

Abstract
The unique vulnerability of the olfactory system to Alzheimer's disease (AD) provides a quintessential translational tool for understanding mechanisms of synaptic dysfunction and pathological progression in the disease. Using the Tg2576 mouse model of β-amyloidosis, we show that aberrant, hyperactive olfactory network activity begins early in life, before detectable behavioral impairments or comparable hippocampal dysfunction and at a time when amyloid-β (Aβ) deposition is restricted to the olfactory bulb (OB). Hyperactive odor-evoked activity in the piriform cortex (PCX) and increased OB–PCX functional connectivity emerged at a time coinciding with olfactory behavior impairments. This hyperactive activity persisted until later in life when the network converted to a hyporesponsive state. This conversion was Aβ-dependent, because liver-X receptor agonist treatment to promote Aβ degradation rescued the hyporesponsive state and olfactory behavior. These data lend evidence to a novel working model of olfactory dysfunction in AD and, complimentary to other recent works, suggest that disease-relevant network dysfunction is highly dynamic and region specific, yet with lasting effects on cognition and behavior.
Introduction
Alzheimer's disease (AD) is a progressive brain disease wherein patients suffer from sensory, cognitive, and motor loss (Bishop et al., 2010). Olfactory system function is especially vulnerable to AD. Deficits in odor detection, identification, and recognition occur early in the disease (Mesholam et al., 1998; Murphy, 1999) as opposed to effects on other modalities, such as vision (Gilbert and Murphy, 2004) or cognition (Bacon et al., 1998). These findings are consistent with the concept that AD preferentially impacts specific vulnerable neural networks in the brain (Arnold et al., 1991; Buckner et al., 2009; Seeley et al., 2009). Molecular and network-level mechanisms for this vulnerability remain unresolved yet likely hold major promise in understanding disease progression.
Multiple pathogenic features accumulate throughout the course of AD. The amyloid-β (Aβ) peptide is a particularly potent modulator of synaptic transmission (Kamenetz et al., 2003; Abramov et al., 2009) and thus synchronous neural oscillations (Palop and Mucke, 2010). Neural oscillations are critical for a range of local circuit and regional network functions, including sensory coding, sensorimotor integration, synaptic plasticity necessary for information storage, and information transfer between brain regions (Freeman, 1975; Varela et al., 2001; Buzsaki, 2006). In agreement with this, numerous neurological disorders, including AD, exhibit atypical neural synchrony and oscillations (Jeong, 2004; Uhlhaas and Singer, 2006).
Olfactory perception relies on the hierarchal processing of odor information throughout a distributed network. In this system, the spatiotemporal pattern of odor-evoked activity in the olfactory bulb (OB) is transferred by means of temporally structured spike trains and coinciding synchronous oscillations into highly plastic olfactory cortical areas, including the piriform cortex (PCX). From the PCX, odor information travels into the entorhinal cortex and hippocampus for consolidation and future retrieval. Proper information processing within the OB and PCX is critical for the spread of information into higher-order areas, and disruption of either structure alone impairs perception (Nusser et al., 2001; Wilson, 2001; Doucette et al., 2007) in manners analogous to that observed in AD (Murphy, 1999). AD pathogenic factors, including Aβ aggregation, are found within the olfactory epithelium, OB, and PCX in both humans with AD and rodent models (Price et al., 1991; Arnold et al., 2010; Wesson et al., 2010). Interestingly, in the AD rodent models, Aβ first deposits in the OB well before deposition in the PCX, entorhinal cortex, or hippocampus (Wesson et al., 2010). Additionally, the magnitude of olfactory behavioral dysfunction strongly correlates with the level of Aβ deposition (Wesson et al., 2010).
The above findings, along with the ability for Aβ to disrupt synaptic transmission, posits the apparent vulnerability of the olfactory system to Aβ as a major factor underlying olfactory impairments in AD. The impact of Aβ on olfactory network function, however, remains unresolved. This is important even beyond the scope of understanding olfactory impairments because, through the vast interconnectedness of this network, atypical olfactory activity may induce widespread changes in brain function, pathogen spread, and other disruptions in cell function throughout the brain that may exacerbate disease progression (Kapogiannis and Mattson, 2011).
Materials and Methods
Mice
Mice bred and maintained within the Nathan S. Kline Institute for Psychiatric Research animal facility were used. Tg2576 [amyloid precursor protein (APP)] mice were generated previously (Hsiao et al., 1996) by overexpressing the 695 aa isoform of human APP containing the K670N–M671L mutation. Age-matched nontransgenic [wild-type (WT)] littermates on B6SJLF1/J background were used as controls.
Multiple, separate cohorts of mice were used in this study. In all experiments, we kept animal numbers to the minimum required for sufficient statistical test power. First, one group of 3- to 4 month-old (seven WT, four male and three female; six APP, three male and three female) and 6- to 7-month-old (five WT, two male and three female; five APP, two male and three female) mice were used for behavior (see Fig. 1A), OB and PCX single-site electrophysiology experiments (see Figs. 1,2), and Aβ quantification (data not shown). Second, a group of 3- to 4-month-old (six WT, four male and two female; six APP, five male and one female) mice were used for hippocampal CA1 electrophysiology (see Fig. 1G,H). A third group of 3- to 4-month-old (four WT, two male and two female; four APP, three male and one female) and 6- to 7-month-old (five WT, two male and three female; five APP, two male and three female) mice were used for simultaneous OB and PCX recordings (see Fig. 3). A fourth cohort of 14- to 16-month-old mice (eight WT, three male and five female; nine APP, five male and four female) were used for behavior, electrophysiology, GW3965 (3-[3-[N-(2-chloro-3-trifluoromethylbenzyl)-(2,2-diphenylethyl)amino]propyloxy]phenylacetic acid hydrochloride) (GW) treatment (see below), and Aβ quantification (see Figs. 4, 5). Finally, a fifth cohort of 14- to 16-month-old mice (six WT, all male; nine APP, eight male and one female) were used for GW3965 withdrawal behavior experiments.
Figure 1.
