

Abstract
Testicular testosterone produced during a critical perinatal period is thought to masculinize and defeminize the male brain from the inherent feminization program and induce male-typical behaviors in the adult. These actions of testosterone appear to be exerted not through its androgenic activity, but rather through its conversion by brain aromatase into estrogen, with the consequent activation of estrogen receptor (ER)-mediated signaling. Thus, the role of androgen receptor (AR) in perinatal brain masculinization underlying the expression of male-typical behaviors remains unclear because of the conversion of testosterone into estrogen in the brain. Here, we report a null AR mutation in mice generated by the Cre-loxP system. The AR-null mutation in males (ARL-/Y) resulted in the ablation of male-typical sexual and aggressive behaviors, whereas female AR-null homozygote (ARL-/L-) mice exhibited normal female sexual behaviors. Treatment with nonaromatizable androgen (5α-dihydrotestosterone, DHT) was ineffective in restoring the impaired male sexual behaviors, but it partially rescued impaired male aggressive behaviors in ARL-/Y mice. Impaired male-typical behaviors in ERα-/- mice were restored on DHT treatment. The role of AR function in brain masculinization at a limited perinatal stage was studied in ARL-/L- mice. Perinatal DHT treatment of females led to adult females sensitive to both 17β-estradiol and DHT in the induction of male-typical behaviors. However, this female brain masculinization was abolished by AR inactivation. Our results suggested that perinatal brain masculinization requires AR function and that expression of male-typical behaviors in adults is mediated by both AR-dependent and -independent androgen signaling.
It is thought that local production of estradiol, converted from testicular testosterone by brain aromatase, and the subsequent activation of estrogen receptor (ER)-mediated signaling is sufficient to induce brain masculinizaton in the male fetus and eventual expression of male typical behaviors in the adult (1–3). This hypothesis is supported by findings that mice deficient in either ER or aromatase display severely reduced male-typical behaviors (2–4). However, studies have indicated that when androgens are given to the female fetus, male-typical behaviors can be induced on further androgen treatment in adulthood (5, 6). Thus, although androgen receptor (AR)-mediated androgen actions are thought to be important in the induction of male-typical behaviors, the activity of locally converted estrogen from aromatizable androgens in the brain has prevented the assessment of the role of AR in perinatal brain masculinization and expression of male-typical behaviors.
Naturally occurring mutations in mammalian AR genes, located on the X chromosome and therefore present as a single copy in males, result in AR dysfunction that can lead to androgen-insensitive testicular feminization mutation (Tfm) (7, 8). Tfm is characterized by a variety of phenotypic abnormalities along with species-specific effects (8). However, Tfm mice appear to express a truncated AR protein (9). Most male animals with severe androgen insensitivity exhibit female-like external sexual organs and are infertile (8). This fact makes it impossible to generate female animals homozygous for AR deficiency and precludes the establishment of AR-null mutant (AR knockout, ARKO) lines either in nature or by conventional gene disruption techniques. Therefore, to define AR function we used the Cre-loxP system to allow the AR-null mutation (10) to be passed to both male and female offspring (11, 12). We report here that perinatal brain masculinization requires AR function and that expression of male-typical behaviors in adults is mediated by both receptor-dependent and -independent androgen signaling.
Materials and Methods
Generation of ARKO Mice. AR genomic clones were isolated from a TT2 embryonic stem cell genomic library by using human AR A/B domain cDNA as a probe. The targeting vector consisted of a 7.6-kb 5′ homologous region containing exon 1, a 1.3-kb 3′ homologous region, a single loxP site, and the neo cassette with two loxP sites (10). Two targeted clones (FB-18 and FC-61), identified by Southern analysis using probes A and B (Fig. 1b), were aggregated with single eight-cell embryos from CD-1 mice. Floxed AR mice (C57BL/6) were then crossed with the CMVCre transgenic mice. The two lines exhibited the same phenotypic abnormalities. The chromosomal sex of each pup was determined by genomic PCR amplification of the Sry gene on the Y chromosome.
Fig. 1.

