

Abstract
Bcl-2 and Bcl-xL are key apoptosis regulators and attractive cancer therapeutic targets. We have designed and optimized a class of small-molecule inhibitors of Bcl-2 and Bcl-xL containing a 4,5-diphenyl-1H-pyrrole-3-carboxylic acid core structure. A 1.4 Å resolution crystal structure of a lead compound, 12, complexed with Bcl-xL has provided a basis for our optimization. The most potent compounds, 14 and 15, bind to Bcl-2 and Bcl-xL with subnanomolar Ki values and are potent antagonists of Bcl-2 and Bcl-xL in functional assays. Compounds 14 and 15 inhibit cell growth with low nanomolar IC50 values in multiple small-cell lung cancer cell lines and induce robust apoptosis in cancer cells at concentrations as low as 10 nM. Compound 14 also achieves strong antitumor activity in an animal model of human cancer.
Introduction
Apoptosis resistance is a hallmark of human cancer,1–4 and targeting key anti-apoptosis regulators with the goal of restoring apoptosis in cancer cells is a promising new therapeutic approach to cancer.5, 6
The Bcl-2 proteins are key regulators of apoptosis and consist of both anti- and pro-apoptotic members.2, 7 The anti-apoptotic proteins include Bcl-2, Bcl-xL, Bcl-w, Mcl-1 and A1, while the pro-apoptotic members consist of BID, BIM, BAD, BAK, BAX and NOXA, among others.7 The balance and their interaction between the pro-apoptotic members and the anti-apoptotic members within the Bcl-2 family of proteins control cell fate. 2, 7 Upon receipt of apoptotic stimuli, the pro-apoptotic proteins BAX and/or BAK disrupt the integrity of the outer mitochondrial membrane by forming oligomers in this membrane, and this leads to the release of the pro-apoptotic signaling proteins cytochrome c and Smac, and activation of downstream caspases. Overexpression of the anti-apoptotic Bcl-2 and Bcl-xL proteins in cancer cells inhibits apoptosis by blocking activation of BAX and/or BAK and confers cancer cells resistance to a variety of chemotherapeutic agents.8, 9 It has been proposed that small molecules that block the interaction of Bcl-2/Bcl-xL proteins with pro-apoptotic Bcl-2 proteins can antagonize the anti-apoptotic function of Bcl-2/Bcl-xL proteins and overcome apoptosis resistance mediated by overexpression of Bcl-2/Bcl-xL in tumor cells.8, 9 Design of potent, cell-permeable small-molecule Bcl-2/Bcl-xL inhibitors has been the subject of intense efforts in the last decade.10 Compound 1 (ABT-737, Figure 1) and its analogue 2 (ABT-263, Figure 1) are probably the two most potent Bcl-2/Bcl-xL inhibitors reported to date.11–13 Both compounds bind to Bcl-2 and Bcl-xL with high affinities and have a weak affinity for Mcl-1. Compound 2 has been advanced into Phase I/II clinical trials for the treatment of human cancer.14, 15 The recently synthesized quinazoline sulfonamide 3 (Figure 1) also shows high binding affinities to Bcl-2 and Bcl-xL and a weak affinity to Mcl-1.16
Figure 1.

Chemical structures of previously reported Bcl-2/Bcl-xL inhibitors.
We recently reported the design of a new class of Bcl-2/Bcl-xL inhibitors, exemplified by compounds 4, 5 and 6 (Figure 1), with 6 being the most potent one among them.17 Although compound 6 binds to Bcl-2 and Bcl-xL with subnanomolar affinities (Ki < 1 nM) and potently inhibits cell growth in the H146 small-cell lung cancer cell line with an IC50 value of 60 nM,17 it fails to induce robust apoptosis in the H146 xenograft tumor tissues at its maximum tolerated dose (data not shown). In this paper, we report further structure-based design, synthesis and evaluation of a set of new Bcl-2/Bcl-xL inhibitors. The best new compound not only binds to Bcl-2/Bcl-xL with subnanomolar affinities and effectively induces apoptosis in tumor cells, but also achieves strong antitumor activity in an animal model of human cancer.
Results and Discussion
We have previously determined the crystal structure of compound 7 complexed with Bcl-xL (Figure 2A) and used the crystal structure for the design of compounds 4, 5 and 6. Analysis of this crystal structure (Figure 2A) suggested that the 3,4-diphenyl-1H-pyrrole in 7 can be changed to 4,5-diphenyl-1H-pyrrole for effective interaction with Bcl-xL. Because the dihydroxybutyl side chain in 7 lacks any specific interaction with the Bcl-xL protein in the crystal structure, it was truncated to a methyl group. The piperazine group was retained because it enhances the aqueous solubility of compounds containing it. These changes led to compound 8 (Figure 3). Compound 8 binds to Bcl-2 and Bcl-xL with Ki values of 38 μM and 88 μM, respectively, 2 times more potent than 7 and was used as the lead structure in subsequent design. Using the crystal structure of 7 complexed with Bcl-xL as a starting point, we developed a model of compound 8 complexed with Bcl-xL (Figure 2B).
Figure 2.

(A) Co-crystal structure of 7 in complex with Bcl-xL (1.7 Å). (B) Predicted binding model of 8 in complex with Bcl-xL.
Figure 3.

Chemical structures of 7 and newly designed Bcl-2/Bcl-xL inhibitors.
In our previous study, we designed compound 4 by tethering 7 with a portion of 1, which occupies two different binding pockets in Bcl-xL.17 Using a similar strategy, we have designed 9 by tethering 8 with the same portion of 1 used for the design of 4. Compound 9 binds to Bcl-2 and Bcl-xL proteins, with Ki values of 7 nM and 1.3 nM, respectively (Table 1). Our previous study further showed that removal of the carbonyl group in the linker region in 4, which yielded 5, improves both binding affinities to Bcl-2 and Bcl-xL, and cellular activity. Thus, we synthesized 10 by removal of the corresponding carbonyl group in compound 9. Compound 10 binds to Bcl-2 (Ki <1 nM) and Bcl-xL (Ki = 2.4 nM) and is more potent than compound 9 in binding to Bcl-2. Both compounds 9 and 10 also show a high specificity over Mcl-1, with IC50 values >10 μM (Table 1).
Table 1.
Binding affinities of designed and reference compounds to Bcl-2, Bcl-xL and Mcl-1 proteins and inhibition of cell growth in three small-cell lung cancer cell lines.
| CPDS | Binding Affinities | Cell Growth Inhibition (IC50 ± SD, nM) | ||||||
|---|---|---|---|---|---|---|---|---|
| Bcl-2 | Bcl-xL | Mcl-1 | ||||||
| IC50 ± SD (nM) | Ki ± SD (nM) | IC50 ± SD (nM) | Ki ± SD (nM) | IC50 ± SD | H146 | H1417 | H1963 | |
| 6 | 4.1 ± 0.7 | 0.82 ± 0.19 | 7.5 ± 1.3 | < 1 | > 2 (μM) | 61 ± 9 | 90 ±3 | n.t. |
| 7 | 213±16 (μM) | 78 ±6 (μM) | 453 ±25 (μM) | 138±8 (μM) | >100 (μM) | >10,000 | >10,000 | >10,000 |
| 8 | 103 ±26 (μM) | 38 ±10 (μM) | 291 (μM) | 88 (μM) | >100 (μM) | >10,000 | >10,000 | >10,000 |
| 9 | 29 ± 8 | 7 ± 2 | 5 ± 2 | < 1 | > 100 (μM) | >10,000 | >10,000 | >10,000 |
| 10 | 2 ± 0.6 | < 0.6 | 9 ± 2 | < 1 | >10 (μM) | 110 ± 42 | 258 ± 43 | 72 ± 6 |
| 1 | 2 ± 0.2 | < 0.6 | 6 ± 2 | < 1 | > 1 (μM) | 37±29 | 412±100 | 59±15 |
| 11 | 5 ± 1 | 1.1 ± 0.2 | 6 ± 3 | < 1 | > 10 (μM) | 36 ± 26 | 109 ± 10 | 30 ± 12 |
| 12 | 1.3 ±0.2 | < 0.6 | 6 ±1 | < 1 | > 2 (μM) | 61±39 | 93±27 | 19±7 |
| 13 | 99 ±5 | 25 ±2 | 11 ±6 | 1.2 ±0.6 | > 10 (μM) | >10,000 | 9229±793 | 6475±5269 |
| 14 | 2 ± 1.6 | < 0.6 | 6.6 ± 2.3 | < 1 | > 2 (μM) | 8.1±3.5 | 17.7±9.5 | 4.5±2.2 |
| 15 | 1.4 ± 0.5 | < 0.6 | 4.8 ± 0.1 | < 1 | > 2 (μM) | 3.0±2.4 | 3.4±1.1 | 1.9± 0.9 |
| 16 | 17.6 ± 2.2 | 4.3 ± 0.6 | 7.7 ± 0.9 | < 1 | > 2 (μM) | 69 ±21 | 154 ±19 | 60±5 |
| BIM | < 1 | < 0.6 | < 1 | < 1 | 5 ± 1 (nM) | n.t. | n.t. | n.t. |
| BAD | 40 ± 8 | 10 ± 2 | 5 ± 0.3 | < 1 | 32 ± 2 (μM) | n.t. | n.t. | n.t. |
| NOXA | 16.7±0.8 (μM) | 3.6 (μM) | 11.0±1.7 (μM) | 3.4 (μM) | 37 ± 3 (nM) | n.t. | n.t. | n.t. |
We evaluated compounds 8, 9 and 10 for their activity in cell growth inhibition in three small-cell lung cancer cell lines (H146, H1417 and H1963), which have been shown to be sensitive to potent and specific Bcl-2/Bcl-xL inhibitors such as compounds 1 and 2. Compound 10 inhibits cell growth in these three small-cell lung cancer lines with IC50 values of 110, 258 and 72 nM, respectively (Table 1). In comparison to 10, compound 9 is much less effective against these three cell lines (IC50 > 10 μM). Consistent with its weak binding affinities to Bcl-2 and Bcl-xL, compound 8 has minimal cellular activity (Table 1). Compound 1 has IC50 values of 37, 412 and 59 nM, respectively, in these three cancer cell lines (Table 1).
To provide direct evidence that compound 10 antagonizes Bcl-2 and Bcl-xL but not Mcl-1, we have established cell-free functional assays using purified mitochondria, recombinant Bcl-2/Bcl-xL/Mcl-1 proteins and the BIM BH3 peptide, which binds to Bcl-2, Bcl-xL and Mcl-1 with very high affinities (Table 1). We employed these cell-free functional assays to determine the functional antagonism of compounds 1 and 10, and also of the BAD and the NOXA BH3 peptides.
