

Abstract
Cutaneous markers of systemic disease are vital for clinicians to recognize. This chapter outlines familial lentiginosis syndromes that include Peutz-Jeghers syndrome, Carney Complex, the PTEN hamartomatous syndromes, and LEOPARD/Noonan syndrome. The inheritance of these syndromes is autosomal dominant; they also share characteristic skin findings that offer a clue to their recognition and treatment. We will discuss the clinical presentation of these disorders, with a focus on the dermatological manifestations, and will provide an update on the molecular mechanisms involved. Recognition of cutaneous markers associated with these rare familial cancer syndromes provides the opportunity to pursue early surveillance for malignancies, as well as genetic counseling.
Keywords: hamartoma, lentigines, mamallian target of rapamycin, tumor suppressor
Introduction
Malignancies may occur in a number of inherited conditions with associated dermatologic manifestations. In this review, we discuss the clinical presentation of disorders associated with lentigines and provide an update on the molecular mechanisms involved. The common feature of lentigines in these familial syndromes is not only a clinical feature but represents an underlying convergence of related signaling pathways. It is now known that most of these disorders (but not all) are caused by mutations in components of the rat sarcoma - mitogen-actived protein kinase (Ras-MAP kinase) and the mammalian target of rapamycin (mTOR) signaling pathway. The presence of lentigines therefore provides a window to underlying cellular mechanisms that control embryonic development and neural crest differentiation.
Lentigines (from the Latin lentigo, ‘small lentil’) consist of flat-pigmented macules on the skin and mucosa. Lentigines are characterized by their small size (< 0.5 cm), irregular borders, and discrete markings of different shades of brown and black. Familial lentiginosis syndromes are associated with an increased incidence of neural, endocrine, and mesenchymal tumors 1. Histological examination of lentigines reveals prominent epidermal thickening and basal cell hyperpigmentation associated with melanocyte hyperplasia. This feature is distinct from the histological appearance of freckles which contain a normal number of melanocytes and are pigmented due to increased melanin in basal keratinocytes (Figure 1) 1. While freckles are found almost exclusively on sun-exposed areas of the body, lentigines may occur on all parts of the body and typically do not darken with sun exposure (as compared to freckles). Specific sites of pigmentation, such as the labia majora, palms and soles, conjunctivae, and vermillion border of the lips, are characteristic locations for lentigines that provide important clues to the presence of an underlying syndrome and associated systemic disease. Peutz-Jeghers syndrome (PJS) is the most well known of the lentiginoses. A number of related disorders are also associated with lentigines and may be confused with PJS, including Carney Complex (CNC), Laugier-Hunziker syndrome (LHS), Ruvalcaba-Myhre-Smith, Bannayan-Zonnana syndrome (BRRS), Cowden disease (CD), and LEOPARD/Noonan syndrome (Table 1) 2. Most of these syndromes are inherited in an autosomal dominant manner, have a relatively high rate of de novo cases, and predispose to a variety of neoplasms 3. An algorithm for the approach of patients presenting with the three most prevalent syndromes associated with lentigines is presented in Figure 2. The downstream signaling pathways involved in these disorders regulate protein kinase A (PKA), Ras-MAP kinase, and the mammalian target of rapamycin (mTOR) 4. These pathways converge to the regulation of growth, proliferation, and differentiation of many cell types. Mutations affecting these signaling pathways may include conditions ranging from benign lentigines to aggressive malignancies.
Figure 1.
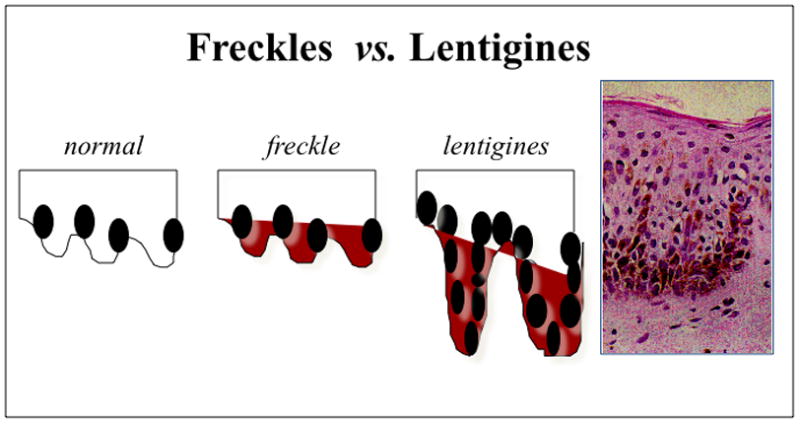
Histological appearance of freckles vs lenigines Magnification of the lentigen showing melanocytic hyperplasia, characteristic of the lesion (x 200). This differs from common freckles, in which the number of melanocytes is normal but the amount of melanin is increased.
Table 1.
| Disease | MIM | Clinical Manifestations | Inheritance | Locus | Gene | Prevalence |
|---|---|---|---|---|---|---|
| Peutz-Jeghers Syndrome | 175200 | lentigines, GI polyps, neoplasia (GI tract, pancreas, breast, ovary, uterus) | AD | 19p13.3 | LKB1/STK11 | 2.2/100,000 |
| Carney Complex | 160980 | lentigines, PPNAD cardiac and skin myxoma schwannomas, acromegaly, breast and testicular tumors | AD | 17q22–24 | PRKAR1A | >500cases described |
| Lentiginoses | 151001 151000 |
lentigines (centrofacial palmoplantar, trunk), As above in addition to mental retardation | AD AD/sporadic |
unknown unknown |
unknown unknown |
unknown |
| PTEN Hamartoma Tumor Syndrome (BRRS + Cowden Disease) | 153480 158350 |
macrocephaly, lipomatosis, pigmentation of the glans penis, mental retardation, vascular malformations | AD | 10q23.31 | PTEN | >500 cases described |
| LEOPARD Syndrome | 151100 611554 |
lentigines, cardiac conduction abnormalities, aneurysms, pulmonic stenosis, cephalo-facial dysmorphism, short stature, sensorineural deafness, mental retardation, skeletal abnormalities | AD AD |
12q24.1 3p25 |
PTPN11
RAF1 |
200 cases described |
Figure 2.
