

Abstract
Streptococcus pneumoniae (the pneumococcus) continues to be responsible for a high level of global morbidity and mortality resulting from pneumonia, bacteremia, meningitis, and otitis media. Here we have used a novel technique involving niche-specific, genome-wide in vivo transcriptomic analyses to identify genes upregulated in distinct niches during pathogenesis after intranasal infection of mice with serotype 4 or 6A pneumococci. The analyses yielded 28 common, significantly upregulated genes in the lungs relative to those in the nasopharynx and 25 significantly upregulated genes in the blood relative to those in the lungs in both strains, some of which were previously unrecognized. The role of five upregulated genes from either the lungs or the blood in pneumococcal pathogenesis and virulence was then evaluated by targeted mutagenesis. One of the mutants (ΔmalX) was significantly attenuated for virulence in the lungs, two (ΔaliA and ΔilvH) were significantly attenuated for virulence in the blood relative to the wild type, and two others (ΔcbiO and ΔpiuA) were completely avirulent in a mouse intranasal challenge model. We also show that the products of aliA, malX, and piuA are promising candidates for incorporation into multicomponent protein-based pneumococcal vaccines currently under development. Importantly, we suggest that this new approach is a viable complement to existing strategies for the discovery of genes critical to the distinct stages of invasive pneumococcal disease and potentially has broad application for novel protein antigen discovery in other pathogens such as S. pyogenes, Haemophilus influenzae type b, and Neisseria meningitidis.
INTRODUCTION
Streptococcus pneumoniae (the pneumococcus) continues to be responsible for a high level of global morbidity and mortality, causing a spectrum of diseases such as pneumonia, bacteremia, meningitis, and otitis media. In developing countries, up to 1 million children under 5 years of age die each year from pneumonia, of which S. pneumoniae is the single commonest cause (30). In these countries, pneumonia is responsible for 20% of all deaths in this age group. The prevalence of antibiotic-resistant pneumococci is increasing rapidly, and vaccination represents the best prospect for managing pneumococcal disease in the 21st century (42). However, currently available vaccines are expensive and have major shortcomings with respect to immunogenicity and/or strain coverage. Therefore, it is imperative that future decisions regarding vaccination strategies be based on a deep knowledge of the complex and dynamic interactions between the pneumococcus and its host.
Asymptomatic colonization of the upper respiratory tract (carriage) is an essential first step in the pathogenesis of pneumococcal disease. Individuals may be colonized by multiple strains or serotypes of S. pneumoniae, and these presumably compete for occupation of the nasopharyngeal niche (20). Individuals can be colonized by pneumococci asymptomatically for many weeks. However, in a proportion of carriers, the organism is able to penetrate host defenses and cause disease, either by translocation to sites such as the lungs or middle ear cavity or by direct invasion of the blood. The precise events associated with progression from asymptomatic carriage to invasive disease are poorly understood. The transition involves a major alteration in the microenvironment to which the pneumococcus is exposed, which requires alteration in the expression of complex sets of genes. Until recently, progress on the systematic examination of pneumococcal gene expression patterns in the distinct environmental niches was hampered by technical difficulties associated with isolating sufficient quantities of pure and intact bacterial RNA from infected host tissues to perform accurate, quantitative mRNA analyses. To overcome these difficulties, we pioneered techniques for rapid extraction of bacterial mRNA from infected mice and demonstrated that certain pneumococcal virulence-related genes are differentially expressed in vivo and in vitro (33). RNA extraction, enrichment, and linear amplification techniques have been further refined, permitting the first systematic examination of transcription patterns of key pneumococcal virulence genes in distinct host niches from the same animal, including sites such as the nasopharynx, where pneumococci exist in very low numbers (19, 22, 31, 35). The results from those studies showed that key pneumococcal virulence genes were differentially expressed in distinct in vivo niches, and simultaneous gene expression patterns of selected host immunomodulatory molecules were also different between host niches (22). These studies have opened a new vista into the study of pneumococcal gene expression and innate host responses during pathogenesis of invasive disease. Nevertheless, several other strategies have also been employed to screen in vivo induced genes for the discovery and appraisal of novel candidate proteins for inclusion in a multicomponent pneumococcal protein vaccine under development. The strategies include, but are not limited to, in vivo expression technology (IVET) (27), signature-tagged mutagenesis (STM) (12, 18, 44), differential fluorescence induction (DFI) (4, 23), microarray transcriptomics (40), reverse vaccinology (3, 45), antigenome technology (10, 26), genomic array footprinting (GAF) (29), and protein expression library screening (28).
To gain novel insights into global expression of pneumococcal virulence genes during pathogenesis of invasive disease, we conducted a comprehensive microarray comparison of gene expression kinetics between pneumococci in the nasopharynx, lungs, and blood of mice infected with two virulent S. pneumoniae strains. We hypothesized that progression from carriage to invasive disease will require niche-specific alterations in virulence gene expression and that transcriptomic analysis will identify common virulence factors critical for disease progression. We also hypothesized that the virulence factors that are upregulated during progression from carriage to disease, if surface exposed, are likely to be protective immunogens. Accordingly, we tested and validated these hypotheses by characterizing the role of these upregulated genes in pathogenesis using targeted mutagenesis, and we confirmed their vaccine potential in mouse challenge models.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The pneumococcal strains used in this study are serotype 2 (D39), serotype 4 (WCH43), serotype 6A (WCH16), and their respective isogenic mutant derivatives (Table 1). Serotype-specific capsule production was confirmed by Quellung reaction, as described previously (5). Opaque-phase variants of the three strains, selected on Todd-Hewitt broth supplemented with 1% yeast extract (THY)-catalase plates (50), were used in all animal experiments. Before infection, the bacteria were grown statically at 37°C in serum broth (10% heat-inactivated horse serum in nutrient broth) to an A600 of 0.16 (equivalent to approximately 5 × 107 CFU/ml).
Table 1.
S. pneumoniae strains used in this study
| Strain | Description (sequence type) | Source or reference |
|---|---|---|
| D39 | Capsular serotype 2 (595) | NCTC 7466 |
| D39::ΔcbiO | cbiO deletion mutant of D39 [Eryr] | Present study |
| D39::ΔpiuA | piuA deletion mutant of D39 [Eryr] | Present study |
| WCH16 | Capsular serotype 6A clinical isolate (4966) | Women's and Children's Hospital, North Adelaide, Australia |
| WCH43 | Capsular serotype 4 clinical isolate (205) | Women's and Children's Hospital, North Adelaide, Australia |
| WCH43::Δadh2 | adh2 deletion mutant of WCH43 [Spcr] | Present study |
| WCH43::ΔaliA | aliA deletion mutant of WCH43 [Eryr] | Present study |
| WCH43::ΔcbiO | cbiO deletion mutant of WCH43 [Eryr] | Present study |
| WCH43::ΔcbpE | cbpE deletion mutant of WCH43 [Eryr] | Present study |
| WCH43::ΔcglB | cglB deletion mutant of WCH43 [Spcr] | Present study |
| WCH43::ΔdltB | dltB deletion mutant of WCH43 [Spcr] | Present study |
| WCH43::ΔilvH | ilvH deletion mutant of WCH43 [Eryr] | Present study |
| WCH43::ΔmalX | malX deletion mutant of WCH43 [Spcr] | Present study |
| WCH43::ΔnrdD | nrdD deletion mutant of WCH43 [Spcr] | Present study |
| WCH43::ΔpiuA | piuA deletion mutant of WCH43 [Eryr] | Present study |
Mice.
