

Abstract
Numerous cytokines, including vascular endothelial growth factor (VEGF), are implicated in the pathogenesis of sepsis. While overexpression of VEGF produces pulmonary capillary leak, the role of VEGF in sepsis is less clear. We investigated VEGF in sepsis, utilizing a VEGF trap (VEGFT). Polymicrobial sepsis was induced in C57BL/6 mice by cecal ligation and puncture (CLP) and resulted in significantly increased plasma VEGF levels (234 vs. 46 pg/mL; p = 0.03). Inhibition of VEGF had no effect on mortality or lung leak but did attenuate plasma IL-6 (120 vs. 236 ng/mL; p = 0.02) and IL-10 (16 vs. 41 ng/mL; p = 0.03). These alterations in inflammatory cytokines were associated with increased levels of the dominant negative inhibitory C/EBPβ. In vitro, VEGF stimulated IL-6, IL-10 and reduced the inhibitory isoform of C/EBPβ in cultured macrophages. Together these data suggest VEGF can regulate inflammatory cytokine production in murine polymicrobial sepsis, via regulation of C/EBPβ.
Keywords: VEGF, sepsis, IL-6, IL-10, C/EBPβ
Introduction
Sepsis is defined as the “systemic inflammatory response syndrome that occurs during an infection” (1). The incidence of sepsis has been increasing, and has recently been estimated as 660,000 cases per year (2). Sepsis carries with it an economic burden that is estimated at 17 billion US dollars and is the 2nd leading cause of death in the noncoronary ICU (2).
Approximately 30% of sepsis patients progress to multiorgan organ dysfunction syndrome (MODS) with 18% developing acute lung injury (ALI) and its more severe form acute respiratory distress syndrome (ARDS). ALI/ARDS is characterized by exudation of fluid into the alveoli and interstitium secondary to increased permeability of the lung's natural barriers.
The development of MODS is in part determined by the balance between pro and anti-inflammatory cytokines. This balance has been studied extensively in both serum and bronchoalveolar lavage (BAL) samples. Recent work in sepsis has focused on mediators leading to lung injury. There is a complex interplay between pro- and anti-inflammatory cytokines in the lung. Early in ARDS there is a significant increase in anti-inflammatory IL-10 (3). Also simply blocking TNF-α or IL-1β, both important mediators of the proinflammatory cascade, fail to improve survival in the septic patient (4).
Transcription factor activity is one method by which host cells control the balance between pro- and anti-inflammatory cytokine production. C/EBPβ is a known regulator of numerous pro- and anti-inflammatory cytokines including IL- 6 and IL-10 (5). C/EBPβ exists in two isoforms, a long 37-kDa stimulating and a short 16-kDa dominant negative inhibitory isoform. The degree of inflammation is controlled by the ratio of the two isoforms (S/I). Recent studies document that in human tuberculosis and murine sepsis induced lung injury in mice, there is an increase in the S/I ratio with a net increase in inflammatory cytokine production (6, 7).
The balance in inflammatory cytokines also controls production of numerous bioactive mediators, which may further modulate the degree of remote organ dysfunction in sepsis. Vascular endothelial growth factor (VEGF) is a cytokine postulated to regulate the degree of capillary leak. The VEGFA gene is organized as 8 exons separated by seven introns yielding four VEGF isoforms (121, 165, 189, and 206 kDa). VEGF165 is the predominate human isoform (8). VEGF is an endothelial cell-specific growth factor, plays a role in migration and proliferation of endothelial cells and has procoagulant activity (9). It promotes vascular dilatation in a dose dependent factor and has been found to be 50,000 times more potent than histamine at inducing vascular permeability (10, 11).
The role of VEGF in ALI and sepsis has only recently been the focus of investigation. VEGF levels are increased in the plasma of patients with ARDS while, levels in the epithelial lining fluid inversely correlated with the patients severity of lung injury (12). This may be due to reduced VEGF production by alveolar macrophages from patients with ARDS (12). In addition overexpression of VEGF in the lung causes a dose-dependent increase in lung capillary permeability (13). Finally IL-6, a proinflammatory cytokine, which is significantly upregulated in sepsis and is a marker of disease severity was shown to be a potent inducer of VEGF expression (14–16).
Together, this data suggests a potential role for VEGF in regulating the host response and development of ALI in polymicrobial sepsis. The availability of a VEGF cytokine trap (VEGFT) which has been shown to neutralize the biological activity of VEGF in vivo permitted us to test the role of VEGF in our mouse model of sepsis (17, 18). Surprisingly, blocking of VEGF did not alter lung leak or mortality but did reduce production of IL-6 and IL-10. These observations suggest that VEGF plays a role in modulating both proinflammatory and anti-inflammatory cytokines during lung injury.