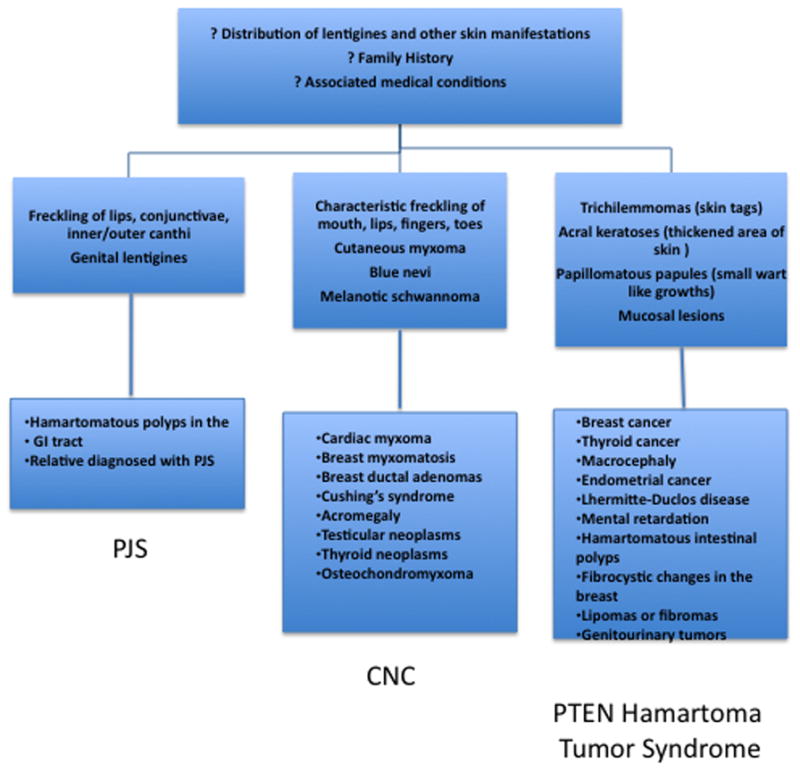
Algorithm of approach to patients presenting with lentigines (includes the three most prevalent lentigines outlined in this review).
Peutz-Jeghers syndrome
Peutz-Jeghers syndrome (PJS) is a dominantly inherited syndrome that incurs an increased risk of malignancy. The association of lentigines with intestinal polyposis in ten cases from different families was named for Drs. Peutz and Jeghers 5, 6. PJS is characterized by an increased susceptibility to tumors, including benign ovarian sex cord tumors, calcifying Sertoli tumors of the testis, cervical cancer, breast cancer, gastrointestinal cancer, pancreatic cancer, and endometrial cancer 7, 8. Thyroid cancer has been associated with PJS, and differentiated thyroid cancer may occur earlier in life and behave more aggressively in patients with PJS9–11. However, the association between PJS and thyroid cancer is not strong enough to warrant routine screening without clinical suspicion. Key diagnostic features of PJS include hamartomatous polyps and mucosal hyperpigmentation. The characteristic pigmentation in PJS consists of dark brown-blue macules, but other lesions are not uncommon. The typical brown-blue macules are commonly found on the border of the lips (Figure 3) and oral and bowel mucosa, as well as on the palms and soles, eyes, nares, and peri-anal region 12. The pigmented macules are first visible in early childhood, and tend to fade in late adulthood 12. The fact that pigmentation may become less prominent over time can contribute to the difficulty in diagnosis of PJS 13. Other syndromes may mimic the pigmentation of PJS and are outlined later in this chapter. Malignant melanoma is not a general feature of Peutz-Jeghers syndrome and has only been reported in a few cases 14, 15. The origin of the lentigines in PJS remains poorly understood. One theory is that the lentigines are benign neoplasms of melanocytes with limited growth potential 16. The most recent surveillance recommendations for PJS patients do not include dermatologic evaluation and these lesions are rarely biopsied 17. Individuals with PJS may desire cosmetic removal of lentigines, which may be successfully treated with laser therapies 18.
Figure 3.
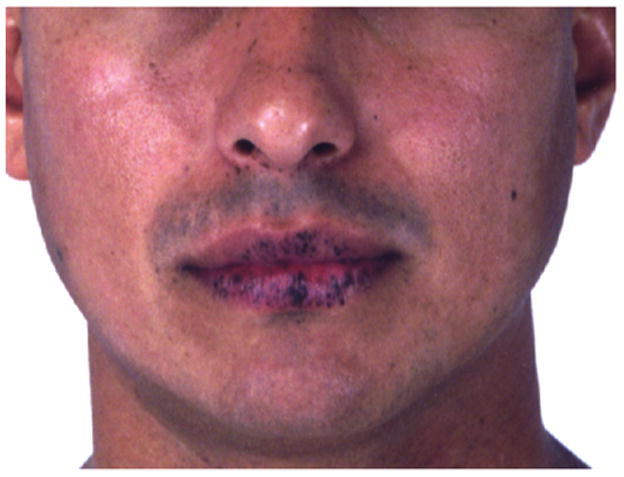
Typical oral pigmentation of a patient with PJS.
Epidemiology
The estimated prevalence of PJS is 2.2 individuals per 100,000. Estimates have included 1 in 8,500 to 23,000 live births19, 1 in 50,000 to 1 in 100,000 in Finland20
Molecular Mechanism
The majority of cases of PJS are due to heterozygous mutations in the serine-threonine kinase STK11/LKB1 tumor suppressor gene located on chromosome 19p13.3 21, 22. To date mutations in LKB1 can be found in only up to 80% of patients; linkage to other loci, including 19q13.4 has also been reported but the causative gene(s) have not been identified 13, 23. Alhopuro et al identified a mutation in the MYH11 gene in 1 of 33 PJS patients who did not have STK11 mutations, and the mutation was not identified in 1,015 controls 24. In most cases, elimination of the kinase activity of STK11 underlies the molecular cause of the phenotype 25. STK11 is a classic tumor suppressor gene, as evidenced by loss of heterozygosity (LOH) of markers in hamartomas and adenocarcinomas from patients with PJS; gastrointestinal polyps in PJS patients develop via germ line mutations in LKB1/STK11 in combination with somatic mutation or LOH of the normal allele 26. Patients with PJS are not known to have an excess of malignant skin tumors; however, the lentigines of PJS patients likely represent small, benign tumors 16, 27. Somatic mutations in LKB1/STK11 were identified in cell lines and tumor samples from 35 patients with sporadic malignant melanoma 16.