Outbred 5- to 6-week-old female CD1 (Swiss) mice, obtained from the Laboratory Animal Services breeding facility of The University of Adelaide, were used in all experiments. The Animal Ethics Committee of The University of Adelaide approved all animal experiments. The study was conducted in compliance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (7th edition, 2004) and the South Australian Animal Welfare Act 1985.
Intranasal challenge of mice for gene expression analyses.
For each triplicate WCH16 and WCH43 challenge experiment, groups of 40 mice were used. The mice were anesthetized by intraperitoneal (i.p.) injection of pentobarbital sodium (Nembutal; Rhone-Merieux) at a dose of 66 mg per g of body weight and challenged intranasally (i.n.) with 50 μl of bacterial suspension containing approximately 1 × 107 to 2 × 107 CFU in serum broth. The challenge dose was confirmed retrospectively by serial dilution and plating of the inocula on blood agar. In each experiment, at 48, 72, and 96 h postchallenge, at least 12 mice were sacrificed by CO2 asphyxiation, and samples from the nasopharynx, lungs, and blood were processed as described previously (19, 33). Before processing, lung samples were further rinsed in 25 ml sterile, ice-cold phosphate-buffered saline (PBS) before homogenization to remove contaminating blood from the surface. A 40-μl aliquot of each sample was serially diluted in serum broth and plated on blood agar to enumerate pneumococci present in each niche. Blood plates were incubated at 37°C in 95% air, 5% CO2 overnight. Samples were then stored at −80°C until further processing was done. The experiment was performed three times for each strain.
Extraction of total RNA from mouse tissues.
RNA for microarray experiments was isolated from the various niches with acid-phenol–chloroform–isoamyl alcohol (125:24:1; pH 4.5; Ambion). Purified RNA samples were checked for purity and integrity as described previously (19, 22). Bacterial RNAs from a minimum of five mice that satisfied these criteria from each specific niche were pooled. The RNA was then purified further using a Qiagen RNeasy minikit. RNA was further enriched for prokaryotic RNA using the MicrobEnrich kit (Ambion). The amount of RNA recovered following purification and enrichment was determined by A260/A280 measurements.
Microarray analysis of bacterial RNA.
Microarray experiments were performed on whole-genome S. pneumoniae PCR arrays obtained from the Bacterial Microarray Group at St George's Hospital Medical School, London, England (http://bugs.sghms.ac.uk/). The array was designed using TIGR4 base strain annotation (49) and extra target genes from strain R6 (15). The array design is available in BμG@Sbase (accession no. A-BUGS-14; http://bugs.sgul.ac.uk/A-BUGS-14) and also ArrayExpress (accession no. A-BUGS-14).
Microarray probes were generated using the 3DNA Array 900 MPX labeling kit (Genisphere) following the manufacturer's guidelines. Total amplified RNA of S. pneumoniae obtained from the nasopharynx, lungs, and blood was used, and pairwise comparisons were made between the nasopharynx and lung and between the lung and blood RNA samples from the 48-, 72-, and 96-h time points. RNA samples were reverse transcribed using Superscript III (Invitrogen) and then labeled with either Alexa Fluor 546 or Alexa Fluor 647 dye. The fluorescently labeled cDNAs for each pairwise comparison were then combined and hybridized to the surface of the microarray, essentially as described previously (25, 36).
Slides were scanned at 10-μm resolution using a Genepix 4000B scanner (Molecular Devices, USA). Detector photomultiplier tube (PMT) voltages were adjusted individually for each slide so that the total red (Alexa Fluor 647) and green (Alexa Fluor 546) fluorescence signals for each channel were approximately equal while minimizing the total number of features with signal above the maximum detectable. Foreground and background mean pixel intensity values were extracted from the scanned images for both channels (Alexa Fluor 546, Alexa Fluor 647) using the Spot plugin (CSIRO, Australia) within the R statistical software package (http://www.R-project.org). The Limma plugin for R (47) was used for data processing and statistical analysis. After background subtraction, the foreground intensities were log2 transformed and a single ratio (Alexa Fluor 647/Alexa Fluor 546) value was obtained for each probe. Ratio values were normalized using the print-tip Loess normalization routine (48). The replicate arrays were normalized to each other to give similar ranges of mRNA expression values. For each probe across the arrays a linear model was fitted to determine a final expression value for each mRNA probed and associated statistics (46). These statistics were used to rank the mRNAs from those most likely to be differentially expressed to the least likely using false-discovery rate values of P < 0.05. Microarray analysis examining RNA from infected nasopharynx versus lungs and lungs versus blood was performed on at least 9 independent hybridizations for each pairwise comparison from three separate assays, including at least one dye reversal per comparison for each strain.
Relative quantitation real-time reverse transcription-PCR (RT-PCR).
For a subset of selected genes that showed significant levels of upregulation in the lungs or blood by microarray analysis, gene expression was validated using SuperScript III one-step RT-PCR kit (Invitrogen) in a LightCycler480 II (Roche) as described previously (22). The relative gene expression was analyzed using the 2−ΔΔCT method (21). The reference gene was 16S rRNA. The primer pairs used for gene expression analysis are listed in Table S1 in the supplemental material. All data were obtained from three biological replicates.
Construction of mutants and assessment of bacterial growth in vitro.
Defined, nonpolar mutants of genes of interest were constructed in WCH43 (serotype 4) and, for ΔcbiO and ΔpiuA mutants, also in D39. Mutants were constructed by overlap extension PCR as described previously (14) and validated by PCR and sequencing to be in-frame deletion mutation replacements. All PCR procedures were performed with the Phusion High Fidelity kit (FINNZYMES). The primer pairs used for construction and validation of the mutants are listed in Table S2 in the supplemental material. In order to evaluate the growth rate of the mutants in comparison to the wild type, bacterial strains were grown in serum broth (SB) and the A600 was monitored overnight on a Spectramax M2 spectrophotometer (Millennium Science).