Methods
Mice
C57BL/6 female mice (5–6 weeks at the time of delivery) were obtained from Taconic (Germantown, NY) and allowed to acclimatize for 1 week prior to use. Mice were provided with free access to food and water and 12 h light and dark cycles in accordance with animal care guidelines. The animal use committee of New York University approved all studies.
Pretreatment of VEGFT or Control
Each mouse was given 25 mg/kg (500 μg/mouse) VEGFT or vehicle control (in equal volumes) i.p., 6 h prior to cecal ligation and puncture. VEGFT and vehicle control were provide by Regeneron Pharmaceuticals (Tarrytown, NY). The VEGFT consists of ligand-binding elements taken from the extracellular domains of VEGF-R 1 & 2 attached to the Fc portion of IgG1. This dose of VEGFT was shown to completely block the hypotension that occurs after an intravenous dose of exogenous VEGF in vivo for up to 3 days and result in inhibition of the effects of endogenous VEGF in prior experiments (17-19).
Cecal Ligation and Puncture (CLP)
CLP was done using a modification of the procedure as previously described (20). Briefly, mice were anesthetized using 2% isoflourane anesthesia with supplemental oxygen. Abdomen was shaven and cleaned with betadine, prior to a 1–2 cm midline incision. Cecum was then isolated and ligated below the ileocecal valve with a 3.0 silk and punctured once through and through with 19 gauge needle. Incision was then sutured with 3.0 silk. Postoperatively all mice were given 1 cc of room temperature sterile normal saline subcutaneously and were monitored until recovery from anesthesia. Plasma and BALF were collected at 18 h as previously described (7). Prior data from our laboratory has shown no difference between sham and unoperated controls and therefore, only unoperated controls were used (7).
Evans Blue Assay
Evans blue permeability was determined as previously described (7). Briefly, each animal was injected with 160 mg/kg of Evans blue dye (EBD) dissolved in sterile PBS (Sigma) intraperitoneally. After 1 h the plasma and lungs were harvested. Lungs were homogenized in PBS and 2 vol. formamide (Sigma, St. Louis, MO) and incubated for 18 h at 60°C. Homogenates were centrifuged at 5000 × g for 30 min and supernatant removed for Evans blue concentration. Spectrophotometric determination of EBD concentration was measured based on standard absorbance curves and a correction for heme pigments was made using the following calculation:
The degree of capillary leak was defined as the ratio of the amount of EBD in lung homogenate to the amount of EBD in plasma.
Liver Homogenates
After cardiac puncture the right ventricle was perfused with 1 cc cold PBS (used to flush liver and lung vasculature of any remaining blood). Organs were excised and snap frozen in liquid N2 and stored at −70°C until use. Livers were homogenized (Dounce tissue homogenizer, 7 mL Bellcoglass, Vineland, NJ) and whole cells extracts were obtained using NP40 extraction buffer (0.5%NP-40, 10% Glycerol, 0.1 mM EDTA, 20 mM HEPES (PH 7.9), 10 mM NaF, 10 mM NaPpi, 300 mM NaCl, 3 μg/mL Aprotinin, 2 μg/mL Leupeptin, 2 μg/mL Pepstatin, 1 mM DTT, 1 mM PMSF, 1 mM Na3 VO4) on ice. Aliquots were stored at −70°C for transcription factor analysis.
Murine Peritoneal Macrophages
Murine peritoneal macrophages were obtained as previously described. Mice were injected with 2 cc of 3% Brewers Thioglycolate intraperitoneally (Sigma) 3–5 days prior to use and subsequently euthanized as previously described and peritoneal cavity lavaged with 10 cc sterile saline. This procedure results in >95% macrophages. Cells were washed and resuspended in RPMI w/(Fungizone, streptomycin and) at 5 × 105/mL. Recombinant murine VEGF164 was purchased from R&D.
Transcription Factor Analysis (Western Blot)
For analysis of transcription factor activity and cytokine production, experiments were repeated in six-well plates (Falcon). Supernatant was collected at the designated time points, centrifuged at 1000 rpm × 10 min (μSpeedfuge SFR13K, Savant Instruments, Inc., Farmingdale, NY) to remove cellular debris and stored at −70°C. Cells were harvested and lysed. Briefly, cells were harvested by gentle scraping and lysed in NP40 lysis buffer (as above) on ice for 30 min with vigorous agitation. Cells were centrifuged at13,000 rpm × 15 min (μSpeedfuge SFR13K, Savant Instruments, Inc., Farmingdale, NY) and supernatants removed and stored for future analysis. Primary antibody for C/EBPβ used at a concentration of 1:2,500 and secondary goat anti-rabbit was used at a concentration of 1:5,000. Both were obtained from Santa Cruz. Briefly, an equal amount of protein (50 μg) was loaded onto a 15% polyacrylamide gel for C/EBPβ. Membranes were blocked in PBS with 0.1% tween and 5% milk, followed by overnight incubation at 4°C. Membranes were developed with ECL Plus (Amersham, Piscataway, NJ).
