

Abstract
Hematopoietic myeloid progenitors released into the circulation are able to promote vascular remodeling through endothelium activation and injury. Endothelial injury is central to the development of pulmonary arterial hypertension (PAH), a proliferative vasculopathy of the pulmonary circulation, but the origin of vascular injury is unknown. In the present study, mice transplanted with BM-derived CD133+ progenitor cells from patients with PAH, but not from healthy controls, exhibited morbidity and/or death due to features of PAH: in situ thrombi and endothelial injury, angioproliferative remodeling, and right ventricular hypertrophy and failure. Myeloid progenitors from patients with heritable and/or idiopathic PAH all produced disease in xenografted mice. Analyses of hematopoietic transcription factors and colony formation revealed underlying abnormalities of progenitors that skewed differentiation toward the myeloid-erythroid lineage. The results of the present study suggest a causal role for hematopoietic stem cell abnormalities in vascular injury, right ventricular hypertrophy, and morbidity associated with PAH.
Introduction
Vascular endothelial injury with in situ thrombi is a typical pathologic finding in pulmonary arterial hypertension (PAH). Progressive disease is characterized by complex vascular malformations composed of disorganized proliferating monoclonal endothelial cells with neointima formation.1 Although endothelial injury is hypothesized to account for the origins of PAH, the underlying mechanism of the vascular injury is unknown. Hematopoietic myeloid proangiogenic progenitors play a central role in endothelial injury and repair. We and others have reported that distinct2–4 or indolent5 myeloid abnormalities are present in the BM of the majority, if not all, of patients with PAH and even in unaffected family members5 in familial cases of the disease. These findings and the unexplained high incidence of PAH among patients with myeloproliferative diseases6,7 suggest a myelopulmonary pathophysiologic link. In support of this concept, competent hematopoietic progenitors are required for disease development in the monocrotaline- or hypoxia-induced murine models of pulmonary hypertension, and BM transplantation can transfer disease to healthy naive mice.8 In contrast to developmental origins of blood cells and vascular endothelium from the common hemangioblast, hematopoietic stem cells in the adult do not differentiate into endothelium, but rather promote postnatal angiogenesis and homeostasis in a paracrine fashion.9–12 In the hierarchy of adult hematopoietic stem cell differentiation, a small pool of pluripotent, BM-resident stem cells exhibit self-renewal and long-term survival.13 These stem cells proliferate and differentiate into relatively short-lived multipotent progenitors that give rise to common lymphoid and common myeloid progenitors. Common lymphoid progenitors further differentiate into B- or T cell–committed precursors. The common myeloid progenitors proliferate and differentiate into bipotent common erythroid/megakaryocyte progenitors and into multipotent monocyte/granulocyte progenitors. These lineage-restricted myeloid progenitors differentiate into mature blood cells via unilineage-committed intermediate precursors. The hierarchy of proliferation and differentiation is at each bifurcation strictly regulated by lineage-specific transcription factors.13 In the BM, the cell-surface glycoprotein CD133 is highly expressed on immature progenitors, allowing CD133 expression to define the population of hematopoietic progenitors. Functional analysis of human BM-derived CD133+ cells indicates that this fraction is enriched in primitive multilineage hematopoietic stem cells.14 Early outgrowth proangiogenic progenitors or colony forming-unit-endothelial cells (CFU-ECs), which are enriched in myeloid progenitors,9 also express CD133.10 Intriguingly, CD133+ cells are detected consistently and in greater numbers in vascular lesions in PAH compared with control lung tissue15,16 and in endarterectomized tissue from patients with chronic thromboembolic pulmonary hypertension.17 Furthermore, PAH patients are characterized by higher than normal levels of circulating subsets of the BM-derived CD133+ progenitors.5,10,18,19 However, mobilization and recruitment of BM progenitors also occurs in response to vascular injury as part of the repair process. Therefore, it is unclear whether the BM-derived progenitor cells are mobilized in response to vascular injury to participate in vascular repair or if the cells are part of the underlying pathogenesis of vascular injury. We hypothesized that CD133+ BM stem cells promote vascular injury and tested the hypothesis in the present study by transplanting BM from PAH patients and healthy controls to mice using a xenograft model.
Methods
Study population
Patients with idiopathic PAH (Class 1.1) or a familial form of PAH (Class 1.2) were enrolled in the study. Healthy, nonsmoking volunteers with no known disease and receiving no medications were recruited as controls. BM aspirates were collected and processed within 6 hours of collection. Germline BMPR2 and Caveolin-1 mutations were analyzed as described previously.10,20 Clinical information on all PAH patients, including pulmonary artery pressures from right heart catheterization, were available from medical history and records. The study was approved by the institutional review board of the Cleveland Clinic, and written informed consent was obtained from all individuals in accordance with the Declaration of Helsinki.
Hematopoietic transcription factor expression analysis
RNA was extracted using the RNeasy kit (QIAGEN), then labeled and hybridized to Illumina HT-12 arrays. Data were analyzed using Genespring GX software (Agilent Technologies). For real-time PCR validation, 500 ng of RNA was reverse transcribed using random primers and Superscript III (Invitrogen). Quantitative PCR was performed in triplicate reactions using QuantiTect SYBR Green mastermix (QIAGEN) and results were normalized to GAPDH.
NOD-SCID engraftment
Mononuclear cells were obtained from BM aspirates by centrifugation on Ficoll-Paque Plus gradient (Amersham Biosciences) and CD133 cells were purified by double MACS separation for CD133 using MACS CD133 isolation kit (Miltenyi Biotec). Purity of sorted cells was checked by flow cytometry after staining with PE–antibiotin Abs (Miltenyi Biotec) and was > 95%. Female immunodeficient NOD-SCID mice (The Jackson Laboratory) were injected with approximately 400 × 103 sorted CD133 cells after whole-body irradiation (325cGy) using a cesium source of radioactivity. After 6 weeks or when animals became morbid (hunched posture, ruffled fur, and hypoactive for > 48 hours), mice were bled and organs were collected for analysis. BM cells were isolated and used for flow cytometry after RBC lysis. The occurrence of adverse events was judged by independent veterinary technicians blinded to study groups. Animals were housed in pathogen-free conditions using micro-isolator cages. All animal experiments were performed in accordance with applicable regulations after approval of the institutional animal care and use committee of the Cleveland Clinic.
Flow cytometry
Engraftment and differentiation of CD133+ cells into NOD-SCID BM were analyzed using anti–human CD45-PE, CD117-PE, CD19-PE, CD33-PE, and CD71-FITC (eBiosciences) and CD34-FITC and CD41a-FITC (BD Biosciences) Abs. BM cells were isolated from NOD-SCID vertebrae and hind legs. RBCs were lysed using ammonium chloride lysis solution. Cells were preincubated with Fc block to eliminate nonspecific Ab binding, which was followed by staining for human CD45, CD34, CD133, CD41a, CD33, and CD14. At least 100 000 events were acquired on a FACScan flow cytometer and stored as list mode files. Mononuclear cells were gated in a lymphoblastoid region on forward/side scatter dot plots and percentages of human cells in this gate were determined on histograms. Naive, nonengrafted NOD-SCID mice BM cells were stained as negative controls for gating.