Virulence assessment of mutants.
To assess the virulence potential of mutants, groups of 12 mice were challenged i.n. with approximately 1 × 107 CFU of either mutant or wild-type bacteria in 50 μl SB, under deep anesthesia. The survival of mice was monitored four times daily for the first 5 days, twice daily for the next 5 days, and then daily until 21 days after challenge. Differences in median survival times for mice between groups were analyzed by the Mann-Whitney U test (one and two tailed).
Competition experiments.
Mutant and wild-type strains were grown separately in SB to an A600 of 0.16 (approximately 5 × 107 CFU/ml). For each competition experiment, mutant and wild-type bacteria were mixed at an input ratio of 1:1, and 10 mice were challenged i.n. with 50 μl bacterial suspension containing approximately 1 × 107 CFU under deep anesthesia. At 48 h postchallenge, mice from each mixed-infection experiment were sacrificed, and samples from the nasopharynx, lungs, and blood were processed as described previously (24). A 40-μl aliquot of each sample was serially diluted in serum broth and plated on blood agar and on blood agar with a selective antibiotic to determine the ratio of mutant to wild-type bacteria. Competitive indices were calculated as the ratio of mutant to wild-type bacteria recovered from each niche adjusted by the input ratio.
Cloning and expression of His6-tagged proteins.
Based on the results of the i.n. challenge of mice with gene deletion mutants, the open reading frame of each gene chosen for further analysis (aliA, malX, and piuA) was PCR amplified from WCH43 genomic DNA with forward and reverse primers [aliA: SphI (F), CTACTTTAGCTGCATGCTCTGGATC; SalI (R), CTTTCTTATATTTGTCGACAGTTATTTCAC; malX: SphI (F), CTACACTTGCTAGCATGCTTTTGGTAGCTT; SalI (R), AGGGGGATTTGATGTCGACCCCCCTTGAAC; piuA: BamHI (F), GCCAGCTTCTGGATCCTACTTGGTG; PstI (R), TAAATTGAGTCTGCAGTGGCTTATTTC], with incorporated restriction sites underlined. Each PCR fragment was digested with the same enzymes and cloned into the corresponding restriction sites in either pQE-30 (for aliA) or pQE32 (for malX and piuA) (Qiagen Inc.) to generate recombinant plasmid constructs. The cloning and expression of His6-tagged recombinant pneumolysin toxoid (PdT) was described previously (11). High-level expression of all recombinant proteins in a lipid A (lpxM) mutant of Escherichia coli BL21(DE3) (9) transformed with pREP4 (Qiagen) was achieved by induction with 2 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 3 h at 37°C. The cells were harvested by centrifugation at 6,000 × g for 10 min, and the recombinant, soluble proteins were purified on a nickel-nitrilotriacetic acid column essentially as described previously (32). Each protein was judged to be >95% pure by SDS-PAGE and staining with Coomassie brilliant blue R-250.
Active immunization challenge experiments.
For active immunization challenge, groups of 5- to 6-week-old female CD1 mice (12 per group) were immunized i.p. with purified recombinant AliA, MalX, PiuA, and PdT, in alum adjuvant (Imject Alum no. 77161; Pierce, Rockford, IL). Each mouse received three doses of 10 μg of each antigen in 100 μg of alum at 14-day intervals. Mice given the placebo received an identical course of saline plus alum. Sera were collected from individual mice by retro-orbital bleeding 1 week after the third immunization. Antigen-specific enzyme-linked immunosorbent assay (ELISA) titers were determined as the reciprocal of the dilution of serum giving 50% of the highest absorbance reading above the background at 405 nm. Two weeks after the third immunization, mice were challenged i.p. with approximately 3 × 104 CFU (in 100 μl) of WCH43. Mice were closely monitored for survival over 21 days. Differences in median survival time of mice between groups were analyzed by the Mann-Whitney U test (one tailed).
Determination of membrane/surface localization.
Wild-type WCH43 cells were grown in C+Y medium to an A600 of 0.3. After one wash in PBS, bacteria were resuspended in 50 μl of protein-specific polyclonal murine antisera or anti-alum serum (diluted 1:50, vol/vol), for 30 min at 37°C, followed by two more washes and then 50 μl of Alexa 488 donkey anti-mouse IgG, heavy and light chain (H+L) (Invitrogen; diluted 1:200, vol/vol) for 1 h on ice. Three replicates for each serum were analyzed using a BD FACSCanto flow cytometer (BD Biosciences, San Jose, CA) and the software package Weasel (Walter and Eliza Hall Institute of Medical Research). Data shown are representative of three independent experiments. Histograms are representative of fluorescence-activated cell sorter (FACS) data for each protein.
For cell fractionation studies, wild-type WCH43 and its isogenic ΔaliA, ΔmalX, and ΔpiuA mutants were grown as described above. Cell pellets were resuspended in PBS and lysed in a French pressure cell (SLM Aminco Inc.) at 12,000 lb/in2. Lysates were then subjected to ultracentrifugation at 100,000 × g for 1 h to separate soluble (cytoplasmic) and insoluble (membrane and cell wall) fractions. After several washes in PBS, pellets containing the insoluble fractions were resuspended in PBS containing 2% SDS overnight at room temperature. All protein samples were quantitated by A280 measurements on a NanoDrop 1000 Spectrophotometer (Thermo Scientific). Approximately 50 μg of each sample was resuspended in sample loading buffer before electrophoresis on a 4 to 12% Bis Tris NuPAGE gel and then transferred on an iBlot dry blotting (nitrocellulose) system (Invitrogen, Life Technologies). After transfer, each membrane was reacted with a dilution of 1/3,000 polyclonal mouse anti-AliA, MalX, and PiuA sera, using anti-PsaA as a loading control and membrane compartment marker for AliA and MalX blots and anti-AliA serum as a marker for the PiuA blot. Bound antibodies were detected by using a 1/3,000 dilution of alkaline phosphatase-conjugated goat anti-mouse IgG (H+L) (Bio-Rad) with disodium p-nitrophenylphosphate (Sigma) as the substrate.
Microarray data accession numbers.
Fully annotated microarray data have been deposited in BμG@Sbase (accession number E-BUGS-133; http://bugs.sgul.ac.uk/E-BUGS-133) and also ArrayExpress (accession number E-BUGS-133).
RESULTS
Transcriptomic analysis identifies novel, differentially expressed genes in discrete in vivo niches.
Previous mouse intranasal challenge experiments in our laboratory had indicated that D39 and WCH43 are more virulent than WCH16 (A. D. Ogunniyi and J. C. Paton, unpublished data). Furthermore, after infection of mice, D39 causes fulminant bacteremia and WCH43 demonstrates the “classical” disease progression from the nasopharynx to the lungs, followed by dissemination to the blood and then to the brain, while WCH16 demonstrates minimal lung and blood involvement before translocation to the brain (22, 39).