At a concentration of 20 nM, the BIM BH3 peptide induces substantial release of cytochrome c and Smac proteins from mitochondria, and Bcl-2, Bcl-xL or Mcl-1, each at 60 nM, efficiently inhibits this release (Figure 4). In the Bcl-2 functional assay (Figure 4A), compounds 1 and 10 dose-dependently antagonize Bcl-2 and restore BIM-induced release of cytochrome c and Smac proteins from mitochondria, and 10 is 3-times less potent than 1. The BAD BH3 peptide is capable of doing so in a dose-dependent manner, but the NOXA BH3 peptide fails to restore the release of cytochrome c and Smac. In the Bcl-xL functional assay (Figure 4B), compounds 1 and 10 antagonize Bcl-xL and restore the release of cytochrome c and Smac, but the NOXA BH3 peptide is unable to do so. Compounds 1 and 10 are equally effective in antagonizing Bcl-xL, but both compounds are less potent than the BAD BH3 peptide. In the Mcl-1 functional assay (Figure 4C), while the NOXA peptide antagonizes Mcl-1 and restores the release of cytochrome c and Smac induced by the BIM peptide in a dose-dependent manner, the BAD peptide, 1 and 10 at concentrations as high as 10 μM fail to do so. These data show that 1, 10, and the BAD peptide all function as potent antagonists of Bcl-2 and Bcl-xL proteins, but all fail to antagonize Mcl-1. On the other hand, while the NOXA peptide efficiently antagonizes Mcl-1, it fails to do so with both Bcl-2 and Bcl-xL proteins. These functional data are consistent with their binding profiles to these Bcl-2 proteins (Table 1).
Figure 4.

Functional antagonism against Bcl-2 (A), Bcl-xL (B) and Mcl-1 (C) proteins by 10, 1, BAD and NOXA BH3 peptides in cell-free system. While 1, 10, and BAD can effectively antagonize Bcl-2 and Bcl-xL to restore the release of cytochrome c (Cyt c) and Smac proteins from mitochondria induced by the Bim BH3 peptide, Noxa fails to do so. Conversely, while 10 and BAD fail to antagonize Mcl-1 to restore the release of Cyt c and Smac proteins from mitochondria induced by the Bim BH3 peptide, Noxa can effectively do so. COX IV was used as the loading control in (B).
We next embarked on modifications of 10 in order to further improve its binding affinities to Bcl-2 and Bcl-xL and its cellular activity against cancer cells. The results are summarized in Figure 5 and Table 1.
Figure 5.

Chemical structures of new analogues of 10.
The 1-methyl-4-propylpiperazinyl side chain on the pyrrole ring of 10 was intended to enhance the aqueous solubility of the compound. This side chain was modified to examine its influence on binding and cellular activity, which led to the amide analogue 11 and the carboxylic acid analogue 12. These both bind to Bcl-2 and Bcl-xL with very high binding affinities (Table 1), exceeding the lower limits of the assays except that compound 11 binds to Bcl-2 with a Ki value of 1.1 nM. Compound 13, in which the carboxyl group has been removed from the pyrrole ring in 12, binds to Bcl-2 100 times less potently than 12, but it is only slightly less potent than 12 in binding to Bcl-xL. These data indicate that the carboxyl group plays a critical role with respect to the binding to Bcl-2, but makes only a modest contribution to Bcl-xL binding.
Compounds 11 and 12 have similar potencies in the cell growth inhibition assay against the H146, H1417 and H1963 cancer lines and are 2–4 times more potent than compound 10. Consistent with its low binding affinity to Bcl-2, compound 13 shows very weak activities (> 5 μM) in these three cancer cell lines.
To provide a solid structural basis for subsequent modifications, we have attempted to determine the crystal structures for these Bcl-2/Bcl-xL inhibitors (10, 11 and 12) complexed with Bcl-2 and/or Bcl-xL. We determined a crystal structure of 12 complexed with Bcl-xL at a resolution of 1.4 Å (Figure 6 and Table S1 in SI). This crystal structure shows that the diphenylpyrrole core structure of 12 binds in the same pocket and with the same orientation as that of 7. Adjacent to the N-methyl group on the pyrrole ring, there is an unoccupied hydrophobic pocket lined by L108, V126, F146, L112 and L150 which can be exploited for further optimization. In addition to the diphenylpyrrole core structure, the thiophenyl ring of 12 has extensive hydrophobic interactions with the protein. This thiophenyl ring is also involved in π-stacking with the nitrophenyl group in 12, which presumably stabilizes the bound conformation of 12. The dimethylamino group forms two hydrogen bonds with the carboxyl group of E96 but the nitro group on the phenyl ring fails to fill adequately the pocket in Bcl-xL lined by Y195, F191, W197, V141 and A141, suggesting the possibility of additional optimization at this site to further improve binding affinities and cellular activities. The carboxylic acid in 12 forms a direct hydrogen bond with R132 and an indirect hydrogen bond with R139 in Bcl-xL, mediated by a water molecule.
Figure 6.
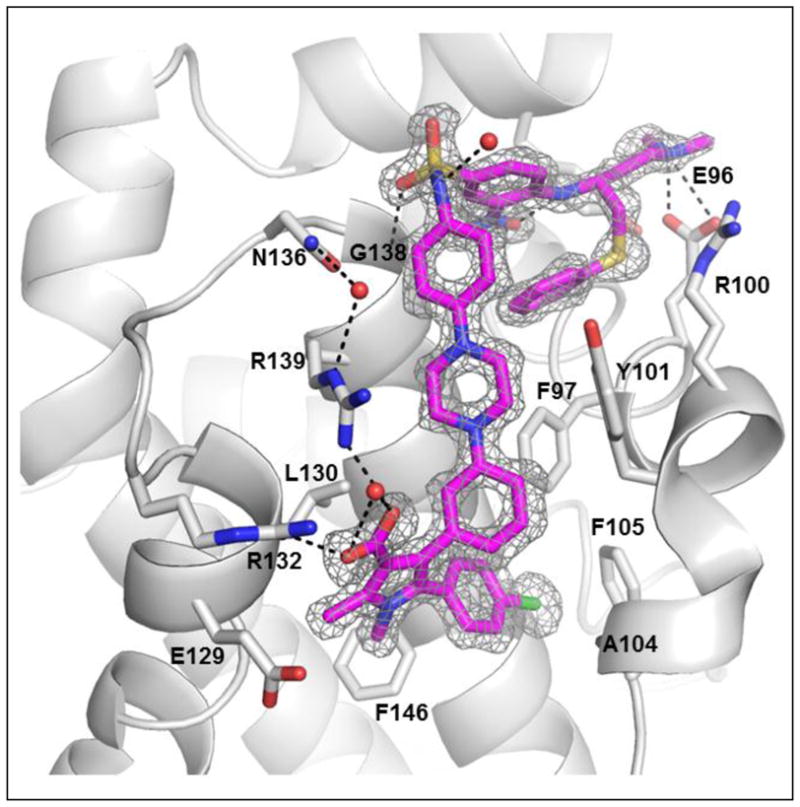
A crystal structure of 12 complexed with Bcl-xL at 1.4 Å resolution. Compound 12 in cyan; key residues interacting in Bcl-xL in grey; oxygens in red; nitrogens in blue, sulphur in yellow and chlorine in green.
Based upon this crystal structure, the N-methyl group on the pyrrole ring binds in a hydrophobic pocket lined by L108, V126, F146, L112 and L150, but there is additional room available to accommodate a larger hydrophobic group. We therefore designed and synthesized three new analogues (compounds 14, 15 and 16) with an N-ethyl, N-isopropyl, or N-cyclopropyl group (Figure 5). Compounds 14 and 15 bind to both Bcl-2 and Bcl-xL with very high affinities (Ki < 1nM), exceeding the lower limits of the assays. While compound 16 binds to Bcl-2 with 10-times lower affinity than 12, they both have similar affinities to Bcl-xL.
Consistent with their high binding affinities to both Bcl-2 and Bcl-xL, both compounds 14 and 15 are very potent in cell growth inhibition in the H146, H1417 and H1963 cancer cell lines. While compound 14 has IC50 values of 8.1, 17.7 and 4.5 nM, respectively, compound 15 achieves IC50 values of 3.0, 3.4 and 1.9 nM (Table 1 and Figure 7A). Compound 16 has much weaker cell growth inhibitory activity than compounds 14 and 15 against these three cancer cell lines, which is consistent with its weaker binding affinity to Bcl-2 than 14 and 15. In direct comparison, compound 15 is 10–100 times more potent than compound 1 against these three cancer cell lines in the cell growth inhibition assay, representing arguably the most potent Bcl-2/Bcl-xL inhibitor reported to date.
Figure 7.

(A). Representative cell growth inhibition of 12, 14, 15 and 1 in three small-cell lung cancer cell lines. (B). Cell death induction by 12, 14, 15 and 1 in H146 cell line. Cells were treated for 24 h and cell death was analyzed by trypan blue assay. (C). Induction of cleavage of PARP and caspase-3 in H146 cell line by 12, 14, 15 and 1. Cells were treated for 24 h and caspase-3 (Cas 3) and PARP were probed by western blotting. Cl PARP, cleaved PARP; Cl Cas 3 (cleaved caspase-3). GAPDH was used as the loading control.
We next evaluated the ability of compounds 12, 14, 15 and 1 to induce cell death in the H146 cell line (Figure 7B). These four compounds all effectively induce cell death in H146 in a dose-dependent manner. While compounds 12 and 1 have similar potencies to each other, 14 and 15 are more potent than both 12 and 1 and, even at 10 nM, induce robust cell death in the H146 cell line. Western blotting analysis further showed that while all these four compounds induce cleavage of caspase-3 and poly (ADP- ribose) polymerase (PARP) in a dose-dependent manner (Figure 7C), compound 15 induces robust cleavage of caspase-3 and PARP at concentrations as low as 10 nM, and is more potent than compounds 12 and 1.
To further probe their cellular mechanism of action, we analyzed release of cytochrome C from mitochondria, which is an early event in apoptosis induction. Treatment of H146 cells with compounds 14 and 15 for as short as 2 h induces a release of cytochrome C in a dose-dependent manner (Figure 8). This release of cytochrome C is accompanied by cleavage of PARP, also in a dose-dependent manner. Release of cytochrome C and cleavage of PARP are robust at concentrations of 30 nM for both 14 and 15.