Cytokine Analysis
IL-6, IL-10, and IL-12 (p40) levels were determined using commercially available enzyme-linked immunosorbent assay (ELISA) kits purchased (R&D systems: Minneapolis, Minnesota). All samples prepared and analyzed as per protocols provided.
Cell Culture
Cells were cultured in RPMI media 1640 (Gibco) with 10% fetal calf plasma (HyClone, USA), Penicillin-Streptomycin (Gibco) and Fungizone-Amphotericin B (Gibco). THP-1 cells (ATCC) at a concentration of 5 × 105 cells/mL were differentiated with 20 ng/mL of 12-O tetradecanoylphorbol 13-acetate (PMA). Recombinant human VEGF165 was purchased (R&D systems: Minneapolis, MN).
Statistical Analysis
All numerical data were expressed as means ± standard error of the mean. P values were derived from a two-tailed Mann-hitney test or a log-rank test for survival analysis. All statistical analysis and graphing were determined using the Graphpad Prism statistical software (Graphpad: San Diego, California, Version IV).
Results
The Role of VEGF Expression in Polymicrobial Sepsis
We first sought to determine whether VEGF was up regulated in polymicrobial sepsis. Eighteen hours after CLP, plasma VEGF levels were increased five fold when compared to unoperated controls (234 pg/mL ± 100 vs. 46pg/mL ± 16; p = 0.03) (Fig. 1). In contrast there were no inductions of VEGF in BALF after CLP compared to unoperated controls (data not shown). CLP produced an intense septic state with 100% mortality and a median survival of 37 h. Neutralization of VEGF with VEGFT did not significantly alter mortality with a median survival of 28 h (Fig. 2). Similar results were obtained when we attempted to modulate the degree of sepsis using a 23 G needle (data not shown). We next sought to determine whether inhibition of VEGF altered the development of pulmonary capillary leak. Evans blue (EB) was used as an indicator of local changes in capillary permeability. CLP increased in ratio of EB dye lung/plasma in control mice (0.75 vs. 0.41, p = 0.05) 18 h after CLP. Mice treated with the VEGFT had no difference in lung leak as shown by EB compared to controls after CLP (0.75 vs. 0.66; p = 0.9).
Fig. 1.
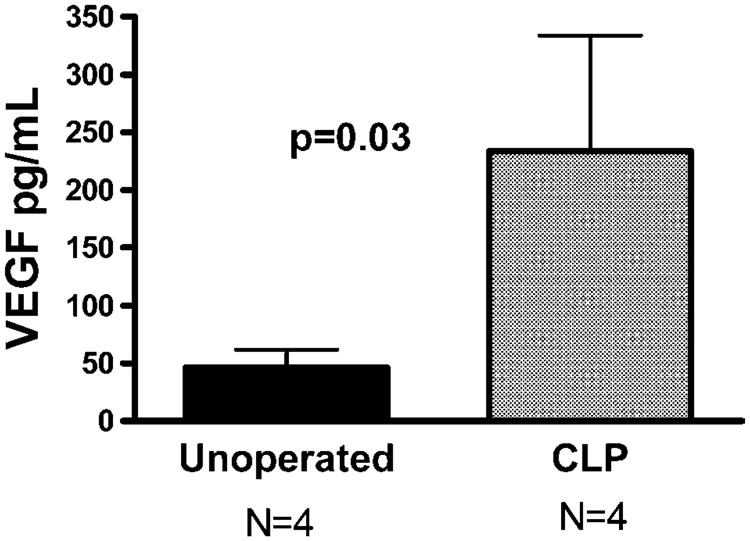
CLP-induced murine polymicrobial sepsis significantly increases plasma levels of VEGF. C57BL/6 female mice underwent CLP or remained as unoperated controls (four animals each). Plasma was collected after 18 h and VEGF levels were measured by ELISA. VEGF levels were increased fivefold when compared to unoperated controls (234 pg/mL ± 100 vs. 46 pg/mL ± 16; p < 0.05. Results expressed as the mean ± standard error of the mean (SEM).
Fig. 2.