Hematopoietic colony-forming cell assay
In vitro hematopoietic colony formation by sorted CD133+ cells was analyzed using complete human colony-forming cell Methocult medium (StemCell Technologies) according to the manufacturer's instructions. Colonies were scored at day 14.
VWF ELISA
Plasma levels of VWF were determined by ELISA. 96-well ELISA plates (BD Biosciences) were coated overnight at 4°C with 15 μg/mL of polyclonal anti-VWF (DAKO) in PBS. Before use, plates were washed with 0.1% Tween 20 in PBS. Plasma from naive C57Bl/6 mice were used as a standard and defined as 1 unit of VWF antigen/mL. Wells were blocked with 3% BSA in PBS, followed by incubation with standards and samples (100 μL/well). Peroxidase-conjugated rabbit–anti-VWF (DAKO) was used as the detection Ab. Each incubation step was for 1 hour at 37°C, followed by 4 washing steps in 0.1% Tween 20 in PBS. Reactivity was visualized by 3,3′,5,5′ tetramethylbenzidine (100 μL/well; Sigma-Aldrich) incubation for 10 minutes in the dark and the reaction was stopped by adding 1N HCl. VWF levels were determined on an ELISA plate reader (Molecular Devices) at a 450-nm wavelength.
Immunohistochemistry
Organs were fixed in 10% formalin and BM samples were decalcified in decalcifying solution (Thermo Scientific) before processing and paraffin embedding. Then, 5-μm sections were stained with H&E, anti–human β2 microglobulin, VWF (DAKO); α smooth muscle-actin (Abcam); and anti–human MHC-I (Epitomics). For VWF, antigen retrieval was performed by pretreatment with proteinase K. Immunoreactivity was detected by diaminobenzidine and hydrogen peroxidase using an automated biotin-avidin peroxidase system (Ventana-320-ES; Ventana Medical Systems). Secondary Ab alone was used as a negative control. Nitrotyrosine was detected using rabbit anti–nitrotyrosine polyclonal Abs (Millipore). Staining was performed on a Benchmark automated immunostainer (Ventana Medical Systems) using a Ventana iVIEW DAB Detection kit minus the provided secondary. Instead, vector goat anti–rabbit biotinylated secondary Ab (Vector Laboratories) was used. Control studies with excess free 3-nitrotyrosine demonstrated nitrotyrosine staining of tissues was specific. All immunostained sections were counterstained with hematoxylin. Slides were masked and then analyzed by a pathologist. Microvessel density was quantified as described previously.21
Protein-bound nitrotyrosine and chlorotyrosine analyses
Protein-bound oxidized amino acids were quantified by stable isotope dilution liquid chromatography–tandem mass spectrometry on a triple quadrupole mass spectrometer (API 4000; Applied Biosystems) interfaced to an Aria LX Series HPLC multiplexing system (Cohesive Technologies) as described previously.22 Briefly, after protein precipitation and desalting/delipidation, known amounts of synthetic [13C6]ring-labeled standards of 3-chlorotyrosine and 3-nitrotyrosine were added to samples and used as internal standards for quantification of natural abundance protein-bound analytes. Simultaneously, known amounts of universal labeled precursor amino acid [13C9,15N1]tyrosine was also added to monitor for possible artifactual formation of nitrotyrosine during sample processing. Proteins were hydrolyzed under argon atmosphere in methane sulfonic acid and hydrolysates passed over mini solid-phase C18 extraction columns (Supelclean LC-C18-SPE mini column; 3 mL; Supelco) before mass spectrometry analysis. Results are reported as the content of the precursor amino acid tyrosine, which was monitored within the same injection. Intrapreparative formation of both 3-nitro[13C9,15N1]tyrosine and 3-chloro[13C9,15N1]tyrosine was routinely monitored and negligible (ie, < 5% of the level of the natural abundance product observed) under the conditions used.
Cardiac myocyte stereologic and planimetric analysis
Formalin-fixed and paraffin-embedded histologic sections of mouse right and left ventricular tissue were stained with fluorescein-labeled wheat germ agglutinin to identify myocyte cell membranes (1:500; Vector Laboratories), tetramethyl rhodamine iso-thiocyanate-labeled lectin from Bandeiraea simplicifolia to identify vascular structures (1:100; Sigma-Aldrich), and DAPI for nuclear identification. The identity of the slides was masked. A total of 12-15 images at 40× magnification were randomly captured for each sample. Each of the 3 component colors of the images were individually thresholded using Metamorph Version 6.3 software (MDS Analytical Technologies) to demarcate structures of interest (ie, myocytes, vessels, and nuclei). Stereologic analysis was performed by projecting the thresholded images onto a grid of 216 points and counting the number of points intersecting with each tissue component. Planimetric analysis of the myocyte cross-sectional area was performed by determining the cross-sectional area of all myocytes within the image, requiring the length-to-breadth ratio for each be ≤ 2 to ensure that transverse cross-sections were close to a circular profile.
Mycoplasma PCR and plasma endotoxin levels
Mycoplasma was evaluated using the VenorGeMR PCB-based Mycoplasma detection kit (Sigma-Aldrich). Plasma endotoxin levels were detected using the Charles River Endosafe Endotoxin Portable Test System consisting of a hand-held spectrophotometer and a cartridge with limulus amebocyte lysate (LAL), control standard endotoxin (CSE), and synthetic substrate for benchtop chromogenic analysis, all dried into a single-use cartridge. Each lot of cartridges had a precalculated control standard endotoxin reaction time curve already programmed into the system. Once the lot number of the cartridge being used was entered into the portable test system, the reader calculated the result based on the preprogrammed standard curve reaction times.
Statistical analysis
The JMP Version 5.1 software program was used for data analysis. The Student t test or ANOVA were used for comparisons of parametric data and the Wilcoxon test was used for comparison of nonparametric data, as appropriate. The Kaplan-Meier method was used for event-free survival analysis. P < .05 was considered significant. Results are expressed as means ± SEM.
Results
Study population
BM was obtained from 13 patients with PAH, including 2 idiopathic PAH and 11 heritable PAH patients from 4 families (age 44 ± 4 years, mean pulmonary artery pressure 64 ± 2 mmHg, 10 female and 3 male, all white; Table 1) and from 6 unrelated healthy control volunteers (age 43 ± 5 years, 4 female and 2 male, 1 Asian, 2 black, and 3 white). Subjects had normal circulating blood cell numbers as described previously.5 The presence of germline bone morphogenetic protein receptor 2 mutation, which is most often present in cases of familial PAH, and germline caveolin-1 mutation were determined as described previously.10,20 Caveolin-1 is a scaffolding tumor suppressor gene that modulates nitric oxide synthesis in endothelial cells. Mice with genetic deletion of caveolin-1 develop pulmonary hypertension, particularly when exposed to hypoxia, but mutations in human PAH have only been identified recently.20 Altogether, 19 BM transplantations were performed to create humanized NOD-SCID mice (The Jackson Laboratory). BM cells were also evaluated ex vivo for their capacity for hematopoietic colony formation and expression of hematopoietic transcription factors.
