresponds to the antisense strand, and the trace of the bottom panel corresponds to the sense strand. (F) Secondary structure of an extended stem-loop motif found at the 3′ end of the ompA mRNA degradation fragment. Base-paired residues in the hairpin region are indicated by horizontal lines. The last four nucleotides of the core degradation fragment are also shown.
A second question of interest was to define the sequence of the degradation fragment. By Northern blot analysis, the size of the major ompA mRNA degradation fragment was estimated to be ∼400 nt. Since the full-length ompA mRNA, including the 5′ and 3′ untranslated regions (UTRs), is 1.2 kb in size, to determine which part of the full-length transcript comprises this fragment, Northern blot analysis was performed on total RNA from strain MG1655*Δrph Δpnp using five oligonucleotide probes that correspond to different regions of the ompA transcript (Fig. 4B). Probe 1 is complementary to the ompA 5′ UTR and binds to a region 53 to 85 nt upstream of the start codon. Probes 2 to 5 are complementary to the coding region, binding to nt 184 to 217, 451 to 481, 744 to 776, and 1008 to 1037 downstream of the translation start site, respectively. With the different oligonucleotide probes, only probe 4 showed the presence of the ompA mRNA degradation fragment (Fig. 4C). From these analyses, it can be inferred that the boundaries of degradation fragment lie between nt 451 and 1037 downstream of the ompA translation start site.
To more precisely define the 5′ and 3′ ends of the ompA mRNA degradation fragment, total RNA derived from MG1655* Δrph Δpnp ΔpcnB, which accumulates high levels of the ompA mRNA degradation fragment, was circularized using RNA ligase. The resulting product was reverse transcribed using an oligonucleotide primer complementary to nt 639 to 658 of the ompA coding region, followed by PCR using this primer and second primer that corresponds to nt 744 to 762 of the ompA sense strand. Given the 85-nt separation between the two primers, it was anticipated that upon ligase treatment, the 5′ and 3′ ends of the ompA mRNA degradation fragment would become joined and allow amplification by this set of otherwise divergent primers to yield a product of ∼0.3 kb. Indeed, a DNA product of the expected size was observed after PCR that was neither observed in the absence of RNA ligation nor present at appreciable levels when RNA from MG1655* was used instead (Fig. 4D). The PCR product was sequenced to provide information about the extent of ompA sequences in the degradation fragment, with the expectation that the sequences at the 5′ end and 3′ ends of this fragment would be juxtaposed. As expected, the sequence of PCR product provided well-defined sequences corresponding to ompA residues 514 to 883 (Fig. 4E). However, instead of a clear sequence transition from the 5′ to the 3′ end of the degradation fragment, as expected due to RNA circularization, the sequence beyond these residues was of mixed composition. This result can be explained if the population of ompA mRNA degradation fragments that is ligated and amplified contains a core region but different members of this population contain terminal sequences of various lengths that causes sequence heterogeneity. This scenario is not unexpected and could be due either to variability in 3′ end trimming or to heterogeneity of the 5′ end cleavage. From these experiments, it can be inferred that the ompA mRNA degradation fragment contains a core 370-nt region that corresponds to nt 514 to 883, with variable extensions among the difference molecules in this population.
Finally, to address the question of whether the accumulation of the ompA mRNA degradation fragment in the absence of the phosphorolytic enzymes is associated with secondary structures, the core region of the degradation product was computationally folded to identify potential secondary structures. Strikingly, a stable extended stem-loop structure (ΔG = −14.6 kcal/mol) was observed near the end of the ompA mRNA degradation fragment, with this motif ending four nucleotides from the 3′ end (Fig. 4F). The presence of this motif near the 3′ terminus of the ompA mRNA degradation fragment strongly suggests that it could be impeding the action of the more single-strand specific RNases, and its removal therefore requires the action of RNases that are effective at digesting through such secondary structures.
DISCUSSION
E. coli harbors more than 20 RNases, whose functions have been characterized to various extents (20). Among the eight known exonucleases, RNase PH and PNPase differ in the mechanism of RNA cleavage, since they both use inorganic phosphate rather than water as a nucleophile to catalyze bond scission (13). Previously, it was found than the absence of both enzymes causes significant growth and ribosomal defects, suggesting that the two enzymes together participate in some important cellular functions. Recently, it has been shown that the overexpression of RNase R or RNase II can partially suppress these growth defects, suggesting that the cellular activities of the phosphorolytic enzymes overlap with the other RNases (19). Nonetheless, the common functions of these phosphorolytic enzymes have remained unclear.