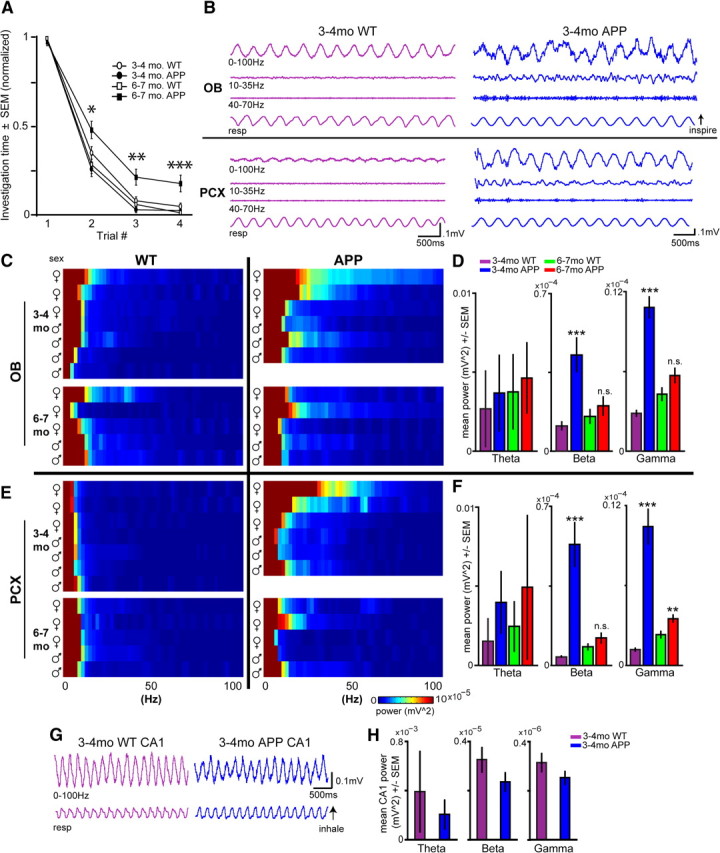
Aberrant, transient spontaneous network activity within the olfactory system precedes behavioral and hippocampal dysfunction in APP mice. A, Odor habituation behavior in 3- to 4-month-old and 6- to 7-month-old Tg2576 (APP) and control mice (WT, n = 12 per age, 6 per genotype). Data represent odor habituation (normalized; see Materials and Methods) across four successive odor presentation trials. *p < 0.05, **p < 0.01, ***p < 0.001, 6-month-old WT versus APP, ANOVA. B, Spontaneous LFP activity in the OB (top) and PCX (bottom) from 3- to 4-month-old mice. Data are filtered (second-order bandpass) to display full band (0–100 Hz), beta (10–35 Hz), and gamma (40–70 Hz). C, Two-dimensional histograms of spontaneous OB LFP power from each mouse organized by age, genotype, and sex (∼2 Hz bins). D, Spontaneous OB LFP power (as seen in C). **p < 0.01, ***p < 0.001, two-tailed t test, age-matched WT versus APP. E, Two-dimensional histograms of spontaneous PCX LFP power from each mouse organized by age, genotype, and sex (∼2 Hz bins). F, Spontaneous PCX LFP power (as seen in E). **p < 0.01, ***p < 0.001, two-tailed t test, age-matched WT versus APP. n.s., p > 0.05, age-matched WT versus APP. Example full band (0–100 Hz) traces (G) and average power spectrum data of spontaneous LFP recordings from the hippocampal CA1 (cornu ammonis region 1) (H). n = 6 mice per genotype, 5 min of data per mouse. Although APP CA1 LFP power was qualitatively less than age-matched WTs, no significant differences were found between groups within given frequency bands (p > 0.06, two-tailed paired t tests) at this age.
Figure 2.

Abnormal cortical odor responsivity in 6- to 7-month-old APP mice coincides with the onset of olfactory behavioral dysfunction. A, OB (top) and PCX (bottom) LFPs from a 3- to 4-month-old WT mouse in response to a 2 s odor presentation. Data are filtered (second-order bandpass) to display full band (0–100 Hz), beta (10–35 Hz), and gamma (40–70 Hz). B, Odor-evoked response magnitudes in the OB (top) and PCX (bottom) (same mice as used in Fig. 1, 2 odors per mouse). Response magnitude = power 2 s during odor/power 2 s pre-odor. *p < 0.05, **p < 0.01, ***p < 0.001, two-tailed t test, age-matched WT versus APP. Note different scale values between bands. C, Percentage adaptation of odor-evoked response magnitudes on trial 4 as a function of the magnitude on trial 1. *p < 0.05, two-tailed t test, age-matched WT versus APP.
Figure 3.

Atypical OB–PCX functional connectivity in 6- to 7-month-old APP mice. A, Simultaneous OB and PCX LFP in a 3- to 4-month-old WT mouse displayed in full band (0–100 Hz). Resp, Respiration. B, Spectral coherence of spontaneous OB–PCX activity. Inset displays mean coherence within groups by individual frequency bands. *p < 0.05, **p < 0.01, two-tailed t tests, APP versus age-matched WT. n = 4 mice per group. C, Odor-evoked spectral coherence of OB–PCX LFP. *p < 0.05, **p < 0.01, two-tailed t tests, APP versus age-matched WT within beta and gamma bands, respectively. n = 4 mice per group, 2 odor trials per mouse. B, C, 5 Hz bins, smoothed for plot only.
Figure 4.

PCX odor-evoked hyperactivity in 6- to 7-month-old APP mice is transient and not displayed by 14- to 16-month-old mice. A, Odor-evoked PCX LFP response magnitudes in 14- to 16-month-old mice (n ≥ 4 mice per group, 2 odors per mouse). Response magnitude = power 2 s during odor/power 2 s pre-odor. *p < 0.05, two-tailed t tests WT versus APP. B, Progression of PCX odor-evoked magnitudes. By 14 months of age, aberrant activity seen in 6- to 7-month-old APPs returns to levels statistically similar to 3- 4-month-old APPs (p > 0.05, two-tailed t tests of average magnitudes for each mouse within each band), significantly different from 6- to 7-month-old mice. *p < 0.05 theta, **p < 0.001 beta, two-tailed t tests, 6–7 versus 14–16 month data. The 3–4 and 6–7 month data are the same data as in Figure 2.
Figure 5.

Pharmacological restoration of network activity in 14- to 16-month-old APP mice coincides with remediation of behavioral dysfunction. A, Design for GW liver-X receptor agonist treatment. B, Average percentage habituation by trial 4 (as a function of maximum investigation during trials 1–3) for each animal (horizontal bar, group average; dots, mean of 4 odors per mouse) and for each group before (baseline) and after treatment with veh or GW. APP + GW behavior (right) improves after GW treatment (p < 0.01, two-tailed t test, baseline vs GW). C, Power spectrograms of PCX LFP power before (pre-odor) and during (odor, 5 s epochs) odor. Similar to that shown in Figure 4, 14–16 month APPs lack normal odor-evoked responsivity in the PCX (**p < 0.01, two-tailed t test of APP + veh vs WT + GW or WT + veh in beta band; 1 averaged LFP power value between 10 and 35Hz per mouse). GW treatment in APPs yielded WT-like beta band PCX LFP power, significantly greater than APP + veh (**p < 0.01, two-tailed t test of APP + GW vs APP + veh in beta band). APP + Veh mice is the same data as shown in Figure 4A. D, Thioflavin-S staining of fibrillar amyloid-β deposits in the OB and PCX in veh- or GW-treated mice. Gr, Granule cell layer; IPl, inner plexiform layer; MT, mitral/tufted cell layer; EPl, external plexiform layer; Gl, glomerular layer. At 14–16 months, the OB and PCX of veh-treated APP mice were mostly defined by the presence of just a few (<3) large dense-core plaques. E, Quantification of percentage area Thioflavin-S-positive and anti-Aβ (4G8+) staining in the OB and PCX. n ≥ 4 sections per mouse, n ≥ 3 mice per group. *p < 0.05, **p < 0.01, two-tailed paired t tests. F, Hemibrain homogenate ELISA quantification of DEA-extracted (non-plaque associated, “soluble”) and FA-extracted Aβ1–40 and Aβ1–42. n ≥ 4 mice per group. *p < 0.05, **p < 0.01, two-tailed paired t tests.