Table 1.
Population of PAH patients
| Variable | Dana point classification | Age, y | Sex | Race | Mutation |
|---|---|---|---|---|---|
| Idiopathic PAH | |||||
| 1 | 1.1 | 61 | Female | White | None identified |
| 2 | 1.1 | 54 | Female | White | None identified |
| Familial PAH | |||||
| Family 1 | 1.2.1 | 38 | Female | White | BMPR2 c.2580delT |
| Family 1 | 1.2.1 | 40 | Male | White | BMPR2 c.2580delT |
| Family 2 | 1.2.3 | 54 | Female | White | None identified |
| Family 2 | 1.2.3 | 28 | Female | White | None identified |
| Family 2 | 1.2.3 | 53 | Female | White | None identified |
| Family 3 | 1.2.1 | 55 | Female | White | BMPR2 Exon-3 deletion |
| Family 6 | 1.2 | 22 | Male | White | Caveolin-1 |
| Family 6 | 1.2 | 43 | Female | White | Caveolin-1 |
| Family 6 | 1.2 | 68 | Male | White | Caveolin-1 |
| Family 6 | 1.2 | 45 | Female | White | Caveolin-1 |
| Family 7 | 1.2.1 | 40 | Female | White | BMPR2 C.189_209del21 |
PAH CD133+ cells are enriched in myeloid colony-forming cells
MACS-purified CD133+ cells were seeded on semisolid hematopoietic colony-forming medium to identify differentiation potential to various lineages. Typical colonies were scored after 14 days. PAH BM CD133+ cells gave rise to more colonies (Table 2). Consistent with previous studies, there were low numbers of unilineage colonies of burst-forming unit–erythroid or CFU-macrophage in the CD133 fractions14 and no significant differences between controls and PAH were found. However, there were significantly higher numbers of multilineage colonies of CFU-granulocyte, erythrocyte, monocyte, megakaryocyte and CFU-granulocyte, monocyte from CD133+ PAH patients compared with healthy controls (Table 2). This indicated that there were more multipotent progenitors and greater myeloid commitment among CD133+ BM stem cells in PAH patients.
Table 2.
Hematopoietic colony formation by CD133+ cells
| Variable | Control | PAH | P |
|---|---|---|---|
| BFU-E | 7.6 ± 2.4 | 16.3 ± 7.2 | NS* |
| CFU-M | 2.0 ± 0.6 | 2.8 ± 1.1 | NS |
| CFU-GM | 25.3 ± 6.7 | 112.3 ± 23.3 | .03 |
| CFU-GEMM | 0.9 ± 1.1 | 6 ± 2.48 | .02 |
| Total CFU | 35.7 ± 8.9 | 137.3 ± 33.7 | .01 |
BFU-E indicates burst-forming unit-erythroid; CFU-M, CFU-macrophage; CFU-GM, CFU-granulocyte, monocyte; CFU-GEMM, CFU-granulocyte, erythrocyte, monocyte, and megakaryocyte.
P > .05.
Increased expression of myeloid-erythroid specific transcription factors in PAH BM
Studies in cell lines have shown that lineage-restricting transcription factor expression and antagonism tightly regulate hematopoietic progenitor cell differentiation.13 Although the presence of GATA-1, and potentially other hypoxia-relevant genes, may influence lineage differentiation, data are lacking for effects in primary cells. Therefore, functional evidence of lineage-skewed differentiation of the human primary stem cells was provided by colony assays. Expression of myeloid transcription factors in PAH was quantified by gene-array analysis (Table 3) and confirmed by real-time PCR (Table 4). GATA1, the common erythroid-megakaryocyte lineage-determining transcription factor, and the erythroid-specific transcription factor EKLF were significantly higher in PAH BM. Increased GATA1/PU.1 ratio in PAH revealed that the common myeloid progenitor in PAH is skewed toward erythroid-megakaryocyte commitment. The greater EKLF/Fli ratio in PAH patients showed further downstream skewed differentiation into erythroid cells. No differences were observed among the PAH subgroups. These data demonstrate higher myeloid-erythroid lineage commitment of hematopoietic progenitor cells in PAH. No differences were observed among the PAH subgroups; however, compared with subjects with BMPR2 mutations, those with caveolin-1 mutations had a significant increase in all the myeloid transcription factors (all P < .05).
Table 3.
Expression of myeloid transcription factors in PAH BM (gene array)
| Transcription factor | Control | PAH | P |
|---|---|---|---|
| GATA1 (erythroid-megakaryocytic) | −0.53 ± 0.23 | 0.34 ± 0.22 | .02 |
| PU.1 (granulocytic-monocytic) | 0.13 ± 0.14 | −0.14 ± 0.12 | NS |
| EKLF (erythroid) | −73 ± 0.30 | 0.37 ± 0.90 | .009 |
| Fli (megakaryocytic) | 0.37 ± 0.09 | −0.32 ± 0.22 | .01 |
| GATA1/PU.1 ratio | 0.14 ± 0.03 | 0.33 ± 0.06 | .01 |
| EKLF/Fli ratio | −2.36 ± 0.70 | 1.05 ± 0.90 | .009 |
Table 4.
Myeloid transcription factors in PAH BM (real-time PCR)
| Transcription factor | Control | PAH | P |
|---|---|---|---|
| GATA1 (erythroid-megakaryocytic) | 0.54 ± 0.08 | 2.32 ± 0.63 | .006 |
| PU.1 (granulocytic-monocytic) | 1.03 ± 0.06 | 1.94 ± 0.30 | NS |
| EKLF (erythroid) | 0.52 ± 0.09 | 3.3 ± 1.1 | .003 |
| Fli (megakaryocytic) | 1.3 ± 0.08 | 5.38 ± 2.1 | NS |
| GATA1/PU.1 ratio | 0.54 ± 0.09 | 1.04 ± 0.14 | .03 |
| EKLF/Fli ratio | 0.41 ± 0.09 | 0.71 ± 0.07 | .02 |
PAH CD133+ progenitors have greater NOD-SCID engraftment
NOD-SCID engraftment was performed to study the in vivo differentiation of PAH progenitors. Total body–irradiated mice were intravenously injected with the purified CD133+ cells from control or PAH BM. NOD-SCID BM engraftment was analyzed by flow cytometry and immunohistochemistry. Recipients of PAH CD133+ cells had a significantly higher percentage of human CD45+ cells in their BM by flow cytometric analyses (Figure 1A), and this was validated by visualization of cells using human-specific MHC-I immunostaining (Figure 1B). Consistent with greater engraftment, human CD133+ progenitors were present at higher levels in the circulation of PAH progenitor–engrafted mice compared with control progenitor–engrafted animals (Figure 1C). CD34, which is expressed on human hematopoietic and vascular progenitors, was also detectable on progenitor cells; the CD34+CD133+ progenitor subset was detected at higher levels in the BM of mice xenografted with PAH samples compared with mice xenografted with control samples (Figure 1C). Among the PAH subgroups, progenitor cell subpopulations were the highest in the mutation-negative group (CD34+CD133+ progenitors: mutation-negative 5.9% ± 1.2%; BMPR2 mutation, 0.56% ± 1.0%; caveolin-1 mutation, 2.4% + 1%; P = .03 by ANOVA; CD34+ progenitors: mutation-negative, 21.2% ± 3.9%; BMPR2 mutation, 5.8% + 3.4%; caveolin-1 mutation, 6.7% ± 3.4%; P = .03 by ANOVA; CD133+ progenitors: mutation-negative, 8.5% + 2%; BMPR2 mutation, 1.5% ± 1.7%; caveolin-1 mutation, 4.9% ± 1.7%; P = .08 by ANOVA). Analysis of hematopoietic lineages showed similar B-cell (CD19) and megakaryopoietic (CD41a) lineages (not shown), but greater differentiation to myelocytic (CD33) and erythroid (CD71) cells in recipients of PAH cells (Figure 1D). There was no difference in the expression of these lineage antigens among the different PAH subgroups (all P > .05).