Figure 8.

Analysis of cytochrome C (Cyto C) release from mitochondria into cytosol and cleavage of PARP (Cl PARP) induced by 14 and 15 in H146 cells in 2 h. GAPDH was used as the loading control.
We evaluated the toxicity of compounds 14 and 15 in severe combined immune deficient (SCID) mice and found that 15 is more toxic than 14. While compound 14 at 25 mg/kg, daily intravenous dosing and five days a week for 2 weeks is well tolerated in SCID mice, 15 at the same dose and schedule is toxic to the animals which had severe weight loss. We therefore focused our subsequent in vivo evaluations on 14.
We tested compound 14 for induction of apoptosis in the H146 xenograft tissues in SCID mice (Figure 9). A single intravenous dose of 14 at 25 mg/kg effectively induced cleavage of PARP and caspase-3 in the H146 tumor tissues as early as 3 h and lasted for at least 24 h.
Figure 9.

Analysis of induction of cleavage of PARP and caspase-3 in the H146 xenograft tumor tissues by 14 (25 mg/kg, i.v.). Mice bearing H146 xenograft tumors were dosed with either 14 or vehicle. Animals were sacrificed at 3, 6 and 24 hr time-points and tumor tissues were analyzed by western blot for cleavage of PARP (Cl PARP) and caspase-3 (Cl caspase-3). GAPDH was used as the loading control.
Compound 14 was next evaluated for its antitumor activity in inhibition of tumor growth in the H146 xenograft model. It was administered at 25 mg/kg daily i.v. 5 days a week for 2 weeks. Compound 14 was very effective in inhibition of tumor growth (Figure 10), consistent with our in vivo pharmacodynamics data (Figure 9). In fact, compound 14 was capable of achieving tumor regression during the treatment phase. The strong antitumor activity of 14 is persistent. At day 92, 2 months after the treatment ended, the tumors treated with 14 had a mean volume of 200 mm3, whereas the vehicle-treated tumors had grown to a mean volume of 800 mm3. Mice treated with 14 had a maximum weight loss <10% during the treatment and animals quickly gained weight after the treatment (Figure S1 in SI). There were no other signs of toxicity observed with 14. Hence, compound 14 has a strong antitumor activity at a well-tolerated dose-schedule.
Figure 10.

Antitumor activity of compound 14 in H146 xenograft model. H146 tumor cells were injected subcutaneously into SCID mice and treatments started when tumors reached a mean volume of 70 mm3. Each group consisted of 8 mice/tumors.
Synthesis
The synthesis of compound 8 is shown in Scheme 1. Condensation of ethyl acetoacetate with benzaldehyde afforded ethyl 2-benzylidene-3-oxobutanoate. A Stetter reaction of this compound with 4-chlorobenzaldehyde gave 17, and the pyrrole 18 was obtained by Paal-Knorr cyclization of 17 with methylamine.18 Compound 8 was prepared by hydrolysis of 18, followed by coupling to 1-(3-aminopropyl)-4-methylpiperazine.
Scheme 1.

Synthesis of compound 8.
Reagents and conditions: a) i. Piperidine, AcOH, Toluene; ii. 4-chlorobenzaldehyde, 3-ethyl-5-(2-hydroxyethyl)-4-methylthiazol-3-ium bromide, Et3N, 70 °C; b) MeNH2, MeOH, then HCI, rt; c) i. NaOH, Dioxane, EtOH, H2O, reflux; ii. 3-(4-methylpiperazin-1-yl)propan-1-amine, EDCl, HOBt, DIEA, DCM.
Scheme 2 shows the general method for the synthesis of compounds 12, 14, 15 and 16 with different substituents on the nitrogen of the pyrrole ring. Compound 19 was prepared in the same way as 17. Paal-Knorr cyclization of 19 with different amines afforded the pyrroles 20a-d which were hydrolyzed to yield the acids 21a-d. An Ullmann-type C-N bond formation reaction19 was employed to prepare intermediate 22a-d from 21a-d. Hydrogenation of the nitro group in 22a-d gave the anilines, treatment of which with 4-fluoro-3-nitrobenzene-1-sulfonyl chloride in pyridine gave 23a-d. Displacement of the fluoro group in 23a-d with (R)-N′,N′-dimethyl-4-(phenylthio)butane-1,3-diamine produced compounds 12, 14, 15 and 16.
Scheme 2.

Synthesis of compounds 12, 14, 15 and 16.
Reagents and conditions: a) i. Piperidine, AcOH, Toluene; ii. 4-chlorobenzaldehyde, 3-ethyl-5-(2-hydroxyethyl)-4-methylthiazol-3-ium bromide, Et3N, 70 °C; b) RNH2, MeOH; c) NaOH, Dioxane, EtOH, H2O, reflux; d) 1-(4-nitrophenyl)piperazine, Cul, L-Proline, K2CO3, 80 °C, overnight; e) i. H2, Pd/C; ii. 4-fluoro-3-nitrobenzene-1-sulfonyl chloride, pyridine; f) (R)-N1,N1-dimethyl-4-(phenylthio)butane-1,3-diamine, DIPEA, DMF
Compound 9 was synthesized as shown in Scheme 3. Coupling 21a to 1-(3-aminopropyl)-4-methylpiperazine afforded 24 and intermediate 25 was prepared as described.20 In the presence of Pd(dba)2 and tri-tert-butylphosphine, Buchwald–Hartwig reaction21 between 24 and 25 produced compound 9.
Scheme 3.

Synthesis of compound 9.
Reagents and conditions: a) 3-(4-methylpiperazin-1-yl)propan-1-amine, EDCl, HOBt, DIEA, DCM; b) Pd(dba)2, tri-tert-butylphosphine, sodium tert-butoxide, DMF, toluene;
Syntheses of compounds 10, 11 and 13 are outlined in Scheme 4. Compounds 10 and 11 were produced by coupling compound 12 to the corresponding amines using EDCI and HOBt. Treatment of compound 12 with TFA afforded the decarboxylated compound 13.
Scheme 4.

Synthesis of compounds 10, 11 and 13.
Reagents and conditions: a) 3-(4-methylpiperazin-1-yl)propan-1-amine, EDCl, HOBt, DIEA, DCM; b) methyl amine, EDCl, HOBt, DIEA, DCM, c) TFA, CH2Cl2;
Summary
Using a structure-based design strategy, we have designed and synthesized new and potent Bcl-2/Bcl-xL inhibitors. The most potent compounds, 14 and 15, bind to Bcl-2 and Bcl-xL with subnanomolar affinities and inhibit cell growth in three lung cancer cell lines with low nanomolar IC50 values. Compounds 14 and 15 induce robust apoptosis in H146 cancer cells at concentrations as low as 10 nM. Furthermore, compound 14 induces robust apoptosis in vivo in the H146 tumor tissues, and strongly inhibits tumor growth and achieves tumor regression during the treatment in the H146 xenograft model in mice at a well-tolerated dose-schedule. Determination of the 1.4 Å resolution crystal structure of a potent analogue, 12 complexed with Bcl-xL, provides a structural basis for its high-affinity binding to Bcl-xL and a solid foundation for our optimization effort. Further optimization of this class of compounds may yield highly potent Bcl-2/Bcl-xL inhibitors with optimized pharmacological properties for the treatment of human cancer.
EXPERIMENTAL SECTION
General Information
Unless otherwise stated, all reactions were performed under a nitrogen atmosphere in dry solvents under anhydrous conditions. Unless otherwise noted, reagents were used as supplied without further purification. NMR spectra were acquired at a proton frequency of 300 MHz and chemical shifts are reported in parts per million (ppm) relative to an internal standard. The final products were purified by a C18 reverse phase semi-preparative HPLC column with solvent A (0.1% of TFA in water) and solvent B (0.1% of TFA in CH3CN) as eluents. The purity was determined by Waters ACQUITY UPLC and all the biologically evaluated compounds were > 95% pure (Table S2 and Figures S2–10).
Ethyl 5-(4-chlorophenyl)-1,2-dimethyl-4-phenyl-1H-pyrrole-3-carboxylate (18)
Ethyl acetoacetate (1.3 g, 10 mmol), benzaldehyde (1.06 g, 10 mmol), piperidine (43 μL), and acetic acid (128 μL) were dissolved in toluene (10 mL) and refluxed with azeotropic removal of water overnight. After the solution was cooled it was diluted with EtOAc, washed with 1.0 M HCl, saturated sodium bicarbonate, brine and dried over sodium sulfate. Removal of the solvent under vacuum gave a crude ethyl 2-benzylidene-3-oxobutanoate, which was used directly in the following step without further purification. To a solution of this compound, 4-chlorobenzaldehyde (1.41 g, 10mmol), and triethylamine (1.0 mL) was added 3-ethyl-5-(2-hydroxyethyl)-4-methylthiazolium bromide (0.38 g, 1.5mmol) and the mixture was stirred and heated at 70 °C overnight. After cooling to room temperature, the mixture was diluted with EtOAc, washed with 1M HCl, saturated sodium bicarbonate and brine then dried over sodium sulfate. The EtOAc was removed in vacuo and the residue was purified by flash chromatography on silica gel (1:5 ethyl acetate:hexane) to provide 17, which was treated with methylamine in MeOH (2M, 8 mL) overnight. The reaction mixture was treated with 2M HCl (8 mL) for 20 min and extracted with EtOAc. The EtOAc solution was washed with brine, dried over sodium sulfate and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (1:4 ethyl acetate:hexane) to give 18 (2.45 g, 73% over three steps). 1H NMR (300 MHz, CDCl3), δ 7.25 (d, J=8.4, 2H), 7.20~7.16 (m, 3H), 7.13~7.06 (m, 4H), 4.10 (q, J=7.1, 2H), 3.44 (s, 3H), 2.65 (s, 3H), 1.03 (t, J=7.1, 3H); 13C NMR (75 MHz, CCl3D), δ 165.8, 136.2, 135.8, 133.4, 132.4, 130.8, 130.3, 130.2, 128.4, 127.2, 125.9, 124.1, 111.3, 59.2, 31.8, 13.9, 11.8; ESI MS: m/z 376.8 (M + Na)+.