PJS is caused by dysregulated signaling in the pathway upstream of mTOR, as loss of function mutations in LKB1/STK11 inhibit AMP-activated protein kinase (AMPK) that signals downstream to inhibit mTOR 28. Many of the regulatory components of this pathway have been elucidated and are outlined in Figure 3. mTOR is a member of the phosphoinositide-3-kinase-related family, and serves as a critical mediator of cell growth and proliferation, responsible for stimulating cell growth and controlling cellular energy levels 29,30. mTOR is highly conserved throughout evolution and regulates ribosomal biogenesis, protein translation, and formation of the actin cytoskeleton 31.
Carney complex (CNC)
Carney complex is a dominantly inherited multiple endocrine neoplasia syndrome first described in 1985 that is characterized by spotty skin pigmentation (lentiginosis), cardiac and peripheral myxomas, schwannomas, and endocrine overactivity 32, 33. The most common cutaneous features of CNC include lentigines, freckling, café-au-lait spots, and blue nevi 34, 35. Typical lentigines of the inner canthi and genital mucosa are presented in Figure 4. The endocrine tumors associated with Carney complex include primary pigmented nodular adrenal cortical disease (PPNAD), growth hormone secreting pituitary adenomas, large-cell calcifying Sertoli cell tumors, Leydig cell tumors, and thyroid neoplasms 36–38. PPNAD, or primary pigmented nodular adrenocortical disease, is a rare cause of corticotrophin (ACTH)-independent Cushing syndrome. Historically, cardiac myxomas have been reported to be responsible for more than 50% of the disease-specific mortality among CNC patients 35. Therefore, identification of affected patients and family members is crucial in order to screen for other features of Carney complex, namely potentially critical cardiac myxomas that require surgical removal 39.
Figure 4.
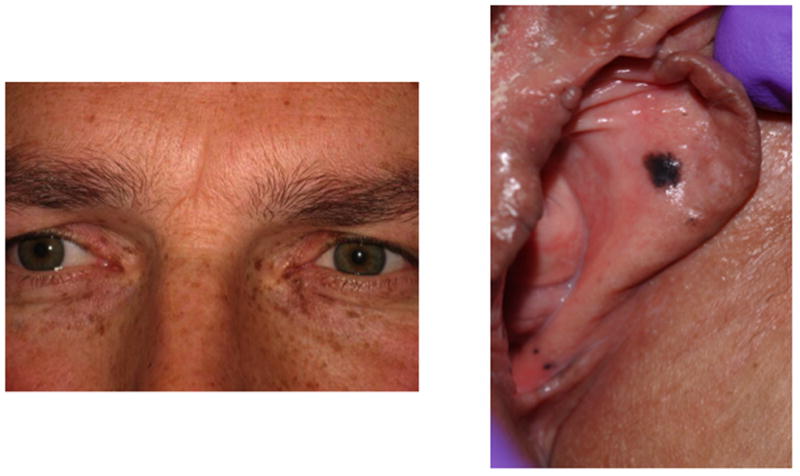
Cutaneous manifestations in CNC: pigmentation at (a) inner canthi; and (b) vaginal mucosa.
Epidemiology
More than 500 patients are known world-wide. In a recent analysis, a total of 353 patients with CNC from 185 families were described35, 40 The majority of patients (68%) had a family history consistent with CNC, whereas 113 cases (32%) had no known affected relatives and were classified as sporadic. A total of 221 patients (63%) were female. The median age of diagnosis was 20 years, although at least 5 patients were identified at birth35, 41
Molecular Mechanism
The molecular cause of disease in most patients with CNC (and isolated PPNAD) has been identified as mutations within the gene PRKAR1A on chromosome 17q22–24 42. PRKAR1A encodes the regulatory subunit type 1-α of Protein Kinase A, a key regulator of the cyclic-AMP-dependent signaling pathway that has been implicated in endocrine tumor formation 43. Inactivating mutations of PRKAR1A have been reported in 45–80% of families with CNC 44, 45. Over 100 pathogenic variants in PRKAR1A have been detected, most leading to R1α haploinsufficiency 46. Loss of R1α leads to increased cAMP-stimulated total kinase activity, however the link to increased tumor formation is still under investigation 42, 47. A number of pathways are involved, but one that links this disease to others covered by this review is that of the mitogen-activated protein kinase (MAPK) ERK 1/2 signaling: lymphocytes isolated from CNC patients with known PRKAR1A mutations showed altered PKA activity and increased ERK 1/2 phosphorylation 48.
Overall PRKAR1A appears to serve as a tumor suppressor gene in which loss of activity is associated with loss of PKA-mediated inhibition of downstream pathways resulting in increased cell proliferation 48. Loss of expression of PRKAR1A has been shown in pigmented epithelioid melanocytoma but not in melanoma or other melanocytic lesions 49. The lentigines in CNC are typically benign; despite the high number of lesions, so far only one case of malignant melanoma in association with CNC has been reported 50, making this tumor incidence among CNC patients lower than that in the general population. On the other hand, high expression (and not loss) of PRKAR1A has been seen in association with increased proliferation of human melanoma cells in vitro 51.
Laugier-Hunziker Syndrome (LHS)
Laugier-Hunziker syndrome (LHS) is a rare, sporadic disorder, originally described in 1970, that is often confused for PJS due to similar appearance and distribution of hyperpigmented cutaneous and mucocutaneous lesions 52. Only 100 cases have been reported thus far in the medical literature 53. However, unlike PJS, LHS is an acquired, benign condition characterized by hyperpigmentation of the nails, palms and soles, and lips and oral mucosa without polyposis 54. Lentigines in LHS typically appear later in life than in those individuals with PJS 52. Interestingly, approximately 50–60% of patients with LHS have longitudinal melanonychia, brown or black pigmentation of the nail unit 55. In LHS there is increased pigmentation in the basal keratinocytes and increased numbers of macrophages, however, the number of melanocytes is not affected55.
Benign lentiginoses
Two separate syndromes, patterned lentiginosis and centrofacial neurodysraphic lentiginosis, are both benign lentiginoses conditions that are inherited in an autosomal dominant manner without systemic involvement 56–58. Patterned lentiginosis was first reported in 1989 by O’Neil et al., who described 10 African-American patients with autosomal dominant transmission of lentigines distributed on the face, lips, extremity, buttock, and palms and soles. None of the patients had lesions of the oral mucosa or internal organ system abnormalities 56. A separate syndrome, centrofacial neurodysraphic lentiginosis, consisting of facial lentigines associated with mental retardation and autosomal dominant inheritance has also been described.58 None of the genes involved in either of these benign lentiginoses syndromes are known to date.
