

Abstract
BACKGROUND AND PURPOSE
Chronic heart failure (CHF) is associated with action potential prolongation and Ca2+ overload, increasing risk of ventricular tachyarrhythmias (VT). We therefore investigated whether ICa blockade was anti-arrhythmic in an intact perfused heart model of CHF.
EXPERIMENTAL APPROACH
CHF was induced in rabbits after 4 weeks of rapid ventricular pacing. Hearts from CHF and sham-operated rabbits were isolated and perfused (Langendorff preparation), with ablation of the AV node. VT was induced by erythromycin and low [K+] (1.5mM). Electrophysiology of cardiac myocytes, with block of cation currents, was simulated by a mathematical model.
KEY RESULTS
Repolarization was prolonged in CHF hearts compared with sham-operated hearts. Action potential duration (APD) and overall dispersion of repolarization were further increased by erythromycin (300 µM) to block IKr in CHF hearts. After lowering [K+] to 1.5mM, CHF and sham hearts showed spontaneous episodes of polymorphic non-sustained VT. Additional infusion of verapamil (0.75 µM) suppressed early afterdepolarizations (EAD) and VT in 75% of sham and CHF hearts. Verapamil shortened APD and dispersion of repolarization, mainly by reducing transmural dispersion of repolarization via shortening of endocardial action potentials. Mathematical simulations showed that EADs were more effectively reduced by verapamil assuming a state-dependent block than a simple block of ICa.
CONCLUSIONS AND IMPLICATIONS
Blockade of ICa was highly effective in suppressing VT via reduction of transmural dispersion of repolarization and suppression of EAD. Such blockade might represent a novel therapeutic option to reduce risk of VT in structurally normal hearts and also in heart failure.
LINKED ARTICLE
This article is commented on by Stams et al., pp. 554–556 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2011.01818.x
Keywords: heart failure, ventricular tachyarrhythmias, ICa-L block, myocardial repolarization
Introduction
Chronic heart failure (CHF) is a common clinical problem that is often the final manifestation of many cardiovascular disorders. Patients with CHF have six to nine times the rate of sudden cardiac death of the general population, and up to 50% of deaths in patients with CHF are sudden and unexpected. The majority of these are due to ventricular tachyarrhythmias (VT) and consecutive ventricular fibrillation (VF) (Rosamond et al., 2008). As regards ionic and molecular changes in CHF, a consistent finding has been prolongation of cardiac repolarization that might lead to a reduced ‘repolarization reserve’ (Roden, 1998), resulting in proarrhythmia. In failing hearts, repolarization reserve may be further diminished by concomitant drug therapy that may prolong repolarization via block of the rapid component of the delayed rectifier potassium current IKr and a variety of other conditions such as hypomagnesemia or hypokalaemia.
One important therapeutic target in the prevention of proarrhythmia is the reduction of dispersion of repolarization. Based on clinical (Lubinski et al., 1998) and experimental (Akar and Rosenbaum, 2003) data, reduction of (transmural) dispersion of repolarization may be effective in preventing proarrhythmia. We recently demonstrated in an experimental intact heart model of acquired long QT syndrome (Milberg et al., 2005a) that blockade of the Ca2+ current ICa by verapamil prevented polymorphic VT by suppressing early afterdepolarizations (EADs) and reducing overall and transmural dispersion of repolarization. Comparable results were obtained in the PAFAC trial (Fetsch et al., 2004), which revealed a higher incidence of proarrhythmia in patients treated with sotalol (2 × 160 mg) than that in patients taking a combination of quinidine (3 × 160 mg) and verapamil (3 × 80 mg). Whether ICa block is also effective in preventing proarrhythmia in CHF is far from being clear. Moreover, the possible underlying electrophysiological mechanisms are completely unknown. Thus, the aim of the present study was to investigate the electrophysiological effects of ICa block, particularly on EAD and dispersion of repolarization, in an intact heart model of CHF.
Methods
All animal care and experimental protocols were approved by the local animal care committee and conformed with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication no. 852-3, revised 1996). The drug/molecular target nomenclature follows Alexander et al., (2011).
Induction of heart failure
CHF was induced in rabbits by three to four weeks of ventricular pacing at 400 beats per minute (Frommeyer et al., 2011). Adult female New Zealand white rabbits (n= 11; 3.5 to 4.5 kg; supplied by Rollié, Oelde) were anaesthetized with ketamine (75 mg·kg−1; Pfizer) and xylazine (5.8 mg·kg−1; Ceva Sante Animale, Tiergesundheit GmbH), and a human transient pacemaker lead was implanted into the right ventricle. One week after the operation, a pacemaker was connected and started stimulating with 400 beats per minute at twice diastolic threshold output. Over a period of 3 to 4 weeks, the rabbits were monitored by clinical examination and echocardiography to observe the progressive process of developing CHF (Stypmann et al., 2007). In addition, 11 sham-operated rabbits received a pacemaker lead, which was not connected to a pacemaker.
Preparation of hearts for perfusion
The method of preparing the hearts has previously been described in detail (Eckardt et al., 1998b; Milberg et al., 2005a). Female New Zealand white rabbits (n= 22) were anaesthetized with sodium thiopental (200–300 mg i.v.; Inresa Freiburg). After midsternal incision and opening of the pericardium, the complete hearts were removed and immediately placed in an ice-cold Krebs–Henseleit solution (composition in mM: CaCl2 1.80, KCl 4.70, KH2PO4 1.18, MgSO4 0.83, NaCl 118, NaHCO3 24.88, Na-pyruvate 2.0 and d-glucose 5.55). The aorta was cannulated, and the spontaneously beating hearts were perfused at constant flow (52 mL·min−1) with warm (36.8 to 37.2°C) Krebs–Henseleit solution. Perfusion pressure was kept stable at 100 mmHg. After cannulation, the hearts were given 10 min to stabilize. The perfusate was equilibrated with 95% O2 and 5% CO2 (pH 7.35; 37°C). The cannulated and perfused hearts were attached to a vertical Langendorff apparatus (Hugo Sachs Elektronic, Medical Research Instrumentation, March-Hugstetten, Germany). A deflated latex balloon was inserted into the left ventricle and connected to a pressure transducer to control haemodynamic stability. The atrioventricular (AV) node was ablated to slow the intrinsic heart rate. This resulted in complete AV block with a ventricular escape rate below 60 beats per minute.
Electrocardiographic and electrophysiological measurements
A volume-conducted ECG was recorded, and signals from a simulated ‘Einthoven’ configuration were amplified by a standard ECG amplifier (filter settings: 0.1–300 Hz). Monophasic action potential (MAP) recordings and stimulation were accomplished simultaneously using contact MAP pacing catheters (EP Technologies, Mountain View, CA, USA). The MAP electrograms were amplified and filtered (low pass 0.1 Hz, high pass 300 Hz). MAPs were analysed using a specifically designed software, permitting precise definition of the amplitude (>5 mV) and duration of the digitized signals. The recordings were considered reproducible and acceptable for analysis only if they had a stable baseline amplitude with a variation of less than 20% and a stable duration measured at 90% repolarization (MAP90). Seven MAPs were evenly spread in a circular pattern around both ventricles; one MAP was recorded from the left endocardium (distribution of MAP catheters; see legend to Figure 4). One of the right-ventricular catheters was used to pace the heart. Pacing at twice diastolic threshold was performed for 1 min at each cycle length (CL) from 900 to 300 ms using a programmable stimulator (Universal Programmable Stimulator, UHS 20, Biotronik, Berlin, Germany), which delivered square-wave pulses of 2 ms pulse width. All data were digitized at a rate of 1 kHz with 12 bit resolution and subsequently stored on a removable hard disk (BARD LabSystem, Bard Electrophysiology, Murray Hill, MA, USA).