The distinct virulence/pathogenic profiles exhibited by WCH16 and WCH43 reflect the spectrum of disease in humans, making these strains suitable for further analysis. Therefore, bacteria harvested from the nasopharynx, lungs, and blood of mice at 48, 72, and 96 h after infection with either WCH16 or WCH43, in three replicate experiments, were subjected to pairwise transcriptomic comparisons, as described in Materials and Methods. Our analysis shows dramatic differences in transcript abundance between the two strains. For WCH16, the comparisons yielded 253 significantly up- or downregulated genes in the lungs relative to the nasopharynx (Fig. 1A), and 171 genes differentially regulated in the blood relative to the lungs (Fig. 1B). For WCH43, 315 genes were differentially regulated in the lungs relative to the nasopharynx (Fig. 1A), while 370 genes were differentially regulated in the blood relative to the lungs (Fig. 1B).
Fig 1.
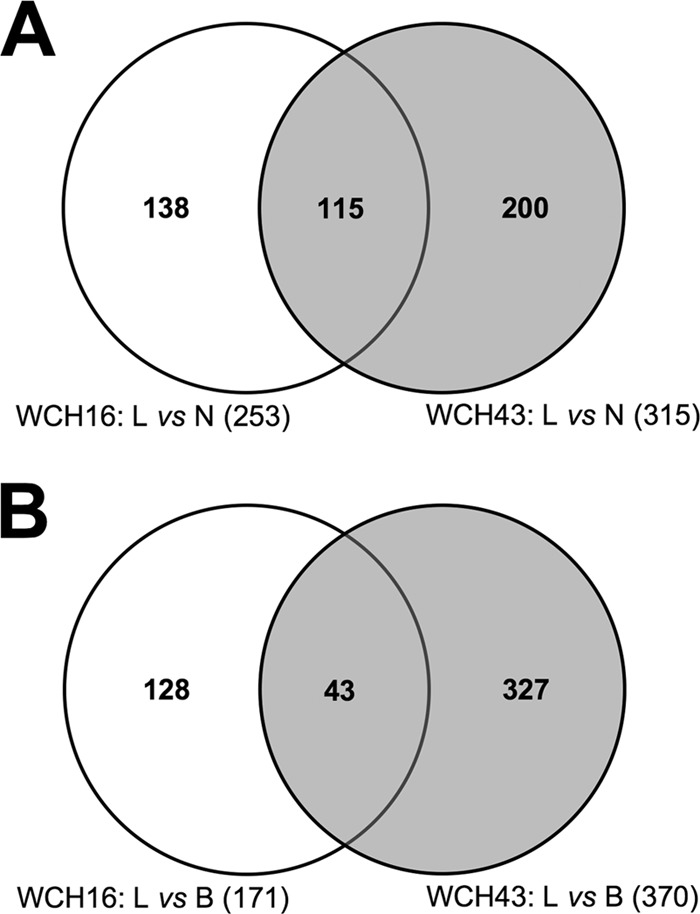
Venn diagram of microarray data comparisons of in vivo-expressed Streptococcus pneumoniae genes. Analysis of interaction of common, differentially expressed genes from microarray comparisons of lungs versus nasopharynx (L vs N) (A) and lungs versus blood (L vs B) (B) in WCH16 and WCH43.
Further, the genes that were differentially expressed in the discrete in vivo niches of mice infected with either WCH16 or WCH43 were cross-compared. In this manner, only genes that were consistently and reproducibly differentially expressed at all three time points, in all three replicate experiments, and in both strains were chosen for analysis. The analyses revealed 115 significantly differentially expressed genes in the lungs versus nasopharynx (Fig. 1A; see Fig. S1 and Table S3 in the supplemental material), and 43 differentially expressed genes in the blood versus lungs (Fig. 1B; see Fig. S2 and Table S3 in the supplemental material), common to both strains. We then used an arbitrary cutoff of ≥1.3-fold regulation for genes for further analysis, as any upregulation below this threshold was not considered to be physiologically relevant. In so doing, we found 28 genes upregulated in the lungs compared to the nasopharynx (Table 2) and 25 genes upregulated in the blood compared to the lungs (Table 3), common to both strains. For each pairwise comparison, 8 of the upregulated genes were randomly selected for validation by RT-PCR for both strains (Tables 2 and 3). Of the 28 genes upregulated in the lungs compared to the nasopharynx, 18 have been identified in other screens, and 14 of the 25 genes upregulated in the blood compared to the lungs have been identified in other screens (Tables 2 and 3).
Table 2.
Differential gene expression profiles of S. pneumoniae WCH16 and WCH43 in the nasopharynx and lungs of mice by microarray analysis
| Gene ID (TIGR4)a | Gene annotation | Identified in previous screensb | Fold change (lungs/nasopharynx)c |
|
|---|---|---|---|---|
| WCH16 | WCH43 | |||
| Sp-0043 | Competence factor transport protein ComB | Yes (4, 18, 23, 40) | 2.0 | 1.7 |
| Sp-0049 | VanZ protein, putative | Yes (12) | 1.6 (7.7) | 1.9 (7.7) |
| Sp-0202 | Anaerobic ribonucleoside triphosphate reductase (NrdD) | Yes (40) | 2.3 (8.2) | 2.0 (7.0) |
| Sp-0463 | Cell wall surface anchor family protein | Yes (10) | 1.3 | 1.6 |
| Sp-0545 | Immunity protein (BlpY) | Yes (40) | 1.6 | 1.8 |
| Sp-0758 | PTS system, IIABC components | No | 1.7 | 1.7 |
| Sp-0785 | HlyD family secretion protein | Yes (10, 12) | 1.7 (1.8) | 2.1 (2.0) |
| Sp-0787 | Antimicrobial peptide ABC transporter permease | Yes (40) | 1.5 | 1.8 |
| Sp-0803 | Bacterial cell division membrane protein (RodA) | No | 1.4 | 1.4 |
| Sp-0804 | 4-Methyl-5(b-hydroxyethyl)-thiazole monophosphate biosynthesis protein | No | 1.3 | 1.7 |
| Sp-1083 | Putative transcriptional regulator | No | 1.4 | 1.5 |
| Sp-1123 | Glycogen biosynthesis protein (GlgD) | Yes (10) | 1.5 | 2.0 |
| Sp-1368 | Psr protein | Yes (23) | 1.3 | 1.4 |
| Sp-1382 | Cytoplasmic alpha-amylase | No | 1.6 | 2.3 |
| Sp-1393 | GntR family transcriptional regulator | Yes (40) | 1.4 | 1.3 |
| Sp-1395 | Phosphate transport system regulatory protein (PhoU) | No | 1.4 | 1.5 |
| Sp-1950 | Bacteriocin formation protein, putative (CylM protein) | No | 1.5 (11.6) | 2.0 (2.3) |
| Sp-2026 | Bifunctional acetaldehyde-coenzyme A/alcohol dehydrogenase (Adh2) | Yes (40) | 3.6 (8.4) | 2.9 (5.6) |
| Sp-2052 | Competence protein CglB | Yes (4, 12) | 1.7 (18.1) | 2.3 (5.8) |
| Sp-2053 | Competence protein CglA | Yes (4, 12) | 1.4 | 1.9 |
| Sp-2106 | Glycogen phosphorylase family protein | No | 2.5 | 3.3 |
| Sp-2107 | 4-Alpha-glucanotransferase | No | 2.8 | 2.8 |
| Sp-2108 | Maltose/maltodextrin ABC transporter, maltose/maltodextrin-binding protein (MalX) | Yes (10, 12, 28) | 2.9 (6.8) | 3.2 (3.8) |
| Sp-2109 | Maltose/maltodextrin transport system permease protein | No | 1.7 | 1.9 |
| Sp-2112 | Maltose operon transcriptional repressor (MalR) | Yes (40) | 1.3 | 1.8 |
| Sp-2173 | DltD protein | Yes (40) | 1.4 | 1.5 |
| Sp-2175 | DltB protein | Yes (12) | 1.4 (2.0) | 2.1 (15.0) |
| Sp-2190 | Choline binding protein A (CbpA) | Yes (12, 40) | 1.4 | 1.7 |
Gene IDs were obtained from the S. pneumoniae TIGR4 (serotype 4) genome as deposited in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database.