Mice were genotyped by PCR analysis of tail DNA using standard methods. Confirmatory PCR of tail DNA was performed after experiments to ensure correct classification of groups. All experiments were conducted in accordance with the guidelines of the National Institutes of Health and were approved by the Nathan S. Kline Institute Institutional Animal Care and Use Committee.
Odor habituation test
As we described previously (Wesson et al., 2010), odors [2-heptanone, isoamyl acetate, (+)-limonene, ethyl valerate; Sigma-Aldrich] were diluted 1 × 10−3 in mineral oil and applied to a cotton-applicator stick. Odors were delivered over four trials, 20 s each, separated by 30 s intervals in the previously listed order. Snout-oriented sniffing was recorded by a single observer blind to genotypes. Mice were tested in a counterbalanced order. For GW3965 experiments (see Fig. 5), the same set of odors described above was used to assess olfactory behavior before and after drug or vehicle (veh) treatment.
In vivo electrophysiology
Within 2 weeks after completion of olfactory testing, mice were anesthetized with urethane (1 g/kg, i.p.) and positioned in a stereotaxic apparatus for in vivo local field potential (LFP) recordings. The stereotaxic frame was outfitted with a water-filled heating pad to maintain core body temperature (38°C). Mice were supplied local anesthetic (1% xylocaine, s.c.) to the cranial surface, and the skin was later removed, exposing the dorsal skull. Small (∼1.5 mm diameter) ipsilateral holes were drilled over the anterior PCX, OB, lateral olfactory tract (LOT), and (in some cases) the hippocampus CA1 region according to Paxinos and Franklin (2000). A Vaseline well was created around the skull surface and filled with warm (38°C) 0.9% NaCl. For local recordings, a tungsten stimulating electrode was lowered onto the LOT to aid in PCX localization under physiologic control by LOT stimulation. For data collection, a recording electrode (catalog # 576000, 0.01 inch; A-M Systems) was lowered first into the PCX (the anterior PCX region), and then, after PCX data collection, the electrode was lowered into the mesial aspect of the OB for OB data acquisition. This order was selected to minimize the effects of experience-dependent changes in odor-evoked activity in the PCX (Wilson et al., 2006). For OB–PCX simultaneous recordings, a bipolar stainless steel recording electrode bundle (catalog #7915, 0.007 inch; A-M Systems) was lowered into the PCX and another into the mesial aspect of the OB for OB data acquisition. Electrode locations were verified with postmortem histology using nuclear staining [4′,6-diamidino-2-phenylindole (DAPI)] of slide-mounted 40 μm coronal sections (as described below, Histology). Recording electrode potentials along with stimulus presentation events were acquired using Spike2 software (Cambridge Electronic Design). Recordings across groups were performed in a pseudorandom order.
Stimulus presentation
Odors were presented to anesthetized mice using an air-dilution olfactometer at 1 L/min flow using medical-grade nitrogen. Stimuli included five monomolecular odorants (1,7-octadiene, 4-methyl-3-penten-2-one, ethyl propionate, heptanal, and isoamyl acetate; Sigma-Aldrich). All odorants were pure in their liquid state except for 1,7-octadiene and 4-methyl-3-penten-2-one, which were diluted 1:1 in mineral oil (all ∼100 ppm concentration). Odors were presented 2 s each, in a cross-habituation paradigm similar to that used for the odor cross-habituation behavioral test (see below, Behavior). Responses were recorded from both the PCX and OB using the same two odors. The paradigm for odor presentation was as follows [stimulus (intertrial interval)]: odor A (2 min) → odor B (2 min) → odor A (30 s) → odor A (30 s) → odor A (30 s) → odor A (30 s) → odor B. Thus, for each brain region per mouse, the first presentation of odors A and B were considered “novel odors” (as used for analysis of basic odor-evoked activity; see Fig. 3), and presentations 2–5 for odor A were used to study cellular adaptation (see Fig. 4). Odor onset was triggered off of the animal's respiration using a piezo-electric foil placed under the animal's chest and a window discriminator to detect peaks of respiration (World Precision Instruments).
In vivo pharmacology
A subset of 14- to 16-month-old mice were gavaged with either the liver-X receptor (LXR) agonist GW (GlaxoSmithKline; 0.033 g · kg−1 · d−1 per mouse; n = 7 WT and 6 APP) or veh (dimethylsulfoxide; n = 6 WT and 5 APP) (Zelcer et al., 2007; Jiang et al., 2008). Several mice died early during treatment, likely as a result of complications from gavage. Final mouse numbers by study completion were n = 3 WT and 4 APP (GW) and n = 5 WT and 5 APP (veh). Total volume gavaged was 0.1 ml/10 g body weight. The average body weight across all mice (33.29 ± 0.26 g) did not significantly differ across groups (p > 0.05, two-way ANOVA), nor did any one group significantly change in body weight over the course of treatment (p > 0.05, repeated-measures ANOVA). An additional cohort of 14- to 16-month-old mice were treated with GW (n = 6 WT and 4 APP) or veh (n = 4 APP) (same protocol as above) and then allowed to rest untreated for 1 week before behavior testing to examine effects of GW withdrawal.
Histology
Tissue collection was performed after urethane anesthesia (1 g/kg, i.p.). Mice were decapitated, and brains were rapidly removed over ice. Hemibrains were placed in 10% Formalin for fixation. A subset of these brains were coronally sectioned (40 μm) on a microtome for Aβ analysis and electrode placement verification.
Electrode verification.
Not all brains were examined for placement of electrodes given a combination of 1) the ease of location (e.g., OB) for electrode guidance and 2) the need for tissue for Aβ analysis versus electrode verification). However, visual inspection of electrode tip locations in Aβ-stained tissue confirmed that electrode placement was accurate. In other tissue, placement analysis occurred after coverslipping with Vectashield hard mount with DAPI (Vector Laboratories) for nuclear counterstain. In all cases, electrode tips were within targeted regions [PCX (anterior PCX layers i, ii, or iii), OB, or hippocampus CA1].
Aβ staining.