Figure 4.
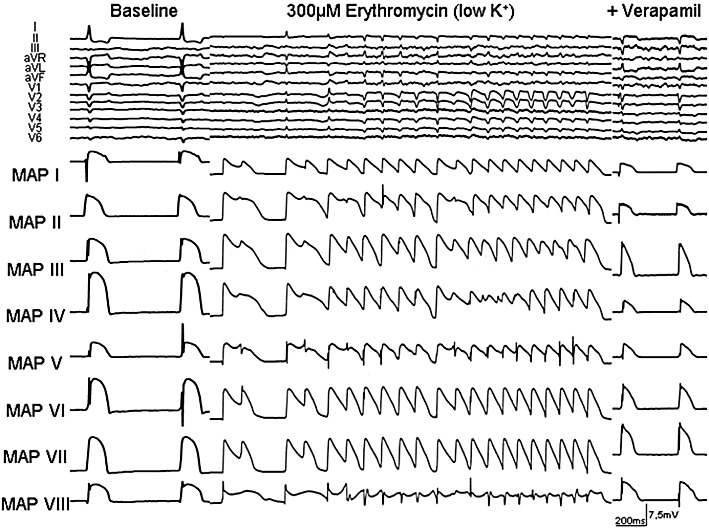
Representative example of EAD and polymorphic VT in the presence of erythromycin and after additional infusion of verapamil during bradycardia (AV block) and hypokalaemia in an isolated Langendorff-perfused heart from a CHF rabbit. ECG characteristics and MAP recordings [distribution of MAP catheters. Left heart: MAP I = base anterior, MAP IV = base posterior, MAP V = between basis and apex (posterolateral), MAP VI = between basis and apex (inferior); MAP VII = apex, MAP VIII = endocardial; right heart: MAP II = apex, MAP III = bases].
Experimental protocol
Cycle length dependence was first investigated under baseline conditions by pacing the hearts at cycle lengths between 900 and 300 ms. Subsequently, the potassium concentration was lowered to 1.5 mM to provoke EAD and VT. Thereafter, potassium concentration was restored to 5.8 mM, and the IKr blocking agent erythromycin (300 µM) was infused just above the heart. Pacing, MAP recording and measurement of ECG parameters were started 10 min after drug infusion. The potassium concentration was lowered again, and the incidence of EAD and VT was quantified. Five minutes later, the potassium concentration was again set back to 5.8 mM, and verapamil (a blocker of the L-type Ca2+ channel; 0.75 µM) was infused in addition to continuous erythromycin infusion. Pacing and MAP measurement were restarted, then potassium concentration was lowered again and the incidence of EAD and VT was registered during simultaneous infusion of erythromycin and verapamil. MAP90 was measured as the average interval between the fastest MAP upstroke and 90% of action potential duration. Dispersion of MAP90 was expressed as the difference between the minimum and the maximum of MAP90, simultaneously recorded in eight endocardial and epicardial catheters (distribution of MAP catheters s; Figure 2A). To ensure reproducibility during the study and to compare the results of different hearts, dispersion was always measured during the pacing part of the protocol. Transmural dispersion was measured between one left endocardial and the mean of four left epicardial catheters, without significant differences between the base and the apex of the left ventricle. EADs were defined as a positive voltage deflection that interrupted the smooth contour of phase 2 or 3 repolarization of the action potential.
Mathematical modelling
We chose to use the mathematical model of rabbit myocytes by Shannon et al. (2004), because of its ability to reproduce the phenomenological behaviour of EAD. To simulate CHF, we applied average changes to overall conductance values as described before: ICa= 63%, CaSRleak= 175%, IK1= 60%, IKr= 68.5%, KV7.1 = 53.5%; Ito= 50.5%. The protocols investigated match the experimental ones, except for neglecting between rabbit variation and modelling the drug interaction by investigating four different levels of block (30%, 50%, 70% and 90% of IKr block), with a stronger effect in the case of CHF due to the lower amount of total channels available. Erythromycin-mediated inhibition of IKr was introduced by a change in conductance. The inhibition of ICa by verapamil was modelled in two different ways: (a) by scaling of the exchange current and (b) by introducing a simplified state-dependent block model based on the paper by Nawrath and Wegener (1997).
 |
f∞ denotes the steady-state probability of inactivation of the ICa voltage gate in the Shannon model. The drug-dependent block rate for verapamil block is given by α (1·ms−1); β (1·ms−1) represents the dissociation rate. Parameters are deduced from the time constants and figures given in Nawrath and Wegener.
Statistical analysis
The observed data were entered onto a computerized database (Microsoft Excel 2003, Microsoft Corporation, Redmont, WA, USA) Statistical analysis was performed using the SPSS Software for Windows, release 18.0.0 (07/30/2009; SPSS Inc., Chicago, IL). Before statistically testing, each continuous variable was analysed exploratively for its normal distribution (‘Kolmogorov–Smirnow test’). Categorical variables are expressed as frequency and percentage, whereas continuous variables are presented as mean ± SD. Before statistical testing, each continuous variable was analysed for its normal distribution using the Kolmogorov–Smirnov test. The non-parametric dependent variables (cycle length influence of erythromycin and verapamil on ECG parameters, MAP duration and dispersion of repolarization) were assessed using Friedman test. Pairwise multiple comparisons following the Friedman test were performed using the procedure proposed by Dunn. The Mann–Whitney U-test was used for comparison of non-parametric variables between independent study groups. The χ2- test was used to compare the occurrence of EAD and VT after infusion of erythromycin and verapamil between study groups. Differences were considered significant at P < 0.05.
Results
Assessment of heart failure
During the period of fast ventricular pacing, rabbits became lethargic, fatigued, showed loss of appetite and developed respiratory distress. To monitor the progress of CHF, two-dimensional echocardiography was carried out periodically and demonstrated a significant decrease in ejection fraction (P < 0.05, Table 1; Figure 1A) after 3 to 4 weeks.
Table 1.
Echocardiographic characteristics before and after rapid pacing
| Pre | Post | P-value | |
|---|---|---|---|
| LVIDD (cm) | 1.64 ± 0.14 | 1.70 ± 0.27 | n.s. |
| LVIDS (cm) | 1.00 ± 0.17 | 1.45 ± 0.20 | P < 0.01 |
| FS (%) | 40 ± 6 | 17 ± 7 | P < 0.001 |
| EDV (cm3) | 4.02 ± 1.02 | 4.55 ± 1.25 | n.s. |
| ESV (cm3) | 1.06 ± 0.50 | 2.86 ± 1.25 | P < 0.02 |
| EF (%) | 75 ± 9 | 30 ± 11 | P < 0.001 |
| SV (cm3) | 2.96 ± 0.67 | 1.87 ± 1.0 | P < 0.037 |
| HR (bpm) | 252 ± 10 | 400 ± 0 | P < 0.001 |
LVIDD, left ventricular internal dimension at diastole; LVIDS, left ventricular internal dimension at systole; FS, fractional shortening; EDV, end-diastolic volume; ESV, end-systolic volume; EF, ejection fraction; SV, stroke volume; HR = heart rate; bpm = beats per minute
Figure 1.