Figure 1.
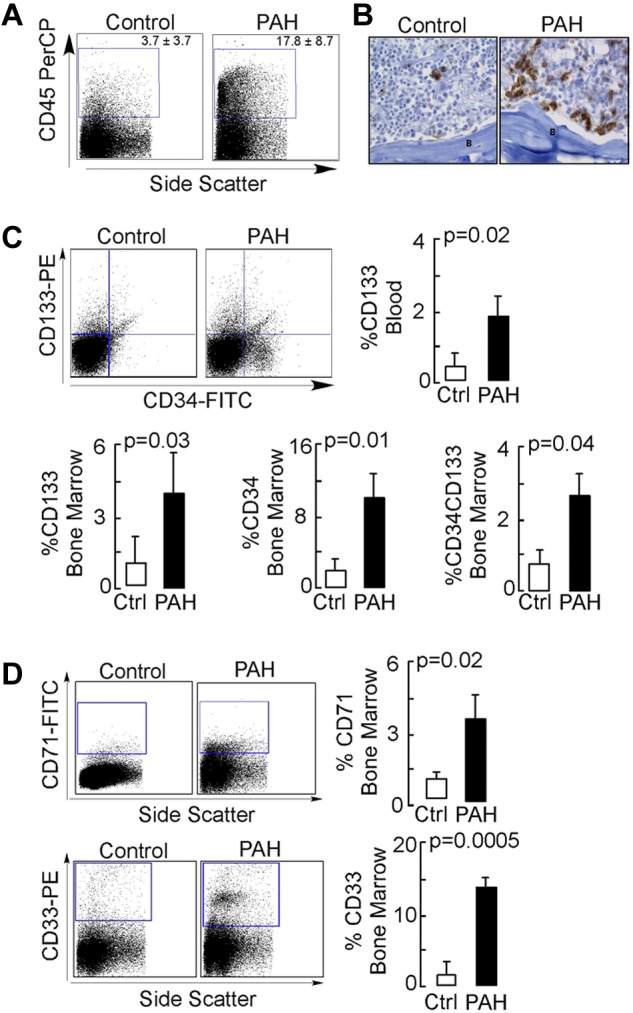
NOD-SCID engraftment potential of CD133+ cells. Purified human BM CD133+ cells were engrafted into NOD-SCID mice by IV injection. After 6 weeks or when animals showed signs of adverse events, BM cells were isolated from NOD-SCID mice and analyzed for the presence of human cells. (A) Flow cytometric analysis for human CD45 expression. (B) Immunohistochemical staining of NOD-SCID BM for human-specific MHC-1 Abs. B indicates bone. Scale bar indicates 100 μm. (C-D) Flow cytometric analysis for human progenitor cell subsets (C) and human hematopoietic lineage engraftment (D).
PAH CD133+ xenograft leads to lung endothelial injury and widespread in situ thrombosis in NOD-SCID mice
Histologic examination of organs revealed widespread vascular injury in 9 of the 13 mice that were engrafted with the PAH progenitors and in none of the 6 mice receiving progenitors from the healthy control group (P = .001 by χ2 test; no significant differences among PAH subgroups, P = .23). Numerous thrombi were present in large and small vessels of the lungs (Figure 2), with pathologic evidence of endothelial cell damage (Figure 2F,H) and organized thrombi with recanalization (Figure 2I-J). The presence of thrombi is consistent with remodeling events in human PAH lungs (Figure 2D-E). Thrombi were also prominent in the ventricle with resultant ischemia and infarction (Figure 2L-M). Six of the PAH-engrafted mice among all PAH subgroups had massive disseminated in situ thrombi formation, mainly in the lungs, but also in other organs such as heart and kidney (Figure 2N). Pulmonary vascular smooth muscle cell hypertrophy, analyzed by planimetric analysis of transverse cross-sectional blood vessels, was observed in some animals (Figure 2O-R), but significance was not reached in a group comparison between control and PAH recipients. Among the PAH subgroups, however, smooth muscle cell hypertrophy was significantly higher in the caveolin-1 mutation group (media thickness/external vessel diameter: mutation-negative, 0.10 ± 0.01; BMPR2 mutation, 0.16 ± 0.01; caveolin-1 mutation, 0.20 ± 0.01, P < .001 by ANOVA). In analogy to the human situation, right ventricular hypertrophy assessed histologically as myocyte volume relative to tissue was increased significantly in PAH recipient mice (Figure 3A-C), which identified pathologic right ventricular hypertrophy and was equally distributed among PAH subgroups. Myocyte/tissue ratio of the left ventricle and septum were similar among PAH and control engrafted mice. Lung, heart, and liver microvessel density of engrafted mice was analyzed to assess angiogenic capacity of the engrafted CD133+ cells. The microvessel density of heart and liver was similar among groups, however, lungs of mice that received the PAH progenitors had more than 2-fold higher numbers of vessels than control engrafted mice (Figure 3D-E).
Figure 2.

Vascular remodeling induced by CD133+ cells. Organs were harvested from CD133+-engrafted mice and fixed in formalin. A pathologist blinded to study groups analyzed paraffin-embedded tissue sections after specific stainings. (A) H&E staining of lungs from a control CD133+-engrafted mouse. (B-C) H&E staining of lungs from a PAH CD133+-engrafted mouse (B); arrows indicate in situ thrombi and recent thrombi (C). (D-E) Human PAH lung. Low- and high-power images of occlusion of tufts of capillaries in plexiform lesion by recent thrombi (arrows). (F-H) Lung endothelial cell damage in PAH CD133+-engrafted mice, H&E and α smooth muscle staining, respectively. (G) High-power field of the boxed region in panel D. (I-J) Lung PAH CD133+-engrafted mouse, organized thrombus in large vessel with recanalization (Masson trichrome). (J) High power of the boxed region in panel I. (K) H&E staining of heart from control CD133+-engrafted mouse. (L-M) H&E staining of heart from PAH CD133+-engrafted mouse, thrombi in the right ventricle with infarction and ischemia. (N) H&E staining of kidney from PAH CD133+-engrafted mouse. Arrows indicate in situ thrombi. (O-R) Lung α smooth muscle actin staining, blood vessels with muscle indicated by arrows. Scale bars indicate 100 μm.