5-(4-Chlorophenyl)-1,2-dimethyl-N-(3-(4-methylpiperazin-1-yl)propyl)-4-phenyl-1H-pyrrole-3-carboxamide (8)
To a solution of ester 18 (0.53 g, 1.5 mmol) in a mixture of dioxane/ethanol/water (1:1:1, 12 mL) was added NaOH (0.3 g, 7.5mmol) and the solution was refluxed until starting material could no longer be observed by TLC. After cooling, the reaction was neutralized with 1M HCl and extracted with EtOAc. The EtOAc solution was washed with brine, dried over sodium sulfate and concentrated under vacuum to provide 5-(4-chlorophenyl)-1,2-dimethyl-4-phenyl-1H-pyrrole-3-carboxylic acid, which was used directly in the following step without further purification. A solution of this acid, 1-(3-aminopropyl)-4-methylpiperazine (0.35 g, 2.3 mmol), EDCI (0.44 g, 2.3mmol), HOBt (0.30 g, 2.3 mmol) and N,N-diisopropylethylamine (0.54 mL, 3.0 mmol) in CH2Cl2 (10 mL) was stirred for 8 h then concentrated. The residue was purified by HPLC to afford 8 (0.62g, 89%).1H NMR (300 MHz, CD3OD), δ 7.21~7.10 (m, 5H), 7.03~7.00 (m, 4H), 3.49~3.45 (m, 4H), 3.39~3.35 (m, 4H), 3.34 (s, 3H), 3.21~3.19 (m, 2H), 2.91~2.89 (m, 2H), 2.87 (s, 3H), 2.38 (s, 3H), 1.78~1.73 (m, 2H);13C NMR (75 MHz, CD3OD), δ 170.5, 136.6, 134.7, 133.7, 133.5, 131.9, 131.7, 131.2, 129.5, 129.2, 127.6, 122.7, 116.1, 55.3, 51.6, 43.4, 37.1, 32.0, 25.3, 11.4; ESI MS: m/z 465.8 (M + H)+.
Ethyl 5-(4-chlorophenyl)-4-(3-iodophenyl)-1,2-dimethyl-1H-pyrrole-3-carboxylate (20a)
Ethyl acetoacetate (6.1 g, 47 mmol), 3-iodobenzaldehyde (10.8 g, 47 mmol), piperidine (200 μL), and acetic acid (600 μL) were dissolved in toluene (20 mL) and refluxed overnight with azeotropic removal of water. After the solution was cooled it was diluted with EtOAc, washed with 1.0 M HCl, saturated sodium bicarbonate, brine and dried over sodium sulfate. Removal of the solvent under vacuum gave a crude ethyl 2-(3-iodobenzylidene)-3-oxobutanoate, which was used directly in the following step without further purification. To a solution of this, ethyl 2-(3-iodobenzylidene)-3-oxobutanoate, 4-chlorobenzaldehyde (6.6 g, 47 mmol), and triethylamine (4.6 mL) was added 3-ethyl-5-(2-hydroxyethyl)-4-methylthiazolium bromide (1.9 g, 7.1 mmol) and the mixture was stirred and heated at 70 °C overnight. After cooling to room temperature, the mixture was diluted with EtOAc, washed with 1M HCl, saturated sodium bicarbonate, brine and dried over sodium sulfate. The EtOAc was removed in vacuo and the residue was purified by flash chromatography on silica gel (1:5 ethyl acetate:hexane) to give 19, which was treated with a solution of methylamine in MeOH (2M, 35 mL) overnight. Then the reaction mixture was treated with 2M HCl (40 mL) for 20 min and extracted with EtOAc. The EtOAc solution was washed with brine, dried over sodium sulfate and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (1:4 ethyl acetate:hexane) to afford 20a (15.8 g, 70% over three steps). 1H NMR (300 MHz, CCl3D), 7.54 (s, 1H), 7.49 (d, J=7.8, 1H), 7.28 (d, J=8.4, 2H), 6.99 (d, J=7.7, 1H), 6.89 (t, J=7.7, 1H), 4.10 (q, J=7.1, 2H), 3.43 (s, 3H), 2.64 (s, 3H), 1.06 (t, 7.1, 3H); 13C NMR (75 MHz, CCl3D), δ 165.5, 139.9, 138.2, 136.7, 134.8, 133.8, 132.4, 130.4, 129.8, 128.9, 128.6, 122.3, 111.1, 93.0, 59.4, 31.8, 14.0, 11.8; ESI MS: m/z 480.3 (M + H)+ .
Ethyl 5-(4-chlorophenyl)-1-ethyl-4-(3-iodophenyl)-2-methyl-1H-pyrrole-3-carboxylate (20b)
Intermediate 20b was prepared from 19 in 86% yield by a procedure similar to that used for 20a. 1H NMR (300 MHz, CCl3D), δ 7.55 (s, 1H), 7.45 (d, J=7.7, 1H), 7.28 (d, J=7.9, 2H), 7.11 (d, J=7.9, 2H), 6.87 (t, J=7.7, 1H), 4.10 (q, J=7.1, 2H), 3.84 (q, J=7.1, 2H), 2.65 (s, 3H), 1.16 (t, J=7.0, 3H), 1.06 (t, J=7.1, 3H); 13C NMR (75 MHz, CCl3D), δ 165.6, 139.9, 138.1, 135.7, 134.6, 134.0, 132.6, 130.2, 129.9, 129.8, 128.9, 128.6, 122.6, 111.2, 93.0, 59.3, 39.1, 16.1, 14.1, 11.5; ESI MS: m/z 494.0 (M + H)+.
Ethyl 5-(4-chlorophenyl)-4-(3-iodophenyl)-1-isopropyl-2-methyl-1H-pyrrole-3-carboxylate (20c)
Ethyl 2-acetyl-4-(4-chlorophenyl)-3-(3-iodophenyl)-4-oxobutanoate 19 (5g, 10.3 mmol) in MeOH (30 mL) was added isopropyl amine (1.8 g, 31 mmol), acetic acid (5 mL). The reaction mixture was heated to 65 °C for 24 h in a sealed tube. Water (40 mL) was added and extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with brine, dried over sodium sulfate and concentrated in vacuo. The residue was purified by flash chromatography on silica gel to provide 20c (3.8g, 72%). 1H NMR (300 MHz, CCl3D), δ 7.51 (s, 1H), 7.42 (d, J=7.8, 1H), 7.26 (d, J=8.1, 2H), 7.10 (d, J=7.9, 2H), 6.96 (d, J=7.2, 1H), 6.83 (t, J=7.7, 1H), 4.42~4.33 (m, 1H), 4.07 (q, J=7.1, 2H), 2.74 (s, 3H), 1.44 (d, J=7.0, 6H), 1.01 (t, J=7.1, 3H); 13C NMR (75 MHz, CCl3D), δ 165.7, 139.8, 138.4, 135.5, 134.5, 134.0, 133.0, 131.0, 130.4, 129.6, 128.8, 128.5, 122.5, 112.2, 93.0, 59.3, 48.7, 22.3, 14.0, 13.1; ESI MS: m/z 508.4 (M + H)+.
Ethyl 5-(4-chlorophenyl)-1-cyclopropyl-4-(3-iodophenyl)-2-methyl-1H-pyrrole-3-carboxyl-ate (20d)
Intermediate 20d was prepared from 19 by a similar procedure as that for 20a in 84% yield. 1H NMR (300 MHz, CCl3D), δ 7.53~7.48 (m, 2H), 7.23 (d, J=8.5, 2H), 7.07 (d, J=8.5, 2H), 6.97 (d, J=7.7, 1H), 6.89 (t, 7.7, 1H), 4.08 (q, J=7.1, 2H), 3.15~3.09 (m, 1H), 2.71 (s, 3H), 1.04 (t, 7.1, 3H), 0.92~0.85 (m, 2H), 0.58~0.55 (m, 2H); 13C NMR (75 MHz, CCl3D), δ 165.4, 140.0, 139.2, 138.3, 134.8, 133.0, 131.9, 131.1, 130.6, 129.9, 129.0, 128.1, 121.9, 111.4, 93.0, 59.3, 27.2, 14.0, 12.8, 9.6; ESI MS: m/z 506.6 (M + H)+.
5-(4-Chlorophenyl)-4-(3-iodophenyl)-1,2-dimethyl-1H-pyrrole-3-carboxylic acid (21a)
Sodium hydroxide (4.2 g, 104mmol) was added to a solution of the ester 20a (10 g, 20.8 mmol) in a mixture of dioxane/ethanol/water (1:1:1, 150 mL) and the solution was refluxed until no starting material was observed by TLC. After cooling the reaction was neutralized with 1M HCl and extracted with EtOAc. The EtOAc solution was washed with brine, dried over sodium sulfate and concentrated in vacuo to provide 21a (9.4 g, 100% yield). 1H NMR (300 MHz, CCl3D), 7.50~7.48 (m, 2H), 7.29 (d, J=8.3, 2H), 7.08~7.02 (m, 3H), 6.92 (t, J=8.0, 1H), 3.42 (s, 3H), 2.64 (s, 3H); 13C NMR (75 MHz, CCl3D), δ 170.1, 139.7, 138.3, 137.5, 135.0, 133.9, 132.4, 131.0, 130.1, 129.7, 129.0, 128.6, 122.8, 109.7, 93.2, 32.0, 12.2; ESI MS: m/z 452.3 (M + H)+.
5-(4-Chlorophenyl)-1-ethyl-4-(3-iodophenyl)-2-methyl-1H-pyrrole-3-carboxylic acid (21b)
Intermediate 21b was prepared from 20b in 97% yield using a similar procedure as that for 21a. 1H NMR (300 MHz, CCl3D) δ 7.51~7.45 (m, 2H), 7.31~7.28 (m, 2H), 7.10~7.05 (m, 3H), 6.90 (t, J=7.8, 1H), 3.83 (q, J=7.1, 2H), 2.65 (s, 3H), 1.17 (t, J=7.1, 3H); 13C NMR (75 MHz, CCl3D), δ 169.9, 139.6, 137.4, 137.3, 134.9, 134.1, 132.6, 130.5, 130.0, 129.0, 128.6, 123.1, 109.9, 93.2, 39.2, 16.0, 11.9; ESI MS: m/z 466.3 (M + H)+.