While characterizing the defects of strains lacking PNPase and RNase PH, an accumulation of fragments derived from ompA mRNA was observed (Fig. 1A). Similar fragments were observed in a strain significantly lacking in PNPase and RNase R activity (Fig. 1B). Since the latter set of enzymes is known to be collectively involved in the degradation of structured RNA fragments, these observations suggested that RNase PH could also be involved in a similar process. Biochemical analysis of RNase PH on duplex substrates confirmed that this enzyme can digest through RNA duplexes of moderate stability that are relatively recalcitrant to RNase II and RNase T (Fig. 3). Additional support for a role for RNase PH in structured RNA degradation comes from the observation that a triple mutant containing Δrph Δrnr pnp(Ts) mutations is much more sensitive to temperature compared to the Δrnr pnp(Ts) strain (Fig. 2). Collectively, the evidence suggests that RNase PH plays a role in structured RNA digestion, which constitutes a novel finding given that RNase PH was previously believed to be primarily involved in digesting single-stranded precursor regions on small RNAs.
Interestingly, two recently identified functions for RNase PH are compatible with a role for this enzyme in structured RNA digestion. First, RNase PH has been found to play a role in initiating the degradation of rRNA upon starvation of E. coli cultures (7). Although the details concerning its role in this process are not known, it is conceivable, given that rRNAs possess high levels of secondary structure, that RNase PH is involved in digestion through structured regions within these molecules. Second, our laboratory has recently shown that RNase PH has a prominent role in the 3′ end maturation of 23S rRNA, a process that includes the removal of five base-paired residues within an extended stem region (17). The ability of RNase PH to digest through secondary structures provides a potential explanation for how this enzyme could be removing these residues.
Despite many years of investigation, very little is known about the naturally occurring structured RNA elements that protect mRNAs from the abundant single-strand specific exoribonucleolytic activities present in the cell. Resistance to digestion by such enzymes has been observed in fragments derived from rRNAs and in mRNAs that contain copies of the commonly occurring palindromic repetitive extragenic sequence (REP) motif that is present in a number of genes (9, 29). As part of these investigations, a prominent ompA mRNA degradation fragment that is stabilized in strains lacking PNPase and RNase PH was characterized by mapping and sequence analysis. Significantly, this mRNA fragment contains a stable hairpin-loop motif at its end, which provides a potential explanation for why the degradation of this fragment requires enzymes that can digest through secondary structures. The ompA mRNA degradation fragment defined here should serve as a useful model mRNA substrate to further explore the role of these enzymes in structured RNA degradation.
An important question that arises is whether the biological functions of these RNases can be explained in terms of their known properties. PNPase and RNase R are collectively required for viability, and high levels of structured RNA fragments are observed in strains containing defects in both enzymes, suggesting that these two enzymes are key factors involved in the turnover of these RNAs (9, 10). However, given the observation that cells lacking PNPase and RNase PH grow poorly and also exhibit an accumulation of such RNA fragments, this suggests that some aspect of structured RNA degradation is carried out by this combination of enzymes that cannot be carried out by RNase R alone. Although RNase R is a potent duplex-degrading enzyme in vitro, it is conceivable that the effectiveness of substrate digestion by this enzyme in vivo is limited by its inability to access certain substrates whose accumulation leads to cellular toxicity. For example, RNase PH and PNPase may be better able to recognize and digest some substrates, such as those that have short 3′ single-stranded overhangs, since RNase R needs a single-stranded “landing pad” of more than 4 nt to initiate digestion (36). A requirement for these phosphorolytic enzymes to digest such transcripts could explain why an absence of these enzymes causes growth defects. Further work on the mechanism and consequences of substrate recognition will be required to elucidate both the distinct and overlapping roles of these different enzymes inside the cell.
ACKNOWLEDGMENTS
I thank Murray Deutscher for comments on an earlier version of the manuscript and Arun Malhotra for the gift of purified RNases.
This study was supported by a grant from the National Institutes of Health (GM81735).