Figure 3.

Right heart hypertrophy, angiogenic remodeling, and morbidity induced by CD133+ cells. (A-B) Right ventricular hypertrophy in human PAH and in mice engrafted with human CD133 cells, respectively. Germ agglutinin staining was performed to visualize the myocyte wall. Scale bar indicates 100 μm. (C) Planimetric analysis of myocyte cross-sectional area of right ventricles per total tissue area. Germ agglutinin and lectin stainings were performed to visualize myocyte wall and blood vessels, respectively. (D) Immunohistochemistry of VWF-positive vessels in lungs of mice engrafted with control or PAH CD133+ cells. Scale bar indicates 200 μm. (E) Quantification of microvessel density of lungs, heart, and liver. *P = .001 compared with control. (F) Plasma levels of VWF quantified by ELISA are higher in mice receiving PAH cells. (G) Immunohistochemistry of nitrotyrosine identified increased oxidative stress in the pulmonary vascular wall among PAH recipients. In the control engrafted mice, gray arrows indicate weak positivity in airway epithelial cells, which normally produce nitric oxide and reactive nitrogen species via expression of nitric oxide synthases,50 and black arrowheads indicate negative immunoreactivity of healthy vascular endothelium. In the PAH-engrafted mice, black arrowheads identify nitrotyrosine-positive vascular cells. Scale bar indicates 100 μm. (H-I) Mass spectrometric quantification of nitrotyrosine and chlorotyrosine in the lungs. (J) Morbidity of CD133+-engrafted mice. NOD-SCID mice were engrafted by IV injection with sorted CD133+ cells after total body irradiation. Animals were monitored for 6 weeks. Morbid events in each group are shown.
Endothelial cell injury was also assessed by measuring plasma VWF, which is rapidly released from endothelial cells after any perturbation. As expected, irradiation led to endothelial injury, as evidenced by the significant increase of plasma levels of VWF after irradiation compared with naive mice (plasma VWF in naive mice, 0.32 ± 0.03 u/mL; 6 hours after irradiation, 0.55 ± 0.08 u/mL, P = .03). The plasma VWF was significantly higher in the PAH recipient mice (Figure 3F) but was not different among PAH subgroups. There was a correlation between lung microvessel density and plasma VWF (R2 = 0.73, P = .003). Lung microvessel density was also correlated with the numbers of PAH CD133+ stem cells engrafted to the NOD-SCID BM (R2 = 0.71, P = .005), as well as the numbers of CD34+CD133+ cells in the BM (R2 = 0.54, P = .02) and circulating CD133+ cells (R2 = 0.55, P = .05).
Endothelial dysfunction is fundamental to the pathogenesis of PAH.1,23 Reactive oxygen species, generated by reactive species produced by activated endothelium and leukocytes, are central to pathogenic mechanisms of vascular diseases.22,24 Myeloperoxidase (MPO), a heme peroxidase stored within the azurophilic granules of leukocytes including neutrophils and monocytes, is a primary enzyme source of leukocyte-generated oxidants and is a participant in vascular injury.24 MPO uses H2O2 as a cosubstrate to oxidize chloride or nitrite, resulting in the formation of halogenating or nitrating oxidants that can react with protein tyrosine residues to produce nitrotyrosine and chlorotyrosine, the latter being a specific marker of MPO-catalyzed oxidation.24 Mass spectrometry quantification of protein bound chlorotyrosine and/or nitrotyrosine has been used as an indicator of chlorinating and reactive nitrogen species formed by activated leukocytes during vasculopathic processes.24 MPO can lead to endothelial dysfunction in part because of consumption of nitrite and depletion of NO.25,26 MPO released by leukocytes infiltrating the vascular wall or blood-derived MPO released by circulating leukocytes can bind to and remain active in the vascular wall for long periods of time, even after leukocytes are no longer present and/or activated.27 Moreover, posttranslational modification of proteins can be long-lived adducts, making detection of MPO-catalyzed oxidation biomarkers such as chlorotyrosine and nitrotyrosine a sensitive and specific way to identify sites of leukocyte activation and potential participation in vascular injury. In the present study, xenograft of PAH BM stem cells led to circulating human leukocytes and the development of endothelial dysfunction evidenced by widespread in situ thromboses, although infiltrating human leukocytes were not appreciable on pathologic sections. Therefore, nitrotyrosine and chlorotyrosine were investigated to determine whether reactive oxidant species formed by activated leukocytes contribute to the molecular mechanisms of the endothelial injury. Recipients of PAH CD133+ stem cells had pulmonary vascular nitrotyrosine positivity on immunostaining, which was in contrast to mice xenografted with healthy control cells (Figure 3G). Quantification of protein nitrotyrosine and chlorotyrosine by mass spectrometric analysis revealed higher levels in the PAH group compared with the mice receiving control BM (Figure 3H-I; nitrotyrosine/tyrosine: healthy control recipients, 17 ± 4 mmol/mol; PAH recipients, 106 ± 27 mmol/mol, P = .04; chlorotyrosine/tyrosine: healthy control recipients, 82 ± 18 mmol/mol; PAH recipients, 168 ± 35 mmol/mol, P = .007). The presence of chlorotyrosine confirmed MPO, and thus leukocytes, as a source of reactive oxidant species formation within the vessel wall in PAH patients.
Engraftment of PAH CD133+ cells leads to morbidity of NOD-SCID mice
In the PAH-engrafted group, 8 of 13 mice developed adverse events (hunched posture, ruffled fur, and hypoactive for > 48 hours) and were euthanized before the 6-week end point of the study. In contrast, none of the 6 healthy control progenitor recipients showed signs of adverse events. Event-free survival was significantly lower for PAH recipient mice (Figure 3I). Among the PAH subgroups, recipients of cells with mutation-negative or with caveolin-1 mutations had the greatest morbidity. Mycoplasma infection as determined by PCR and plasma endotoxin levels were undetectable, indicating that adverse events were not likely to be due to microorganism infection of the immunodeficient mice. To ascertain that morbidity was related to CD133+ cells, some mice were engrafted with PAH BM CD133− cells (n = 3) or injected with 100 μL of PAH BM plasma (n = 3). All of these mice survived with no adverse events over the 6 weeks.
Discussion
Human BM-derived CD133+ stem cells support vascular health, but can also participate in pathologic vascular remodeling in some model systems.28,29 In the present study, transplantation of PAH BM-derived CD133+ cells to immunodeficient mice caused morbidity due to vascular injury, thromboses, and right ventricular hypertrophy. These findings establish a causal role of hematopoietic stem cells in PAH (Figure 4).
Figure 4.