Ruvalcaba-Myhre-Smith or Bannayan-Zonnana syndrome (BRRS), and Cowden disease (CD): PTEN gene-related disorders
BRRS and CD are part of a group of inherited disorders that have been previously been classified as familial hamartoma syndromes. With the discovery of mutations in the tumor suppressor gene PTEN (10q22-q23) in up to 80% of CD patients and up to 60% of BRRS patients, it has been suggested that these conditions should be all listed under the heading “PTEN hamartoma tumor syndromes” (PHTS) 59–67. Approximately 500 individuals with germline pathogenic PTEN mutations have been reported to date.68
Patients with PTEN mutations have an increased risk of developing multiple hamartomas in various organ systems such as the breast, skin, thyroid, central nervous system, and the GI tract 69. BRRS is characterized by delayed motor development, macrocephaly, lipomatosis, as well as the presence of lentigines on the glans penis beginning in childhood 69. Diagnostic criteria for CD include skin findings such as gingival papillomas and acral keratoses. Patients are susceptible to breast cancer, follicular thyroid cancer, and endometrial carcinoma 70. Major criteria include breast cancer, thyroid cancer, macrocephaly, and Lhermitte-Duclos disease (hamartomatous growths of the cerebellum with associated ataxia). Minor criteria include thyroid disease (adenoma and goiter), gastrointestinal hamartomas, lipomas, fibromas, genitourinary tumors, fibrocystic breast disease, and developmental delay 71. Up to 10 percent of patients with CD develop follicular carcinoma, and 50–75% will manifest benign thyroid disease 72, 73. Lentigines develop by age 20 in 90% of patients with CD. In order to diagnose CD patients must have one major and three minor criteria. Most of the cutaneous lesions in CD are benign, however, the association of squamous cell carcinoma with CD has been reported 74–76
Molecular Mechanism
PTEN hamartoma tumor syndromes, including BRRS and CD, all represent additional disorders associated with loss-of-function mutations in a tumor suppressor gene leading to constitutive mTOR activation. A recent overview of the PTEN hamartoma tumor syndromes as well as a newly developed clinical scoring system to guide selection of patients for PTEN testing provide excellent resources for clinicians68, 77. Approximately 80 percent of patients with CD are found to have associated mutations in the phosphate and tensin gene (PTEN) on chromosome 10q23.31, which codes for a neuroendocrine developmental regulator protein 70, 78. PTEN plays a key role in cell growth, apoptosis, differentiation, membrane trafficking, cellular interactions and cellular motility 67, 79, 80. Loss of PTEN activity leads to constitutive activation of the cytosolic signaling protein AKT 81. One of the key downstream targets of AKT is the tuberin-hamartin complex (TSC1/TSC2), mutations of which are associated with the hamartomatous syndrome tuberous sclerosis (TS) 82. In TS, loss of PTEN function results in the constitutive activation of AKT, down regulation of tuberin/TSC2 and mTOR and subsequent promotion of cell-cycle progression and suppression of apoptosis. The discovery of this pathway therefore links the pathogenesis of tumor formation in hamartomatous tumor syndromes as outlined in Figure 5 83. Loss of PTEN may have a critical role in the pathogenesis of melanoma, as widespread PTEN loss has been demonstrated in primary cutaneous melanomas 84. Recent developments have shown PTEN as a key tumor suppressor for the development of basal cell carcinoma, squamous cell carcinoma, and melanoma 74.
Figure 5.
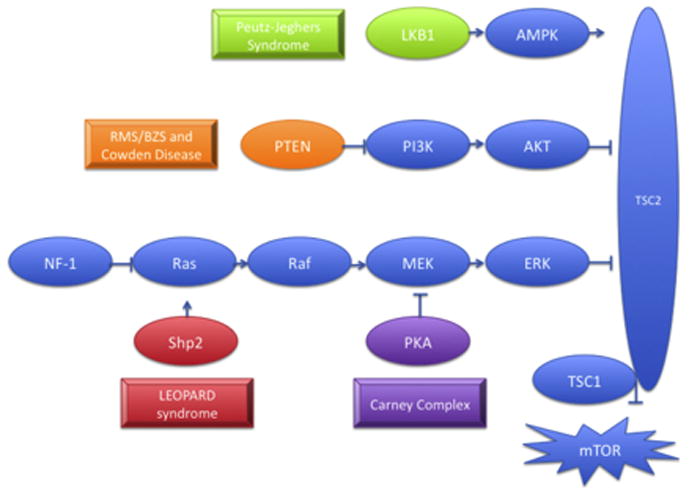
The tumor-suppressor genes LKB1, PTEN, PRKAR1A exert inhibitory effects on signaling through the TSC complex that inactivates mTOR, while SHP2 gain-of-function mutations also lead to downstream mTOR activation. Each of genes is mutated in distinct syndromes that are characterized by the development of lentigines and other skin lesions. Abbreviations: LKB1, serine/threonine kinase 11; AMPK, AMP-activated protein kinase; PTEN, phosphatase and tensin homolog; P13K, 1-phosphatidylinositol 3-kinase; AKT, protein kinase B; NF1; neurofibromatosis type 1MEK, mitogen activated protein kinase kinase; ERK, extracellular signal-regulated kinase; TSC1; tuberous sclerosis 1; TSC2; tuberous sclerosis 2; mTOR, mammalian target of rapamycin.
LEOPARD syndrome
LEOPARD is an acronym that also describes the pattern of pigmentation found in this familial multiple lentigines syndrome. The manifestations of this syndrome include Lentigines, Electrocardiographic conduction defects, Ocular hypertelorism, Pulmonary stenosis, Abnormal genitalia, Retardation of growth, and sensorineural Deafness 85. The diagnosis is established if multiple lentigines are present in association with at least two other features; if lentigines are absent, a first-degree relative affected with LEOPARD syndrome and three of the other six features are needed for diagnosis 86, 87.
Lentigines are often the first clinical manifestation of in LEOPARD syndrome, and they are found primarily on the face and upper trunk, although rarely involve the oral mucosa, extremities, genitalia or conjunctiva 88. Lentigines in LEOPARD syndrome do not cross the vermilion border of the lips, a characteristic that distinguishes this disorder from CNC and PJS 4. It is unknown whether pigmented lesions in LEOPARD syndrome progress to malignancy; to date, only one case of LEOPARD syndrome associated with malignant melanoma has been reported 89.