Establishment of the AR-null mutant mouse line. (a) Diagram of the wild-type AR genomic locus (+), the floxed AR L3 allele (L3), and the AR allele (L-) obtained after Cre-mediated excision of exon 1. K, KpnI; E, EcoRI; H, HindIII; B, BamHI; LoxP sites are indicated as black arrowheads. The targeting vector consisted of a 7.6-kb 5′ homologous region containing exon 1, a 1.3-kb 3′ homologous region, a single loxP site, and the neo cassette with two loxP sites. (b) Southern blot analysis of targeted embryonic stem clones. Genomic DNA from WT TT2 embryonic stem cells (WT) and homologous targeted clones (FB-18 and FC-61) were digested with KpnI for hybridization with probe 1 or with EcoRI for hybridization with probe 2. (c) Strategy for the disruption of the floxed AR gene by using the CMV-Cre transgene. ARL3/Y mice were crossed with CMV-Cre(tg/0)/ARL-/+ mice to generate CMV-Cre(0/0)/ARL-/Y and CMVCre(tg/0)/ARL-/L- mice. (d) Detection of the Y chromosome-specific Sry gene in ARL-/Y mice by PCR of mouse-tail DNA. (e) Lack of AR transcripts in male (ARL-/Y) and female (ARL-/L-) AR-null mutant mouse brains as shown by Northern blot analysis with an AR E-domain cDNA probe. (f) Absence of AR protein in ARL-/Y and ARL-/L- mouse brains by Western blot analysis with a specific C-terminal antibody. (g) Female-like external genitalia in ARL-/Y mice (Upper). Atrophic testis without internal male and female reproductive organs in ARL-/Y mice. Ep, epididymis; Pr, prostate; Sv, seminal vesicle; Te, testis; Vd, vas deferens; Od, oviduct, Ov, ovary; Ut, uterus.
RNA Analysis. Total RNA was isolated by the improved acid-guanidine-phenol-chloroform method and poly(A)+ RNA purified with Dynabeads (Dynal, Oslo). Total and poly(A)+ RNA was separated in formaldehyde-containing agarose gels for Northern blot analysis as described (13, 14). An AR cDNA fragment (353 bp: 2368–2721) or neuronal nitric oxide synthase (nNOS) cDNA fragment (1,814 kbp: 182–1995) was used as a probe. RT-PCR analysis was conducted as described in Supporting Materials and Methods in Supporting Text, which is published as supporting information on the PNAS web site.
Protein Analysis. To detect AR protein expression (7), brain cell lysates were separated by SDS/PAGE and transferred onto nitrocellulose membranes. Membranes were probed with polyclonal AR antibodies (C-19 and N-20; Santa Cruz Biotechnology) and then peroxidase-conjugated second antibody. Blots were visualized by using an ECL detection kit (Amersham Bioscience). Immunohistochemistry was performed as described in Supporting Materials and Methods.
Animal Conditions for Behavior Tests. Gonadally intact experimental mice (8 weeks old) were housed singly under controlled light (light/dark 12:12, lights off at 1800 hours) and temperature (22–24°C) conditions throughout the tests. For adult hormone administration, a Silastic tube [10 mm length, i.d. = 0.062 in (1 in = 2.54 cm), Dow–Corning] containing methyltrienolone (R1881; 5 mg/20 μl) (15), or 60-day time-release pellets (Innovative Research of America) containing either 5α-dihydrotestosterone (DHT; 10 mg per pellet), 17β-estradiol (E2; 0.25 mg per pellet), or placebo (PLA) were implanted s.c. 2 weeks after gonadectomy under avertin anesthesia. All behavioral tests were conducted during the dark phase of the light/dark cycle, starting at 2 h after lights off.
Male Sexual Behavior Tests. Every week experimental mice underwent two 30-min sexual behavioral tests and then a 90-min test in their home cages (3, 4, 16). As stimuli, female mice (C57BL/6J) were ovariectomized and injected with estradiol benzoate (30 μg at 48 h and 15 μg at 24 h before tests) and progesterone (500 μg at 4 h before tests), with only females that showed lordosis in response to mounting by stud males subjected to experimentation. Sexual behaviors of each male were recorded for either 30 or 90 min as follows: number of mounts and intromissive and ejaculatory behaviors. The latency period (seconds) of the first male behavioral response was also measured.