Right ventricular hypertrophy, lung endothelial cell injury, and in situ thrombi are common pathologic characteristics of patients with PAH. Increased numbers of BM-derived CD133+ stem cells are found in PAH pulmonary artery wall, but the lineage differentiation of these cells and whether they contribute to the vascular remodeling or play a reparative role are unknown. Increased expression of the myeloid transcription factor GATA-1 is found in PAH CD133+ hematopoietic stem cells (HSC) and common myeloid progenitors (CMP), which skew differentiation of PAH CD133+ cells into megakaryocyte-erythroid progenitors (MEP). Subsequently, EKLF expression in MEPs induces erythroid commitment, resulting in higher numbers of myeloerythroid progenitors in PAH BM. Engraftment of PAH CD133+ HSCs into NOD-SCID mice confirms the increased myeloerythroid proliferation. In analogy to human PAH, xenografted mice develop lung endothelial cell injury, in situ thrombi, and right ventricular hypertrophy. The data reveal a contributory role of BM CD133+ myeloid-erythroid progenitors in PAH. Illustration is reprinted with permission of the Cleveland Clinic Center for Medical Art & Photography (copyright 2012; all rights reserved).
The in situ thromboses and high levels of VWF suggest that the myeloid cells caused endothelial injury. A multimetric glycoprotein stored in the Weibel-Palade bodies of endothelial cells and, in smaller amounts, in the α granules of platelets, VWF is released from storage granules on endothelial perturbation into the circulation. Therefore, VWF is an important indicator of endothelial injury and is a mediator of thrombus formation via initiation of platelet adhesion, aggregation, and activation of the coagulation cascade.30 The endothelial injury findings in the xenograft model parallel findings in patients. Plasma levels of VWF are increased PAH patients and serve as an independent prognostic factor for reduced patient survival.31,32 However, in contrast to findings of CD133+ cells in vascular lesions in human PAH lungs,15–17 lungs of CD133-engrafted animals had rare or no incorporated human cells, suggesting that PAH CD133+ cells had paracrine effects in this immunodeficient model. The presence and enrichment of protein-bound chlorotyrosine, a specific oxidation product formed by MPO,24 identify activation of myeloid cells in the mechanisms of the vascular injury. However, the subset of cells causing the injury, the progenitors or their progeny, remains to be determined. Likewise, although myeloid cells are implicated by detection of MPO oxidation products, it is currently unknown whether other progenitor-derived molecules or organelles may be transferred, for example, by microvesicles, active transport, or cell fusion.
In animal models of PAH, thrombosis and endothelial cell injury are consistent findings.33 Endothelial cell alterations and endothelial cell injury are currently accepted as one of the underlying mechanisms of PAH.34 However, it is still unclear whether the pulmonary vascular disease arises from underlying myeloproliferative disease or if the pulmonary vascular disease leads to a myeloproliferative BM that fuels endothelial injury and the proliferative angiopathy. In support of the latter notion, BM precursors acquire an aberrant angiogenic phenotype in parallel to development of PAH in mouse models of hypoxia-induced pulmonary hypertension.8,35 Conversely, hypoxia-inducible factor-1α signal transduction in the pulmonary vascular endothelium is a recognized molecular mechanism in PAH,36 and a recent study showed increased levels of hypoxia-inducible myeloid proliferative factors in the circulation of patients with PAH, some of which are produced by the pulmonary vascular endothelium.5 Hypoxia-inducible factors instruct the myeloid-erythroid differentiation of hematopoietic stem cells37 via induction of transcription factor GATA-1.38 Therefore, high levels of myeloid proliferative factors in PAH may select for specific lineages of myeloid progenitors, which then become predominant in the BM. There are limitations to the xenograft model. Inflammation is an important component of vascular remodeling,1,34,39,40 and thus the effects of xenografted PAH CD133+ cells may be blunted in the immunocompromised mouse model. However, under immunocompetent conditions, interactions between the hematopoietic progenitors and inflammatory cells are more likely to cause more profound vascular injury.41 Irradiation, as used in this xenograft model to allow transplantation and engraftment of human cells, can cause injury of pulmonary endothelial cells.42 In 1 case, lung irradiation in rats resulted in disruption of the endothelium in the pulmonary vasculature, followed by vascular remodeling and PAH.43 Interestingly, this effect was not seen in mice receiving control BM cells. The pulmonary vasculature is the first capillary bed that the CD133+ cells encounter after IV injection. Others have reported that hematopoietic stem cells are trapped in the lungs after injection44 and/or that BM-derived proangiogenic cells home specifically to the lungs after IV administration.45 In this context, the control stem cells may have been beneficial to the mice in the present study, whereas the PAH CD133+ cells may have exacerbated endothelial injury caused by irradiation. Chemotherapy- or radiotherapy-induced lung injury is a major complication associated with BM hematopoietic stem cell transplantation.46 More than 1 in 10 patients develop a life-threatening idiopathic pneumonia syndrome related to widespread lung endothelial cell injury. In rare cases, the patients develop PAH.47 Our present data show that healthy control CD133+ progenitors are able to restore the endothelial injury. In contrast, PAH CD133+ progenitors aggravated injury to the lung endothelium. Monocrotaline, an alkaloid used in animals to induce experimental pulmonary hypertension, is an endothelial toxin. In this model, surviving rats eventually die of widespread thromboses and renal failure.48 Indeed, thromboses are an essential part of the natural history of PAH, as evidenced by the first pathologic descriptions of lung tissue from PAH patients obtained at autopsy.49 Therefore, nearly all PAH patients benefit from anticoagulant therapy, which prolongs lifespan. The results of the present study indicate that targeting BM-derived CD133+ cells may also reduce, and perhaps even reverse, the ongoing endothelial injury and thromboses that lead to the progressive pulmonary remodeling and ultimately heart failure and death.
Acknowledgments
The authors thank M. Baaklini, M. Cleggett-Mattox, M. Koo, and J. Sharp for patient recruitment; J. Baran-Smiley, A. Janocha, J. Hanson, L. Mavrakis, and R. Steinle for technical assistance; C. Burns in Central Cell Services for endotoxin testing; L. Vargo, J. Drazba, and A.J. Peterson in the Digital Imaging Core; C. Shemo and S. O'Bryant in the Flow Cytometry Core for technical advice and assistance; A. Vasanji in the Biomedical Imaging & Analysis Core for assistance with α-smooth muscle actin quantification; K. Litwak and the Biologic Resources Unit for assistance with animal experiments; and Chiara Federici for designing the primers for the real-time PCR and cDNA synthesis.