Facial dysmorphism in LEOPARD syndrome includes low-set ears, hypertelorism and palpebral ptosis. These features, combined with an increased incidence of pulmonic stenosis, show significant phenotypic overlap with Noonan syndrome, which goes along with molecular evidence that these two disorders share the same allele 90. Morbidity and mortality associated with LEOPARD syndrome are dependent on the extent of cardiac disease. Multiple congenital heart defects have been reported to include not only pulmonic stenosis (present in 40% of patients) but also subaortic and subpulmonic stenosis, and hypertrophic obstructive cardiomyopathy 88.
Molecular Mechanism
LEOPARD syndrome is caused by mutations in genes that regulate the mTOR signaling pathway 91. LEOPARD syndrome can be caused by mutations in the PTPN11 gene on chromosome 12q24.1 This is the identical allele implicated in Noonan syndrome, and explains the phenotypic overlap between the two conditions. A distinct form of LEOPARD syndrome is caused by RAF1 defects (chromosome 3p25); this form is strongly associated with hypertrophic cardiomyopathy 92, 93.
PTPN11 encodes SHP2, a positive regulator of RAS-MAPK signaling The PTPN11 mutations that have been described in both NS and LEOPARD are believed to be gain-of-function mutations leading to dysregulated phosphatase activity with subsequent increased inhibition of GTPase which in turn leads to increased Ras activity 90.
The lentiginoses and the mTOR Pathway
The genetic defects of most of the disorders associated with lentigines are linked directly or indirectly to a common oncogenic pathway, that of the mammalian target of rapamycin (mTOR). mTOR is a highly conserved serine/threonine kinase that mediates cellular growth, and is also thought to play key roles in cancer, diabetes and ageing 83, 94. Dysregulated activation of mTOR is hypothesized to be the unifying underlying mechanism responsible for the formation of lentigines and neoplasia in PJS and related conditions. Activation of mTOR is strongly associated with malignant melanocytic lesions in vivo, suggesting that mTOR inhibition may have clinical benefit in patients with melanoma and/or other malignant lesions associated with the lentiginoses 95. Rapamycin inhibits mTOR and results in decreased expression of mRNAs necessary for cell cycle progression and arresting cells in the G1 phase of the cell cycle 96. A clinical trial using the mTOR inhibitor Everolimus was initiated in 2008 as a pilot-open label phase II study for patients with PJS and gastrointestinal polyps (clinical trials.gov identifier NCT00811590). As dysregulation of mTOR has been demonstrated in several types of cancers, inhibition of mTOR may be of benefit to these patients. Targeting of the mTOR pathway with rapamycin together with the RAF inhibitor Sorafenib as been investigated in melanoma cell lines in vitro 97. A clinical trial investigating the use of Sorafenib and Temsirolimus (an analog of rapamycin) in treating patients with metastatic, recurrent, or unresectable melanoma is currently underway (clinical trials.gov identifier NCT00349206).
Conclusions
Familial lentiginosis syndromes represent a large phenotypic spectrum ranging from a benign inherited predisposition to develop lentigines alone, to associations with several syndromes that carry an elevated risk of neoplasia. The etiologic molecular pathways have been defined over the past 20 years, including the PKA pathway in CNC the Ras/Erk MAP kinase pathway in LEOPARD/Noonan syndromes, and mTOR in both Peutz-Jeghers syndrome and the diseases caused by PTEN mutations. Patients presenting with lentigines that may be associated with syndromes linked to increased risk of malignancy should be evaluated to confirm the suspected diagnosis. This will allow the patient to be appropriately screened for malignancy and other disease associated sequellae.
References
- 1.Fitzpatrick TB, Wolff K. Fitzpatrick’s dermatology in general medicine. 7. New York: McGraw-Hill; 2008. [Google Scholar]
- 2.Marsh DJ, Stratakis CA. Hamartoma and lentiginosis syndromes: clinical and molecular aspects. Front Horm Res. 2001;28:167–213. doi: 10.1159/000061045. [DOI] [PubMed] [Google Scholar]
- 3.Stratakis CA. Genetics of Peutz-Jeghers syndrome, Carney complex and other familial lentiginoses. Horm Res. 2000;54:334–43. doi: 10.1159/000053283. [DOI] [PubMed] [Google Scholar]
- 4.Bauer AJ, Stratakis CA. The lentiginoses: cutaneous markers of systemic disease and a window to new aspects of tumourigenesis. J Med Genet. 2005;42:801–10. doi: 10.1136/jmg.2003.017806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peutz JLA. Very remarkable case of familial polyposis of mucous membrane of intestinal tract and nasopharynx accompanied by peculiar pigmentations of skin and mucous membrane. (Dutch) Nederl Maandschr Geneesk. 1921;10:134–46. [Google Scholar]
- 6.Jeghers H, McKusick V, Katz KH. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N Engl J Med. 1949;241:993. doi: 10.1056/NEJM194912222412501. illust; passim. [DOI] [PubMed] [Google Scholar]
- 7.Hearle N, Schumacher V, Menko FH, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res. 2006;12:3209–15. doi: 10.1158/1078-0432.CCR-06-0083. [DOI] [PubMed] [Google Scholar]
- 8.Stratakis CA. Genetics of Carney complex and related familial lentiginoses, and other multiple tumor syndromes. Front Biosci. 2000;5:D353–66. doi: 10.2741/stratakis. [DOI] [PubMed] [Google Scholar]
- 9.Yalcin S, Kirli E, Ciftci AO, et al. The association of adrenocortical carcinoma and thyroid cancer in a child with Peutz-Jeghers syndrome. J Pediatr Surg. 2011;46:570–3. doi: 10.1016/j.jpedsurg.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 10.Zirilli L, Benatti P, Romano S, et al. Differentiated thyroid carcinoma (DTC) in a young woman with Peutz-Jeghers syndrome: are these two conditions associated? Exp Clin Endocrinol Diabetes. 2009;117:234–9. doi: 10.1055/s-0028-1102920. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto M, Hoshino H, Onizuka T, Ichikawa M, Kawakubo A, Hayakawa S. Thyroid papillary adenocarcinoma in a woman with Peutz-Jeghers syndrome. Intern Med. 1992;31:1117–9. doi: 10.2169/internalmedicine.31.1117. [DOI] [PubMed] [Google Scholar]