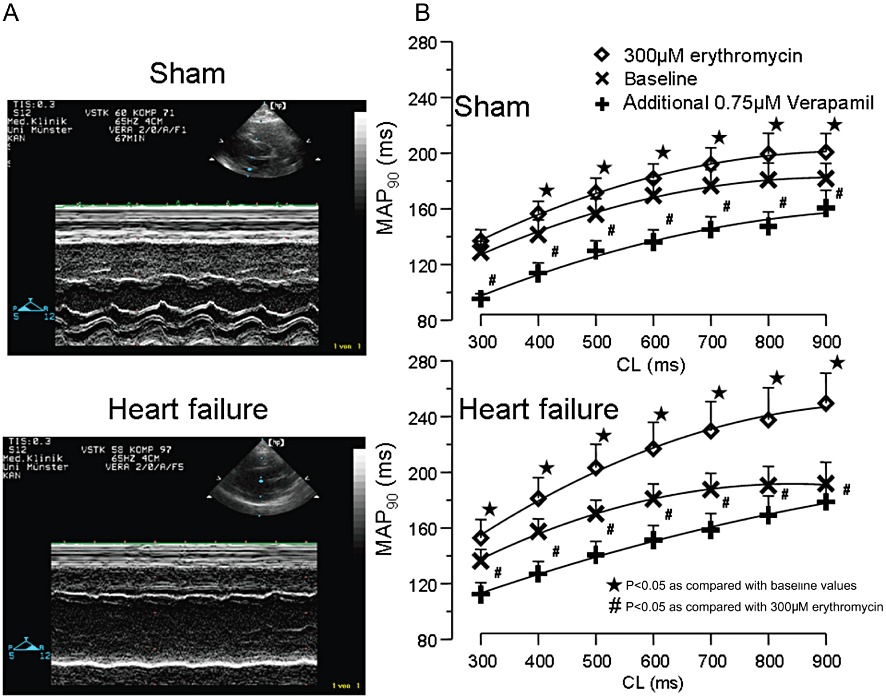
(A) Echocardiography before Langendorff experiment in sham hearts (top) and after 4 weeks of rapid ventricular pacing (bottom). (B) Cycle length (CL)-dependent effects on action potential duration (mean MAP90) under baseline conditions, after infusion of 300 µM erythromycin and after additional infusion of 0.75 µM verapamil. ⋆P < 0.05, significantly different from baseline values; #P < 0.05, significantly different from 300 µM erythromycin).
Effects of erythromycin on action potential duration and dispersion of repolarization in sham-operated and failing hearts
During electrophysiological examination, all electrocardiographic parameters reached equilibrium within 10 min. After this stabilization period, MAP recordings and pacing thresholds (mean threshold 1.7 ± 1.4 mA) remained highly reproducible throughout the experimental protocol. The MAP amplitude and the MAP duration during each protocol part did not change by more than 20% for the subsequent investigation period. Erythromycin (300 µM) led to an increase in MAP90 in sham and failing hearts. During exposure to 300 µM erythromycin, the prolongation in sham hearts ranged between 6% at a cycle length of 300 ms and 10% at a cycle length of 900 ms (P < 0.05 except for 300 ms; Figure 1B). This repolarization prolonging potential was more marked in failing hearts with a prolongation of 12% at a cycle length of 300 ms and 30% at a cycle length of 900 ms in the presence of IKr block, exhibiting pronounced reverse-use dependence in failing hearts (Figure 1B, P < 0.05, compared with sham hearts). The increase in action potential duration was paralleled by an increase in QT interval. Dispersion of repolarization under baseline conditions was identical in sham-operated and in failing hearts (Figure 2A). After administration of erythromycin, dispersion of repolarization increased in sham-operated hearts by 35% during 300 µM erythromycin infusion, whereas in failing hearts the increase was 72% (P < 0.05, Figure 2A). To assess regional differences, the mean values of action potentials recorded from the left ventricular epicardium were compared with the means of the right epicardial action potential recordings as well as the endocardial recordings. All hearts demonstrated a significant increase in left ventricular transmural dispersion of repolarization with a more marked increase in failing hearts (P < 0.01; Figure 2B) as compared with sham hearts. The significant increase of transmural dispersion of repolarization in failing hearts resulted from marked left endocardial MAP prolongation during treatment with erythromycin (Table 2).
Figure 2.
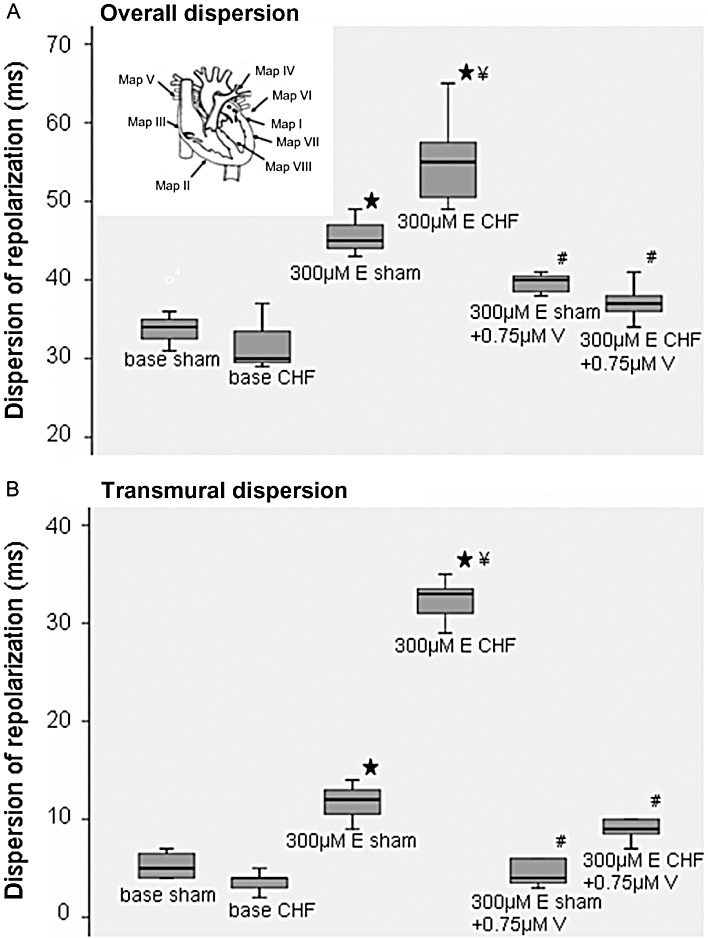
(A) Dispersion of repolarization after erythromycin (E) and additional verapamil (V) administration in sham and heart failure (CHF) hearts (⋆P < 0.05, significantly different from baseline; ¥P < 0.05, significantly different from erythromycin treated sham hearts) and significant decrease after additional infusion of verapamil (#P < 0.01, significantly different from erythromycin-treated hearts; left top, distribution of MAP catheters). (B) Transmural dispersion of repolarization after erythromycin and additional verapamil administration (⋆P < 0.05, significantly different from baseline; ¥P < 0.05, significantly different from erythromycin-treated sham hearts; #P < 0.01, significantly different from erythromycin-treated hearts).
Table 2.