Male Aggressive Behavior Test. Each mouse was tested three times in his home cage (as a resident) against an olfactory bulbectomized male (C57BL/6J) intruder mouse for 15 min (3, 4). For each experiment, percent of mice showing aggressive behaviors, total number of aggressive bouts, and latency (seconds) to first aggressive behavioral act by resident males were scored. An aggressive bout was defined as a consecutive series of behavioral acts separated by <3 sec, consisting of tail rattling, chasing, boxing, biting, offensive attacks, and wrestling.
Female Sexual Behavior Test. Gonadally intact or hormone-primed mice were introduced into a vigorous WT male's home cage (17). Lordosis in response to mount was defined as receptive posture with dorsoflexion of the back and elevation of the hindquarters. The lordosis quotient was calculated as the number of lordosis events per 10 mounts × 100. Observation continued for 20 min after introduction of the experimental animals, or until they received 10 mounts.
Perinatal DHT Treatment. Pregnant mice were implanted with 21-day time-release pellets (Innovative Research of America) containing DHT (35 mg per pellet) from days 14 to 19 of pregnancy. All offspring were injected with DHT (300 μg/30 μl) on days 0, 2, 4, 6, and 8 after birth. PLA and sesame oil were administered to control animals in the same schedule. These ARL-/L- and male and female WT mice were gonadectomized at 50 days of age, and behavioral tests were performed after hormone treatments as described above.
Statistical Analysis. Behavioral test data are shown as mean (±SE) and analyzed by using Student's t test or two-way ANOVA followed by post hoc comparison with the Fisher's protected least significant difference test. The nonparametric Kruskal–Wallis test and Mann–Whitney U test were used when variances were not equal among groups. Differences in incidences of behaviors were analyzed by the χ2 test or Fisher's exact probability test.
Results
Generation of AR-Deficient Mice by Using the Cre-loxP System. To circumvent male infertility due to AR inactivation, we introduced three loxP sites around exon 1, which encodes the transcription start site (Fig. 1a), to generate AR-floxed (flox/Y) mice (Fig. 1b). AR-floxed male (ARL3/Y) and female (ARL3/+) mice exhibited no overt abnormalities in terms of sexual behaviors, with normal reproduction frequencies and AR mRNA expression levels (Fig. 1e). AR-floxed mice were then crossed with CMV-Cre transgenic mice (tg/0) to generate female AR heterozygotes (ARL-/+) for further generation of male ARL-/Y and female ARL-/L- null mutant mice (Fig. 1c) as described (10). The genetic sex of male ARL-/Y and female ARL-/L- mice was confirmed by detection of the Y chromosome Sry gene (Fig. 1d). No AR transcripts in the brain were detected by Northern blotting (Fig. 1e), and RT-PCR using primer pairs designed to amplify exons encoding the AR ORF also failed to detect AR mRNA (data not shown). Immunoblotting experiments using N- and C-terminal antibodies for AR showed no AR protein expression in the brain and confirmed the inactivation of the AR gene (Fig. 1f).
Tfm Abnormalities in Male ARKO (ARL-/Y) Mice. From birth, both ARL-/Y and ARL-/L- mice were externally indistinguishable from normal female WT littermates (Fig. 1g) in terms of anogenital distance (ARL-/Y, 6.0 ± 0.6 mm; ARL-/L-, 5.8 ± 0.5 mm; AR+/Y, 16.2 ± 1.3 mm; and ARL3/+, 5.9 ± 0.4 mm) and growth curve up to 10 weeks (Fig. 5c, which is published as supporting information on the PNAS web site). ARL-/Y mice exhibited female-typical external appearance, including a vagina with a blind end, and a clitoral-like phallus instead of a penis and scrotum (Fig. 1g), similar to that observed in another mice line (18). Male reproductive organs, including seminal vesicles, vas deferens, epididymis, and prostate, were absent in ARL-/Y mice (Fig. 1g). Although no ovaries or uteri were observed, small inguinal testes were present. Histological examination of these testes showed that spermatogenesis was severely arrested (Fig. 5a). Testicular androgen levels were very low [testosterone, t(24) = 3.1, P < 0.01; and DHT, t(15) = 3.1, P < 0.01], whereas serum estrogen levels appeared normal in 10-week-old mutant mice (Fig. 5b). To our surprise, no phenotypic abnormalities in female reproductive organs were found in female ARL-/L- mice up to 10 weeks of age (H. Shiina, J. Miyamoto, T. Sato, T. Matsumoto, and S. Kato, unpublished results).