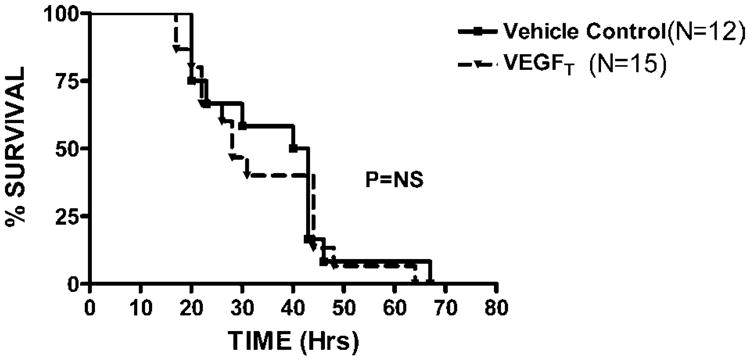
Survival not improved with VEGFT treatment in murine sepsis. C57BL/6 female mice underwent CLP 6 h after receiving either VEGFT 500 μg/mouse (▴, N = 15) or vehicle control (■, N = 12) IP and in equal volumes. Mice were observed at regular time intervals for a total of 72 h. CLP induced a 100% mortality with a median survival of 37 h. Treatment with VEGFT did not significantly alter mortality with a median survival of 28 h. Kaplan–Meier survival analysis was performed.
While mortality was not altered by VEGFT, treated mice were more mobile and had less piloerection. We wished to determine whether inhibition of VEGF lead to this alteration in clinical appearance by modulating inflammatory cytokine production. There was a significant decrease in plasma levels of IL-6 in the VEGFT-treated mice compared to vehicle-treated controls (119.5 ng/mL vs. 236.2 ng/mL; p = 0.02) after CLP. In contrast VEGFT had no affect on BAL IL-6 (850 pg/mL vs. 1520 pg/mL; p = ns). Similar results were obtained with plasma levels of IL-10. VEGFT-treated mice had a threefold reduction compared to controls (16 ng/ml vs. 41 ng/mL; p = 0.03) with a trend toward a similar effect in BALF (93 pg/mL vs. 311 pg/mL; p = ns). A similar trend was observed with IL-12, although this did not reach statistical significance (Fig. 3).
Fig. 3.
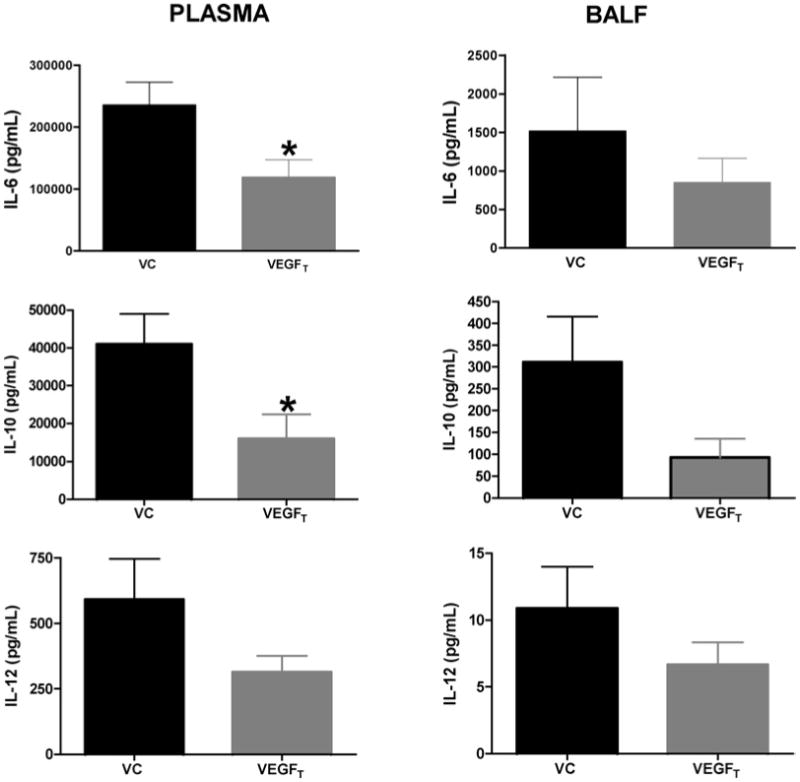
Plasma levels of IL-6, IL-10 significantly decreased after VEGFT treatment in murine sepsis. Plasma levels of IL-6 in the VEGFT-treated mice (N = 11) was decreased compared to vehicle-treated (N = 8) controls, (119.5 ng/mL vs. 236.2 ng/mL*) after CLP. In contrast VEGFT had no affect on BAL IL-6 (850 pg/mL vs. 1520 pg/mL). Similar results were seen for IL-10 (16 ng/ml vs. 41 ng/mL*) in plasma and in BALF (93 pg/mL vs. 311 pg/mL). A similar trend was observed with IL-12, although this also did not reach statistical significance. Data represented as mean ± SEM with asterisk indicating a significant p-value.