Regional differences of MAP duration (MAP90; ms) after infusion of erythromycin (E) and after additional infusion of verapamil (V) in failing hearts
| CL (ms) | LV (MAP90; ms) | RV (MAP90; ms) | Endo (MAP90; ms) | LV difference (ms) | Endo difference (ms) |
|---|---|---|---|---|---|
| Baseline | |||||
| 900 | 189 ± 28 | 194 ± 21 | 191 ± 42 | ||
| 800 | 188 ± 26 | 193 ± 27 | 187 ± 28 | ||
| 700 | 184 ± 25 | 191 ± 21 | 186 ± 29 | ||
| 600 | 177 ± 25 | 183 ± 18 | 188 ± 29 | ||
| 500 | 168 ± 21 | 174 ± 13 | 168 ± 21 | ||
| 400 | 155 ± 15 | 162 ± 17 | 156 ± 14 | ||
| 300 | 134 ± 17 | 139 ± 11 | 135 ± 11 | ||
| 300 µM E | Prolongation to base | Prolongation to base | |||
| 900 | 244 ± 29* | 256 ± 27* | 283 ± 25* | +55 | +92¥ |
| 800 | 233 ± 26* | 245 ± 29* | 277 ± 31* | +46 | +90¥ |
| 700 | 225 ± 22* | 237 ± 28* | 267 ± 25* | +41 | +80¥ |
| 600 | 213 ± 26* | 225 ± 23* | 246 ± 28* | +35 | +58¥ |
| 500 | 198 ± 25* | 211 ± 25* | 230 ± 26* | +30 | +62¥ |
| 400 | 176 ± 28* | 190 ± 22* | 204 ± 23* | +21 | +48¥ |
| 300 | 149 ± 21* | 161 ± 20* | 168 ± 21* | +15 | +32¥ |
| 300 µM E + 0.75 µM V | Shortening to 300 µM E | Shortening to 300 µM E | |||
| 900 | 178 ± 21# | 177 ± 28# | 207 ± 27# | −66 | −76ж |
| 800 | 170 ± 29# | 166 ± 21# | 188 ± 25# | −64 | −89ж |
| 700 | 160 ± 22# | 156 ± 27# | 169 ± 23# | −65 | −98ж |
| 600 | 151 ± 24# | 150 ± 23# | 156 ± 11# | −61 | −89ж |
| 500 | 141 ± 19# | 140 ± 21# | 141 ± 18# | −57 | −89ж |
| 400 | 127 ± 15# | 126 ± 18# | 129 ± 16# | −49 | −75ж |
| 300 | 112 ± 15# | 114 ± 15# | 112 ± 14# | −37 | −56ж |
LV left ventricle; RV, right ventricle.
P < 0.05, significantly different from baseline;
P < 0.05, significantly different from erythromycin-treated hearts;
P < 0.05, significantly different from prolongation in LV;
P < 0.01, significantly different from shortening in LV.
Effects of verapamil on action potential duration and dispersion of repolarization in sham operated and failing hearts
When verapamil (0.75 µM) was administered in the presence of erythromycin, a significant decrease in QT interval and MAP duration was observed in sham hearts and in failing hearts (Figure 1B). Remarkably, verapamil led to a significantly greater decrease of MAP90 in endocardial cells, compared with left epicardial ventricle (Table 2), thereby reducing transmural dispersion of repolarization (Figure 2B). There was no difference between sham and failing hearts after administration of verapamil.
EAD and VT
Under baseline conditions, proarrhythmia was not observed, even after lowering of K+ concentration in perfusate to 1.5mM. In the presence of erythromycin, EADs and triggered activity were a frequent finding in sham operated and in failing hearts (Figure 3A). MAP recordings showed EADs after lowering of potassium concentration. In the majority of cases, EADs were followed by VT within a few seconds. Polymorphic VTs were always associated with EAD (P < 0.05, McNemar test). After 300 µM erythromycin, the number of single VT episodes in failing hearts was almost four times that in sham hearts (Figure 3A,B). Verapamil (0.75 µM) was anti-arrhythmic in sham and failing hearts after erythromycin, with only one sham heart showing five VT episodes and two hearts of the heart failure group showing altogether seven VT. Again, no difference between sham and failing hearts was observed after verapamil application. Figure 4 shows an experimental trace of action potential recordings, under baseline conditions, a typical episode of EAD and VT following erythromycin with low K+ perfusion and the action potential after adding verapamil.
Figure 3.
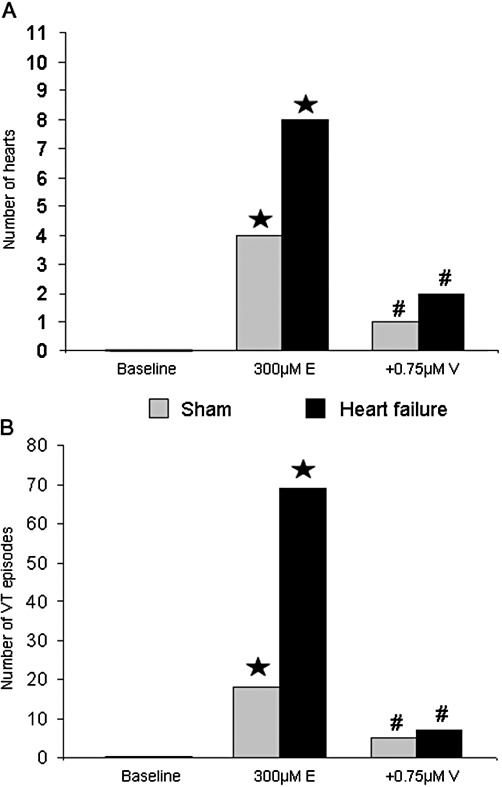
(A) VT incidence in sham and failing hearts after infusion of erythromycin (⋆P < 0.05, significantly different from baseline) and additional infusion of verapamil (#P < 0.05, significantly different from erythromycin treatment). (B) Number of single VT episodes in sham and failing hearts after infusion of erythromycin (⋆P < 0.05, significantly different from baseline) and additional infusion of verapamil (#P < 0.05, significantly different from erythromycin treatment).
Mathematical modelling
The model yielded results comparable to those obtained experimentally. IKr block (by erythromycin) together with hypokalaemia induced EAD in the mathematical model of single rabbit myocytes. Action potential duration was substantially increased in simulations of the failing heart and even more by IKr.blockade. As in the experiments, the block of ICa reduced the incidence of EAD. Mathematical modelling was able to elucidate another important factor: state-dependent block rather than a simple reduced conduction of ICa. Modelling the simple, state-independent block was not as effective in reducing the number of EAD as modelling a state-dependent block (as is observed with verapamil), even though both types of block showed similar effects without IKr block. Interestingly, the difference between the two types of block was apparent only when the action potentials became long enough – and especially with respect to the appearance of EAD. The model using voltage/state-dependent block showed greater inhibition of EAD because the simple reduction in conductance was not as effective in preventing ICa from re-opening and eliciting EAD (see Figure 5). However, modelling either type of ICa blockers showed a reduction of the duration of the action potentials and thereby showed lower numbers of EAD, than without block.
Figure 5.