Impaired Male-Typical Behaviors in ARKO Male Mice. On matching with WT stimulus females (3, 4, 16), gonadally intact 10-week-old ARL-/Y mice that had been housed singly exhibited no male sexual behaviors (Fig. 2a), including mounts [χ2(1) = 17.0, P < 0.001], intromissions [χ2(1) = 17.0, P < 0.001], and ejaculation [χ2(1) = 17.0, P < 0.001], despite having normal levels of serum E2 (Fig. 5b) and normal levels of ER α and β protein expression in the hypothalamus (Fig. 2g). To restore the reduced serum androgen levels, gonadectomized ARL-/Y mice were given nonaromatizable androgen DHT. Although DHT treatment of WT male littermates enhanced the manifestation of male sexual behaviors [mounts and intromissions, χ2(1) = 8.2, P < 0.01, and ejaculation, χ2(1) = 7.5, P < 0.05], it was totally ineffective in ARL-/Y mice (Fig. 2a). Male aggressive behaviors (3, 4) were also severely impaired in ARL-/Y mice [χ2(1) = 10.8, P < 0.01] as measured by decreased numbers of aggressive bouts (F7,79 = 6.71, P < 0.01) with longer latency periods (F7,79 = 6.55, P < 0.01). Unexpectedly, treatment with either DHT or a nonaromatizable synthetic AR ligand (R1881) (15) enhanced, to at least some extent, the impaired male aggressive behaviors of ARL-/Y mice [DHT, χ2(1) = 4.0, P < 0.05, and R1881, χ2(1) = 5.5, P < 0.05; Fig. 2 b–d]. Our results suggested that both AR-independent and -dependent pathways mediated the effects of DHT on male aggressive behaviors.
Fig. 2.

Ablation of AR in male mice resulted in the lack of both male and female sexual behaviors. (a) Loss of all components of male sexual behavior in intact (gonads: +) 10-week-old ARL-/Y mice. E2 treatment of gonadectomized (gonads: -) ARL-/Y mice partially induced mounts and intromissions but not ejaculation. Results of 90-min sexual behavioral tests performed after 30-min tests performed twice every week for 2 weeks are shown and were basically identical with those of the 30-min tests (data not shown). *, P < 0.05; **, P < 0.01; ***, P < 0.001. (b) Percentage of intact (gonads: +) and gonadectomized (gonads: -) 10-week-old mice exhibiting aggressive behaviors (Incidence of male aggressive behavior) toward olfactory bulbectomized (OBX) male intruder mice. Three tests in total were carried out every week, and results of the third test are shown. *, P < 0.05; **, P < 0.01. (c) Number of bouts with attacks. **, P < 0.01; ***, P < 0.001. (d) Latency to the first attack during resident–intruder tests. Aggressive behavioral acts were markedly reduced in intact ARL-/Y mice and were partially recovered after DHT treatment. *, P < 0.05; ***, P < 0.001. (e) Absence of female sexual behaviors in intact and E2- and progesterone-primed gonadectomized 10-week-old ARL-/Y mice. Twenty days after implantation of E2 pellets, progesterone (P; 500 μg) was administered to ARL-/Y and WT mice 4 h before the behavioral test. Normal expression of female sexual behaviors was observed in intact 10-week-old ARL-/L- mice. (f) Lordosis was not induced in gonadectomized ARL-/Y mice after treatment with E2. (g) Absence of AR protein and no reduction in ERα and ERβ protein levels in the medial preoptic area of ARL-/Y mice analyzed by immunohistochemistry.
In agreement with previous findings (2, 19), E2 also potently induced male sexual behaviors in WT male mice [χ2(1) = 10.5, P < 0.01], whereas E2-treated ARL-/Y mice showed recovery of mounts and intromissions [χ2(1) = 5.2, P < 0.05] but not ejaculation. However, where behavioral recovery did occur, it was only to ≈50% of that observed in WT mice (Fig. 2a). The impaired aggressive behaviors of male ARKO mice were effectively restored by E2 treatment [χ2(1) = 7.9, P < 0.01; Fig. 2 b–d].