Footnotes
Published ahead of print 18 May 2012
REFERENCES
- 1. Aiba H. 2007. Mechanism of RNA silencing by Hfq-binding small RNAs. Curr. Opin. Microbiol. 10:134–139 [DOI] [PubMed] [Google Scholar]
- 2. Aiba H, Adhya S, de Crombrugghe B. 1981. Evidence for two functional gal promoters in intact Escherichia coli cells. J. Biol. Chem. 256:11905–11910 [PubMed] [Google Scholar]
- 3. Andrade JM, Pobre V, Silva IJ, Domingues S, Arraiano CM. 2009. The role of 3′-5′ exoribonucleases in RNA degradation. Prog. Mol. Biol. Transl. Sci. 85:187–229 [DOI] [PubMed] [Google Scholar]
- 4. Arnold TE, Yu J, Belasco JG. 1998. mRNA stabilization by the ompA 5′ untranslated region: two protective elements hinder distinct pathways for mRNA degradation. RNA 4:319–330 [PMC free article] [PubMed] [Google Scholar]
- 5. Arraiano CM, Yancey SD, Kushner SR. 1988. Stabilization of discrete mRNA breakdown products in ams pnp rnb multiple mutants of Escherichia coli K-12. J. Bacteriol. 170:4625–4633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baba T, et al. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006–2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Basturea GN, Zundel MA, Deutscher MP. 2011. Degradation of rRNA during starvation: comparison to quality control during steady-state growth and a role for RNase PH. RNA 17:338–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blattner FR, et al. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1474 [DOI] [PubMed] [Google Scholar]
- 9. Cheng ZF, Deutscher MP. 2005. An important role for RNase R in mRNA decay. Mol. Cell 17:313–318 [DOI] [PubMed] [Google Scholar]
- 10. Cheng ZF, Deutscher MP. 2003. Quality control of rRNA mediated by polynucleotide phosphorylase and RNase R. Proc. Natl. Acad. Sci. U. S. A. 100:6388–6393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coburn GA, Mackie GA. 1996. Overexpression, purification, and properties of Escherichia coli ribonuclease II. J. Biol. Chem. 271:1048–1053 [DOI] [PubMed] [Google Scholar]
- 12. Coburn GA, Miao X, Briant DJ, Mackie GA. 1999. Reconstitution of a minimal RNA degradosome demonstrates functional coordination between a 3′ exonuclease and a DEAD-box RNA helicase. Genes Dev. 13:2594–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deutscher MP, Marshall GT, Cudny H. 1988. RNase PH: an Escherichia coli phosphate-dependent nuclease distinct from polynucleotide phosphorylase. Proc. Natl. Acad. Sci. U. S. A. 85:4710–4714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deutscher MP, Li Z. 2001. Exoribonucleases and their multiple roles in RNA metabolism. Prog. Nucleic Acids Res. Mol. Biol. 66:67–105 [DOI] [PubMed] [Google Scholar]
- 15. Diwa A, Bricker AL, Jain C, Belasco JG. 2000. An evolutionarily conserved RNA stem-loop functions as a sensor that directs feedback regulation of RNase E gene expression. Genes Dev. 14:1249–1260 [PMC free article] [PubMed] [Google Scholar]
- 16. Donovan WP, Kushner SR. 1986. Polynucleotide phosphorylase and ribonuclease II are required for cell viability and mRNA turnover in Escherichia coli K-12. Proc. Natl. Acad. Sci. U. S. A. 83:120–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gutgsell NS, Jain C. 2012. Role of precursor sequences in the ordered maturation of Escherichia coli 23S rRNA. RNA 18:345–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hajnsdorf E, Steier O, Coscoy L, Teysset L, Regnier P. 1994. Roles of RNase E, RNase II, and PNPase in the degradation of the rpsO transcripts of Escherichia coli: stabilizing function of RNase II and evidence for efficient degradation in an ams pnp rnb mutant. EMBO J. 13:3368–3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jain C. 2009. Identification and characterization of growth suppressors of Escherichia coli strains lacking phosphorolytic ribonucleases. J. Bacteriol. 191:5622–5627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Janssen BD, Hayes CS. 2012. The tmRNA ribosome-rescue system. Adv. Protein Chem. Struct. Biol. 86:151–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kelly KO, Reuven NB, Li Z, Deutscher MP. 1992. RNase PH is essential for tRNA processing and viability in RNase-deficient Escherichia coli cells. J. Biol. Chem. 267:16015–16018 [PubMed] [Google Scholar]