- 12.Tomlinson IP, Houlston RS. Peutz-Jeghers syndrome. J Med Genet. 1997;34:1007–11. doi: 10.1136/jmg.34.12.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boardman LA, Couch FJ, Burgart LJ, et al. Genetic heterogeneity in Peutz-Jeghers syndrome. Hum Mutat. 2000;16:23–30. doi: 10.1002/1098-1004(200007)16:1<23::AID-HUMU5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 14.Wong SS, Rajakulendran S. Peutz-Jeghers syndrome associated with primary malignant melanoma of the rectum. Br J Dermatol. 1996;135:439–42. [PubMed] [Google Scholar]
- 15.Braitman M. Subungual malignant melanoma. Cutis. 1979;23:617–23. [PubMed] [Google Scholar]
- 16.Rowan A, Bataille V, MacKie R, et al. Somatic mutations in the Peutz-Jeghers (LKB1/STKII) gene in sporadic malignant melanomas. J Invest Dermatol. 1999;112:509–11. doi: 10.1046/j.1523-1747.1999.00551.x. [DOI] [PubMed] [Google Scholar]
- 17.van Lier MG, Wagner A, Mathus-Vliegen EM, Kuipers EJ, Steyerberg EW, van Leerdam ME. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol. 2010;105:1258–64. doi: 10.1038/ajg.2009.725. author reply 65. [DOI] [PubMed] [Google Scholar]
- 18.DePadova-Elder SM, Milgraum SS. Q-switched ruby laser treatment of labial lentigines in Peutz-Jeghers syndrome. J Dermatol Surg Oncol. 1994;20:830–2. doi: 10.1111/j.1524-4725.1994.tb03714.x. [DOI] [PubMed] [Google Scholar]
- 19.Mallory SB, Stough DBt. Genodermatoses with malignant potential. Dermatol Clin. 1987;5:221–30. [PubMed] [Google Scholar]
- 20.Hemminki A. The molecular basis and clinical aspects of Peutz-Jeghers syndrome. Cell Mol Life Sci. 1999;55:735–50. doi: 10.1007/s000180050329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jenne DE, Reimann H, Nezu J, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998;18:38–43. doi: 10.1038/ng0198-38. [DOI] [PubMed] [Google Scholar]
- 22.Hemminki A, Markie D, Tomlinson I, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–7. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 23.Hearle N, Lucassen A, Wang R, et al. Mapping of a translocation breakpoint in a Peutz-Jeghers hamartoma to the putative PJS locus at 19q13. 4 and mutation analysis of candidate genes in polyp and STK11-negative PJS cases Genes Chromosomes. Cancer. 2004;41:163–9. doi: 10.1002/gcc.20067. [DOI] [PubMed] [Google Scholar]
- 24.Alhopuro P, Phichith D, Tuupanen S, et al. Unregulated smooth-muscle myosin in human intestinal neoplasia. Proc Natl Acad Sci U S A. 2008;105:5513–8. doi: 10.1073/pnas.0801213105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mehenni H, Gehrig C, Nezu J, et al. Loss of LKB1 kinase activity in Peutz-Jeghers syndrome, and evidence for allelic and locus heterogeneity. Am J Hum Genet. 1998;63:1641–50. doi: 10.1086/302159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyaki M, Iijima T, Hosono K, et al. Somatic mutations of LKB1 and beta-catenin genes in gastrointestinal polyps from patients with Peutz-Jeghers syndrome. Cancer Res. 2000;60:6311–3. [PubMed] [Google Scholar]
- 27.Boardman LA, Thibodeau SN, Schaid DJ, et al. Increased risk for cancer in patients with the Peutz-Jeghers syndrome. Ann Intern Med. 1998;128:896–9. doi: 10.7326/0003-4819-128-11-199806010-00004. [DOI] [PubMed] [Google Scholar]
- 28.Zbuk KM, Eng C. Hamartomatous polyposis syndromes. Nat Clin Pract Gastroenterol Hepatol. 2007;4:492–502. doi: 10.1038/ncpgasthep0902. [DOI] [PubMed] [Google Scholar]
- 29.Kwiatkowski DJ, Manning BD. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet. 2005;14(Spec No 2):R251–8. doi: 10.1093/hmg/ddi260. [DOI] [PubMed] [Google Scholar]
- 30.Rosner M, Hanneder M, Siegel N, Valli A, Fuchs C, Hengstschlager M. The mTOR pathway and its role in human genetic diseases. Mutat Res. 2008;659:284–92. doi: 10.1016/j.mrrev.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 31.Sandsmark DK, Pelletier C, Weber JD, Gutmann DH. Mammalian target of rapamycin: master regulator of cell growth in the nervous system. Histol Histopathol. 2007;22:895–903. doi: 10.14670/HH-22.895. [DOI] [PubMed] [Google Scholar]
- 32.Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VL. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine (Baltimore) 1985;64:270–83. doi: 10.1097/00005792-198507000-00007. [DOI] [PubMed] [Google Scholar]
- 33.Carney JA, Hruska LS, Beauchamp GD, Gordon H. Dominant inheritance of the complex of myxomas, spotty pigmentation, and endocrine overactivity. Mayo Clin Proc. 1986;61:165–72. doi: 10.1016/s0025-6196(12)61843-6. [DOI] [PubMed] [Google Scholar]
- 34.Chrousos GP, Stratakis CA. Carney complex and the familial lentiginosis syndromes: link to inherited neoplasias and developmental disorders, and genetic loci. J Intern Med. 1998;243:573–9. doi: 10.1046/j.1365-2796.1998.00341.x. [DOI] [PubMed] [Google Scholar]
- 35.Stratakis CA, Kirschner LS, Carney JA. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab. 2001;86:4041–6. doi: 10.1210/jcem.86.9.7903. [DOI] [PubMed] [Google Scholar]
- 36.Stratakis CA, Carney JA, Lin JP, et al. Carney complex, a familial multiple neoplasia and lentiginosis syndrome. Analysis of 11 kindreds and linkage to the short arm of chromosome 2. J Clin Invest. 1996;97:699–705. doi: 10.1172/JCI118467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Premkumar A, Stratakis CA, Shawker TH, Papanicolaou DA, Chrousos GP. Testicular ultrasound in Carney complex: report of three cases. J Clin Ultrasound. 1997;25:211–4. doi: 10.1002/(sici)1097-0096(199705)25:4<211::aid-jcu10>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 38.Stratakis CA, Courcoutsakis NA, Abati A, et al. Thyroid gland abnormalities in patients with the syndrome of spotty skin pigmentation, myxomas, endocrine overactivity, and schwannomas (Carney complex) J Clin Endocrinol Metab. 1997;82:2037–43. doi: 10.1210/jcem.82.7.4079. [DOI] [PubMed] [Google Scholar]