Absence of Female-Typical Behaviors in Male ARKO Mice. We then tested the female sexual behaviors of gonadally intact ARL-/Y mice by placing these mice into the home cages of WT males (17). ARL-/Y mice did not perform lordosis in response to WT male mounts (Fig. 2e). To further verify the inability of ARL-/Y mice to display lordosis, ovarian hormones were administered to stimulate female sexual behaviors. In WT females, the levels of lordosis gradually increased after administration of E2, whereas no lordosis was induced in ARL-/Y mice even when progesterone was given together with E2 (Fig. 2 e and f). In contrast, female intact ARL-/L- mice exhibited normal levels of lordosis, similar to that of WT females (Fig. 2e), and fertility (data not shown) but not male-typical behaviors.
Partial Recovery of Impaired Male-Typical Behaviors in ERα-/- Mice after DHT Treatment. As male-typical behaviors have been shown to be impaired in male ERα-/- mice (2, 4), but not in ERβ-/- mice (2, 17), we attempted to verify the role of AR in male-typical behaviors by testing the effects of DHT in male ERα-/- mice (20). In male WT mice, all three components of male sexual behaviors were enhanced after DHT treatment. In male ERα-/- mice, DHT treatment restored impaired mount and intromission behaviors [χ2(1) = 5.8, P < 0.05], but not ejaculation (Fig. 3a). Similarly, the impaired aggressive behaviors of male ERα-/- mice were partially recovered by DHT treatment as measured by increased incidences of aggression [χ2(1) = 6.2, P < 0.05] with shorter latencies (F3,26 = 5.41, P < 0.05; Fig. 3 b–d). Thus, male behavioral responses to DHT in null ERα-/- mice further confirmed the physiological importance of androgen signaling in male-typical behaviors.
Fig. 3.

Partial recovery of impaired male-typical behaviors in ERα-/- mice after DHT treatment. (a) Treatment with DHT, but not E2, restored the impaired mount and intromission but not ejaculation of gonadectomized 10-week-old male ERα-/- mice. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (b) Impaired male aggressive behaviors of male ERα-/- mice were restored by DHT treatment. *, P < 0.05. (c) Number of bouts with attacks. *, P < 0.05. (d) Latency to the first attack during resident–intruder tests. *, P < 0.05.
DHT-Induced Perinatal Brain Masculinization in Female Mice Was Abolished by AR Inactivation. Serum levels of testosterone in newborn ARL-/Y mice were still within the normal range compared with WT males (WT, 618.8 ± 233.1 pg/ml; and ARKO, 404.9 ± 171.7 pg/ml; Fig. 4f). However, we could not exclude the possibility that the lowered estrogen conversion due to impaired testosterone production from atrophic testis was insufficient to cause masculinization of the ARL-/Y fetal brain. Therefore, to precisely define the role of AR function in brain masculinization, we assessed AR-mediated androgen activity in the female fetus. Intact female brains physiologically undergo the innate feminization program due to the absence of sex hormone activity. It is well known that perinatal exposure of female rodents to androgens results in behavioral masculinization, observed as increased male-typical behavioral patterns after ovariectomy and further hormonal treatment in adulthood (5, 21). Only 30–33% of control WT females showed mount and intromissive patterns, but not ejaculatory patterns, even after either DHT or E2 were administered in adulthood (Fig. 4a). In contrast, perinatal DHT treatment (see Fig. 6, which is published as supporting information on the PNAS web site) induced mount and intromissive patterns in WT females on further postpubertural DHT [mount and intromissions, χ2(1) = 9.7, P < 0.01] or E2 [mount and intromissions, χ2 (1) = 10.5, P < 0.05] treatment (Fig. 4a). Perinatal DHT exposure enabled 2 of 10 WT females to exhibit ejaculatory patterns in response to postpubertural DHT treatment. Similarly, male aggressive behaviors were also induced in WT females exposed perinatally to DHT after either DHT [χ2(1) = 6.5, P < 0.05] or E2 [χ2(1) = 9.8, P < 0.01] treatment in adulthood (Fig. 4 b–d). However, such male-typical patterns were completely abolished in female ARKO (ARL-/L-) mice. Similar to control WT females, ARL-/L- mice exposed to perinatal DHT treatments exhibited normal female sexual behaviors in response to E2 and progesterone (Fig. 4e). Thus, it is likely that full and normal brain masculinization and defeminization require both androgen and estrogen signaling.
Fig. 4.