VEGF Increases Cytokines and Decreases Inhibitory C/EBPβ
We next sought to determine whether the alteration in cytokines were associated with alterations in transcription factor activity. CLP was found to induce both forms of C/EBPβ (Fig. 4A, Lane 1) with an overall increase in the ratio of stimulatory/inhibitory isoforms (S/I) (Fig. 4B). In contrast, VEGFT-treated mice had a reduction in the S/I ratio of CEBPβ (S/I) compared to control mice after CLP. (Figure 4A, Lane 2) The ratio of S/I forms was quantified by densitometry and was found be significantly increased in the VEGFT-treated mice; p = 0.05 (Fig. 4B) Together our data suggest a role for VEGF in regulating inflammatory cytokine production in vivo We subsequently wished to directly test this hypothesis in vitro.
Fig. 4.
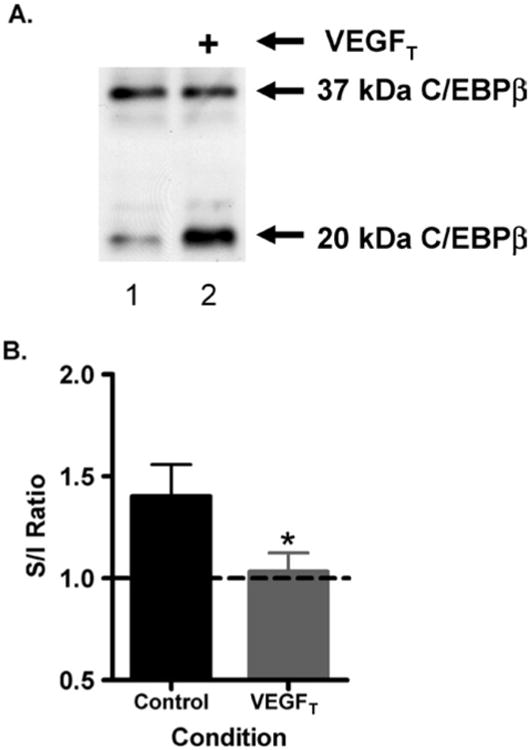
Dominant negative (short) form of C/EBPβ has increased expression when VEGF is inhibited. (A) Representative examples of a Western blot for C/EBPβ protein expression of liver homogenates/whole cell extract of vehicle control (lane 1) and VEGFT-treated (lane 2) samples. (B) Ratio of Stimulatory/Inhibitory (S/I) form of C/EBPβ for vehicle control (N = 4) vs. VEGFT-treated mice (N = 9); *significant p-value.
Recombinant VEGF stimulated induction of IL-6 and IL-10 in a dose-dependent manner in murine macrophages and caused a significant fold induction at a dose of 10–1000 ng/mL (Fig. 5A) with a maximal effect observed at 1 μg/mL at 2 h. In contrast, IL-12 production was completely unaffected (data not shown). We observed similar results in THP-1 cells, a human monocytic cell line. Exogenous VEGF at a dose of 1 μg/mL caused a sixfold induction of IL-6 with a maximal effect at 2 h and a return to baseline at 24 h (Fig. 5B).
Fig. 5.
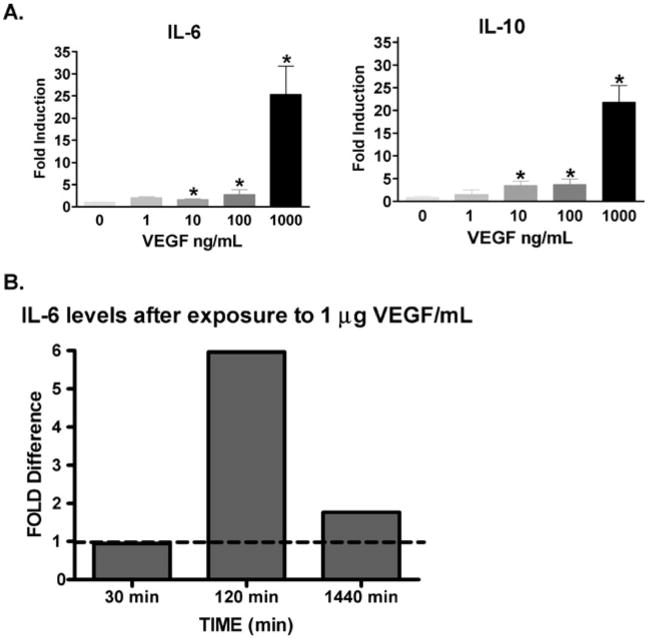
IL-6 and IL-10 expression in macrophages after VEGF stimulation. (A) Levels of both IL-6 and IL-10 had a graded dose response to exogenous murine VEGF. Murine thioglycolate induced peritoneal macrophages were used. Results expressed as the mean ± SEM. *Significant p-value (B) time course of cytokine production in THP-1 derived macrophages after VEGF stimulation. Exogenous VEGF at a dose of 1 μg/mL caused a sixfold induction of IL-6 at 2 h.