Reference(s) for previous screens are in parentheses.
Data in parentheses represent corresponding real-time RT-PCR values for the indicated genes.
Table 3.
Differential gene expression profiles of S. pneumoniae WCH16 and WCH43 in the lungs and blood of mice by microarray analysis
| Gene ID (TIGR4)a | Gene annotation | Identified in previous screensb | Fold change (blood/lungs)c |
|
|---|---|---|---|---|
| WCH16 | WCH43 | |||
| Sp-0056 | Adenylosuccinate lyase | Yes (12, 40) | 1.5 | 1.5 |
| Sp-0217 | 50S ribosomal protein L29 (RpmC) | No | 1.6 | 1.7 |
| Sp-0282 | PTS system, mannose-specific IID component | Yes (18) | 1.5 | 1.3 |
| Sp-0290 | Dihydrofolate synthetase | Yes (40) | 1.4 | 1.4 |
| Sp-0346 | Capsular polysaccharide biosynthesis protein (Cps4A) | Yes (40) | 1.4 | 1.7 |
| Sp-0347 | Capsular polysaccharide biosynthesis protein (Cps4B) | No | 1.5 | 1.8 |
| Sp-0366 | Oligopeptide ABC transporter, oligopeptide-binding protein (AliA) | Yes (18, 40) | 1.6 (6.8) | 1.4 (5.1) |
| Sp-0369 | Penicillin-binding protein 1A (Pbp1A) | Yes (10, 18, 23) | 1.8 (2.1) | 1.7 (7.0) |
| Sp-0435 | Elongation factor P (EfP) | No | 1.4 | 1.3 |
| Sp-0443 | Dihydroxyacetone kinase family protein | No | 1.4 | 1.8 |
| Sp-0446 | Acetolactate synthase 3 regulatory subunit (IlvH) | Yes (12, 40) | 1.7 (7.5) | 1.9 (10.6) |
| Sp-0447 | Ketol-acid reductoisomerase | Yes (40) | 1.5 | 2.0 |
| Sp-0448 | Hypothetical protein | No | 1.4 | 1.9 |
| Sp-0652 | SAM-dependent methyltransferase | No | 1.4 | 1.8 |
| Sp-0765 | DNA polymerase III subunit delta | No | 1.5 | 1.6 |
| Sp-0846 | Sugar ABC transporter ATP-binding protein | No | 1.4 | 1.3 |
| Sp-0930 | Choline binding protein E (CbpE) | Yes (10, 18) | 1.5 (3.8) | 1.4 (7.0) |
| Sp-0989 | MutT/nudix family protein; ADP-ribose pyrophosphatase | No | 1.4 | 1.3 |
| Sp-1024 | Serine hydroxymethyltransferase | No | 1.5 | 1.3 |
| Sp-1573 | Lysozyme (LytC) | Yes (10) | 1.6 (3.8) | 1.5 (6.7) |
| Sp-1872 | Iron-compound ABC transporter, iron-compound-binding protein (PiuA) | Yes (12, 18) | 1.2 (2.6)d | 2.6 (13.1) |
| Sp-1877 | Site-specific tyrosine recombinase XerD-like protein | Yes (40) | 1.4 | 1.3 |
| Sp-1939 | MATE efflux family protein (DinF) | Yes (12) | 1.3 (6.7) | 2.1 (10.8) |
| Sp-2009 | 50S ribosomal protein L33 | No | 1.5 | 2.1 |
| Sp-2219 | Cobalt/nickel transport system permease protein | Yes (18) | 1.4 | 1.3 |
| Sp-2220 | Cobalt transporter ATP-binding subunit (CbiO) | Yes (18) | 1.7 (1.7) | 1.6 (10.4) |
Gene IDs were obtained from the S. pneumoniae TIGR4 (serotype 4) genome as deposited in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database.
Reference(s) for previous screens are in parentheses.
Data in parentheses represent corresponding real time RT-PCR values for the indicated genes.
Value did not reach statistical significance by microarray analysis, but genes were significantly upregulated by real-time RT-PCR.
Pneumococcal genes upregulated in the lungs and blood contribute to pathogenesis and virulence.
To evaluate the role in pathogenesis and/or virulence of genes preferentially upregulated in either the lungs or blood, five genes specifically upregulated in either niche were subjected to targeted mutagenesis in which each deleted target gene is replaced by a nonpolar antibiotic resistance cassette using overlap PCR (see Materials and Methods). Selection criteria included, but were not limited to, in silico bioinformatics prediction of a role in metabolism, nutrient uptake for survival in the lungs and/or blood, and putative contribution to pathogenesis. The mutation was carried out in WCH43 because it is more virulent than WCH16. Mutation of the 10 genes chosen for further analysis did not adversely affect their growth in vitro in serum broth (data not shown).