Floating sections containing the OB or PCX were stained with either Thioflavin-S or 4G8 (anti-Aβ). For Thioflavin-S staining, floating sections were left standing in 1% filtered Thioflavin-S solution for 10 min and then rinsed in ddH2O (three times) for 1 min. The remaining sections were left floating in Tris-buffered saline (TBS) for 4G8 (anti-Aβ) immunohistochemistry (Mi et al., 2007). Sections were washed (three times for 5 min) in TBS, after which they were treated with 85% formic acid for 5 min to enhance amyloid staining. Sections were then washed in blocking buffer (0.05 m Tris-HCl, pH 7.6, 0.9% NaCl, 0.25% Triton X-100, 20% normal goat serum, and 0.2% bovine serum albumin) three times (10 min each) before incubating for 12 h in 4G8 primary antibody at 4°C (Signet Labs; 1:200 in blocking buffer). Sections were rinsed (three times for 5 min) in blocking buffer before incubating for 2 h at room temperature in Alexa Fluor 488 secondary antibody (Invitrogen). Finally, sections were rinsed in TBS (three times for 5 min), placed on slides, and coverslipped.
A control section (containing the cortex and hippocampus of a 16-month-old Tg2576 mouse) was concurrently stained as a positive control (used within all staining wells for the GW3965 experiment) (see Fig. 5). Optical scans of brain sections were collected within 7 d after staining by use of a Carl Zeiss Axioscope microscope (model 200M) and a Carl Zeiss digital camera. Sections were excited at 488 nm and captured with a fluorescein-5-isothiocyanate filter.
Aβ sandwich ELISAs
Human Aβ levels were determined by solid-phase sandwich ELISAs to the Aβ1–40 and Aβ1–42 sequence as described previously (Schmidt et al., 2005) using a manufactured ELISA kits (Invitrogen). Ten percent (weight/volume) homogenates were prepared from flash-frozen hemibrains (n ≥ 4 mice per group) lacking the OBs and cerebellum in a homogenization buffer containing 250 mm sucrose, 20 mm Tris base, 1 mm EDTA, 1 mm EGTA, and protease inhibitors. Diethylamine (DEA) extracted supernatant was used for soluble Aβ quantification, and the remaining DEA pellet was sonicated with formic acid (FA) for measures of plaque-associated Aβ. Both soluble and plaque-associated fractions were plated in duplicate along with WT mouse control homogenates and kit-provided Aβ1–40 and Aβ1–42 standards.
Data analysis
Behavior.
For analysis of olfactory behavior data (as described previously by Wesson et al., 2010), all raw investigatory values (seconds) were pooled within animals and organized according to odor presentation (trial) number. As a measure of odor habituation, the raw investigatory values were normalized to the maximum investigatory duration per animal for each odor (maximum during trials 1–4). These normalized data were selected for analysis because of subtle group differences in trial 1 odor investigation behavior. The maximum investigation duration was assigned a value of “1” and the lesser investigation times a proportion of 1. These normalized investigation durations of subjects for each odor were analyzed. In some cases (see Fig. 5B), “percentage habituation” values were created on trial 4 data as a function of the maximum investigation time (“1”; only data from trial 4 was used in the analysis).
Histology.
Histological analysis of Aβ levels was performed in NIH ImageJ (http://rsbweb.nih.gov/ij). Fluorescence levels of anti-Aβ 4G8 and Thioflavin-S were thresholded, and regions of interest (ROIs) were determined with the guidance of the DAPI counterstain and standard anatomical coordinates (Paxinos and Franklin, 2000). Aβ deposition area (%) was quantified within the OB and PCX. Individual layers in both structures were manually outlined as described previously (Wesson et al., 2010). Aβ deposition (percentage area) was defined as the cumulative area of fluorescent pixels above threshold within each ROI. At least three coronal brain sections (range of three to four) containing each ROI per mouse were used for analysis. Percentage area values were in some cases analyzed using one-way ANOVAs for independent groups followed by post hoc group comparisons using Fisher's PLSD. Percentage area values for each ROI within each section were treated as independent measures for analysis.
Electrophysiology.
Analysis of electrophysiological data was performed in Spike2 (Cambridge Electronic Design) using Spike2 scripts. In most cases, raw data were processed with an offline fast Fourier transform (FFT) analysis to allow classification of data within components of different LFP frequencies (0.49 s Hanning window, 2.035 Hz resolution). A power spectrum was extracted containing the LFP power from 0 to 100 Hz. For odor-evoked data, we normalized the LFP power spectrum before to that during odor presentation to calculate the odor-evoked response magnitude. OB–PCX simultaneous recordings were analyzed by first subtracting the signals of each bipolar electrode to create one OB and one PCX channel that would more closely reflect localized events. From this, we then performed waveform correlations or analyzed olfactory LFP coherence (Chabaud et al., 1999) using the COHER openware script freely available (http://www.ced.co.uk) as described previously (Wilson and Yan, 2010). For the COHER performance, the OB was set as the reference signal. All values are reported as mean ± SEM unless otherwise stated.
Results
We previously reported that, in mice overexpressing human mutations of the APP (Tg2576) (Hsiao et al., 1996), Aβ first deposits in the OB well before deposition in the PCX, entorhinal cortex, or hippocampus (Wesson et al., 2010). Additionally, the magnitude of olfactory behavioral dysfunction, which emerges early in life, strongly correlates with the level of Aβ deposition (Wesson et al., 2010). Here we used neural synchrony measures along with olfactory behavioral and pathological assays to examine the influence of progressive β-amyloidosis on olfactory network activity and function.
Aberrant, transient spontaneous network activity within the olfactory system precedes behavioral dysfunction in APP mice
First, we confirmed olfactory behavioral impairments before recordings by testing 3- to 4-month-old and 6- to 7-month-old WT and APP mice in the odor habituation test (Wesson et al., 2010) (n > 12 per age, n > 5 per genotype). In this test, the time mice spend sniffing odors is measured across successive odor presentations as a gross behavioral index of olfactory function. Odor habituation was similar between WT and APP mice at 3–4 months of age (p > 0.05, two-tailed t test within each odor trial). However, 6- to 7-month-old APP mice displayed statistically less habituation to repeated odor presentation at trials 2 (F(1,49) = 9.502; p = 0.0034), 3 (F(1,49) = 12.21; p = 0.001), and 4 (F(1,49) = 13.978; p = 0.0005) compared with WTs (Fig. 1A). Given the importance of the PCX and OB to odor habituation (Wilson and Linster, 2008), these results suggest that odor information processing is disrupted in these regions of APP mice and highlight the potential benefits of examining network activity at these two ages (3–4 and 6–7 months), because they uniquely represent time points wherein olfactory behavior is respectively either functional or dysfunctional.
Next we recorded 10 min of spontaneous OB and PCX LFP activity from the same mice used for behavior (Fig. 1A) and used FFT to allow analysis of different LFP frequency components (Fig. 1B,C). Olfactory LFP activity can be organized into theta (1–10 Hz), beta (10–35 Hz), and gamma (40–70 Hz) frequency bands. Olfactory theta activity mostly reflects large synchronous volleys of afferent input from the nasal epithelium (Adrian, 1951). Gamma oscillations reflect local circuit interactions within both the OB and PCX (Rall and Shepherd, 1968; Neville and Haberly, 2003). Beta oscillations are believed to reflect more large-scale network activity (Kopell et al., 2000), for example, between the OB and PCX (Neville and Haberly, 2003; Kay and Beshel, 2010). Each of these bands reflect distinct circuit activities contributing unique aspects to odor-perception and odor-guided behavior (Kay et al., 2009). Importantly, all recordings were performed under anesthesia to control for changes in behavioral state that may exert top-down influences on olfactory function.