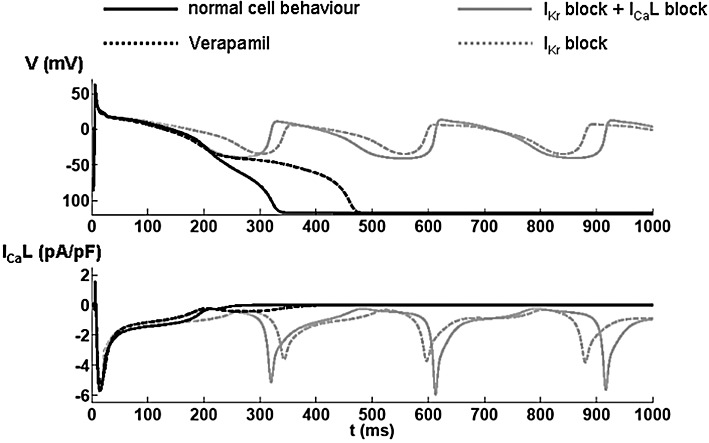
Results from a mathematical model of the electrophysiology of single cardiac myocytes. The upper panel shows the simulated membrane potential (V) and the lower panel shows the modelled Ca2+ current through the L-type Ca2+ channels (ICaL). Each trace represents the first action potentials after a switch to hypokalaemic ([K+] 1.5 mM) conditions in the model. The solid black line represents changes in membrane potential and ICa under normal conditions. In the presence of a substantial block of IKr, EADs appear when switching to hypokalaemic conditions, as shown by the dashed grey line. When adding a simple ICa pore blocker to the IKr block, the action potential repolarization is faster, but due to reactivation of the L-type Ca2+ channels, EADs are still elicited (grey solid line). However, assuming a state-dependent block (for verapamil) substantially inhibits the reactivation and therefore only leads to a prolongation of the action potential (black dashed line).
When simulating myocytes under heart failure conditions, the Ca2+ cycling was reduced, and it was easier to elicit EAD than in normal myocytes (see Figure 6). Nevertheless, application of either ICa blocking mechanisms reduced the number of EAD, and again, assuming a state/voltage-dependent block (as with verapamil) was more effective than a simple reduction in conductance.
Figure 6.
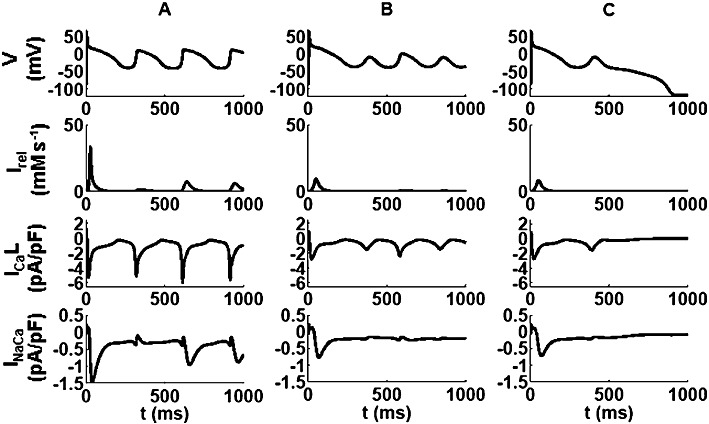
The panels in columns A, B and C show simulations of the effects of combining substantial IKr block together with ICa block on currents through the Na/Ca exchanger (INaCa), through L-type Ca2+ channels (ICaL) and the ryanodine release current (Irel), as well as the membrane potential (V) under conditions modelling (A) sham-operated hearts and (B and C) CHF hearts. Traces in A and B show results from a simple pore-blocker, whereas those in C denote the results for the state/voltage-dependent ICa block by verapamil. The reduction in Ca2+ cycling in the failing hearts is apparent as well as the benefit of verapamil state-dependent block over the other blocking type. Note the difference in the ICaL dynamics during the EAD between the three traces.
Discussion
The main finding of the present study is that ICa block by verapamil in hearts from rabbits with CHF, leads to suppression of EADs, which represent the causal trigger for the initiation of VT and to reduction of dispersion of repolarization, the required substrate for the maintenance of reentry circuits. Verapamil is known to exhibit an anti-arrhythmic profile inhibiting ventricular arrhythmias and EADs in humans without structural heart disease and in a number of experimental models. To our knowledge, this is the first experimental study in a whole heart model that showed the beneficial effect of verapamil was preserved in CHF.
Our experimental data disclosed a new anti-arrhythmic mechanism in heart failure, in which Ca2+ channel block leads to a reduction of dispersion especially at the transmural level, because of a marked effect on endocardial tissue. We suggest these electrophysiological effects are the result of preventing intracellular Ca2+ overload, leading to the inhibition of triggered arrhythmias and that such effects provide a novel therapeutic strategy in chronically failing hearts.
CHF and repolarization reserve
The mechanisms of arrhythmogenesis in failing hearts are more complex than in ischaemic heart disease (Eckardt et al., 2000). The development of proarrhythmia is an individual response to a repolarization prolonging drug depending on the ‘so-called’ repolarization reserve, first reported by Roden (1998). When multiple subclinical changes influence the repolarization process because of clinical risk factors such as CHF, superimposition of IKr blocking drugs may produce marked action potential prolongation, resulting in proarrhythmia (Roden, 2006). In the present study, we used erythromycin, which is known to induce proarrhythmia as a life-threatening side effect in patients (Shaffer, 2001) and in a preparation that reproducibly induced VT in rabbit isolated hearts (Milberg et al., 2002).
Although the extent of QT prolongation does not necessarily correlate with the degree of proarrhythmia (Milberg et al., 2004), it acts as an important condition for the generation of EAD and may be associated with increased dispersion of repolarization. Thus, to some degree, action potential prolongation in the failing heart may be regarded as a form of ‘acquired LQT syndrome’. This is underlined by the occurrence of polymorphic VT, resembling torsade de pointes in the present study, and in patients suffering from CHF (Middlekauff et al., 1995; Lehmann et al., 1996; Vecchia et al., 1999; Sweeney, 2001).
Effect of ICa block on EADs and transmural dispersion of repolarization
The preventative effect of verapamil on EAD and dispersion of repolarization in the present study supports the concept that Ca2+ influx via ICa channels plays not only a pivotal role in arrhythmogenesis in normal hearts (Milberg et al., 2005a) but also in the setting of heart failure. In particular, the reduction in transmural dispersion of repolarization mainly due to shortening of endocardial action potential prolongation in failing hearts has not been demonstrated before. With Our use of electrophysiological techniques in an experimental whole-heart model, where cell-to-cell coupling effects can be studied, allowed us to identify the causal trigger (EAD) and the underlying substrate (transmural dispersion) that was affected by ICa block in heart failure. The importance of EAD in CHF is underlined by the work of Pogwizd et al. (1998) in human failing hearts. They hypothesized that focal initiation of VT could be due to triggered activity that can easily be initiated in failing myocardium of dilated cardiomyopathy. They found that ventricular premature beats arose primarily in the subendocardium by a focal mechanism. EADs are the probable triggers for proarrhythmia in the presence of a substrate (dispersion of repolarization) appropriate for the initiation and perpetuation of proarrhythmia. Our mathematical model indicated that not all ICa blockers would show similar efficacy and the state-dependent block of verapamil was shown to be more effective in reducing EADs. Verapamil is known to suppress EADs in short segments of sheep and canine cardiac Purkinje fibres (January et al. (1988).
Shimizu et al. (1995) used MAP recordings in humans to support the hypothesis that polymorphic VT is maintained by a re-entrant mechanism due to increased dispersion of repolarization. We were able to show a more marked increase in overall dispersion of repolarization in CHF hearts, compared with sham hearts. This finding is in agreement with an increased QT dispersion in patients with heart failure (Bonnar et al., 1999), which has been linked to an increased risk of sudden cardiac death (Barr et al., 1994). In our study, transmural dispersion increased more in failing hearts than in sham hearts, due to a marked increase in endocardial APD. Previously, we found that transmural heterogeneities of cellular repolarization play a critical role in the genesis of torsade de pointes in a model of acquired long QT syndrome (Milberg et al., 2005b). The present data demonstrate that transmural heterogeneities are enhanced in heart failure and play a significant role in the mechanisms of proarrhythmia.