- 39.Bertherat J. Carney complex (CNC) Orphanet J Rare Dis. 2006;1:21. doi: 10.1186/1750-1172-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bertherat J, Horvath A, Groussin L, et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5′-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab. 2009;94:2085–91. doi: 10.1210/jc.2008-2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boikos SA, Stratakis CA. Carney complex: the first 20 years. Curr Opin Oncol. 2007;19:24–9. doi: 10.1097/CCO.0b013e32801195eb. [DOI] [PubMed] [Google Scholar]
- 42.Kirschner LS, Carney JA, Pack SD, et al. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet. 2000;26:89–92. doi: 10.1038/79238. [DOI] [PubMed] [Google Scholar]
- 43.Amieux PS, McKnight GS. The essential role of RI alpha in the maintenance of regulated PKA activity. Ann N Y Acad Sci. 2002;968:75–95. doi: 10.1111/j.1749-6632.2002.tb04328.x. [DOI] [PubMed] [Google Scholar]
- 44.Kirschner LS, Sandrini F, Monbo J, Lin JP, Carney JA, Stratakis CA. Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with the carney complex. Hum Mol Genet. 2000;9:3037–46. doi: 10.1093/hmg/9.20.3037. [DOI] [PubMed] [Google Scholar]
- 45.Groussin L, Kirschner LS, Vincent-Dejean C, et al. Molecular analysis of the cyclic AMP-dependent protein kinase A (PKA) regulatory subunit 1A (PRKAR1A) gene in patients with Carney complex and primary pigmented nodular adrenocortical disease (PPNAD) reveals novel mutations and clues for pathophysiology: augmented PKA signaling is associated with adrenal tumorigenesis in PPNAD. Am J Hum Genet. 2002;71:1433–42. doi: 10.1086/344579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rothenbuhler A, Stratakis CA. Clinical and molecular genetics of Carney complex. Best Pract Res Clin Endocrinol Metab. 2010;24:389–99. doi: 10.1016/j.beem.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 47.Casey M, Vaughan CJ, He J, et al. Mutations in the protein kinase A R1alpha regulatory subunit cause familial cardiac myxomas and Carney complex. J Clin Invest. 2000;106:R31–8. doi: 10.1172/JCI10841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robinson-White A, Hundley TR, Shiferaw M, Bertherat J, Sandrini F, Stratakis CA. Protein kinase-A activity in PRKAR1A-mutant cells, and regulation of mitogen-activated protein kinases ERK1/2. Hum Mol Genet. 2003;12:1475–84. doi: 10.1093/hmg/ddg160. [DOI] [PubMed] [Google Scholar]
- 49.Zembowicz A, Knoepp SM, Bei T, et al. Loss of expression of protein kinase a regulatory subunit 1alpha in pigmented epithelioid melanocytoma but not in melanoma or other melanocytic lesions. Am J Surg Pathol. 2007;31:1764–75. doi: 10.1097/PAS.0b013e318057faa7. [DOI] [PubMed] [Google Scholar]
- 50.Ryan MW, Cunningham S, Xiao SY. Maxillary sinus melanoma as the presenting feature of Carney complex. Int J Pediatr Otorhinolaryngol. 2008;72:405–8. doi: 10.1016/j.ijporl.2007.10.022. [DOI] [PubMed] [Google Scholar]
- 51.Mantovani G, Bondioni S, Lania AG, et al. High expression of PKA regulatory subunit 1A protein is related to proliferation of human melanoma cells. Oncogene. 2008;27:1834–43. doi: 10.1038/sj.onc.1210831. [DOI] [PubMed] [Google Scholar]
- 52.Lampe AK, Hampton PJ, Woodford-Richens K, Tomlinson I, Lawrence CM, Douglas FS. Laugier-Hunziker syndrome: an important differential diagnosis for Peutz-Jeghers syndrome. J Med Genet. 2003;40:e77. doi: 10.1136/jmg.40.6.e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rangwala S, Doherty CB, Katta R. Laugier-Hunziker syndrome: A case report and review of the literature. Dermatol Online J. 2010;16:9. [PubMed] [Google Scholar]
- 54.Veraldi S, Cavicchini S, Benelli C, Gasparini G. Laugier–Hunziker syndrome: a clinical, histopathologic, and ultrastructural study of four cases and review of the literature. J Am Acad Dermatol. 1991;25:632–6. doi: 10.1016/0190-9622(91)70244-v. [DOI] [PubMed] [Google Scholar]
- 55.Ko JH, Shih YC, Chiu CS, Chuang YH. Dermoscopic features in Laugier-Hunziker syndrome. J Dermatol. 2011;38:87–90. doi: 10.1111/j.1346-8138.2010.01077.x. [DOI] [PubMed] [Google Scholar]
- 56.O’Neill JF, James WD. Inherited patterned lentiginosis in blacks. Arch Dermatol. 1989;125:1231–5. [PubMed] [Google Scholar]
- 57.Xing Q, Chen X, Wang M, et al. A locus for familial generalized lentiginosis without systemic involvement maps to chromosome 4q21.1-q22. 3. Hum Genet. 2005;117:154–9. doi: 10.1007/s00439-005-1284-1. [DOI] [PubMed] [Google Scholar]
- 58.Dociu I, Galaction-Nitelea O, Sirjita N, Murgu V. Centrofacial lentiginosis. A survey of 40 cases. Br J Dermatol. 1976;94:39–43. doi: 10.1111/j.1365-2133.1976.tb04339.x. [DOI] [PubMed] [Google Scholar]
- 59.Nelen MR, Padberg GW, Peeters EA, et al. Localization of the gene for Cowden disease to chromosome 10q22–23. Nat Genet. 1996;13:114–6. doi: 10.1038/ng0596-114. [DOI] [PubMed] [Google Scholar]
- 60.Nelen MR, van Staveren WC, Peeters EA, et al. Germline mutations in the PTEN/MMAC1 gene in patients with Cowden disease. Hum Mol Genet. 1997;6:1383–7. doi: 10.1093/hmg/6.8.1383. [DOI] [PubMed] [Google Scholar]
- 61.Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 62.Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997;57:2124–9. [PubMed] [Google Scholar]
- 63.Steck PA, Pershouse MA, Jasser SA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23. 3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–62. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 64.Liaw D, Marsh DJ, Li J, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64–7. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 65.Marsh DJ, Coulon V, Lunetta KL, et al. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum Mol Genet. 1998;7:507–15. doi: 10.1093/hmg/7.3.507. [DOI] [PubMed] [Google Scholar]