5-(4-Chlorophenyl)-4-(3-iodophenyl)-1-isopropyl-2-methyl-1H-pyrrole-3-carboxylic acid (21c)
Intermediate 21c was prepared from 20c in 98% yield using a similar procedure as that for 21a. 1H NMR (300 MHz, CCl3D) δ 7.46~7.43 (m, 2H), 7.30~7.27 (m, 2H), 7.08~7.02 (m, 3H), 6.88 (t, J=7.7, 1H), 4.41~4.32 (m, 1H), 2.73 (s, 3H), 1.44 (d, J=7.1, 6H); 13C NMR (75 MHz, CCl3D) δ 169.0, 139.6, 137.7, 137.1, 134.8, 134.1, 133.0, 131.0, 130.7, 129.9, 128.9, 128.4, 123.0, 110.6, 93.1, 48.8, 13.4; ESI MS: m/z 502.5 (M + Na)+.
5-(4-Chlorophenyl)-1-cyclopropyl-4-(3-iodophenyl)-2-methyl-1H-pyrrole-3-carboxylic acid (21d)
Intermediate 21d was prepared in 98% yield from 20d by a similar procedure as that for 21a. 1H NMR (300 MHz, CCl3D) δ 7.52~7.49 (m, 2H), 7.24 (d, J=8.5, 2H), 7.06~7.03 (m, 3H), 6.92 (t, J=7.7, 1H), 3.16~3.08 (m, 1H), 2.72 (s, 3H), 0.93~0.86 (m, 2H), 0.59~0.53 (m, 2H);13C NMR (75 MHz, CCl3D) δ 169.4, 140.8, 139.8, 137.5, 135.1, 133.1, 131.9, 131.7, 130.4, 130.1, 129.1, 128.2, 122.4, 109.9, 93.2, 27.3, 13.1, 9.6.
5-(4-Chlorophenyl)-1,2-dimethyl-4-(3-(4-(4-nitrophenyl)piperazin-1-yl)phenyl)-1H-pyrrole-3-carboxylic acid (22a)
21a (3.1g, 6.86 mmol), 1-(4-nitrophenyl)piperazine (2.84g, 13.7 mmol), CuI (131 mg, 0.69 mmol), L-proline (158 mg, 1.37mmol) and K2CO3 (1.89 g, 13.7mmol) were dissolved in DMSO (20 mL). This mixture was heated to 80 °C overnight under nitrogen. After the solution was cooled, saturated ammonium chloride solution was added and the solution was extracted with CH2Cl2. The combined organic layers were washed with brine and dried over sodium sulfate and concentrated. Purification of the residue by flash chromatography on silica gel (1:1 ethyl acetate:hexane) afforded 22a (2.48 g, 68%). 1H NMR (300 MHz, CCl3D) δ 8.13 (d, J=9.2, 2H), 7.28 (d, J=8.2, 2H), 7.08~7.06 (m, 3H), 6.84~6.78 (m, 4H), 6.68 (d, J=7.4, 1H), 3.50 (br. 4H), 3.44 (s, 3H), 3.18 (br. 4H), 2.63 (s, 3H); 13C NMR (75 MHz, CCl3D), δ 170.0, 154.6, 138.6, 135.8, 133.7, 132.5, 130.9, 130.1, 128.6, 128.3, 126.0, 124.2, 120.2, 114.7, 112.7, 109.6, 49.3, 46.6, 32.0, 12.2; ESI MS: m/z 531.5 (M + H)+.
5-(4-Chlorophenyl)-1-ethyl-2-methyl-4-(3-(4-(4-nitrophenyl)piperazin-1-yl)phenyl)-1H-pyrrole-3-carboxylic acid (22b)
Intermediate 22b was prepared in 70% yield from 21b using a similar procedure as that for 22a. 1H NMR (300 MHz, CCl3D) δ 8.13 (d. J=9.2, 2H), 7.28 (d, J=8.2, 2H), 7.12 (d, J=8.3, 2H), 7.05 (t, J=7.8, 1H), 6.83 (d, J=9.3, 2H), 6.73~6.66 (m, 3H), 3.85 (q, J=7.1, 2H), 3.46~3.45 (m, 4H), 3.17~3.15 (m, 4H), 2.64 (s, 3H), 1.18 (t, J=7.0, 3H); 13C NMR (75 MHz, CCl3D), δ 170.8, 154.8, 149.4, 138.4, 137.1, 135.7, 133.8, 132.7, 130.6, 130.3, 128.6, 128.1, 126.0, 124.7, 123.2, 119.9, 114.2, 112.6, 109.9, 48.9, 46.7, 39.2, 16.0, 12.0; ESI MS: m/z 545.5 (M + H)+.
5-(4-Chlorophenyl)-1-isopropyl-2-methyl-4-(3-(4-(4-nitrophenyl)piperazin-1-yl)phenyl)-1H-pyrrole-3-carboxylic acid (22c)
Intermediate 22c was prepared from 21c in 73% yield by a similar procedure as that for compound 22a. 1H NMR (300 MHz, CCl3D) δ 11.8 (br. 1H), 8.13 (d, J=9.1, 2H), 7.27 (d, J=8.6, 2H), 7.10 (d, J=8.2, 2H), 7.03 (t, J=8.2, 1H), 6.83 (d, J=9.2, 2H), 6.67~6.53 (m, 3H), 4.44~4.35 (m, 1H), 3.46 (br. 4H), 3.14 (br. 4H), 2.73 (s, 3H), 1.45 (d, J=7.0, 6H); 13C NMR (75 MHz, CCl3D), δ 170.9, 154.8, 149.4, 138.4, 137.0, 136.0, 133.8, 133.1, 131.4, 130.8, 128.4, 128.0, 126.0, 124.6, 123.2, 119.8, 114.1, 112.6, 110.9, 48.9, 48.8, 46.7, 22.3, 13.5; ESI MS: m/z 559.6 (M + H)+.
5-(4-Chlorophenyl)-1-cyclopropyl-2-methyl-4-(3-(4-(4-nitrophenyl)piperazin-1-yl)phenyl)-1H-pyrrole-3-carboxylic acid (22d)
Intermediate 22d was prepared from 21d in 76% yield by a similar procedure as that for compound 22a. 1H NMR (300 MHz, CCl3D) δ 8.13 (d, J=9.3, 2H), 7.22 (d, J=8.4, 2H), 7.12~7.05 (m, 3H), 6.83 (d, J=8.4, 2H), 6.74~6.70 (m, 2H), 6.64 (d, J=7.5, 1H), 3.48~3.45 (m, 4H), 3.18~3.15 (m, 5H), 2.70 (s, 3H), 0.93~0.86 (m, 2H), 0.56~0.53 (m, 2H); 13C NMR (75 MHz, CCl3D), δ 170.2, 154.8, 149.6, 140.6, 138.4, 135.8, 132.9, 132.0, 131.5, 131.0, 128.2, 128.1, 125.9, 123.9, 123.3, 120.0, 114.4, 112.6, 110.1, 48.9, 46.7, 27.3, 13.2, 9.7; ESI MS: m/z 557.9 (M + H)+.
5-(4-Chlorophenyl)-4-(3-(4-(4-(4-fluoro-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-1,2-dimethyl-1H-pyrrole-3-carboxylic acid (23a)
To a solution of compound 22a (1.2 g, 2.3 mmol) in a mixture of CH2Cl2 (10 mL) and MeOH (10 mL) was added 10% Pd-C (120 mg). The solution was stirred under 1 atm of H2 at room temperature for 0.5 hour before filtering through celite and being concentrated. The resulting aniline was used in the next step without purification. To this aniline in pyridine (20 mL), 4-fluoro-3-nitrobenzene-1-sulfonyl chloride (0.54 g, 2.3 mmol) was added at 0 °C. The mixture was stirred at 0 °C for 30 minutes. The pyridine was removed under vacuum and the residue was purified by flash chromatography on silica gel (3:2 ethyl acetate:hexane) to give 23a (1.18 g, 74% in two steps). 1H NMR (300 MHz, CCl3D) δ 8.43 (dd, J=2.1, 6.7, 1H), 7.89~7.86 (m, 1H), 7.33 (d, J=9.6, 1H), 7.25 (d, J=8.4, 2H), 7.16 (br. 1H), 7.08~7.05 (m, 3H), 6.80 (d, J=8.9, 2H), 6.73 (s, 2H), 6.66 (d, J=7.4, 1H), 3.44 (s, 3H), 3.17~3.11 (m, 8H), 2.64 (s, 3H); 13C NMR (75 MHz, CCl3D:CD3OD=5:1), δ 167.7, 149.8, 149.5, 137.0, 136.7, 136.6, 135.9, 134.2, 134.1, 133.3, 132.3, 130.4, 130.3, 128.3, 127.9, 127.5, 125.6, 124.6, 124.1, 123.3, 119.7, 119.3, 119.0, 116.7, 114.5, 110.2, 49.7, 48.1, 31.7, 11.8;ESI MS: m/z 704.6 (M + H)+.
5-(4-Chlorophenyl)-1-ethyl-4-(3-(4-(4-(4-fluoro-3-nitrophenylsulfonamido)phenyl)-piperazin-1-yl)phenyl)-2-methyl-1H-pyrrole-3-carboxylic acid (23b)
Intermediate 23b was prepared from 22b in 67% yield in two steps using a similar procedure as that for 23a. 1H NMR (300 MHz, CCl3D) δ 8.44 (dd, J=2.2, 6.8, 1H), 7.89~7.86 (m, 1H), 7.34~7.25 (m, 3H), 7.11 (d, J=8.4, 2H), 7.04 (t, J=8.0, 1H), 6.95 (d, J=8.9, 2H), 6.80 (d, J=8.9, 2H), 6.72~6.64 (m, 3H), 3.85 (q, J=7.1, 2H), 3.18~3.10 (m, 8H), 2.65 (s, 3H), 1.18 (t, J=7.1, 3H); 13C NMR (75 MHz, CCl3D), δ 169.8, 150.1, 149.9, 137.14, 137.07, 136.4, 135.7, 134.4, 134.3, 133.8, 132.7, 130.6, 130.3, 128.5, 128.1, 126.5, 125.9, 125.5, 124.7, 123.1, 119.8, 119.6, 119.3, 116.6, 114.4, 109.8, 49.3, 48.7, 39.2, 16.0, 12.0; ESI MS: m/z 740.3 (M + Na)+.