The virulence characteristic of each mutant relative to that of the isogenic wild-type strain (WCH43) was assessed by intranasal (i.n.) challenge of mice. In this assay, two of the mutants (ΔcbiO and ΔpiuA) were completely avirulent (Fig. 2). These mutants were also completely avirulent in this challenge model in a D39 background (not shown). Other mutants, such as the ΔmalX, ΔaliA, and ΔilvH strains, were also significantly attenuated (Fig. 2). To further examine the contribution of each differentially expressed gene to pathogenesis, we carried out competition experiments in which groups of mice were challenged i.n. with approximately equal numbers of mutant and wild-type pneumococci. This enabled evaluation of fine differences between wild type and mutant by calculating the competitive index for each mutant in the various niches at 48 h postinfection. The ΔcbiO and ΔpiuA mutants were excluded from this assay because of their complete attenuation in the challenge model. Compared to the wild type, the ΔmalX mutant was significantly attenuated in the lungs (Fig. 3A) and the ΔaliA mutant was significantly attenuated in the lungs and blood (Fig. 3B), while the ΔilvH mutant was significantly attenuated in the blood (Fig. 3C). However, the numbers of the ΔdltB mutant were not significantly different from those of the wild type in any of the niches (Fig. 3D). These results are complementary to the virulence data (Fig. 2), and they further validate the in vivo transcriptomic approach used to identify these genes.
Fig 2.
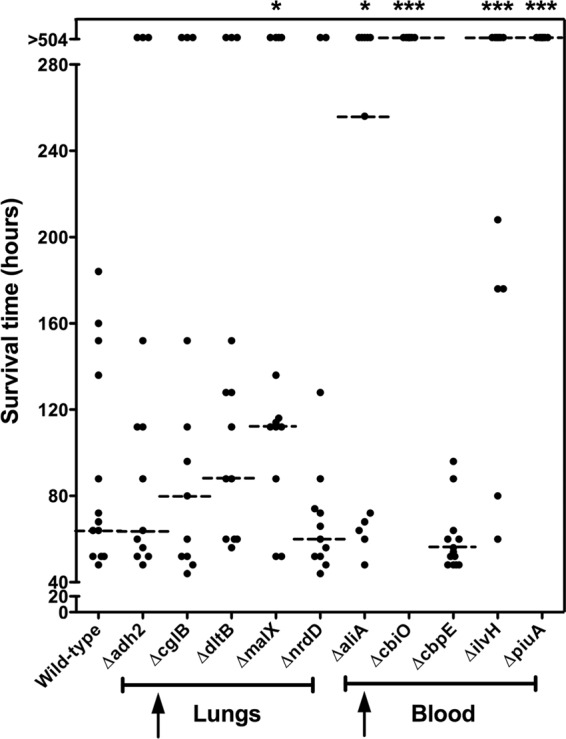
Survival times for mice after i.n. challenge with WCH43 and isogenic mutant derivatives. Groups of 12 or 13 CD1 mice were challenged i.n. with approximately 1 × 107 CFU of the indicated strains. Each datum point represents one mouse. Upward arrows represent cohorts of pneumococcal genes upregulated in the lungs or blood. The horizontal broken lines denote the median survival time for each group. *, P < 0.05; ***, P < 0.001; Mann-Whitney U test; one and two tailed.
Fig 3.

Competition experiments between wild-type WCH43 and its isogenic mutants in the nasopharynx, lungs, and blood of mice at 48 h postinfection. (A) Wild type versus ΔmalX mutant; (B) wild type versus ΔaliA mutant; (C) wild type versus ΔilvH mutant; (D) wild type versus ΔdltB mutant. Ten CD1 mice were infected i.n. with equal numbers (approximately 1 × 107 CFU each) of wild type and mutant in each experiment. Each datum point represents the ratio of recovered mutant bacteria to wild-type bacteria. The horizontal solid line represents a 1:1 ratio of recovered mutant bacteria. The horizontal broken line denotes the geometric mean value of the ratio of recovered mutant bacteria for each comparison. *, P < 0.05; ***, P < 0.001; one sample t test; two tailed.
Differentially expressed virulence factors are protective against pneumococcal sepsis.
The promising pathogenesis and virulence data obtained for ΔcbiO, ΔpiuA, ΔmalX, ΔaliA, and ΔilvH mutants led us to carry out in silico bioinformatics analysis to predict cellular localization. Of the aforementioned factors, PiuA, MalX, and AliA had signal peptidase II recognition sequences and were predicted to be lipoproteins. Accordingly, the respective recombinant products were expressed and purified as His6-tagged fusion proteins in E. coli as detailed in Materials and Methods. Flow cytometry of whole cells with protein-specific murine polyclonal antisera revealed a very low level of surface exposure of PiuA, MalX, and AliA compared to Pht proteins, which are known to be exposed on the pneumococcal surface (Fig. 4A and B). However, the difference in fluorescence for these antigens compared to negative-control anti-alum serum was similar to that of PsaA, a proven lipoprotein and vaccine candidate (25). We then used Western blotting of soluble and membrane compartments of fractionated cells (Fig. 4C to E) to show that like PsaA, PiuA, MalX, and AliA were almost exclusively present in the membrane fraction.
Fig 4.

Membrane/surface localization studies. (A) Histograms showing marginal surface labeling of lipoproteins AliA, MalX, PiuA, and PsaA but substantial labeling of surface-localized PhtABDE with respective antisera. (B) Mean fluorescence data for AliA, MalX, PiuA, PsaA, and PhtABDE labeling with respective antisera, using alum-immunized serum as a negative control (**, P < 0.01; ***, P < 0.0001; unpaired t test; one tailed). (C, D, and E), Western blots of soluble (S) and membrane (M) fractions of S. pneumoniae WCH43 and its isogenic ΔaliA (C), ΔmalX (D), and ΔpiuA (E) mutants showing the reactivities of the antibodies raised against the various protein antigens. The samples were reacted with specific antisera generated from mice immunized with the various antigens; PsaA was used as a loading control and membrane compartment marker for AliA and MalX blots, and AliA was used as a marker for the PiuA blot. The expected mobilities and molecular mass of each test and marker protein are also indicated.
We then evaluated the potential of purified recombinant PiuA, MalX, and AliA to protect against fatal sepsis in a mouse intraperitoneal (i.p) immunization challenge experiment, using the nontoxic pneumolysin derivative PdT as a control (see Materials and Methods). First, we found that strong, antigen-specific antibody responses were generated in mice to each of the immunogens (total IgG titer for AliA = 16,000 ± 1,000, for MalX = 48,000 ± 2,000, for PiuA = 16,000 ± 1,000, and for PdT = 12,000 ± 1,000). Second, we found that mice immunized with recombinant PiuA or AliA alone were significantly protected against challenge with WCH43 compared to mice in the placebo group (P = 0.004 and P = 0.010, respectively) (Fig. 5), while protection imparted by immunization with either MalX or PdT alone was not significant (P = 0.060 and P = 0.051, respectively).