Finally, we sought to determine whether the alterations in IL-6 and IL-10 were associated with changes in C/EBPβ. In human macrophages, VEGF resulted in an increase in the S/I ratio of C/EBPβ at 2 h (Fig. 6, Lane 3) with complete loss of the inhibitory isoform by 24 h (Fig. 6, Lane 4). These results were specific for VEGF as treatment with LPS resulted in up regulation of both isoforms.
Fig. 6.
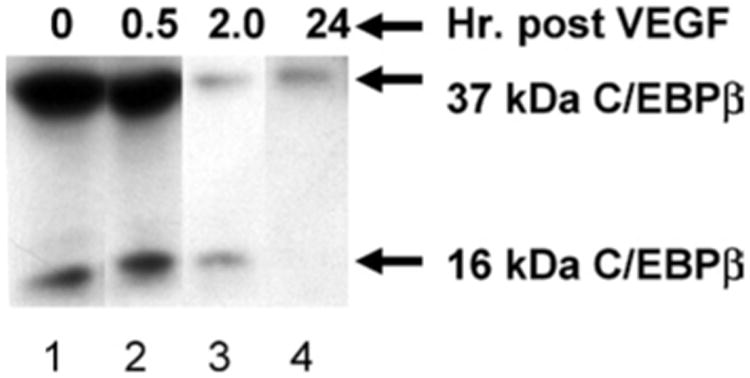
In vitro dose response and time course assay shows a reduction in inhibitory C/EBPβ at 2 h and complete loss of this isoform by 24 h. THP-1 derived macrophages were exposed to recombinant VEGF at a dose of 1 μg/mL and C/EBPβ protein expression analyzed by Western blot at 0.5, 2, and 24 h. All lanes were normalized for equal protein content.
Discussion
In this study, we demonstrated that VEGF is up regulated in polymicrobial sepsis. Inhibition of VEGF with a VEGFT attenuated IL-6 and IL-10 induction during sepsis. In vitro VEGF was capable of inducing both IL-6 and IL-10 in human and murine macrophages. This was associated with the loss of an inhibitory C/EBPβ transcription factor. Together this data suggests a role for VEGF in regulating inflammatory cytokine production in polymicrobial sepsis.
The finding of increased levels of VEGF in our model of polymicrobial sepsis suggests a potentially important role in the host inflammatory response. While, VEGF is typically described as a potent angiogenic factor, and a significant inducer vascular permeability, numerous properties of VEGF make it a likely candidate to regulate the inflammatory response in vivo. VEGF is made by and stimulates epithelial cells, macrophages, monocytes and endothelial cells which play a pivotal role in regulating the host inflammatory response in sepsis (21, 22).
Our study demonstrates that circulating levels of VEGF are significantly increased in the plasma of septic mice. This is similar to findings in human studies. Levels of VEGF have been shown to be increased in the plasma of septic patient that have ARDS. In addition studies from the same group have shown that the addition of plasma from ARDS patients significantly increased the flux of albumin across human pulmonary endothelial monolayers (23). However, VEGF has been shown to be decreased in epithelial lining fluid (ELF) taken from ARDS patients compared to that collected from patients at risk for lung injury. Subsequent measurements showed that an increase in the ELF levels of VEGF was associated with recovery. Also of interest was that alveolar macrophages from patients with ARDS produced less VEGF than those collected from at risk patients (12). This result is in concert with our finding of increased levels of VEGF only in the plasma and not in the BALF.
Our study showed that VEGF inhibition had no effect on survival or lung leak as determined by Evans blue dye. This is contrast to an in vivo murine model of VEGF over expression in the lung which resulted in increased pulmonary capillary leak (13). The most likely explanation for these differences lie in the degree of VEGF expression in the lung, with no alterations of VEGF observed in BALF from septic mice. Therefore, the utility of VEGF inhibition on regulating capillary leak in ALI may be most dependent on the degree of VEGF expression in the lung.
One interesting finding was that VEGF inhibition did result in a decrease in IL-6 and IL-10 after CLP. This attenuation was most marked in plasma (as opposed to BALF) which is consistent with the finding of increased VEGF levels only in plasma after CLP. Furthermore, the less pronounced effect on IL-12 suggests this is not a global effect of VEGF inhibition but rather specific to certain inflammatory pathways. The ability of recombinant VEGF to stimulate production of these cytokines in vitro as early as 2 h provides further evidence that VEGF can be a direct inducer of specific inflammatory cytokines. Numerous studies document inflammatory cytokines and specifically IL-6 can regulate VEGF production. Specifically, VEGF is up regulated in conjunction with IL-6 in patients with rheumatoid arthritis. In addition, when levels of IL-6 were effectively decreased using anti-interleukin-6 receptor antibody therapy, levels of VEGF also decreased (16).