- 22. Li Z, Deutscher MP. 1996. Maturation pathways for Escherichia coli tRNA precursors: a random multienzyme process in vivo. Cell 86:503–512 [DOI] [PubMed] [Google Scholar]
- 23. Li Z, Deutscher MP. 1994. The role of individual exoribonucleases in processing at the 3′ end of Escherichia coli tRNA precursors. J. Biol. Chem. 269:6064–6071 [PubMed] [Google Scholar]
- 24. Li Z, Pandit S, Deutscher MP. 1998. 3′ exoribonucleolytic trimming is a common feature of the maturation of small, stable RNAs in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 95:2856–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lin PH, Lin-Chao S. 2005. RhlB helicase rather than enolase is the beta-subunit of the Escherichia coli polynucleotide phosphorylase (PNPase)-exoribonucleolytic complex. Proc. Natl. Acad. Sci. U. S. A. 102:16590–16595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liou GG, Chang HY, Lin CS, Lin-Chao S. 2002. DEAD-box RhlB RNA helicase physically associates with exoribonuclease PNPase to degrade double-stranded RNA independent of the degradosome-assembling region of RNase E. J. Biol. Chem. 277:41157–41162 [DOI] [PubMed] [Google Scholar]
- 27. McLaren RS, Newbury SF, Dance GSC, Causton HC, Higgins CF. 1991. mRNA degradation by processive 3′-5′ exoribonucleases in vitro and the implications for prokaryotic mRNA decay in vivo. J. Mol. Biol. 221:81–95 [PubMed] [Google Scholar]
- 28. Melefors Ö, von Gabain A. 1988. Site-specific endonucleolytic cleavages and the regulation of stability of Escherichia coli ompA mRNA. Cell 52:893–901 [DOI] [PubMed] [Google Scholar]
- 29. Newbury SF, Smith NH, Robinson EC, Hiles ID, Higgins CF. 1987. Stabilization of translationally active mRNA by prokaryotic REP sequences. Cell 48:297–310 [DOI] [PubMed] [Google Scholar]
- 30. Py B, Higgins CF, Krisch HM, Carpousis AJ. 1996. A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature 381:169–172 [DOI] [PubMed] [Google Scholar]
- 31. Régnier P, Hajnsdorf E. 2009. Poly(A)-assisted RNA decay and modulators of RNA stability. Prog. Mol. Biol. Transl. Sci. 85:137–185 [DOI] [PubMed] [Google Scholar]
- 32. Sobrero P, Valverde C. 2012. The bacterial protein Hfq: much more than a mere RNA-binding factor. Crit. Rev. Microbiol., in press. doi:10.3109/1040841X.2012.664540 [DOI] [PubMed] [Google Scholar]
- 33. Spickler C, Mackie GA. 2000. Action of RNase II and polynucleotide phosphorylase against RNAs containing stem-loops of defined structure. J. Bacteriol. 182:2422–2427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Udekwu KI, et al. 2005. Hfq-dependent regulation of OmpA synthesis is mediated by an antisense RNA. Genes Dev. 19:2355–2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vincent HA, Deutscher MP. 2009. Insights into how RNase R degrades structured RNA: analysis of the nuclease domain. J. Mol. Biol. 387:570–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vincent HA, Deutscher MP. 2006. Substrate recognition and catalysis by the exoribonuclease RNase R. J. Biol. Chem. 281:29769–29775 [DOI] [PubMed] [Google Scholar]
- 37. Withey J, Friedman D. 1999. Analysis of the role of trans-translation in the requirement of tmRNA for lambdaimmP22 growth in Escherichia coli. J. Bacteriol. 181:2148–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yancey SD, Kushner SR. 1990. Isolation and characterization of a new temperature-sensitive polynucleotide phosphorylase mutation in Escherichia coli K-12. Biochimie 72:835–843 [DOI] [PubMed] [Google Scholar]
- 39. Zhou Z, Deutscher MP. 1997. An essential function for the phosphate-dependent exoribonucleases RNase PH and polynucleotide phosphorylase. J. Bacteriol. 179:4391–4395 [DOI] [PMC free article] [PubMed] [Google Scholar]