Failure of ARL-/L- mice to express male-typical behaviors after DHT-induced perinatal brain masculinization. (a) WT female mice, but not ARL-/L- mice, showed male sexual behaviors in response to either DHT or E2 treatment as adults after perinatal exposure to DHT. Behavioral tests were performed as described in Fig. 2a. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (b) Percentage of mice exhibiting aggressive behaviors (incidence of male aggressive behavior) toward olfactory bulbectomized (OBX) male intruder mice. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (c) Number of bouts with attacks. *, P < 0.05. (d) Latency to the first attack during resident–intruder tests. *, P < 0.05; **, P < 0.01. (e) Expression of female sexual behaviors is independent of AR/androgen signaling during the perinatal stage. Behavioral tests were performed after E2 and progesterone treatment (E2/P) as described in Fig. 2e. (f) No clear reduction in serum testosterone levels in ARL-/Y neonate mice (1 day after birth). (g) Reduced expression levels of nNOS transcript in ARL-/Y hypothalamus by Northern blot analysis. Densitometric analysis of the relative expression level by semiquantitative RT-PCR, expressed as fold WT after normalization to GAPDH. *, P < 0.05.
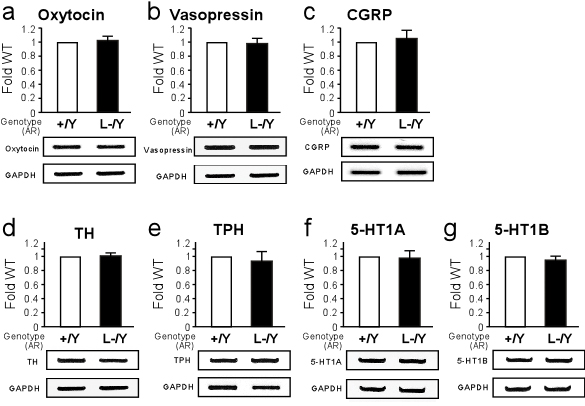
Alteration in Gene Expression of Neuronal Nitric Oxide Synthase by AR Inactivation. Finally, to gain insight into the molecular basis of impaired male-typical behaviors in male ARKO mice, we analyzed the expression of several genes known to be involved in the control of male-typical behaviors (as described in Supporting Results in Supporting Text). No significant alterations in neuropeptide (oxytocin, vasopressin, and calcitonin gene-related peptide) and monoaminergic-specific gene [tyrosine hydroxylase, tryptophan hydroxylase, and 5-hydroxytryptamine (5-HT) receptor 5-HT1A and 5-HT1B] mRNA levels were detected by semiquantitative RT-PCR analysis of ARL-/Y mouse hypothalamus (Fig. 7 a–g, which is published as supporting information on the PNAS web site). In contrast, a clear reduction in nNOS mRNA expression in the hypothalamus of ARL-/Y mice was observed by Northern blotting and semiquantitative RT-PCR (Fig. 4g). Thus, our results suggested the possible involvement of NO neurotransmission in male-typical behaviors regulated by AR.
Discussion
The present study revealed that AR gene inactivation in intact male mice caused the complete loss of male sexual behaviors and severely reduced male aggressive behaviors. Furthermore, ARL-/Y mice displayed significantly reduced male-typical behaviors regardless of sex steroid hormone treatment. Thus, these results confirmed that AR function is critical for male-typical behaviors. Nevertheless, it was still impossible to determine whether impaired male-typical behavior in ARL-/Y mice was due to the lack of AR function in adulthood or the failure of masculinization because of AR deficiency during the perinatal stage.
To address this issue, we used female AR-null mutant mice and found that DHT-induced brain masculinization at the perinatal stage was abolished in ARL-/L- mice. These data provide genetic evidence that liganded-AR function alone is sufficient for the sexual conversion of perinatal female brains. Moreover, as both E2- and DHT-induced male-typical behaviors in adult WT females treated perinatally with DHT, it is likely that once the brain is perinatally masculinized through activated AR function, the sexually developed brain becomes sensitive to both androgens and estrogens with regard to the expression of male-typical behaviors in adulthood. Thus, our results revealed that both brain masculinization during a limited perinatal period and expression of male-typical behaviors in adults require AR-mediated androgen signaling. Furthermore, the finding of significantly reduced nNOS mRNA levels after AR inactivation suggested that the nNOS gene may be one of the genes downstream of AR-mediated androgen signaling.