- 66.Gorlin RJ, Cohen MM, Jr, Condon LM, Burke BA. Bannayan-Riley-Ruvalcaba syndrome. Am J Med Genet. 1992;44:307–14. doi: 10.1002/ajmg.1320440309. [DOI] [PubMed] [Google Scholar]
- 67.Waite KA, Eng C. Protean PTEN: form and function. Am J Hum Genet. 2002;70:829–44. doi: 10.1086/340026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tan MH, Mester J, Peterson C, et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am J Hum Genet. 2011;88:42–56. doi: 10.1016/j.ajhg.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marsh DJ, Kum JB, Lunetta KL, et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8:1461–72. doi: 10.1093/hmg/8.8.1461. [DOI] [PubMed] [Google Scholar]
- 70.Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22:183–98. doi: 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- 71.Stratakis CA, Kirschner LS, Taymans SE, et al. Carney complex, Peutz-Jeghers syndrome, Cowden disease, and Bannayan-Zonana syndrome share cutaneous and endocrine manifestations, but not genetic loci. J Clin Endocrinol Metab. 1998;83:2972–6. doi: 10.1210/jcem.83.8.5042. [DOI] [PubMed] [Google Scholar]
- 72.Nose V. Familial non-medullary thyroid carcinoma: an update. Endocr Pathol. 2008;19:226–40. doi: 10.1007/s12022-008-9045-z. [DOI] [PubMed] [Google Scholar]
- 73.Blumenthal GM, Dennis PA. PTEN hamartoma tumor syndromes. Eur J Hum Genet. 2008;16:1289–300. doi: 10.1038/ejhg.2008.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ming M, He YY. PTEN: new insights into its regulation and function in skin cancer. J Invest Dermatol. 2009;129:2109–12. doi: 10.1038/jid.2009.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Camisa C, Bikowski JB, McDonald SG. Cowden’s disease. Association with squamous cell carcinoma of the tongue and perianal basal cell carcinoma. Arch Dermatol. 1984;120:677–8. doi: 10.1001/archderm.120.5.677. [DOI] [PubMed] [Google Scholar]
- 76.Nuss DD, Aeling JL, Clemons DE, Weber WN. Multiple hamartoma syndrome (Cowden’s disease) Arch Dermatol. 1978;114:743–6. [PubMed] [Google Scholar]
- 77.Hobert JA, Eng C. PTEN hamartoma tumor syndrome: an overview. Genet Med. 2009;11:687–94. doi: 10.1097/GIM.0b013e3181ac9aea. [DOI] [PubMed] [Google Scholar]
- 78.Eng C. Will the real Cowden syndrome please stand up: revised diagnostic criteria. J Med Genet. 2000;37:828–30. doi: 10.1136/jmg.37.11.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol. 2004;22:2954–63. doi: 10.1200/JCO.2004.02.141. [DOI] [PubMed] [Google Scholar]
- 80.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999;96:4240–5. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–8. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 82.Manning BD, Cantley LC. United at last: the tuberous sclerosis complex gene products connect the phosphoinositide 3-kinase/Akt pathway to mammalian target of rapamycin (mTOR) signalling. Biochem Soc Trans. 2003;31:573–8. doi: 10.1042/bst0310573. [DOI] [PubMed] [Google Scholar]
- 83.Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. 2004;23:3151–71. doi: 10.1038/sj.onc.1207542. [DOI] [PubMed] [Google Scholar]
- 84.Tsao H, Mihm MC, Jr, Sheehan C. PTEN expression in normal skin, acquired melanocytic nevi, and cutaneous melanoma. J Am Acad Dermatol. 2003;49:865–72. doi: 10.1016/s0190-9622(03)02473-3. [DOI] [PubMed] [Google Scholar]
- 85.Gorlin RJ, Anderson RC, Blaw M. Multiple lentigenes syndrome. Am J Dis Child. 1969;117:652–62. doi: 10.1001/archpedi.1969.02100030654006. [DOI] [PubMed] [Google Scholar]
- 86.Legius E, Schrander-Stumpel C, Schollen E, Pulles-Heintzberger C, Gewillig M, Fryns JP. PTPN11 mutations in LEOPARD syndrome. J Med Genet. 2002;39:571–4. doi: 10.1136/jmg.39.8.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chong WS, Klanwarin W, Giam YC. Generalized lentiginosis in two children lacking systemic associations: case report and review of the literature. Pediatr Dermatol. 2004;21:139–45. doi: 10.1111/j.0736-8046.2004.21211.x. [DOI] [PubMed] [Google Scholar]
- 88.Abdelmalek NF, Gerber TL, Menter A. Cardiocutaneous syndromes and associations. J Am Acad Dermatol. 2002;46:161–83. doi: 10.1067/mjd.2002.120928. quiz 83–6. [DOI] [PubMed] [Google Scholar]
- 89.Seishima M, Mizutani Y, Shibuya Y, Arakawa C, Yoshida R, Ogata T. Malignant melanoma in a woman with LEOPARD syndrome: identification of a germline PTPN11 mutation and a somatic BRAF mutation. Br J Dermatol. 2007;157:1297–9. doi: 10.1111/j.1365-2133.2007.08229.x. [DOI] [PubMed] [Google Scholar]
- 90.Digilio MC, Conti E, Sarkozy A, et al. Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am J Hum Genet. 2002;71:389–94. doi: 10.1086/341528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zenker M. Genetic and pathogenetic aspects of Noonan syndrome and related disorders. Horm Res. 2009;72 (Suppl 2):57–63. doi: 10.1159/000243782. [DOI] [PubMed] [Google Scholar]
- 92.Razzaque MA, Nishizawa T, Komoike Y, et al. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet. 2007;39:1013–7. doi: 10.1038/ng2078. [DOI] [PubMed] [Google Scholar]
- 93.Pandit B, Sarkozy A, Pennacchio LA, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007;39:1007–12. doi: 10.1038/ng2073. [DOI] [PubMed] [Google Scholar]
- 94.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Karbowniczek M, Spittle CS, Morrison T, Wu H, Henske EP. mTOR is activated in the majority of malignant melanomas. J Invest Dermatol. 2008;128:980–7. doi: 10.1038/sj.jid.5701074. [DOI] [PubMed] [Google Scholar]
- 96.Shaw RJ, Bardeesy N, Manning BD, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6:91–9. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 97.Lasithiotakis KG, Sinnberg TW, Schittek B, et al. Combined inhibition of MAPK and mTOR signaling inhibits growth, induces cell death, and abrogates invasive growth of melanoma cells. J Invest Dermatol. 2008;128:2013–23. doi: 10.1038/jid.2008.44. [DOI] [PubMed] [Google Scholar]