However, our data now suggests VEGF in turn regulates inflammatory cytokines production placing it in a positive feedback loop to regulate its own production. The mechanism by which VEGF regulates inflammatory cytokine production may be in part due to regulation of C/EBPβ. C/EBPβ exists in two isoforms, a stimulatory 37 kDa and an inhibitory 16–20 kDa dominant negative inhibitory isoform. The ratio of these two isoform can regulate transcription of genes with CEBP sites in their promoters including IL-6 and IL-10 (6). The finding that VEGF inhibition is associated with a reduction in the ratio of S/I isoform of C/EBPβ compared to control mice after CLP is consistent with the cytokine data. This was further confirmed by the observation that VEGF increased the S/I ratio with near complete loss of the inhibitory isoform by 24 h. Again, this effect was specific for VEGF as LPS resulted in an increase in both isoforms as previously described. The mechanism by which VEGF regulates C/EBPβ remains unclear. However, in a recent article human gingival fibroblast when exposed to hyperglycemia, had demonstrated IL-6 induced phosphorylation of p44/p42 MAPK with subsequent CEBP nuclear translocation and VEGF165 expression (24).
In conclusion, our data suggests a role for VEGF in regulating inflammatory cytokine production in polymicrobial sepsis, specifically IL-6 and IL-10. This regulation is in part mediated by C/EBPβ. While our data suggests inhibition of VEGF may be of limited clinical utility in the treatment of fulminant polymicrobial sepsis and ALI, the ability of VEGF inhibition to attenuate inflammatory cytokine production may have important clinical sequela in the treatment of more chronic inflammatory disease such as rheumatoid arthritis and neoplasia. Our data supports a novel role for VEGF in the inflammatory cytokine cascade of murine polymicrobial sepsis, possibly via regulation of C/EBPβ.
Acknowledgments
This work was supported by NIH NCRR GCRC MO1 RR0096, K08 HL070710, RO1 HL57879. VEGFT provided by Regeneron Pharmaceuticals, Inc. Tarrytown, NY.
References
- 1.Bone RC, Sibbald WJ, Sprung CL. The ACCP-SCCM consensus conference on sepsis and organ failure. Chest. 1992;101(6):1481–1483. doi: 10.1378/chest.101.6.1481. [DOI] [PubMed] [Google Scholar]
- 2.Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348(16):1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 3.Park WY, Goodman RB, Steinberg KP, Ruzinski JT, Radella F, II, Park DR, Pugin J, Skerrett SJ, Hudson LD, Martin TR. Cytokine balance in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;164(10 Pt 1):1896–1903. doi: 10.1164/ajrccm.164.10.2104013. [DOI] [PubMed] [Google Scholar]
- 4.Eichacker PQ, Parent C, Kalil A, Esposito C, Cui X, Banks SM, Gerstenberger EP, Fitz Y, Danner RL, Natanson C. Risk and the efficacy of antiinflammatory agents: retrospective and confirmatory studies of sepsis. Am J Respir Crit Care Med. 2002;166(9):1197–1205. doi: 10.1164/rccm.200204-302OC. [DOI] [PubMed] [Google Scholar]
- 5.Akira S, Isshiki H, Sugita T, Tanabe O, Kinoshita S, Nishio Y, Nakajima T, Hirano T, Kishimoto T. A nuclear factor for IL-6 expression NF-IL6 is a member of a C/EBP family. Embo J. 1990;9(6):1897–1906. doi: 10.1002/j.1460-2075.1990.tb08316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiden M, Tanaka N, Qiao Y, Zhao BY, Honda Y, Nakata K, Canova A, Levy DE, Rom WN, Pine R. Differentiation of monocytes to macrophages switches the Mycobacterium tuberculosis effect on HIV-1 replication from stimulation to inhibition: Modulation of interferon response and CCAAT/enhancer binding protein beta expression. J Immunol. 2000;165(4):2028–2039. doi: 10.4049/jimmunol.165.4.2028. [DOI] [PubMed] [Google Scholar]
- 7.Gold JA, Parsey M, Hoshino Y, Hoshino S, Nolan A, Yee H, Tse DB, Weiden MD. CD40 contributes to lethality in acute sepsis: In vivo role for CD40 in innate immunity. Infect Immun. 2003;71(6):3521–3528. doi: 10.1128/IAI.71.6.3521-3528.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tischer E, Mitchell R, Hartman T, Silva M, Gospodarowicz D, Fiddes JC, Abraham JA. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J Biol Chem. 1991;266(18):11947–11954. [PubMed] [Google Scholar]