Male sexual behaviors were abolished in intact male ARKO mice, whereas treatment with E2, but not DHT, rescued male sexual behaviors without the induction of female sexual behaviors. This finding indicated that the brains of ARL-/Y mice were defeminized and partially masculinized. However, human patients with severe Tfm are usually socially adopted as females, irrespective of castration and E2 treatments. Together with the findings that prenatal treatment of female rhesus monkeys with DHT had clear and significant defeminizing and masculinizing effects on behavior (22), it appears that AR may serve a more critical role in male-typical behaviors than ER in higher mammals, including humans. Alternatively, because human sexuality is a complex behavioral trait formed through the combined function of multiple factors, the influence of social learning in humans might generate a species difference in terms of AR function in male sexual behaviors.
Examination of hormonal responses in ARL-/Y and ERα-/- mice suggested that androgen and estrogen receptor functions are convergent with regard to male sexual behaviors. However, as ejaculation was not restored in single-receptor knockout mice by any of the hormone treatments, both AR and ER function appears to be essential for normal male sexual behaviors. Along with mapping the sites of receptor activity in the brain and identification of target genes, evaluation of the critical developmental stages for AR function and its downstream factors is of particular interest and will be greatly assisted by the spatiotemporal control of receptor gene inactivation (10).
Unlike male-typical behaviors, the expression of female sexual behaviors was unaffected by AR inactivation. Furthermore, perinatal DHT exposure had no effect on female sexual behaviors in ARL-/L- or WT female mice, and no female sexual behaviors were induced in ARL-/Y mice in response to any hormone treatment. These findings suggested that the defeminization process is totally independent from masculinization in the brains of both sexes and that the function of liganded ER, but not AR, appears to be indispensable for brain defeminization.
In contrast to male sexual behaviors, male aggressive behaviors appeared to be mediated at least in part by AR-independent signaling and were distinct from ER-mediated estrogen signaling. Although the molecular basis for this receptor-independent DHT action in males is unclear at present, DHT may exert its effects through an as-yet-unknown cell-membrane receptor (23). This hypothesis is supported by recent reports of the identification of cell membrane receptors for progesterone (24). Another possibility is that a currently unidentified DHT metabolite may be responsible for activating a nuclear receptor (25, 26) involved in male aggressive behaviors. Thus, AR-null mutant mice will be useful in further investigating novel androgen-signaling pathways that are independent of AR.
Supplementary Material
Acknowledgments
We thank G. K Schütz and K. S. Korach for helpful discussions, S. Aizawa for TT2 embryonic stem cells, J. Miyazaki for CAG-tester mice, and H. Higuchi and R. Nakamura for manuscript preparation. This work was supported in part by funding from the Human Frontier Science Program (to P.C. and S.K.) and a Grant-in-Aid for Priority Areas from the Ministry of Education, Science, Sports and Culture of Japan (to S.K.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: AR, androgen receptor; ARKO, AR knockout; DHT, 5α-dihydrotestosterone; ER, estrogen receptor; Tfm, testicular feminization mutation; PLA, placebo; E2, 17β-estradiol; nNOS, NO synthase.
References
- 1.Couse, J. F. & Korach, K. S. (1999) Endocr. Rev. 20, 358-417. [DOI] [PubMed] [Google Scholar]
- 2.Ogawa, S., Chester, A. E., Hewitt, S. C., Walker, V. R., Gustafsson, J. A., Smithies, O., Korach, K. S. & Pfaff, D. W. (2000) Proc. Natl. Acad. Sci. USA 97, 14737-14741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsumoto, T., Honda, S. & Harada, N. (2003) Neuroendocrinology 77, 416-424. [DOI] [PubMed] [Google Scholar]
- 4.Ogawa, S., Lubahn, D. B., Korach, K. S. & Pfaff, D. W. (1997) Proc. Natl. Acad. Sci. USA 94, 1476-1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Plapinger, L. & McEwen, B. S. (1978) in Biological Determinants of Sexual Behaviour, ed. Hutchison, J. B. (Wiley, New York).
- 6.Olsen, K. L. (1985) in Neurobiology, eds. Gilles, R. & Balthazart, J. (Springer, Heidelberg), pp. xv, 149-164.