At just 3–4 months of age, APP mice display abnormal spontaneous OB and PCX LFP activity, with prominent increases in both beta and gamma band power (Fig. 1B, right). This finding is fairly consistent across APP animals, with perhaps greater expression among females (Fig. 1C,E). Increases in spontaneous beta and gamma band power were observed within both the OB and PCX in 3- to 4-month-old APP mice compared with age-matched WT (p < 0.001, two-tailed t tests) (Fig. 1D,F). No differences in spontaneous theta power were observed between genotypes at this age. Similar recordings performed from the CA1 of the hippocampus from a separate cohort of 3- to 4-month-old mice (n = 6 per genotype) revealed a near-complete preservation of hippocampal LFP activity at this age (p > 0.05, within theta, beta, and gamma frequency bands) (Fig. 1G). Thus, the aberrant spontaneous activity in the OB and PCX is selectively localized.
Surprisingly, the enhanced spontaneous activity in the OB and PCX of 3- to 4-month-old APP mice was transient (Fig. 1D,F). In 6- to 7-month-old mice, only within PCX gamma band activity was there a significant increase in APP mice (p < 0.01). Instead, APP beta and gamma band power significantly decreased from 3–4 to 6–7 months within both the OB (p < 0.05, beta; p < 0.001, gamma) and PCX (p < 0.001, beta and gamma). Theta band power showed a progressive trend across age within both genotypes; however, no significant differences were observed (p > 0.05). To determine whether these changes affect the power of specific subfrequency bands, we performed an analysis of peak LFP frequencies within each band. No group differences within peak theta, beta, or gamma band activity within the OB or PCX were found (p > 0.05; data not shown).
Heightened responsivity to odors in the PCX of 6- to 7-month-old APP mice coincides with the onset of olfactory behavioral dysfunction
One advantage the olfactory system brings to the study of AD is that network function can be probed during the processing of discrete stimuli (odors), thus allowing conclusions regarding behaviorally relevant information processing. We recorded odor-evoked LFP activity (Fig. 2A) in the PCX and then later in the OB (two odors per region; same mice as in Fig. 1). All groups showed significant odor-evoked responses (relative changes) compared with the 2 s pre-odor (p < 0.05, two-tailed t tests within frequency bands and groups), however, as detailed below, with significant differences between groups (Fig. 2B).
As shown in Figure 2B, heightened odor-evoked beta and gamma band response magnitudes were found in the PCX of 6- to 7-month-old but not at 3- to 4-month-old APP mice. In contrast, whereas odor-evoked gamma activity in the OB was not affected in any age group of APP mice, odor-evoked beta (in 3- to 4-month-old and 6- to 7-month-old mice) and theta (in 6- to 7-month-old mice) activity were significantly suppressed in the OB. No group effects were observed within peak LFP bands of odor-evoked recordings (p > 0.05; data not shown). Thus, the display of aberrant local circuit and network function at 6–7 months in the PCX corresponds with the emergence of impaired olfactory behavior at this time.
Adaptation to odor is a normal feature of olfactory circuit function, which is important to odor perception (Dalton and Wysocki, 1996). Because 6- to 7-month-old APP mice show deficient odor habituation behavior, we hypothesized that cortical odor adaptation may be compromised in these animals. To test this, we compared the percentage of adaptation in odor-evoked response magnitudes (percentage adaptation) across four trials of the same odor (n = 4 odors per animal). Six- to 7-month-old APP mice showed enhanced odor-evoked response adaptation specifically within the PCX but not OB (Fig. 2C). This was significant within the beta frequency band (p < 0.05, 6–7 month WT vs APP). No other within-age effects were found. Thus, one factor that may contribute to the modified behavioral odor habituation is atypical cortical adaptation.
Altered OB–PCX functional connectivity in 6- to 7-month-old APP mice
Information transfer between the OB and PCX is thought to be critically dependent on beta oscillations (Neville and Haberly, 2003; Kay and Beshel, 2010). Disruption of inter-regional communication may provide additional mechanisms for network dysfunction in AD (Buckner et al., 2009). Therefore, we performed simultaneous OB and PCX LFP recordings with bipolar electrodes in an additional cohort of mice (Fig. 3A) to provide a test of this hypothesis.
First, we recorded 10 min of spontaneous OB–PCX activity to examine resting-state OB–PCX dynamics. Although waveform correlation analysis did not detect an effect of age or genotype (data not shown), spectral coherence measures found heightened OB–PCX coherence within beta and gamma band frequencies in 6- to 7-month-old APP mice compared with age-matched WTs (Fig. 3B) (p < 0.05). Similar to that found during spontaneous state, waveform correlation analysis did not detect an effect of age or genotype on odor-evoked data (data not shown). However, spectral coherence of odor-evoked activity within the beta (p < 0.05) and also low gamma (p < 0.01) band was greatly enhanced within 6- to 7-month-old APP mice compared with age-matched WTs (Fig. 3C). Thus, an additional contributing mechanism to olfactory behavioral dysfunction in APP mice may be impaired, perhaps hyperactive, OB–PCX functional connectivity.
PCX hyperactivity in 6- to 7-month-old APP mice is transient and not displayed later in life
Does hyperactivity in the PCX of 6- to 7-month-old APP mice become progressively greater with age? We examined PCX odor-evoked activity in 14- to 16-month-old APP and WT mice (same methods used for 3- to 4-month-old and 6- to 7-month-old mice). Strikingly, in 14- to 16-month-old APP mice, we found significantly hypoactive odor responsivity (p < 0.05 in beta and gamma separately, WT vs APP) (Fig. 4A). Spontaneous PCX activity was not affected in APP mice of this age (p > 0.05, two-tailed t tests in each band; data not shown). Thus, whereas WT PCX activity is relatively consistent throughout age (3–16 months), odor-evoked activity in APP mice shows a transient dramatic increase that later decreases to hypoactive levels by 14–16 months of age (Fig. 4B) (theta, p < 0.05; beta, p < 0.001; 6- to 7-month-old vs 14- to 16-month-old APP, n ≥ 4 mice per group).