Fig 5.
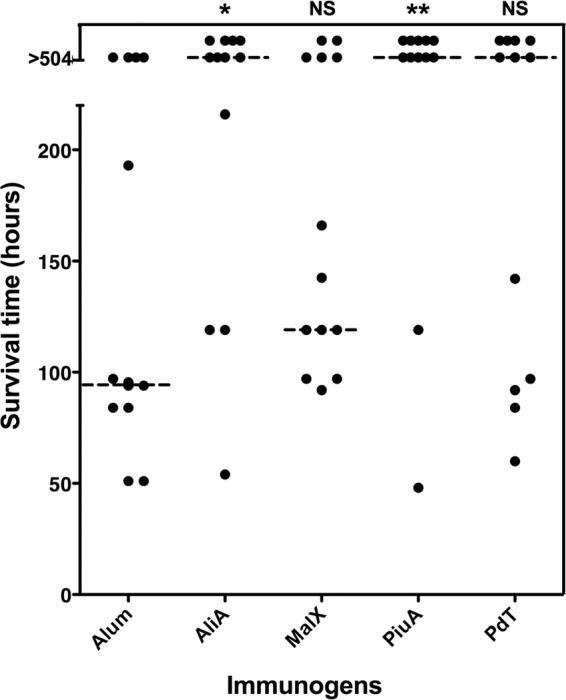
Survival times for mice after i.p. challenge. Groups of 12 CD1 mice were immunized i.p. with the indicated antigens and challenged 2 weeks after the third immunization with approximately 3 × 104 CFU of serotype 4 strain WCH43. The broken lines denote the median survival time for each group. *, P < 0.05; **, P < 0.01; Mann-Whitney U test; one tailed.
DISCUSSION
The increase in the prevalence of antibiotic-resistant pneumococci and the existing and emerging shortcomings associated with current vaccine formulations have underscored the need for alternative approaches to management of pneumococcal disease. One of these strategies involves the development of vaccines based on pneumococcal proteins that contribute to pathogenesis and are common to all serotypes, to provide affordable, effective, and broad protection. Foremost among these antigens are choline binding protein A (CbpA, also known as PspC or SpsA), pneumococcal surface protein A (PspA), and pneumolysin (Ply) toxoid (2, 41, 43). These proteins have been shown to provide significant protective immunity alone; they work in synergy, and they serve as the gold standard for pneumococcal protein vaccine candidates (2, 34, 41–43). Nevertheless, protection using the current combination is by no means complete, and protein vaccine formulations are still being optimized.
An earlier study (40) used either in vitro surrogates for given in vivo niches (e.g., coculture with Detroit 562 cells in lieu of nasopharyngeal colonization; coculture with type II pneumocytes in lieu of lung infection), or cross-species comparisons of gene expression between distinct niches (e.g., blood of mice infected with D39 and adherence assay with Detroit 562 cells using a rough derivative of TIGR4), neither of which are appropriate models to investigate pneumococcal pathogenesis. Our mouse intranasal infection model mimics natural progression of invasive pneumococcal disease in humans, underscoring the superiority of our technique for identifying novel genes that might be critical to pneumococcal pathogenesis. The genome-wide transcriptomic comparisons of gene expression during transition of pneumococci from the nasopharynx to the lungs, and from there to the blood in the same animal, provide an alternative approach to characterizing niche-specific roles of virulence genes during pathogenesis. Such assessment gives insight into the optimization, design, and composition of surface protein combinations that might be more appropriate as vaccine antigens against carriage or invasive pneumococcal disease.
The analysis of gene expression profiles of pneumococci harvested from the nasopharynx compared with expression profiles of those harvested in the lungs revealed two important observations. First, the expression of many genes was higher in the nasopharynx relative to the lungs for WCH43, consistent with our previous observation of gene expression profiles of a few selected pneumococcal virulence proteins in various host tissues (22). It also provides further molecular evidence in support of the hypothesis advanced by others that many pneumococcal virulence factors have dual roles in pathogenesis: establishment and maintenance of carriage, as well as involvement in certain stages of invasive disease (6, 12, 13, 38). Second, the relative expression profiles of many genes of WCH16 in the nasopharynx compared to the lungs were not as distinct as those seen for WCH43. This is in concordance with our previous observation that WCH16 seems to progress to the bloodstream with minimal lung involvement and then to the brain without causing fulminant bacteremia (22). Mutation in only one of the five pneumococcal genes upregulated in the lungs—namely, Sp-2108, which encodes the maltose/maltodextrin-binding protein MalX—resulted in attenuation in the i.n. challenge model. To corroborate this finding, a mouse intranasal competition assay with wild-type WCH43 versus its isogenic ΔmalX mutant showed a massive attenuation of the ΔmalX mutant in the lungs but not in the nasopharynx or blood. Consistent with these findings, MalX was previously identified by STM as one of the genes essential for lung infection (12). Recent structural and biochemical analyses have also provided evidence for a critical role for MalX in the uptake of degradation products of α-glucans, such as glycogen, which is abundant in the lungs (1). Furthermore, other workers have found antibodies to MalX in convalescent-phase sera (10), and the protein was recently shown to be protective against nasopharyngeal colonization through a TH-17 (IL-17A)-dependent mechanism (28). Thus, we suggest that MalX is likely to play a role at mucosal surfaces: in nasopharyngeal colonization and in progression to, and in, lung disease.
We also observed that in the transcriptomic comparisons of lung- versus blood-borne pneumococci, fewer genes were differentially regulated than in the scenario seen for the nasopharynx-versus-lung comparisons. This strongly suggests limited regulation of pneumococcal gene expression during translocation from the lungs to the blood. Nevertheless, mutation in four of the five genes that were significantly upregulated in the blood relative to the lungs demonstrated either significant (for ΔaliA and ΔilvH mutants) or complete (for ΔcbiO and ΔpiuA mutants) attenuation relative to the otherwise isogenic wild-type strain WCH43 in a mouse i.n. challenge model. Significant attenuation of the ΔaliA and ΔilvH mutants was further confirmed in a mouse i.n. competition assay, strongly suggesting a requirement for all these genes for optimal survival of pneumococci in the blood. The significant attenuation of the ΔaliA mutant in the blood is in contrast to that observed earlier in a D39 background (17), suggesting a strain-dependent contribution to virulence.