Akar and Rosenbaum (2003) demonstrated, in a model of pacing-induced heart failure, a more marked prolongation of action potential recordings in M cells compared with epicardial cells, leading to a heterogeneous action potential prolongation across the ventricular wall. The spatial distribution of repolarization leads to conduction block necessary for reentrant circuits. In our study, the area with longest repolarization was in endocardial and subendocardial cells. As recently reported (Milberg et al., 2005b), the endocardial MAP catheter in our experimental setup might record endocardial and M-cell potentials simultaneously due to a very thin endocardium tissue in rabbits, especially as these cells are often localized close to endocardial cells in the subendocardium or even penetrate to the endocardium (Yan et al., 1998). This finding is in agreement with experimental data of other groups that found a preferential response of M cells to the class III action of erythromycin (Antzelevitch et al., 1996). In addition, the importance of M cells for the pathogenesis of proarrhythmia was emphasized by results from a dog model of pacing induced heart failure, where midmyocardial ventricular cells exhibited an increased APD and more EADs than cells from controls, thereby underlying the development of VT in heart failure (Nuss et al. (1999).
Verapamil has been used in patients to suppress EAD and torsade de pointes in acquired and congenital long QT syndrome (Liao et al., 1996; Komiya et al., 2004). The important effect on EAD is supported by our mathematical model that showed a reduction of EAD in sham and failing hearts, particularly due to a state- or voltage-dependent block of verapamil. In addition, verapamil decreased the dispersion of repolarization and suppressed EADs in patients with congenital long QT syndrome (Shimizu et al. (1995). In the present study, verapamil shortened especially the endocardial APD, leading to the hypothesis of a heterogeneous distribution of Ca2+ channels across the ventricular wall and this finding is in agreement with the greater L-type Ca2+ current in endocardial than in epicardial isolated canine myocytes, reported by Wang and Cohen (2003).
Potential Ca2+-dependent anti-arrhythmic effects in heart failure
Hondeghem et al. (2001) and our own group (Milberg et al., 2002) have speculated on the role of action potential prolongation within the L-type Ca2+‘window’ voltage range, thus permitting Ca2+ channel reactivation as the basis for the generation of EAD and thereby VT. Prolongation of the action potential increases the amplitude of the [Ca2+]i transient, activating CaM kinase, thereby promoting arrhythmogenic EAD (Roden et al., 2002). Thus, a reasonable explanation for the anti-arrhythmic potential of verapamil might be that suppression of the L-type Ca2+ current (January and Riddle, 1989) leads not only to a reduction of transmural dispersion via shortening of endocardial MAP but also to suppression of EAD (Liao et al., 1996) by preventing intracellular Ca2+-overload. Verapamil produces potent Ca2+ channel block by acting as an intracellular pore blocker (Striessnig et al., 1990), thus directly interfering with Ca2+ movement through the pore.
Independent of the electrophysiological effects of direct inhibition of ICa, secondary alterations of Ca2+ handling may follow reduced Ca2+ uptake into cardiac myocytes. Spontaneous Ca2+ release from the sarcoplasmic reticulum (SR) may trigger a sudden activation of the Na+/Ca2+ exchanger (NCX). By extruding one Ca2+ ion from the cell in exchange for three Na+ ions, NCX is electrogenic, generating an inward electrical current (Pott et al., 2011). This again may re-depolarize the myocyte to a potential from which a new – premature – action potential or EAD is triggered (see Pogwizd and Bers 2004). By reducing Ca2+ uptake, verapamil may prevent SR overload, which is thought to cause spontaneous release (Kimura et al., 1984). As NCX expression and activity are increased in human failing hearts, this mechanism may be especially important in the clinical setting of heart failure (Hasenfuss et al., 1999). Additionally, reduced Ca2+ channel activity will reduce dyadic cleft Ca2+ concentration and thus Ca2+ concentration near the ryanodine receptor. Thus, verapamil may reduce the probability of Ca2+ binding to the ryanodine receptor and spontaneous ryanodine receptor opening with subsequent release from the SR.
Conclusion
Blockade of Ca2+ channels leads to a reduction of dispersion of repolarization, especially on the transmural level, following a particular effect on endocardial repolarization, resulting in a suppression of VT in an experimental model of CHF. Moreover, it leads to a suppression of EAD, confirmed by a mathematical model. The present data suggest an anti-arrhythmic effect of Ca2+ channel block in CHF. In addition, most CHF patients require polypharmacy, such as treatment with IKr blocking drugs and concomitant Ca2+ channel block may exert a beneficial effect on drug-induced reduction of myocardial repolarization reserve.
Limitations of the study
Of note, the beneficial effects of Ca2+ channel block reported here have to be seen in light of the limitations in its' clinical use in CHF. The applicability of Ca2+ channel block in the case of an advanced phase of systolic cardiac dysfunction may be limited due to its negative inotropic effects. Findings from large trials suggest that Ca2+ channel blockers may have detrimental effects upon survival and cardiovascular events in patients with severe heart failure (Elkayam et al., 1990; Goldstein et al., 1991). However, this may not necessarily be a ‘class’ effect as there is considerable heterogeneity in the chemical structure of individual agents (Mahe et al., 2003). First-generation Ca2+ channel antagonists such as nifedipine, verapamil or diltiazem have direct negative inotropic effects, but second-generation Ca2+ channel blockers (amlodipine, felodipine) have little or no negative inotropic activity at the usual therapeutic doses and may be better tolerated in patients with severe CHF (Packer et al., 1996). In fact, they appear to have a greater effect in vascular smooth muscle cells than in myocardial cells. However, they do block ICa (Furukawa et al., 1999), thus possibly representing a basis for further drug development. As regards the use of verapamil in heart failure, trials showed at least no increase in mortality rates, compared with placebo (Danish Verapamil Infarction Trial, 1990). In addition, in small trials in patients with CHF due to diastolic dysfunction (Hung et al., 2001) or only mild CHF (Wojnicz et al., 2010), verapamil had neutral clinical effects or even improved CHF. According to the present data, it might be reasonable to re-evaluate Ca2+ channel blockade at least in stable CHF patients.
Acknowledgments
This study was supported by the Dr Peter Osypka Foundation. Lars Eckardt holds the Peter Osypka Professorship of Experimental and Clinical Electrophysiology.
The work in Oxford is supported by the BHF (PG/08/019). Martin Fink is now with Modeling & Simulation at Novartis Pharma AG, Basel, Switzerland. CP was supported by a grant by the Innovative Medical Research Program of the Medical Faculty (IMF Po 12 06 07).
Glossary
- APD
action potential duration
- AV
atrioventricular
- CHF
chronic heart failure
- CL
cycle length
- EAD
early afterdepolarizations
- MAP
monophasic action potential
- NCX
Na+/Ca2+ exchanger
- SR
sacoplasmic reticulum
- VF
ventricular fibrillation
- VT
ventricular tachyarrhythmias
Conflict of interest
All authors declare no conflicts of interest.