This work was supported by the National Institutes of Health (grants HL60917, P01 HL103453, P01HL076491, and M01 RR018390); the American Thoracic Society/Pulmonary Association Research (grant PH-07-003); and the Hematopoietic Stem Cell Core Facility of the Case Comprehensive Cancer Center (P30 CA43703). Figure 4 illustration is by David Schumick, BS, CMI and is reprinted with the permission of the Cleveland Clinic Center for Medical Art & Photography (copyright 2012; all rights reserved). K.A. is a scholar of the International Society for Advancement of Cytometry.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: K.A. conducted the study, designed and performed the research, analyzed and interpreted the data, and wrote the manuscript; S.F., A.L., and D.G. conducted the study and recruited the subjects; B.G., M.A., and R.T. performed the research, analyzed and interpreted the data, and wrote the manuscript; S.L.H. performed the research, analyzed and interpreted the data, and reviewed the manuscript; J.L. performed the research and reviewed the manuscript; and S.C.E. designed the research, analyzed the data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Kewal Asosingh, MS, PhD, or Serpil Erzurum, MD, Lerner Research Institute, Cleveland Clinic, Department of Pathobiology, NC22, 9500 Euclid Ave, OH 44195; e-mail: asosink@ccf.org or erzurus@ccf.org.
References
- 1.Tuder RM, Chacon M, Alger L, et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J Pathol. 2001;195(3):367–374. doi: 10.1002/path.953. [DOI] [PubMed] [Google Scholar]
- 2.Popat U, Frost A, Liu E, et al. New onset of myelofibrosis in association with pulmonary arterial hypertension. Ann Intern Med. 2005;143(6):466–467. doi: 10.7326/0003-4819-143-6-200509200-00017. [DOI] [PubMed] [Google Scholar]
- 3.Popat U, Frost A, Liu E, et al. High levels of circulating CD34 cells, dacrocytes, clonal hematopoiesis, and JAK2 mutation differentiate myelofibrosis with myeloid metaplasia from secondary myelofibrosis associated with pulmonary hypertension. Blood. 2006;107(9):3486–3488. doi: 10.1182/blood-2005-08-3319. [DOI] [PubMed] [Google Scholar]
- 4.Zetterberg E, Popat U, Hasselbalch H, Prchal J, Palmblad J. Angiogenesis in pulmonary hypertension with myelofibrosis. Haematologica. 2008;93(6):945–946. doi: 10.3324/haematol.12426. [DOI] [PubMed] [Google Scholar]
- 5.Farha S, Asosingh K, Xu W, et al. Hypoxia-inducible factors in human pulmonary arterial hypertension: a link to the intrinsic myeloid abnormalities. Blood. 2011;117(13):3485–3493. doi: 10.1182/blood-2010-09-306357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adir Y, Humbert M. Pulmonary hypertension in patients with chronic myeloproliferative disorders. Eur Respir J. 2010;35(6):1396–1406. doi: 10.1183/09031936.00175909. [DOI] [PubMed] [Google Scholar]
- 7.Guilpain P, Montani D, Damaj G, et al. Pulmonary hypertension associated with myeloproliferative disorders: a retrospective study of ten cases. Respiration. 2008;76(3):295–302. doi: 10.1159/000112822. [DOI] [PubMed] [Google Scholar]
- 8.Launay JM, Herve P, Callebert J, et al. Serotonin 5-HT2B receptors are required for bone-marrow contribution to pulmonary arterial hypertension. Blood. 2012;119(7):1772–1780. doi: 10.1182/blood-2011-06-358374. [DOI] [PubMed] [Google Scholar]
- 9.Yoder MC. Defining human endothelial progenitor cells. J Thromb Haemost. 2009;7(Suppl 1):49–52. doi: 10.1111/j.1538-7836.2009.03407.x. [DOI] [PubMed] [Google Scholar]
- 10.Asosingh K, Aldred MA, Vasanji A, et al. Circulating angiogenic precursors in idiopathic pulmonary arterial hypertension. Am J Pathol. 2008;172(3):615–627. doi: 10.2353/ajpath.2008.070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sieveking DP, Buckle A, Celermajer DS, Ng MK. Strikingly different angiogenic properties of endothelial progenitor cell subpopulations: insights from a novel human angiogenesis assay. J Am Coll Cardiol. 2008;51(6):660–668. doi: 10.1016/j.jacc.2007.09.059. [DOI] [PubMed] [Google Scholar]
- 12.Hirschi KK, Ingram DA, Yoder MC. Assessing identity, phenotype, and fate of endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2008;28(9):1584–1595. doi: 10.1161/ATVBAHA.107.155960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132(4):631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yin AH, Miraglia S, Zanjani ED, et al. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997;90(12):5002–5012. [PubMed] [Google Scholar]
- 15.Majka SM, Skokan M, Wheeler L, et al. Evidence for cell fusion is absent in vascular lesions associated with pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2008;295(6):L1028–L1039. doi: 10.1152/ajplung.90449.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toshner M, Voswinckel R, Southwood M, et al. Evidence of dysfunction of endothelial progenitors in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2009;180(8):780–787. doi: 10.1164/rccm.200810-1662OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yao W, Firth AL, Sacks RS, et al. Identification of putative endothelial progenitor cells (CD34+CD133+Flk-1+) in endarterectomized tissue of patients with chronic thromboembolic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2009;296(6):L870–L878. doi: 10.1152/ajplung.90413.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piaggio G, Rosti V, Corselli M, et al. Endothelial colony-forming cells from patients with chronic myeloproliferative disorders lack the disease-specific molecular clonality marker. Blood. 2009;114(14):3127–3130. doi: 10.1182/blood-2008-12-190991. [DOI] [PubMed] [Google Scholar]
- 19.Massa M, Rosti V, Ramajoli I, et al. Circulating CD34+, CD133+, and vascular endothelial growth factor receptor 2-positive endothelial progenitor cells in myelofibrosis with myeloid metaplasia. J Clin Oncol. 2005;23(24):5688–5695. doi: 10.1200/JCO.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 20.Austin ED, Ma L, Leduc C, et al. Whole exome sequencing to identify a novel gene (Caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet. 2012;5(3):336–343. doi: 10.1161/CIRCGENETICS.111.961888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asosingh K, Swaidani S, Aronica M, Erzurum SC. Th1- and Th2-dependent endothelial progenitor cell recruitment and angiogenic switch in asthma. J Immunol. 2007;178(10):6482–6494. doi: 10.4049/jimmunol.178.10.6482. [DOI] [PubMed] [Google Scholar]