LXR-mediated rescue of Aβ-induced behavioral- and circuit-level olfactory dysfunction
To more directly test whether Aβ plays a role in olfactory network dysfunction, we used acute pharmacotherapy with the LXR agonist GW (Zelcer et al., 2007; Jiang et al., 2008) (see Materials and Methods), to promote Aβ proteolytic degradation. Before treatment, WT and APP mice were randomly divided into two groups (Fig. 5A) for repeated-measures testing, one for veh treatment and the other for GW (WT + veh, n = 5; APP + veh, n = 5; WT + GW, n = 3; APP + GW, n = 4) and screened for baseline odor habituation behavior (Fig. 5B). Both groups of APP mice performed significantly worse than WT controls, showing deficient odor habituation (as measured with percentage habituation) (p < 0.01 in both groups). After testing, mice were orally administered either GW or vehicle for 2 weeks. Mice were then again tested for odor habituation behavior. Odor habituation was significantly improved in APP + GW mice versus baseline (p < 0.01, two-tailed t test) and versus the APP + veh group (p < 0.01, two-tailed t test) (Fig. 5B), reflecting functional improvement of a PCX-dependent olfactory behavior with GW treatment. In an additional cohort of mice, we explored whether GW treatment had long-lasting effects after withdrawal by performing baseline odor habituation testing, then administering GW or veh as described above, and then allowing mice to rest untreated for 1 week (see Materials and Methods). After 1 week of withdrawal, APP + GW mice returned to APP + veh mouse levels (p > 0.05, two-tailed t test) (data not shown) and were significantly different from WT + GW mice (p < 0.05, two-tailed t test), suggesting no long-lasting effect of the GW treatment after its withdrawal.
After the second round of behavior during GW treatment, we performed PCX LFP recordings to explore whether PCX circuit function in APP + GW mice might be restored parallel to the observed recovery of olfactory behavior. Figure 5C shows averaged PCX LFP power spectrograms from APP and WT groups. Average theta, beta, and gamma band powers were calculated within each mouse similar to that performed above (Figs. 3, 4). As mentioned previously (data not shown), no significant differences in spontaneous activity were found between groups at this age (APP + veh vs APP + GW mice showed a trend in spontaneous beta band activity; p = 0.0502, two-tailed t test). Like that shown in Figure 4, we found the PCX of 14–16 month APPs to be hyporesponsive to odors (Fig. 5C). No significant effect of odor presentation was observed in APP + veh mice (p > 0.05 in either beta or gamma bands). In contrast, in both WT groups, a significant odor-evoked enhancement of LFP power within the beta band was observed (p < 0.01, two-tailed t test of odor vs pre-odor; Fig. 5C). Remarkably, GW treatment partially restored PCX odor-evoked activity in APP mice toward WT levels, particularly within the beta frequency band (p < 0.01, APP + GW vs APP + veh) (Fig. 5C).
In confirmation of the efficacy of the GW treatment, we observed GW-dependent reductions in Aβ deposition (Fig. 5D,E) and in soluble and insoluble Aβ1–40 and Aβ1–42 as assayed with ELISA (Fig. 5F). Together, these data demonstrate that GW-dependent reductions in Aβ (Fig. 5D–F) or perhaps modulation of other factors that co-occur with over-accumulation of Aβ in the OB and PCX can restore olfactory behaviors and the underlying function of the network required to perform the behavior.
Discussion
Many studies have examined the impact of Aβ on network activity and synaptic transmission (for review, see Selkoe, 2008; Palop and Mucke, 2010). By probing neural circuit function in the olfactory system of APP transgenic mice, we were able to examine the influence of low levels of Aβ on behaviorally relevant perturbations in network function throughout several processing centers and during Aβ progression. Our results demonstrate a nonlinear emergence of network dysfunction in APP mice and support a novel working model for how the olfactory system becomes disrupted throughout the course of Alzheimer's β-amyloidosis (Fig. 6).
Figure 6.

Working model for how β-amyloidosis contributes to olfactory network dysfunction and perceptual impairments. Based on the findings uncovered throughout this study, we propose the following working model for how progressive β-amyloidosis throughout the olfactory system contributes to olfactory network dysfunction and perceptual impairments. Early life (3 months) Aβ deposition within the initial synaptic layer of the OB results in hyperactivity within the OB network, which through either direct (enhanced synaptic input) or indirect [altering of PCX inhibitory (GABAergic) circuits] mechanisms increases activity in the PCX, thereby rendering the system unable to process odor information normally. Through established mechanisms over time, this hyperactivity upregulates Aβ production within both structures subsequently, resulting in a weakened, hyporesponsive state. Thus, different network states (hyperactive or hypoactive) can contribute to olfactory behavioral impairments more or less equally yet may indicate differential accumulation or involvement of Aβ.
Aβ, network excitability, and disease progression
Abnormal network excitability is a prevalent feature of AD (Palop and Mucke, 2010; Santos et al., 2010). Aβ deposition is positively associated with hyperexcitability within the default network of humans (Sperling et al., 2009) and in the neocortex and hippocampus of aged APP mice (Palop et al., 2007; Busche et al., 2008). In a recent model proposed by Palop and Mucke (2010), moderate levels of Aβ may enhance presynaptic activity and potentiate synaptic transmission. At high levels, however, Aβ may have inverse effects, including enhancing long-term depression and loss of dendritic spines. Here, in the earliest parts of the olfactory system of APP transgenic mice, we revealed early-life hyperactivity that coincides with only modest levels of Aβ [that found solely in the OB glomerular layer (Wesson et al., 2010)]. Although in 3-month-old mice elevations in Aβ likely contribute [via enhancing synaptic transmission at low levels (Puzzo et al., 2008)] to network hyperexcitability that is maintained, if not in some cases enhanced, in 6-month-old mice, this hyperexcitability is not sufficient to disrupt behavior (at least to the extent detectable by the odor habituation assay). Later in life, however (>14 months), the “hyperactive” state subsides and, in effect, renders the PCX hypoactive. Indeed, even brief periods of hyperactivity are thought sufficient to elicit a cascade of later-life system hypoactivity and dysfunction (Kapogiannis and Mattson, 2011). Thus, hyperexcitability in the olfactory network emerges at a time point coinciding with behavioral dysfunction and later recedes, leaving a hypoactive system. Importantly, at both stages (hyperactive and hypoactive), behavioral dysfunction persists, suggesting that even a transient change in local circuit and/or network-level activity (that observed at 6 months of age) is sufficient to render the system incapable of properly responding to odor information, perhaps through disrupting plasticity among the ensembles required for normal odor coding (Barnes et al., 2008). Similar mechanisms may also underlie the observed spectral coherence results (Fig. 3). Future use of more precise olfactory and behavioral assays may help link specific physiological dysfunction with specific behavioral abnormalities.
We used acute pharmacotherapy with the LXR agonist GW3965 to enhance the degradation of Aβ and thereby compare behavior and network activity of APP mice with high versus those with low levels of Aβ. It was shown recently that GW3965 promotes proteolytic degradation of soluble Aβ (Jiang et al., 2008). Similarly, we found that GW administration for 2 weeks reduced Aβ deposition in the OB and PCX, improved behavior, and recovered odor-evoked responsivity in the PCX of aged APP mice. Withdrawal from GW treatment for 1 week resulted in a return of behavioral impairment. These findings suggest that Aβ-dependent effects on synaptic dysfunction are transient, consistent with the ability to reverse these effects with γ-secretase inhibitors or anti-Aβ methods (Janus et al., 2000; Kounnas et al., 2010). The transient improvement may be associated with changes in cell excitability, synaptic function, and/or reversible anatomical changes in synaptic or dendritic spine structure. In addition, although GW3965 lowered Aβ deposition within both the OB and PCX (and across other structures), alternative factors that are impacted by LXR treatment may have contributed to the restoration of behavior and olfactory network function. LXR activation drives macrophages into M2 or alternative activation states that are anti-inflammatory and facilitate phagocytosis (A-Gonzalez et al., 2009). This restoration of phagocytic competence with LXRs is primarily attributable to the transrepression of cytokine expression (Koenigsknecht and Landreth, 2004). An ideal paradigm for future studies would be one wherein Aβ degradation and cytokine levels could be differentially regulated to assess impacts of both factors, independently, in behavior and network activity.