5-(4-Chlorophenyl)-4-(3-(4-(4-(4-fluoro-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-1-isopropyl-2-methyl-1H-pyrrole-3-carboxylic acid (23c)
Intermediate 23c was prepared in 71% yield in two steps using a similar procedure as that for 23a. 1H NMR (300 MHz, CCl3D) δ 8.45 (dd, J=2.1, 6.7, 1H), 7.90~7.85 (m, 1H), 7.39 (br., 1H), 7.34~7.23 (m, 3H), 7.10~7.01 (m, 3H), 6.95 (d, J=8.8, 2H), 6.81 (d, J=8.9, 2H), 6.71~6.63 (m, 3H), 4.43~4.34 (m, 1H), 3.18~3.10 (m, 8H), 2.73 (s, 3H), 1.45 (d, J=7.0, 6H); 13C NMR (75 MHz, CCl3D), δ 169.7, 150.1, 149.9, 137.2, 137.0, 136.9, 136.5, 136.4, 135.9, 134.4, 134.3, 133.8, 133.1, 131.3, 130.8, 128.3, 128.1, 126.5, 125.9, 125.5, 124.6, 123.1, 119.6, 119.3, 116.6, 114.4, 110.8, 49.3, 48.8, 48.7, 22.2, 13.5; ESI MS: m/z 754.2 (M + Na)+.
(R)-5-(4-Chlorophenyl)-4-(3-(4-(4-(4-((4-(dimethylamino)-1-(phenylthio)butan-2-yl)amino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-1,2-dimethyl-1H-pyrrole-3-carboxylic acid (12)
DIEA (70 μL, 0.4 mmol) was added to a solution of 23a (158 mg, 0.22mmol) and (R)-N1,N1-dimethyl-4-phenylthio)butane-1,3-diamine (51 mg, 0.22mmol) in DMF. The solution was stirred overnight and concentrated. The residue was purified by HPLC to afford 12 (145 mg, 71%). 1H NMR (300 MHz, CD3OD), δ 8.30 (d, J=2.2 Hz, 1H), 7.59 (dd, J=2.2, 9.2, 1H), 7.26~7.23 (m, 2H), 7.18~7.13 (m, 3H), 7.09~6.88 (m, 13H), 4.10~4.07 (m, 1H), 3.40 (s, 3H), 3.36-3.29 (m, 9H), 3.21-3.15(m, 3H), 2.83 (s, 6H), 2.58 (s, 3H), 2.26-2.10 (m, 2H); 13C NMR (75 MHz, CD3OD), δ 169.1, 148.5, 148.0, 146.9, 139.0, 138.4, 136.2, 134.8, 134.4, 134.0, 132.3, 132.2, 132.1, 131.9, 131.6, 130.1, 129.6, 129.5, 129.0, 128.0, 127.9, 127.6, 124.8. 124.3, 122.9, 118.9, 117.2, 116.2, 111.5, 55.9, 53.2, 52.4, 50.1, 43.5, 39.5, 32.2, 30.1, 12.1; ESI MS: m/z 908.9 (M + H)+.
(R)-5-(4-Chlorophenyl)-4-(3-(4-(4-(4-((4-(dimethylamino)-1-(phenylthio)butan-2-yl)amino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-1-ethyl-2-methyl-1H-pyrrole-3-carboxylic acid (14)
Compound 14 was prepared from 23b in 81% yield using a similar procedure as that for compound 12. 1H NMR (300 MHz, CD3OD) δ 8.24 (d, J=2.1, 1H), 7.54 (dd, J=2.1, 9.1, 1H), 7.20 (d, J=8.4, 2H), 7.12~6.85 (m, 16H), 4.06~4.03 (m, 1H), 3.80 (q, J=7.1, 2H), 3.28~3.24 (m, 9H), 3.16~3.09 (m, 3H), 2.78 (s, 6H), 2.54 (s, 3H), 2.22~2.06 (m, 2H), 1.03 (t, J=7.1, 3H); 13C NMR (75 MHz, CD3OD), δ 169.1, 148.1, 148.0, 145.9, 139.1, 137.4, 136.2, 135.1, 134.4, 134.3, 132.6, 132.1, 132.0, 131.7, 131.6, 130.1, 129.64, 129.58, 128.0, 127.9, 127.4, 125.1, 124.1, 123.1, 119.0, 117.5, 116.4, 111.7, 55.9, 53.6, 52.4, 50.0, 43.5, 40.2, 39.5, 30.1, 16.2, 12.0; ESI MS: m/z 922.6 (M + H)+.
(R)-5-(4-Chlorophenyl)-4-(3-(4-(4-(4-((4-(dimethylamino)-1-(phenylthio)butan-2-yl)amino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-1-isopropyl-2-methyl-1H-pyrrole-3-carboxylic acid (15)
Compound 15 was prepared from 22c in 79% yield using a similar procedure as that for 12. 1H NMR (300 MHz, CD3OD), δ 8.29 (d, J=2.2, 1H), 7.60 (dd, J=2.2, 9.2, 1H), 7.26~6.90 (m, 18H), 4.41~4.36 (m, 1H), 4.10~4.08 (m, 1H), 3.38~3.31 (m, 9H), 3.21~3.15 (m, 3H), 2.83 (s, 6H), 2.68 (s, 3H), 2.24~2.14 (m, 2H), 1.39 (d, J=7.1, 6H); 13C NMR (75 MHz, CD3OD), δ 169.2, 148.2, 148.0, 145.8, 139.4, 137.1, 136.2, 135.2, 134.8, 134.4, 132.9, 132.6, 132.2, 132.1, 131.6, 130.1, 129.9, 129.6, 129.4, 128.0, 127.9, 127.6, 125.1. 124.2, 122.9, 119.0, 117.5, 116.2, 112.8, 55.9, 53.8, 52.4, 50.3, 50.0, 43.5, 39.5, 30.1, 22.5, 13.5; ESI MS: m/z 936.8 (M + H)+.
(R)-5-(4-Chlorophenyl)-1-cyclopropyl-4-(3-(4-(4-(4-((4-(dimethylamino)-1-(phenylthio)-butan-2-yl)amino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-2-methyl-1H-pyrrole-3-carboxylic acid (16)
Compound 16 was prepared from 22d in 78% yield by a similar procedure as that for compound 12. 1H NMR (300 MHz, CD3OD), δ 8.29 (d, J=2.3, 1H), 7.60 (dd, J=2.3, 9.2, 1H), 7.24~6.91 (m, 18H), 4.12~4.07 (m, 1H), 3.39~3.31 (m, 9H), 3.24~3.14 (m, 4H), 2.83 (s, 6H), 2.67 (s, 3H), 2.24~2.15 (m, 2H), 0.89~0.82 (m, 2H), 0.50~0.44 (m, 2H); 13C NMR (75 MHz, CD3OD), δ 169.0, 148.5, 147.9, 140.8, 139.0, 136.2, 134.4, 134.1, 133.6, 132.8, 132.7, 132.3, 131.6, 130.1, 129.6, 129.1, 128.0, 127.9, 127.7, 124.5, 124.3, 122.9, 118.9, 117.3, 116.2, 112.0, 55.9, 53.2, 52.4, 50.1, 43.5, 39.6, 30.1, 28.3, 13.2, 10.3; ESI MS: m/z 935.3 (M + H)+.
5-(4-Chlorophenyl)-4-(3-iodophenyl)-1,2-dimethyl-N-(3-(4-methylpiperazin-1-yl)propyl)-1H-pyrrole-3-carboxamide (24)
A solution of 21a (1.0 g, 2.2 mmol), 1-(3-aminopropyl)-4-methylpiperazine (0.52g, 3.3 mmol), EDCI (0.64g, 3.3mmol), HOBt (0.43 g, 3.3 mmol) and N,N-diisopropylethylamine (0.77 mL, 4.4 mmol) in CH2Cl2 (15 mL) was stirred for 8 h then concentrated. The residue was purified by flash chromatography on silica gel (2:1 ethyl acetate:methanol) to afford 24 (1.1 g, 86%).1H NMR (300 MHz, CCl3D), δ7.42 (s, 1H), 7.39 (d, J=7.9, 1H), 7.16 (d, d, J=8.4, 2H), 6.95~6.92 (m, 3H), 6.82 (t, J=7.7, 1H), 5.41 (t, J=5.3, 1H), 3.28 (s, 3H), 3.19~3.11 (m, 2H), 2.44 (s, 3H), 2.24~2.11 (m, 11H), 2.04 (t, J=7.0, 2H), 1.43~1.34 (m, 2H); 13C NMR (75 MHz, CCl3D), δ165.7, 139.3, 137.1, 135.4, 133.7, 133.4, 132.2, 129.9, 129.8, 129.6, 128.6, 119.0, 115.1, 94.1, 55.9, 55.0, 53.0, 46.0, 37.9, 31.6, 26.3, 11.3; ESI MS: m/z 591.7 (M + H)+.
(R)-5-(4-Chlorophenyl)-4-(3-(4-(4-(((4-((4-(dimethylamino)-1-(phenylthio)butan-2-yl)amino)phenyl)sulfonyl)carbamoyl)phenyl)piperazin-1-yl)phenyl)-1,2-dimethyl-N-(3-(4-methylpiperazin-1-yl)propyl)-1H-pyrrole-3-carboxamide (9)
Pd(dba)2 (3.5 mg, 0.006 mmol), tri-tert-butylphosphine in toluene (1M, 4.8 μL), and sodium tert-butoxide (18 mg, 0.18 mmol) were added to a stirred slurry of 24 (71mg, 0.12 mmol) and 25 (88 mg, 0.14 mmol) in a mixture of toluene/DMF (1:1, 4 mL) at room temperature under nitrogen. The mixture was heated to 70 °C and monitored by thin-layer chromatography. After complete consumption of starting materials, the reaction mixture was filtered through celite and concentrated. Purification of the residue by HPLC yielded 9 (37 mg, 29%). 1H NMR (300 MHz, CD3OD), δ 8.59 (d, J=2.3, 1H), 7.83 (dd, J=2.3, 9.2, 1H), 7.68 (d, d, J=8.9, 2H), 7.21~7.06 (m, 5H), 7.03~6.84 (m, 9H), 6.75~6.74 (m, 2H), 4.09~4.08 (m, 1H), 3.50~3.25 (m, 16H), 3.20~3.14 (m, 9H), 2.95~2.79 (m, 11H), 2.34 (s, 3H), 2.20~2.12 (m, 2H), 1.82~1.77 (m, 2H);13C NMR (75 MHz, CD3OD), δ 170.3, 167.1, 155.5, 148.5, 137.8, 136.2, 135.4, 134.8, 133.9, 133.4, 132.1, 132.0, 131.5, 131.3, 130.4, 130.2, 130.1, 129.8, 129.6, 127.9, 127.3, 122.5, 121.9, 120.7, 119.8, 116.7, 116.2, 116.0, 115.055.9, 55.4, 52.5, 52.3, 51.7, 47.6, 43.6, 43.5, 39.3, 37.3, 32.1, 30.1, 25.4, 11.5; ESI MS: m/z 1076.3 (M + H)+ .