- 22.Zheng L, Nukuna B, Brennan ML, et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J Clin Invest. 2004;114(4):529–541. doi: 10.1172/JCI21109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duong HT, Comhair SA, Aldred MA, et al. Pulmonary artery endothelium resident endothelial colony-forming cells in pulmonary arterial hypertension. Pulm Circ. 2011;1(4):475–486. doi: 10.4103/2045-8932.93547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hazen SL, Heinecke JW. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J Clin Invest. 1997;99(9):2075–2081. doi: 10.1172/JCI119379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abu-Soud HM, Hazen SL. Nitric oxide is a physiological substrate for mammalian peroxidases. J Biol Chem. 2000;275(48):37524–37532. doi: 10.1074/jbc.275.48.37524. [DOI] [PubMed] [Google Scholar]
- 26.Eiserich JP, Baldus S, Brennan ML, et al. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science. 2002;296(5577):2391–2394. doi: 10.1126/science.1106830. [DOI] [PubMed] [Google Scholar]
- 27.Zhang C, Yang J, Jacobs JD, Jennings LK. Interaction of myeloperoxidase with vascular NAD(P)H oxidase-derived reactive oxygen species in vasculature: implications for vascular diseases. Am J Physiol Heart Circ Physiol. 2003;285(6):H2563–H2572. doi: 10.1152/ajpheart.00435.2003. [DOI] [PubMed] [Google Scholar]
- 28.Friedrich EB, Walenta K, Scharlau J, Nickenig G, Werner N. CD34-/CD133+/VEGFR-2+ endothelial progenitor cell subpopulation with potent vasoregenerative capacities. Circ Res. 2006;98(3):e20–e25. doi: 10.1161/01.RES.0000205765.28940.93. [DOI] [PubMed] [Google Scholar]
- 29.Bartunek J, Vanderheyden M, Vandekerckhove B, et al. Intracoronary injection of CD133-positive enriched bone marrow progenitor cells promotes cardiac recovery after recent myocardial infarction: feasibility and safety. Circulation. 2005;112(9 Suppl):I178–183. doi: 10.1161/CIRCULATIONAHA.104.522292. [DOI] [PubMed] [Google Scholar]
- 30.Valentijn KM, Sadler JE, Valentijn JA, Voorberg J, Eikenboom J. Functional architecture of Weibel-Palade bodies. Blood. 2011;117(19):5033–5043. doi: 10.1182/blood-2010-09-267492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lopes AA, Maeda NY. Circulating von Willebrand factor antigen as a predictor of short-term prognosis in pulmonary hypertension. Chest. 1998;114(5):1276–1282. doi: 10.1378/chest.114.5.1276. [DOI] [PubMed] [Google Scholar]
- 32.Kawut SM, Horn EM, Berekashvili KK, Widlitz AC, Rosenzweig EB, Barst RJ. von Willebrand factor independently predicts long-term survival in patients with pulmonary arterial hypertension. Chest. 2005;128(4):2355–2362. doi: 10.1378/chest.128.4.2355. [DOI] [PubMed] [Google Scholar]
- 33.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol. 2009;297(6):L1013–L1032. doi: 10.1152/ajplung.00217.2009. [DOI] [PubMed] [Google Scholar]
- 34.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2008;118(7):2372–2379. doi: 10.1172/JCI33452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marsboom G, Pokreisz P, Gheysens O, et al. Sustained endothelial progenitor cell dysfunction after chronic hypoxia-induced pulmonary hypertension. Stem Cells. 2008;26(4):1017–1026. doi: 10.1634/stemcells.2007-0562. [DOI] [PubMed] [Google Scholar]
- 36.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol. 2008;294(2):H570–H578. doi: 10.1152/ajpheart.01324.2007. [DOI] [PubMed] [Google Scholar]
- 37.Yoon D, Pastore YD, Divoky V, et al. Hypoxia-inducible factor-1 deficiency results in dysregulated erythropoiesis signaling and iron homeostasis in mouse development. J Biol Chem. 2006;281(35):25703–25711. doi: 10.1074/jbc.M602329200. [DOI] [PubMed] [Google Scholar]
- 38.Zhang FL, Shen GM, Liu XL, Wang F, Zhao YZ, Zhang JW. Hypoxia-inducible factor 1-mediated human GATA1 induction promotes erythroid differentiation under hypoxic conditions [published online ahead of print November 3, 2011]. J Cell Mol Med. doi: 10.1111/j.1582-4934.2011.01484.x. doi: 10.1111/j.1582-4934.2011.01484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burke DL, Frid MG, Kunrath CL, et al. Sustained hypoxia promotes the development of a pulmonary artery-specific chronic inflammatory microenvironment. Am J Physiol Lung Cell Mol Physiol. 2009;297(2):L238–L250. doi: 10.1152/ajplung.90591.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hassoun PM, Mouthon L, Barbera JA, et al. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol. 2009;54(1 Suppl):S10–S19. doi: 10.1016/j.jacc.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 41.Barcelos LS, Duplaa C, Krankel N, et al. Human CD133+ progenitor cells promote the healing of diabetic ischemic ulcers by paracrine stimulation of angiogenesis and activation of Wnt signaling. Circ Res. 2009;104(9):1095–1102. doi: 10.1161/CIRCRESAHA.108.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Onoda JM, Kantak SS, Diglio CA. Radiation induced endothelial cell retraction in vitro: correlation with acute pulmonary edema. Pathol Oncol Res. 1999;5(1):49–55. doi: 10.1053/paor.1999.0049. [DOI] [PubMed] [Google Scholar]
- 43.Ghobadi G, Bartelds B, van der Veen SJ, et al. Lung irradiation induces pulmonary vascular remodelling resembling pulmonary arterial hypertension. Thorax. 2012;67(4):334–341. doi: 10.1136/thoraxjnl-2011-200346. [DOI] [PubMed] [Google Scholar]
- 44.Dooner M, Cerny J, Colvin G, et al. Homing and conversion of murine hematopoietic stem cells to lung. Blood Cells Mol Dis. 2004;32(1):47–51. doi: 10.1016/j.bcmd.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 45.Smits PA, Kleppe LS, Witt TA, Mueske CS, Vile RG, Simari RD. Distribution of circulation-derived endothelial progenitors following systemic delivery. Endothelium. 2007;14(1):1–5. doi: 10.1080/10623320601177254. [DOI] [PubMed] [Google Scholar]
- 46.Cooke KR, Yanik G. Acute lung injury after allogeneic stem cell transplantation: is the lung a target of acute graft-versus-host disease? Bone Marrow Transplant. 2004;34(9):753–765. doi: 10.1038/sj.bmt.1704629. [DOI] [PubMed] [Google Scholar]
- 47.Limsuwan A, Pakakasama S, Hongeng S. Reversible course of pulmonary arterial hypertension related to bone marrow transplantation. Heart Vessels. 2011;26(5):557–561. doi: 10.1007/s00380-010-0100-6. [DOI] [PubMed] [Google Scholar]
- 48.Zhao YD, Courtman DW, Deng Y, Kugathasan L, Zhang Q, Stewart DJ. Rescue of monocrotaline-induced pulmonary arterial hypertension using bone marrow-derived endothelial-like progenitor cells: efficacy of combined cell and eNOS gene therapy in established disease. Circ Res. 2005;96(4):442–450. doi: 10.1161/01.RES.0000157672.70560.7b. [DOI] [PubMed] [Google Scholar]
- 49.Fuster V, Steele PM, Edwards WD, Gersh BJ, McGoon MD, Frye RL. Primary pulmonary hypertension: natural history and the importance of thrombosis. Circulation. 1984;70(4):580–587. doi: 10.1161/01.cir.70.4.580. [DOI] [PubMed] [Google Scholar]
- 50.Guo FH, De Raeve HR, Rice TW, Stuehr DJ, Thunnissen FB, Erzurum SC. Continuous nitric oxide synthesis by inducible nitric oxide synthase in normal human airway epithelium in vivo. Proc Natl Acad Sci U S A. 1995;92(17):7809–7813. doi: 10.1073/pnas.92.17.7809. [DOI] [PMC free article] [PubMed] [Google Scholar]