Relation to sensory dysfunction in Alzheimer's and other disorders
The present results greatly extend our understanding of AD-related olfactory impairments in several notable ways. First, we found that Aβ strongly impacts the activity and function of the OB and PCX. Although PCX activity is clearly compromised in AD (Li et al., 2010), the role of the OB has remained elusive. Our results demonstrate that the OB may be a major culprit behind olfactory dysfunction in AD. Early-life aberrant activity in the OB activity may disrupt normal coding of odors as they spread into the PCX, thereby resulting in PCX disruption. Alternatively, given the relevance of synaptic activity in the postsynaptic upregulation of Aβ levels (Cirrito et al., 2005) and its spread between regions (Lazarov et al., 2002), these data suggest that early-life hyperactivity within the OB may effectively contribute to dysfunction in the PCX as observed here and previously in humans (Li et al., 2010) by enhancing pathological accumulation of Aβ. This accumulation may ultimately elicit synaptic loss (Spires et al., 2005) and thus result in a hyporesponsive network as indeed we observed later in life. Second, our work shows that olfactory network dysfunction appears early in life, before the appearance of significant behavioral impairments. These findings suggest that more refined methods to examine olfactory network activity clinically may serve to identify prodromal AD populations. Additionally, the improvement of olfactory behavioral and network function by enhancing Aβ proteolysis suggests that olfactory loss may be reversible. Future clinical studies examining whether similar methods to reduce Aβ can prevent olfactory loss entirely or even just reduce it will be important in understanding the mechanisms of sensory loss in AD and other Aβ-related disorders.
Conclusion
The results of these studies provide novel evidence for a relationship between the onset of sensory dysfunction commonly reported in AD and early-stage sensory network hyperactivity. These findings lend evidence to a model whereby early-life elevations in Aβ are associated with dramatic increases in high-frequency network oscillatory activity that, because of its own persistent activity, contributes to subsequent hyporesponsivity and dysfunction later in life (Fig. 6). In light of these results, it is interesting to consider that olfactory dysfunction may exacerbate AD through activity-dependent modulation of Aβ and possibly other pathogens. Future studies examining the role of other APP-related neurotoxic factors in combination with in vivo neural network activity measures will be valuable in establishing a complete understanding of the basis of neural dysfunction in AD.
Footnotes
This work was supported by National Institutes of Health Grants DC003906 (D.A.W.), AG037693 (D.A.W, E.L., R.A.N.), AG017617 (R.A.N.), and AG030482 (G.E.L.). We thank Julie Chapuis for helpful comments on a previous version of this manuscript, Paige Cramer and Paul Mathews for advice on ELISAs, and Adrienn Varga-Wesson for assistance performing ELISAs.
References
- Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid-β as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci. 2009;12:1567–1576. doi: 10.1038/nn.2433. [DOI] [PubMed] [Google Scholar]
- Adrian ED. The role of air movement in olfactory stimulation. J Physiol. 1951;114:4–5. [PubMed] [Google Scholar]
- A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, Deniz J, Ramirez C, Díaz M, Gallardo G, de Galarreta CR, Salazar J, Lopez F, Edwards P, Parks J, Andujar M, Tontonoz P, Castrillo A. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31:245–258. doi: 10.1016/j.immuni.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer's disease. Cereb Cortex. 1991;1:103–116. doi: 10.1093/cercor/1.1.103. [DOI] [PubMed] [Google Scholar]
- Arnold SE, Lee EB, Moberg PJ, Stutzbach L, Kazi H, Han LY, Lee VM, Trojanowski JQ. Olfactory epithelium amyloid-beta and paired helical filament-tau pathology in Alzheimer disease. Ann Neurol. 2010;67:462–469. doi: 10.1002/ana.21910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacon AW, Bondi MW, Salmon DP, Murphy C. Very early changes in olfactory functioning due to Alzheimer's disease and the role of apolipoprotein E in olfaction. Ann N Y Acad Sci. 1998;855:723–731. doi: 10.1111/j.1749-6632.1998.tb10651.x. [DOI] [PubMed] [Google Scholar]
- Barnes DC, Hofacer RD, Zaman AR, Rennaker RL, Wilson DA. Olfactory perceptual stability and discrimination. Nat Neurosci. 2008;11:1378–1380. doi: 10.1038/nn.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop NA, Lu T, Yankner BA. Neural mechanisms of ageing and cognitive decline. Nature. 2010;464:529–535. doi: 10.1038/nature08983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Sepulcre J, Talukdar T, Krienen FM, Liu H, Hedden T, Andrews-Hanna JR, Sperling RA, Johnson KA. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer's disease. J Neurosci. 2009;29:1860–1873. doi: 10.1523/JNEUROSCI.5062-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008;321:1686–1689. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- Buzsaki G. Rhythms of the brain. New York: Oxford UP; 2006. [Google Scholar]
- Chabaud P, Ravel N, Wilson DA, Gervais R. Functional coupling in rat central olfactory pathways: a coherence analysis. Neurosci Lett. 1999;276:17–20. doi: 10.1016/s0304-3940(99)00773-9. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-β levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Dalton P, Wysocki CJ. The nature and duration of adaptation following long-term odor exposure. Percept Psychophys. 1996;58:781–792. doi: 10.3758/bf03213109. [DOI] [PubMed] [Google Scholar]
- Doucette W, Milder J, Restrepo D. Adrenergic modulation of olfactory bulb circuitry affects odor discrimination. Learn Mem. 2007;14:539–547. doi: 10.1101/lm.606407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman WJ. New York: Academic; 1975. Mass action in the nervous system: examination of the neurophysiological basis of adaptive behavior through the EE. [Google Scholar]