(R)-5-(4-Chlorophenyl)-4-(3-(4-(4-(4-((4-(dimethylamino)-1-(phenylthio)butan-2-yl)amino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-1,2-dimethyl-N-(3-(4-methylpiperazin-1-yl)propyl)-1H-pyrrole-3-carboxamide (10)
A mixture of 12 (88 mg, 0.097mmol), 1-(3-aminopropyl)-4-methylpiperazine (22.8 mg, 0.145 mmol), EDCI (27.9 mg, 0.145 mmol), HOBt (18.7 mg, 0.145 mol) and DIEA (52 μL, 0.29 mmol) in CH2Cl2 (5 mL) was stirred for 8 h and concentrated. The residue was purified by HPLC to provide 10 (86 mg, 85%). 1H NMR (300 MHz, CD3OD), δ 8.29 (d, J=2.1, 1H), 7.58 (dd, J=2.1, 9.0, 1H), 7.26 (d, J=8.3, 2H), 7.17~6.89 (m, 14H), 6.73~6.70 (m, 2H), 4.10~4.05 (m, 1H), 3.38~3.31 (m, 10H), 3.24~3.18 (m, 15H), 2.82~2.87 (m, 11H), 2.42 (s, 3H), 2.20~2.16 (m, 2H), 1.76~1.71 (m, 2H); 13C NMR (75 MHz, CD3OD), δ 170.3, 150.3, 148.5, 148.0, 137.7, 136.2, 134.8, 134.4, 133.8, 133.3, 132.5, 132.2, 132.1, 131.6, 131.2, 130.2, 130.1, 129.6, 128.0, 127.9, 127.6, 125.1, 124.2, 122.6, 120.6, 118.9, 116.3, 116.2, 55.9, 55.3, 52.6, 52.4, 51.1, 50.9, 50.4, 43.6, 43.5, 39.5, 37.4, 32.0, 30.1, 25.9, 11.4; ESI MS: m/z 1047.8(M + H)+.
(R)-5-(4-Chlorophenyl)-4-(3-(4-(4-(4-((4-(dimethylamino)-1-(phenylthio)butan-2-yl)amino)-3-nitrophenylsulfonamido)phenyl)piperazin-1-yl)phenyl)-N,1,2-trimethyl-1H-pyrrole-3-carboxamide (11)
A mixture of 12 (37 mg, 0.039 mmol), methylamine in THF (2M, 39 μM, 0.078 mmol), EDCI (15.0 mg, 0.078 mmol), HOBt (10.1mg, 0.078mol) and DIEA (22 μL, 0.12 mmol) in CH2Cl2 (3 mL) was stirred for 8 h then concentrated. The residue was purified by HPLC to provide 11 (30 mg, 81%). 1H NMR (300 MHz, CD3OD), δ 8.36 (d, J=2.1, 1H), 7.65 (dd, J=2.1, 9.2, 1H), 7.33 (d, J=8.4, 2H), 7.35~7.01 (m, 13H), 6.97 (d, J=9.2, 1H), 6.91 (s, 1H), 6.85 (d, J=7.4, 1H), 4.15~4.14 (m, 1H), 3.43~3.36 (m, 12H), 3.27~3.20 (m, 3H), 2.89 (s, 6H), 2.73 (s, 3H), 2.45 (s, 3H), 2.30~2.15 (m, 2H); 13C NMR (75 MHz, CD3OD), δ 170.2, 148.3, 147.9, 147.5, 137.8, 136.2, 134.8, 134.4, 133.9, 133.2, 132.9, 132.24, 132.16, 131.6, 131.1, 130.3, 130.1, 129.6, 128.0, 127.9, 127.6, 126.9, 124.1, 121.9, 121.2, 119.3, 117.0, 116.7, 116.2, 55.9, 52.4, 52.3, 50.8, 43.5, 39.5, 31.9, 30.1, 26.6, 11.3; ESI MS: m/z 921.3 (M + H)+.
(R)-N-(4-(4-(3-(2-(4-Chlorophenyl)-1,5-dimethyl-1H-pyrrol-3-yl)phenyl)piperazin-1-yl)phenyl)-4-((4-(dimethylamino)-1-(phenylthio)butan-2-yl)amino)-3-nitrobenzenesulfonamide (13)
TFA (0.5 mL) was added to a solution of 12 (45mg, 0.050 mmol) in CH2Cl2 (2 mL). The solution was stirred for 15 min and evaporated. The residue was purified by HPLC to afford compound 13 (38 mg, 88%). 1H NMR (300 MHz, CD3OD), δ 8.28 (d, J=2.2, 1H), 7.59 (dd, J=2.2, 9.1, 1H), 7.34 (d, J=8.4, 2H), 7.18~6.89 (m, 17H), 4.09~4.05 (m, 1H), 3.36~3.27 (m, 1H), 3.20~3.14 (m, 4H), 2.82 (s, 6H), 2.24 (s, 3H), 2.16~2.14 (m, 2H); 13C NMR (75 MHz, CD3OD), δ 148.02, 148.95, 147.2, 140.1, 136.2, 134.7, 134.4, 134.0, 133.7, 132.7, 132.2, 131.6, 131.2, 130.6, 130.1, 130.0, 128.0, 127.9, 127.5, 125.5, 124.1, 122.0, 119.0, 118.9, 116.2, 55.9, 53.2, 52.4, 50.2, 43.5, 39.5, 31.6, 30.1, 12.4; ESI MS: m/z 865.3 (M + H)+.
Fluorescence polarization-based (FP) binding assays
The expression and purification of Bcl-2, Bcl-xL and Mcl-1 proteins and the FP binding assay methods are the same as previously described.17 Briefly, IC50 and Ki values of our synthesized compounds to Bcl-2/Bcl-xL/Mcl-1 proteins were determined in competitive binding experiments. Mixtures of serial dilutions of tested compounds and pre-incubated protein/probe complex were incubated at room temperature for 1–2 hours with gentle shaking. The final concentrations of proteins and fluorescent probes were 1.5 nM and 1 nM for the Bcl-2 assay, 10 nM and 2 nM for the Bcl-xL assay, and 20 nM and 2 nM for the Mcl-1 assay, respectively. Final DMSO concentration is 4%. Fluorescence polarization (mP) values were measured and plotted over total compound concentrations. IC50 values were determined by nonlinear regression fitting of the binding curves. Ki values were calculated as described previously.22
Molecular modeling
Crystal structures of Bcl-xL with 124 (PDB entry: 2YXJ) and 7 were used to model the binding stances of our designed compounds with Bcl-xL with the GOLD program (version 4.0.1).25, 26 In the docking simulation, the center of the binding sites for Bcl-xL was set at F97 and the radius of the binding site was defined as 12 Å, large enough to cover all the binding pockets. For each genetic algorithm (GA) run, a maximum of 200,000 operations were performed on a population of 5 islands of 100 individuals. Operator weights for crossover, mutation and migration were set to 95, 95 and 10 respectively. The docking simulation was terminated after 20 runs for 8 and designed compounds. ChemScore, implemented in Gold, was used as the fitness function to evaluate the docked conformations. The 20 conformations ranked highest by each fitness function were saved for analysis of the predicted docking modes. All the ligand modifications were performed using the Sybyl program.27
Cell-free mitochondrial functional assays
The MDA-MB-231 (subclone 2LMP) cancer cell line was used in the functional assays. Cells were cultured in 150 mm cell culture dishes up to 80% confluent in a healthy state. After harvesting, cell pellets were washed by cold PBS.
Cell pellets were reconstituted in the mitochondrial isolation buffer (MIB, 10mM Tris, 0.1mM EDTA, 250mM sucrose with protease inhibitors and 1mM PMSF added immediately prior to the assay, pH = 7.4). Mixtures were incubated at 4 °C for 20 min with occasional gentle shaking. Following homogenizing the cells with glass cell homogenizer on ice, mitochondria were isolated by differential centrifugation steps followed by two washes in MIB and the third wash in mitochondrial reaction buffer (MRB, 20mM HEPES, 100mM KCl, 2.5mM MgCl2, 100mM sucrose, 1mM DTT, with protease inhibitors and 1mM PMSF added immediately prior to the assay, pH = 7.5).
Enriched mitochondria pellets were resuspended in the mitochondria resuspending buffer. Aliquots of the mitochondria suspensions (total protein 100–150μg for each aliquot) were incubated with Bcl-2 or Bcl-xL or Mcl-1 protein, together with BIM peptide, and different concentrations of an inhibitor at 37 °C for 1 h. Mitochondria treated with DMSO were included as the negative control. Cytochrome c and Smac released to the supernatant and remaining in the mitochondria pellets were analyzed by Western Blotting using primary anti-Cytochrome c and anti-Smac antibodies from Cell Signaling (Danvers, MA) and Calbiochem (Darmstadt, Germany), respectively. The results were shown in Figure 4.
Assays of cell growth and cell death and Western blotting
These assays were performed same as previously described.17 Human small cell lung cancer cell lines H146 and H1417 were purchased from American Type Culture Collection (ATCC) and were maintained in RPMI-1640 medium containing 10% FBS. Rabbit antibodies against PARP and caspase-3 are from Cell Signaling Technology. Rabbit anti-GAPDH is from Santa Cruz Biotechnology.
Assay to assess release of cytochrome C from mitochondria in cells
To examine release of cytochrome C from mitochondria into cytosol, H146 cancer cells were treated with different concentrations of compounds 14 and 15 for 2 h. Cells were collected. Release of cytochrome C from mitochondria into cytosol was analyzed using the Cytochrome C Assay kit (Millipore).
In vivo pharmacodynamic (PD) and efficacy studies in the H146 xenograft model
For in vivo PD and efficacy studies, the H146 small-cell lung cancer xenograft model was employed. To develop xenograft tumors, 5 × 106 H146 cancer cells with matrigel were injected subcutaneously on the dorsal side of the SCID mice (from Charles River), one tumor per mouse.
For PD studies, mice bearing H146 xenograft tumors were administered with a single dose of 14, 1 or vehicle when tumor volume was approximately 100 mm3. Tumor tissues were harvested at indicated time points. Tumor tissues were analyzed using Western blotting to examine levels of PARP and caspase-3, as well as cleaved PARP and caspase-3 in the tumor tissues.