- 7.Takeyama, K., Ito, S., Yamamoto, A., Tanimoto, H., Furutani, T., Kanuka, H., Miura, M., Tabata, T. & Kato, S. (2002) Neuron 35, 855-864. [DOI] [PubMed] [Google Scholar]
- 8.Quigley, C. A., De Bellis, A., Marschke, K. B., el-Awady, M. K., Wilson, E. M. & French, F. S. (1995) Endocr. Rev. 16, 271-321. [DOI] [PubMed] [Google Scholar]
- 9.Gaspar, M. L., Meo, T., Bourgarel, P., Guenet, J. L. & Tosi, M. (1991) Proc. Natl. Acad. Sci. USA 88, 8606-8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li, M., Indra, A. K., Warot, X., Brocard, J., Messaddeq, N., Kato, S., Metzger, D. & Chambon, P. (2000) Nature 407, 633-636. [DOI] [PubMed] [Google Scholar]
- 11.Sato, T., Matsumoto, T., Yamada, T., Watanabe, T., Kawano, H. & Kato, S. (2003) Biochem. Biophys. Res. Commun. 300, 167-171. [DOI] [PubMed] [Google Scholar]
- 12.Kato, S. (2002) Clin. Pediatr. Endocrinol. 11, 1-7. [Google Scholar]
- 13.Ohtake, F., Takeyama, K., Matsumoto, T., Kitagawa, H., Yamamoto, Y., Nohara, K., Tohyama, C., Krust, A., Mimura, J., Chambon, P., et al. (2003) Nature 423, 545-550. [DOI] [PubMed] [Google Scholar]
- 14.Suzawa, M., Takada, I., Yanagisawa, J., Ohtake, F., Ogawa, S., Yamauchi, T., Kadowaki, T., Takeuchi, Y., Shibuya, H., Gotoh, Y., et al. (2003) Nat. Cell Biol. 5, 224-230. [DOI] [PubMed] [Google Scholar]
- 15.Cologer-Clifford, A., Simon, N. G., Richter, M. L., Smoluk, S. A. & Lu, S. (1999) Physiol. Behav. 65, 823-828. [DOI] [PubMed] [Google Scholar]
- 16.Matsumoto, T. & Yamanouchi, K. (2000) Neurosci. Lett. 291, 143-146. [DOI] [PubMed] [Google Scholar]
- 17.Ogawa, S., Chan, J., Chester, A. E., Gustafsson, J. A., Korach, K. S. & Pfaff, D. W. (1999) Proc. Natl. Acad. Sci. USA 96, 12887-12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeh, S., Tsai, M. Y., Xu, Q., Mu, X. M., Lardy, H., Huang, K. E., Lin, H., Yeh, S. D., Altuwaijri, S., Zhou, X., et al. (2002) Proc. Natl. Acad. Sci. USA 99, 13498-13503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meisel, R. L. & Sachs, B. D. (1994) in The Physiology of Reproduction, eds. Knobil, E. & Neill, J. D. (Raven, New York), pp. 3-105.
- 20.Dupont, S., Krust, A., Gansmuller, A., Dierich, A., Chambon, P. & Mark, M. (2000) Development (Cambridge, U.K.) 127, 4277-4291. [DOI] [PubMed] [Google Scholar]
- 21.Simon, N. G. & Whalen, R. E. (1987) Horm. Behav. 21, 493-500. [DOI] [PubMed] [Google Scholar]
- 22.Thornton, J. & Goy, R. W. (1986) Horm. Behav. 20, 129-147. [DOI] [PubMed] [Google Scholar]
- 23.Revelli, A., Massobrio, M. & Tesarik, J. (1998) Endocr. Rev. 19, 3-17. [DOI] [PubMed] [Google Scholar]
- 24.Zhu, Y., Rice, C. D., Pang, Y., Pace, M. & Thomas, P. (2003) Proc. Natl. Acad. Sci. USA 100, 2231-2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mangelsdorf, D. J., Thummel, C., Beato, M., Herrlich, P., Schutz, G., Umesono, K., Blumberg, B., Kastner, P., Mark, M., Chambon, P., et al. (1995) Cell 83, 835-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glass, C. K. & Rosenfeld, M. G. (2000) Genes Dev. 14, 121-141. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}