- 9.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9(6):669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 10.Senger DR, Connolly DT, Van de Water L, Feder J, Dvorak HF. Purification and NH2-terminal amino acid sequence of guinea pig tumor-secreted vascular permeability factor. Cancer Res. 1990;50(6):1774–1778. [PubMed] [Google Scholar]
- 11.Roberts WG, Palade GE. Increased microvascular permeability and endothelial fenestration induced by vascular endothelial growth factor. J Cell Sci. 1995;108(Pt 6):2369–2379. doi: 10.1242/jcs.108.6.2369. [DOI] [PubMed] [Google Scholar]
- 12.Thickett DR, Armstrong L, Millar AB. A role for vascular endothelial growth factor in acute and resolving lung injury. Am J Respir Crit Care Med. 2002;166(10):1332–1337. doi: 10.1164/rccm.2105057. [DOI] [PubMed] [Google Scholar]
- 13.Kaner RJ, Ladetto JV, Singh R, Fukuda N, Matthay MA, Crystal RG. Lung overexpression of the vascular endothelial growth factor gene induces pulmonary edema. Am J Respir Cell Mol Biol. 2000;22(6):657–664. doi: 10.1165/ajrcmb.22.6.3779. [DOI] [PubMed] [Google Scholar]
- 14.Meduri GU, Headley S, Kohler G, Stentz F, Tolley E, Umberger R, Leeper K. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS. Plasma IL-1 beta and IL-6 levels are consistent and efficient predictors of outcome over time. Chest. 1995;107(4):1062–1073. doi: 10.1378/chest.107.4.1062. [DOI] [PubMed] [Google Scholar]
- 15.Meduri GU, Kohler G, Headley S, Tolley E, Stentz F, Postlethwaite A. Inflammatory cytokines in the BAL of patients with ARDS. Persistent elevation over time predicts poor outcome. Chest. 1995;108(5):1303–1314. doi: 10.1378/chest.108.5.1303. [DOI] [PubMed] [Google Scholar]
- 16.Nakahara H, Song J, Sugimoto M, Hagihara K, Kishimoto T, Yoshizaki K, Nishimoto N. Anti-interleukin-6 receptor antibody therapy reduces vascular endothelial growth factor production in rheumatoid arthritis. Arthritis Rheum. 2003;48(6):1521–1529. doi: 10.1002/art.11143. [DOI] [PubMed] [Google Scholar]
- 17.Wong AK, Alfert M, Castrillon DH, Shen Q, Holash J, Yancopoulos GD, Chin L. Excessive tumor-elaborated VEGF and its neutralization define a lethal paraneoplastic syndrome. Proc Natl Acad Sci U S A. 2001;98(13):7481–7486. doi: 10.1073/pnas.121192298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saishin Y, Takahashi K, Lima e Silva R, Hylton D, Rudge JS, Wiegand SJ, Campochiaro PA. VEGF-TRAP(R1R2) suppresses choroidal neovascularization and VEGF-induced breakdown of the blood-retinal barrier. J Cell Physiol. 2003;195(2):241–248. doi: 10.1002/jcp.10246. [DOI] [PubMed] [Google Scholar]
- 19.Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, Boland P, Leidich R, Hylton D, Burova E, et al. VEGF-Trap: A VEGF blocker with potent antitumor effects. Proc Natl Acad Sci USA. 2002;99(17):11393–11398. doi: 10.1073/pnas.172398299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wichterman KA, Baue AE, Chaudry IH. Sepsis and septic shock–a review of laboratory models and a proposal. J Surg Res. 1980;29(2):189–201. doi: 10.1016/0022-4804(80)90037-2. [DOI] [PubMed] [Google Scholar]
- 21.Voelkel NF, Cool C, Taraceviene-Stewart L, Geraci MW, Yeager M, Bull T, Kasper M, Tuder RM. Janus face of vascular endothelial growth factor: the obligatory survival factor for lung vascular endothelium controls precapillary artery remodeling in severe pulmonary hypertension. Crit Care Med. 2002;30(5 Suppl):S251–S256. doi: 10.1097/00003246-200205001-00013. [DOI] [PubMed] [Google Scholar]
- 22.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348(2):138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 23.Thickett DR, Armstrong L, Christie SJ, Millar AB. Vascular endothelial growth factor may contribute to increased vascular permeability in acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;164(9):1601–1605. doi: 10.1164/ajrccm.164.9.2011071. [DOI] [PubMed] [Google Scholar]
- 24.Omori K, Naruishi K, Nishimura F, Yamada-Naruishi H, Takashiba S. High glucose enhances interleukin-6-induced vascular endothelial growth factor 165 expression via activation of gp130-mediated p44/42 MAPK-CCAAT/enhancer binding protein signaling in gingival fibroblasts. J Biol Chem. 2004;279(8):6643–6649. doi: 10.1074/jbc.M311688200. [DOI] [PubMed] [Google Scholar]