For efficacy studies, when tumors reached tumor volumes between 40–110 mm3, mice were randomized into different groups, 8 mice per group, with a mean tumor volume of 70 mm3. Mice were treated with 14 at 25 mg/kg, intravenously, daily, 5 days a week for 2 weeks, or 1 at 100 mg/kg, intraperitoneally, daily, 5 days a week for 2 weeks, or vehicle control. Tumor sizes and animal weights were measured 3 times a week during the treatment and twice a week after the treatment. Data are presented as mean tumor volumes ± SEM. Statistical analyses were performed by two-way ANOVA and unpaired two-tailed t test, using Prism (version 4.0, GraphPad, La Jolla, CA). P < 0.05 was considered statistically significant. The efficacy experiment was performed under the guidelines of the University of Michigan Committee for Use and Care of Animals.
Bcl-xL Crystallographic studies
Expression and Purification of Bcl-xL protein was used the same method as described earlier.17 Prior to crystallization, Bcl-xL was incubated with a 5-fold molar excess of compound 12 in the presence of 4% DMSO for 1 h at 4 °C and then concentrated to 7 mg/mL. Crystals of Bcl-xL:12 were grown by vapor diffusion in a sitting drop tray against a well solution of 4% PEG 3000, 0.6 M zinc acetate, and 100 mM sodium acetate pH 4.5. In each experiment, the crystallization drops contained equal volumes of protein and well solution. Prior to data collection, crystals were cryoprotected in well solution with increasing amounts of glycerol to a final concentration of 20%, then flash frozen in liquid nitrogen.
X-ray data was collected at LS-CAT ID-21-F and G lines at the Advanced Photon Source at Argonne National Lab. Data were processed with HKL2000.28 Bcl-xL:12 crystallized in P212121 space group and diffracted to 1.4 Å resolution. The structure contained one molecule in the asymmetric unit. The structure of the complex was solved by molecular replacement with Phaser29 using a structure of Bcl-xL previously solved in our laboratory as a starting model. Iterative rounds of refinement and model building were completed using Buster30 and Coot31 respectively. The initial Fo-Fc electron density map revealed the presence of the compounds in the binding site of Bcl-xL. Every atom of 12 was visible in the Fo-Fc electron density map contoured at 3 sigma. The PRODRG server32 was used to create the starting coordinates and restraint files for the compounds. The current Rfree/Rwork for the Bcl-xL:12 structure is 0.1847/.0.1641. All amino acids fall into the allowed regions of the Ramanchandran plot with 98% in the preferred regions. Data collection and refinement statistics are given in Table S1 in SI.
Supplementary Material
Acknowledgments
This research was supported in part by a grant from the National Cancer Institute, National Institutes of Health (U19CA113317). Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817). Coordinates for Bcl-xL complexed with 12 were deposited into the Protein Data Bank with accession numbers 3SP7.
Footnotes
Supporting Information (SI) Available: Experimental procedures and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324–1337. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–315. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- 4.Evan G, Littlewood T. A matter of life and cell death. Science. 1998;281:1317–1322. doi: 10.1126/science.281.5381.1317. [DOI] [PubMed] [Google Scholar]
- 5.Ziegler DS, Kung AL. Therapeutic targeting of apoptosis pathways in cancer. Curr Opin Oncol. 2008;20:97–103. doi: 10.1097/CCO.0b013e3282f310f6. [DOI] [PubMed] [Google Scholar]
- 6.Reed JC. Apoptosis-based therapies. Nat Rev Drug Discov. 2002;1:111–121. doi: 10.1038/nrd726. [DOI] [PubMed] [Google Scholar]
- 7.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 8.Cory S, Adams JM. Killing cancer cells by flipping the Bcl-2/Bax switch. Cancer Cell. 2005;8:5–6. doi: 10.1016/j.ccr.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 9.Labi V, Erlacher M, Kiessling S, Villunger A. BH3-only proteins in cell death initiation, malignant disease and anticancer therapy. Cell Death Differ. 2006;13:1325–1338. doi: 10.1038/sj.cdd.4401940. [DOI] [PubMed] [Google Scholar]
- 10.Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov. 2008;7:989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- 11.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 12.Petros AM, Dinges J, Augeri DJ, Baumeister SA, Betebenner DA, Bures MG, Elmore SW, Hajduk PJ, Joseph MK, Landis SK, Nettesheim DG, Rosenberg SH, Shen W, Thomas S, Wang X, Zanze I, Zhang H, Fesik SW. Discovery of a potent inhibitor of the antiapoptotic protein Bcl-xL from NMR and parallel synthesis. J Med Chem. 2006;49:656–663. doi: 10.1021/jm0507532. [DOI] [PubMed] [Google Scholar]
- 13.Wendt MD, Shen W, Kunzer A, McClellan WJ, Bruncko M, Oost TK, Ding H, Joseph MK, Zhang H, Nimmer PM, Ng SC, Shoemaker AR, Petros AM, Oleksijew A, Marsh K, Bauch J, Oltersdorf T, Belli BA, Martineau D, Fesik SW, Rosenberg SH, Elmore SW. Discovery and structure-activity relationship of antagonists of B-cell lymphoma 2 family proteins with chemopotentiation activity in vitro and in vivo. J Med Chem. 2006;49:1165–1181. doi: 10.1021/jm050754u. [DOI] [PubMed] [Google Scholar]
- 14.Park CM, Bruncko M, Adickes J, Bauch J, Ding H, Kunzer A, Marsh KC, Nimmer P, Shoemaker AR, Song X, Tahir SK, Tse C, Wang X, Wendt MD, Yang X, Zhang H, Fesik SW, Rosenberg SH, Elmore SW. Discovery of an orally bioavailable small molecule inhibitor of prosurvival B-cell lymphoma 2 proteins. J Med Chem. 2008;51:6902–6915. doi: 10.1021/jm800669s. [DOI] [PubMed] [Google Scholar]
- 15.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, Roberts L, Tahir SK, Xiao Y, Yang X, Zhang H, Fesik S, Rosenberg SH, Elmore SW. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 16.Sleebs BE, Czabotar PE, Fairbrother WJ, Fairlie WD, Flygare JA, Huang DC, Kersten WJ, Koehler MF, Lessene G, Lowes K, Parisot JP, Smith BJ, Smith ML, Souers AJ, Street IP, Yang H, Baell JB. Quinazoline sulfonamides as dual binders of the proteins B-cell lymphoma 2 and B-cell lymphoma extra long with potent proapoptotic cell-based activity. J Med Chem. 2011;54:1914–1926. doi: 10.1021/jm101596e. [DOI] [PubMed] [Google Scholar]
- 17.Zhou H, Chen J, Meagher JL, Yang CY, Aguilar A, Liu L, Bai L, Cong X, Cai Q, Fang X, Stuckey JA, Wang S. Design of Bcl-2 and Bcl-xL Inhibitors with Subnanomolar Binding Affinities Based upon a New Scaffold. J Med Chem. 2012 doi: 10.1021/jm300178u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roth BD, Blankley CJ, Chucholowski AW, Ferguson E, Hoefle ML, Ortwine DF, Newton RS, Sekerke CS, Sliskovic DR, Stratton CD, et al. Inhibitors of cholesterol biosynthesis. 3. Tetrahydro-4-hydroxy-6-[2-(1H-pyrrol-1-yl)ethyl]-2H-pyran-2-one inhibitors of HMG-CoA reductase. 2. Effects of introducing substituents at positions three and four of the pyrrole nucleus. J Med Chem. 1991;34:357–366. doi: 10.1021/jm00105a056. [DOI] [PubMed] [Google Scholar]
- 19.Zhang H, Cai Q, Ma D. Amino acid promoted CuI-catalyzed C-N bond formation between aryl halides and amines or N-containing heterocycles. J Org Chem. 2005;70:5164–5173. doi: 10.1021/jo0504464. [DOI] [PubMed] [Google Scholar]
- 20.Bruncko M, Oost TK, Belli BA, Ding H, Joseph MK, Kunzer A, Martineau D, McClellan WJ, Mitten M, Ng SC, Nimmer PM, Oltersdorf T, Park CM, Petros AM, Shoemaker AR, Song X, Wang X, Wendt MD, Zhang H, Fesik SW, Rosenberg SH, Elmore SW. Studies leading to potent, dual inhibitors of Bcl-2 and Bcl-xL. J Med Chem. 2007;50:641–662. doi: 10.1021/jm061152t. [DOI] [PubMed] [Google Scholar]
- 21.Hartwig JF, Kawatsura M, Hauck SI, Shaughnessy KH, Alcazar-Roman LM. Room-Temperature Palladium-Catalyzed Amination of Aryl Bromides and Chlorides and Extended Scope of Aromatic C-N Bond Formation with a Commercial Ligand. J Org Chem. 1999;64:5575–5580. doi: 10.1021/jo990408i. [DOI] [PubMed] [Google Scholar]
- 22.Nikolovska-Coleska Z, Wang R, Fang X, Pan H, Tomita Y, Li P, Roller PP, Krajewski K, Saito NG, Stuckey JA, Wang S. Development and optimization of a binding assay for the XIAP BIR3 domain using fluorescence polarization. Anal Biochem. 2004;332:261–273. doi: 10.1016/j.ab.2004.05.055. [DOI] [PubMed] [Google Scholar]
- 23.Huang X. Fluorescence polarization competition assay: the range of resolvable inhibitor potency is limited by the affinity of the fluorescent ligand. J Biomol Screen. 2003;8:34–38. doi: 10.1177/1087057102239666. [DOI] [PubMed] [Google Scholar]
- 24.Lee EF, Czabotar PE, Smith BJ, Deshayes K, Zobel K, Colman PM, Fairlie WD. Crystal structure of ABT-737 complexed with Bcl-xL: implications for selectivity of antagonists of the Bcl-2 family. Cell Death Differ. 2007;14:1711–1713. doi: 10.1038/sj.cdd.4402178. [DOI] [PubMed] [Google Scholar]
- 25.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 26.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein-ligand docking using GOLD. Proteins. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 27.Sybyl; a molecular modeling system; is supplied by Tripos, I, St. Louis, MO 63144.
- 28.Otwinowski Z, Minor W. [20] Processing of X-ray diffraction data collected in oscillation mode. In: Carter Charles W., Jr, editor. Methods in Enzymology. Vol. 276. Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 29.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart O, Vonrhein C, Womack T. BUSTER, version 2.9. Global Phasing Ltd; Cambridge, United Kingdom: 2010. [Google Scholar]
- 31.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 32.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr Sect D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.