References
- Akar FG, Rosenbaum DS. Transmural electrophysiological heterogeneities underlying arrhythmogenesis in heart failure. Circ Res. 2003;93:638–645. doi: 10.1161/01.RES.0000092248.59479.AE. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C, Sun ZQ, Zhang ZQ, Yan GX. Cellular and ionic mechanisms underlying erythromycin-induced long QT intervals and torsade de pointes. J Am Coll Cardiol. 1996;28:1836–1848. doi: 10.1016/S0735-1097(96)00377-4. [DOI] [PubMed] [Google Scholar]
- Barr CS, Naas A, Freeman M, Lang CC, Struthers AD. QT dispersion and sudden unexpected death in chronic heart failure. Lancet. 1994;343:327–329. doi: 10.1016/s0140-6736(94)91164-9. [DOI] [PubMed] [Google Scholar]
- Bonnar CE, Davie AP, Caruana L, Fenn L, Ogston SA, McMurray JJ, et al. QT dispersion in patients with chronic heart failure: beta blockers are associated with a reduction in QT dispersion. Heart. 1999;81:297–302. doi: 10.1136/hrt.81.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danish Verapamil Infarction Trial. Effect of verapamil on mortality and major events after acute myocardial infarction (the Danish Verapamil Infarction Trial II–DAVIT II) Am J Cardiol. 1990;66:779–785. doi: 10.1016/0002-9149(90)90351-z. [DOI] [PubMed] [Google Scholar]
- Eckardt L, Haverkamp W, Borggrefe M, Breithardt G. Experimental models of torsade de pointes. Cardiovasc Res. 1998a;39:178–193. doi: 10.1016/s0008-6363(98)00043-1. [DOI] [PubMed] [Google Scholar]
- Eckardt L, Haverkamp W, Mertens H, Johna R, Clague JR, Borggrefe M, et al. Drug-related torsades de pointes in the isolated rabbit heart: comparison of clofilium, d,l-sotalol, and erythromycin. J Cardiovasc Pharmacol. 1998b;32:425–434. doi: 10.1097/00005344-199809000-00013. [DOI] [PubMed] [Google Scholar]
- Eckardt L, Haverkamp W, Johna R, Bocker D, Deng MC, Breithardt G, et al. Arrhythmias in heart failure: current concepts of mechanisms and therapy. J Cardiovasc Electrophysiol. 2000;11:106–117. doi: 10.1111/j.1540-8167.2000.tb00746.x. [DOI] [PubMed] [Google Scholar]
- Elkayam U, Amin J, Mehra A, Vasquez J, Weber L, Rahimtoola SH. A prospective, randomized, double-blind, crossover study to compare the efficacy and safety of chronic nifedipine therapy with that of isosorbide dinitrate and their combination in the treatment of chronic congestive heart failure. Circulation. 1990;82:1954–1961. doi: 10.1161/01.cir.82.6.1954. [DOI] [PubMed] [Google Scholar]
- Fetsch T, Bauer P, Engberding R, Koch HP, Lukl J, Meinertz T, et al. Prevention of atrial fibrillation after cardioversion: results of the PAFAC trial. Eur Heart J. 2004;25:1385–1394. doi: 10.1016/j.ehj.2004.04.015. [DOI] [PubMed] [Google Scholar]
- Frommeyer G, Milberg P, Witte P, Stypmann J, Koopmann M, Lücke M, et al. A new mechanism potential of amiodarone in contrast to sotalol in a model of pacing-induced heart failure. Eur J Heart Fail. 2011;13:1060–1069. doi: 10.1093/eurjhf/hfr107. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Yamakawa T, Midera T, Sagawa T, Mori Y, Nukada T. Selectivities of dihydropyridine derivatives in blocking Ca(2+) channel subtypes expressed in Xenopus oocytes. J Pharmacol Exp Ther. 1999;291:464–473. [PubMed] [Google Scholar]
- Goldstein RE, Boccuzzi SJ, Cruess D, Nattel S. Diltiazem increases late-onset congestive heart failure in postinfarction patients with early reduction in ejection fraction. The Adverse Experience Committee; and the Multicenter Diltiazem Postinfarction Research Group. Circulation. 1991;83:52–60. doi: 10.1161/01.cir.83.1.52. [DOI] [PubMed] [Google Scholar]
- Hasenfuss G, Schillinger W, Lehnart SE, Preuss M, Pieske B, Maier LS, et al. Relationship between Na+-Ca2+-exchanger protein levels and diastolic function of failing human myocardium. Circulation. 1999;99:641–648. doi: 10.1161/01.cir.99.5.641. [DOI] [PubMed] [Google Scholar]
- Hondeghem LM, Carlsson L, Duker G. Instability and triangulation of the action potential predict serious proarrhythmia, but action potential duration prolongation is antiarrhythmic. Circulation. 2001;103:2004–2013. doi: 10.1161/01.cir.103.15.2004. [DOI] [PubMed] [Google Scholar]
- Hung MJ, Cherng WJ, Wang CH, Kuo LT. Effects of verapamil in normal elderly individuals with left ventricular diastolic dysfunction. Echocardiography. 2001;18:123–129. doi: 10.1046/j.1540-8175.2001.00123.x. [DOI] [PubMed] [Google Scholar]
- January CT, Riddle JM. Early afterdepolarizations: mechanism of induction and block. A role for L-type Ca2+ current. Circ Res. 1989;64:977–990. doi: 10.1161/01.res.64.5.977. [DOI] [PubMed] [Google Scholar]
- January CT, Riddle JM, Salata JJ. A model for early afterdepolarizations: induction with the Ca2+ channel agonist Bay K 8644. Circ Res. 1988;62:563–571. doi: 10.1161/01.res.62.3.563. [DOI] [PubMed] [Google Scholar]
- Kimura S, Cameron JS, Kozlovskis PL, Bassett AL, Myerburg RJ. Delayed afterdepolarizations and triggered activity induced in feline Purkinje fibers by alpha-adrenergic stimulation in the presence of elevated calcium levels. Circulation. 1984;70:1074–1082. doi: 10.1161/01.cir.70.6.1074. [DOI] [PubMed] [Google Scholar]
- Komiya N, Tanaka K, Doi Y, Fukae S, Nakao K, Isomoto S, et al. A patient with LQTS in whom verapamil administration and permanent pacemaker implantation were useful for preventing torsade de pointes. Pacing Clin Electrophysiol. 2004;27:123–124. doi: 10.1111/j.1540-8159.2004.00400.x. [DOI] [PubMed] [Google Scholar]
- Lehmann MH, Hardy S, Archibald D, quart B, MacNeil DJ. Sex difference in risk of torsade de pointes with d,l-sotalol. Circulation. 1996;94:2535–2541. doi: 10.1161/01.cir.94.10.2535. [DOI] [PubMed] [Google Scholar]
- Liao WB, Bullard MJ, Kuo CT, Hsiao CT, Chu PH, Chiang CW. Anticholinergic overdose induced torsade de pointes successfully treated with verapamil. Jpn Heart J. 1996;37:925–931. doi: 10.1536/ihj.37.925. [DOI] [PubMed] [Google Scholar]
- Lubinski A, Lewicka-Nowak E, Kempa M, Baczynska AM, Romanowska I, Swiatecka G. New insight into repolarization abnormalities in patients with congenital long QT syndrome: the increased transmural dispersion of repolarization. Pacing Clin Electrophysiol. 1998;21:172–175. doi: 10.1111/j.1540-8159.1998.tb01083.x. [DOI] [PubMed] [Google Scholar]
- Mahe I, Chassany O, Grenard AS, Caulin C, Bergmann JF. Defining the role of calcium channel antagonists in heart failure due to systolic dysfunction. Am J Cardiovasc Drugs. 2003;3:33–41. doi: 10.2165/00129784-200303010-00004. [DOI] [PubMed] [Google Scholar]