- Gilbert PE, Murphy C. The effect of the ApoE e4 allele on recognition memory for olfactory and visual stimuli in patients with pathologically confirmed Alzheimer's disease, probable Alzheimer's disease, and healthy elderly controls. J Clin Exp Neuropsychol. 2004;26:779–794. doi: 10.1080/13803390490509439. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, St George-Hyslop P, Westaway D. Aβ peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- Jeong J. EEG dynamics in patients with Alzheimer's disease. Clin Neurophysiol. 2004;115:1490–1505. doi: 10.1016/j.clinph.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE. ApoE promotes the proteolytic degradation of Aβ. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kapogiannis D, Mattson MP. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer's disease. Lancet Neurol. 2011;10:187–198. doi: 10.1016/S1474-4422(10)70277-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay LM, Beshel J. A beta oscillation network in the rat olfactory system during a 2-alternative choice odor discrimination task. J Neurophysiol. 2010;104:829–839. doi: 10.1152/jn.00166.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay LM, Beshel J, Brea J, Martin C, Rojas-Líbano D, Kopell N. Olfactory oscillations: the what, how and what for. Trends Neurosci. 2009;32:207–214. doi: 10.1016/j.tins.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsknecht J, Landreth G. Microglial phagocytosis of fibrillar β-amyloid through a β1 integrin-dependent mechanism. J Neurosci. 2004;24:9838–9846. doi: 10.1523/JNEUROSCI.2557-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopell N, Ermentrout GB, Whittington MA, Traub RD. Gamma rhythms and beta rhythms have different synchronization properties. Proc Natl Acad Sci USA. 2000;97:1867–1872. doi: 10.1073/pnas.97.4.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kounnas MZ, Danks AM, Cheng S, Tyree C, Ackerman E, Zhang X, Ahn K, Nguyen P, Comer D, Mao L, Yu C, Pleynet D, Digregorio PJ, Velicelebi G, Stauderman KA, Comer WT, Mobley WC, Li YM, Sisodia SS, Tanzi RE, Wagner SL. Modulation of gamma-secretase reduces beta-amyloid deposition in a transgenic mouse model of Alzheimer's disease. Neuron. 2010;67:769–780. doi: 10.1016/j.neuron.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov O, Lee M, Peterson DA, Sisodia SS. Evidence that synaptically released β-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci. 2002;22:9785–9793. doi: 10.1523/JNEUROSCI.22-22-09785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Howard JD, Gottfried JA. Disruption of odour quality coding in piriform cortex mediates olfactory deficits in Alzheimer's disease. Brain. 2010;133:2714–2726. doi: 10.1093/brain/awq209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesholam RI, Moberg PJ, Mahr RN, Doty RL. Olfaction in neurodegenerative disease: a meta-analysis of olfactory functioning in Alzheimer's and Parkinson's diseases. Arch Neurol. 1998;55:84–90. doi: 10.1001/archneur.55.1.84. [DOI] [PubMed] [Google Scholar]
- Mi W, Pawlik M, Sastre M, Jung SS, Radvinsky DS, Klein AM, Sommer J, Schmidt SD, Nixon RA, Mathews PM, Levy E. Cystatin C inhibits amyloid-β deposition in Alzheimer's disease mouse models. Nat Genet. 2007;39:1440–1442. doi: 10.1038/ng.2007.29. [DOI] [PubMed] [Google Scholar]
- Murphy C. Loss of olfactory function in dementing disease. Physiol Behav. 1999;66:177–182. doi: 10.1016/s0031-9384(98)00262-5. [DOI] [PubMed] [Google Scholar]
- Neville KR, Haberly LB. Beta and gamma oscillations in the olfactory system of the urethane-anesthetized rat. J Neurophysiol. 2003;90:3921–3930. doi: 10.1152/jn.00475.2003. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Kay LM, Laurent G, Homanics GE, Mody I. Disruption of GABAA receptors on GABAergic interneurons leads to increased oscillatory power in the olfactory bulb network. J Neurophysiol. 2001;86:2823–2833. doi: 10.1152/jn.2001.86.6.2823. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Amyloid-β-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2000. [Google Scholar]
- Price JL, Davis PB, Morris JC, White DL. The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer's disease. Neurobiol Aging. 1991;12:295–312. doi: 10.1016/0197-4580(91)90006-6. [DOI] [PubMed] [Google Scholar]
- Puzzo D, Privitera L, Leznik E, Fà M, Staniszewski A, Palmeri A, Arancio O. Picomolar amyloid-β positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008;28:14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rall W, Shepherd GM. Theoretical reconstruction of field potentials and dendrodendritic synaptic interactions in olfactory bulb. J Neurophysiol. 1968;31:884–915. doi: 10.1152/jn.1968.31.6.884. [DOI] [PubMed] [Google Scholar]
- Santos SF, Pierrot N, Octave JN. Network excitability dysfunction in Alzheimer's disease: insights from in vitro and in vivo models. Rev Neurosci. 2010;21:153–171. doi: 10.1515/revneuro.2010.21.3.153. [DOI] [PubMed] [Google Scholar]
- Schmidt SD, Nixon RA, Mathews PM. ELISA method for measurement of amyloid-β levels. Methods Mol Biol. 2005;299:279–297. doi: 10.1385/1-59259-874-9:279. [DOI] [PubMed] [Google Scholar]
- Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron. 2009;62:42–52. doi: 10.1016/j.neuron.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Laviolette PS, O'Keefe K, O'Brien J, Rentz DM, Pihlajamaki M, Marshall G, Hyman BT, Selkoe DJ, Hedden T, Buckner RL, Becker JA, Johnson KA. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63:178–188. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W. Neural synchrony in brain disorders: relevance for cognitive dysfunctions and pathophysiology. Neuron. 2006;52:155–168. doi: 10.1016/j.neuron.2006.09.020. [DOI] [PubMed] [Google Scholar]
- Varela F, Lachaux JP, Rodriguez E, Martinerie J. The brainweb: phase synchronization and large-scale integration. Nat Rev Neurosci. 2001;2:229–239. doi: 10.1038/35067550. [DOI] [PubMed] [Google Scholar]
- Wesson DW, Levy E, Nixon RA, Wilson DA. Olfactory dysfunction correlates with β-amyloid plaque burden in an Alzheimer's disease mouse model. J Neurosci. 2010;30:505–514. doi: 10.1523/JNEUROSCI.4622-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DA. Scopolamine enhances generalization between odor representations in rat olfactory cortex. Learn Mem. 2001;8:279–285. doi: 10.1101/lm.42601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DA, Linster C. Neurobiology of a simple memory. J Neurophysiol. 2008;100:2–7. doi: 10.1152/jn.90479.2008. [DOI] [PubMed] [Google Scholar]
- Wilson DA, Yan X. Sleep-like states modulate functional connectivity in the rat olfactory system. J Neurophysiol. 2010;104:3231–3239. doi: 10.1152/jn.00711.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DA, Kadohisa M, Fletcher ML. Cortical contributions to olfaction: plasticity and perception. Semin Cell Dev Biol. 2006;17:462–470. doi: 10.1016/j.semcdb.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Zelcer N, Khanlou N, Clare R, Jiang Q, Reed-Geaghan EG, Landreth GE, Vinters HV, Tontonoz P. Attenuation of neuroinflammation and Alzheimer's disease pathology by liver x receptors. Proc Natl Acad Sci USA. 2007;104:10601–10606. doi: 10.1073/pnas.0701096104. [DOI] [PMC free article] [PubMed] [Google Scholar]