- Middlekauff HR, Stevenson WG, Saxon LA, Stevenson LW. Amiodarone and torsades de pointes in patients with advanced heart failure. Am J Cardiol. 1995;76:499–502. doi: 10.1016/s0002-9149(99)80138-6. [DOI] [PubMed] [Google Scholar]
- Milberg P, Eckardt L, Bruns HJ, Biertz J, Ramtin S, Reinsch N, et al. Divergent proarrhythmic potential of macrolide antibiotics despite similar QT prolongation: fast phase 3 repolarization prevents early afterdepolarizations and torsade de pointes. J Pharmacol Exp Ther. 2002;303:218–225. doi: 10.1124/jpet.102.037911. [DOI] [PubMed] [Google Scholar]
- Milberg P, Ramtin S, Monnig G, Osada N, Wasmer K, Breithardt G, et al. Comparison of the in vitro electrophysiologic and proarrhythmic effects of amiodarone and sotalol in a rabbit model of acute atrioventricular block. J Cardiovasc Pharmacol. 2004;44:278–286. doi: 10.1097/01.fjc.0000129581.81508.78. [DOI] [PubMed] [Google Scholar]
- Milberg P, Reinsch N, Osada N, Wasmer K, Monnig G, Stypmann J, et al. Verapamil prevents torsade de pointes by reduction of transmural dispersion of repolarization and suppression of early afterdepolarizations in an intact heart model of LQT3. Basic Res Cardiol. 2005a;100:365–371. doi: 10.1007/s00395-005-0533-8. [DOI] [PubMed] [Google Scholar]
- Milberg P, Reinsch N, Wasmer K, Monnig G, Stypmann J, Osada N, et al. Transmural dispersion of repolarization as a key factor of arrhythmogenicity in a novel intact heart model of LQT3. Cardiovasc Res. 2005b;65:397–404. doi: 10.1016/j.cardiores.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Nawrath H, Wegener JW. Kinetics and state-dependent effects of verapamil on cardiac L-type calcium channels. Naunyn Schmiedebergs Arch Pharmacol. 1997;355:79–86. doi: 10.1007/pl00004921. [DOI] [PubMed] [Google Scholar]
- Nuss HB, Kaab S, Kass DA, Tomaselli GF, Marban E. Cellular basis of ventricular arrhythmias and abnormal automaticity in heart failure. Am J Physiol. 1999;277:H80–H91. doi: 10.1152/ajpheart.1999.277.1.H80. [DOI] [PubMed] [Google Scholar]
- Packer M, O'Connor CM, Ghali JK, Pressler ML, Carson PE, Belkin RN, et al. Effect of amlodipine on morbidity and mortality in severe chronic heart failure. Prospective Randomized Amlodipine Survival Evaluation Study Group. N Engl J Med. 1996;335:1107–1114. doi: 10.1056/NEJM199610103351504. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM, Bers DM. Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc Med. 2004;14:61–66. doi: 10.1016/j.tcm.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM, McKenzie JP, Cain ME. Mechanisms underlying spontaneous and induced ventricular arrhythmias in patients with idiopathic dilated cardiomyopathy. Circulation. 1998;98:2404–2414. doi: 10.1161/01.cir.98.22.2404. [DOI] [PubMed] [Google Scholar]
- Pott C, Eckardt L, Goldhaber JI. Triple threat: the Na(+)/Ca(2+) exchanger in the pathophysiology of cardiac arrhythmia, ischemia and heart failure. Curr Drug Targets. 2011;12:737–747. doi: 10.2174/138945011795378559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden DM. Taking the ‘idio’ out of ‘idiosyncratic’: predicting torsades de pointes. Pacing Clin Electrophysiol. 1998;21:1029–1034. doi: 10.1111/j.1540-8159.1998.tb00148.x. [DOI] [PubMed] [Google Scholar]
- Roden DM. Long QT syndrome: reduced repolarization reserve and the genetic link. J Intern Med. 2006;259:59–69. doi: 10.1111/j.1365-2796.2005.01589.x. [DOI] [PubMed] [Google Scholar]
- Roden DM, Balser JR, George AL, Jr, Anderson ME. Cardiac ion channels. Annu Rev Physiol. 2002;64:431–475. doi: 10.1146/annurev.physiol.64.083101.145105. [DOI] [PubMed] [Google Scholar]
- Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N, et al. Heart disease and stroke statistics–2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117:e25–146. doi: 10.1161/CIRCULATIONAHA.107.187998. [DOI] [PubMed] [Google Scholar]
- Shaffer DN. 2001. Macrolide antibiotics and Torsade de Pointes. Macrolide and Quinolone TdP reports. Postmarketing Analysis FDA Report 2001.
- Shannon TR, Wang F, Puglisi J, Weber C, Bers DM. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys J. 2004;87:3351–3371. doi: 10.1529/biophysj.104.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu W, Ohe T, Kurita T, Kawade M, Arakaki Y, Aihara N, et al. Effects of verapamil and propranolol on early afterdepolarizations and ventricular arrhythmias induced by epinephrine in congenital long QT syndrome. J Am Coll Cardiol. 1995;26:1299–1309. doi: 10.1016/0735-1097(95)00313-4. [DOI] [PubMed] [Google Scholar]
- Striessnig J, Glossmann H, Catterall WA. Identification of a phenylalkylamine binding region within the alpha 1 subunit of skeletal muscle Ca2+ channels. Proc Natl Acad Sci U S A. 1990;87:9108–9112. doi: 10.1073/pnas.87.23.9108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stypmann J, Engelen MA, Breithardt AK, Milberg P, Rothenburger M, Breithardt OA, et al. Doppler echocardiography and tissue Doppler imaging in the healthy rabbit: differences of cardiac function during awake and anaesthetised examination. Int J Cardiol. 2007;115:164–170. doi: 10.1016/j.ijcard.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Sweeney MO. Sudden death in heart failure associated with reduced left ventricular function: substrates, mechanisms, and evidence-based management, Part I. Pacing Clin Electrophysiol. 2001;24:871–888. doi: 10.1046/j.1460-9592.2001.00871.x. [DOI] [PubMed] [Google Scholar]
- Vecchia L, Ometto R, Finocchi G, Vincenzi M. Torsade de pointes ventricular tachycardia during low dose intermittent dobutamine treatment in a patient with dilated cardiomyopathy and congestive heart failure. Pacing Clin Electrophysiol. 1999;22:397–399. doi: 10.1111/j.1540-8159.1999.tb00461.x. [DOI] [PubMed] [Google Scholar]
- Wang HS, Cohen IS. Calcium channel heterogeneity in canine left ventricular myocytes. J Physiol. 2003;547:825–833. doi: 10.1113/jphysiol.2002.035410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojnicz R, Nowak J, Lekston A, Wilczewski P, Nowalany-Kozielska E, Streb W, et al. Therapeutic window for calcium-channel blockers in the management of dilated cardiomyopathy: a prospective, two-centre study on non-advanced disease. Cardiology. 2010;117:148–154. doi: 10.1159/000320103. [DOI] [PubMed] [Google Scholar]
- Yan GX, Shimizu W, Antzelevitch C. Characteristics and distribution of M cells in arterially perfused canine left ventricular wedge preparations. Circulation. 1998;98:1921–1927. doi: 10.1161/01.cir.98.18.1921. [DOI] [PubMed] [Google Scholar]