We then used an active i.p. immunization/challenge experiment to test the hypothesis that surface-accessible virulence factors that are upregulated in distinct host niches during pathogenesis are likely to be protective immunogens. For this purpose, we chose AliA, MalX, and PiuA for further analysis based on the presence of signal peptidase II cleavage sites at their N termini. We were particularly interested in protection against systemic disease, which is responsible for the significant mortality associated with S. pneumoniae. Significant protection was afforded by immunization with AliA and, particularly, with PiuA alone. PiuA is a lipoprotein first characterized as a highly conserved iron uptake ATP binding cassette (ABC) transporter required for bacterial growth and full virulence by Brown et al. (7) following identification by an STM screen (18). Intraperitoneal immunization challenge experiments showed that PiuA is protective against sepsis (8), and passive protection data obtained later confirmed this to occur via complement-dependent and -independent bacterial opsonophagocytosis, rather than by inhibition of iron transport (16). In this study, the level of protection observed for PiuA is better than what we obtained earlier using D39 as a challenge strain (8). The difference could be due to a higher challenge dose used for the D39 experiment but could also be challenge strain dependent, as we proposed previously (34).
Western blot analysis confirmed that MalX, AliA, and PiuA, like PsaA, are located in the membrane compartment, as expected for lipoproteins. Thus, the likely explanation for the observed protection for PiuA and AliA is that exogenous antibody can penetrate the capsule/cell wall layer to a limited extent and bind the lipoproteins inhibiting their biological activity (nutrient transport), rather than promoting opsonophagocytosis. This is in agreement with the argument advanced by Whalan et al. for PiuA (51). The fact that labeling by FACS is very marginal could be due to limited penetration of both primary antibody as well as secondary antibody conjugate during the brief (30-min) incubation period employed. Nevertheless, labeling with anti-PiuA, anti-MalX, and anti-AliA was significantly greater than that with the negative-control serum when judged either by mean fluorescence intensity (Fig. 4B) or percent positive cells (result not shown).
Although the protection imparted by PdT in this study did not reach significance, immunization with PdT (or PdB, a similar pneumolysoid derivative) has been shown to confer protection against other virulent pneumococci (11, 34), and the protein is able to synergize with other protective immunogens in systemic challenge models (32, 34, 37). Given the fact that PiuA provided solid protection in this study, it would be of interest in a future study to investigate if a combination of PdT and PiuA would afford superior protection over each antigen alone in a high-dose challenge model.
The current study provides further evidence that PiuA is a valid vaccine candidate against systemic pneumococcal disease, and it provides the first evidence that AliA is a promising protein vaccine candidate against systemic disease as well. We also provide proof-of-concept data that validate the in vivo microarray screen used to identify these promising vaccine candidates. The presence of elevated levels of antibody to PiuA in convalescent-phase sera of patients with pneumococcal septicemia, and the demonstration that the immune response elicited is serotype independent (51), supports this notion. This also strengthens our earlier suggestion that decisions on the best protein vaccine formulation that would proceed to clinical trials will require rigorous comparisons of individual antigens and all possible combinations thereof, using multiple mouse models and challenge strains (34). A careful examination of the literature shows that 15 of the 16 randomly selected genes (except the gene for CylM protein [Sp-1950]) listed in our analysis were identified by one or more, but not all, of the current screens for in vivo-expressed genes. This indicates that no single screening method is necessarily a stand-alone procedure but argues strongly in favor of our technique as valid, more robust, and more comprehensive, since it identified more candidate genes than other studies. Our technique also offers the advantage that it is less laborious and identifies key genes that are critical (either for survival or virulence) at each stage of the disease process. The screen also identified several other potentially interesting genes upregulated in discrete niches (at least 10 in the lungs and 8 in the blood) that are yet to be evaluated for their role in pathogenesis, and this presents avenues for future work. Such work would involve a thorough characterization of the critical regulatory pathways employed by the pneumococcus in discrete in vivo niches in accordance with host factors, nutrient availability, etc., and deployment of primary virulence determinants in those niches, leading to an improved understanding of pneumococcal pathogenesis, and potentially, identification of novel targets for intervention. We also propose that identification of the best candidates for an optimal pneumococcal protein vaccine formulation is likely to require a combination of existing and novel strategies (such as the one used in this study) used for antigen discovery. The in vivo transcriptomic technique described here could also be easily applied to novel antigen discovery in other pathogens such as S. pyogenes, Haemophilus influenzae type b, and Neisseria meningitidis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Meningitis Research Foundation (United Kingdom) Research Grant 0802.0 to A.D.O., J.C.P., and L.K.M., as well as by National Health and Medical Research Council of Australia (NHMRC) Program Grant 565526 to J.C.P. and Project Grant 627142 to J.C.P. and A.D.O.
We acknowledge BμG@S (the Bacterial Microarray Group at St George's, University of London) for supply of the microarray and advice and The Wellcome Trust for funding the multicollaborative microbial pathogen microarray facility under its Functional Genomics Resources Initiative. J.C.P. is an NHMRC Australia Fellow.
A.D.O., L.K.M., C.T., and J.C.P. conceived and designed the experiments; A.D.O., L.K.M., C.T., N.V., D.M., and C.D.P. performed the experiments; A.D.O., L.K.M., C. T., N.V., D.M., M.B.V.D.H., C.D.P., and J.C.P. analyzed the data; M.B.V.D.H. contributed analysis tools; A.D.O., L.K.M., C. T., N.V., D.M., M.B.V.D.H., and J.C.P. wrote the paper.
Footnotes
Published ahead of print 9 July 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Abbott DW, et al. 2010. The molecular basis of glycogen breakdown and transport in Streptococcus pneumoniae. Mol. Microbiol. 77:183–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander JE, et al. 1994. Immunization of mice with pneumolysin toxoid confers a significant degree of protection against at least nine serotypes of Streptococcus pneumoniae. Infect. Immun. 62:5683–5688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barocchi MA, Censini S, Rappuoli R. 2007. Vaccines in the era of genomics: the pneumococcal challenge. Vaccine 25:2963–2973 [DOI] [PubMed] [Google Scholar]
- 4. Bartilson M, et al. 2001. Differential fluorescence induction reveals Streptococcus pneumoniae loci regulated by competence stimulatory peptide. Mol. Microbiol. 39:126–135 [DOI] [PubMed] [Google Scholar]
- 5. Berry AM, Paton JC. 2000. Additive attenuation of virulence of Streptococcus pneumoniae by mutation of the genes encoding pneumolysin and other putative pneumococcal virulence proteins. Infect. Immun. 68:133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Briles DE, Novak L, Hotomi M, van Ginkel FW, King J. 2005. Nasal colonization with Streptococcus pneumoniae includes subpopulations of surface and invasive pneumococci. Infect. Immun. 73:6945–6951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brown JS, Gilliland SM, Holden DW. 2001. A Streptococcus pneumoniae pathogenicity island encoding an ABC transporter involved in iron uptake and virulence. Mol. Microbiol. 40:572–585 [DOI] [PubMed] [Google Scholar]
- 8. Brown JS, Ogunniyi AD, Woodrow MC, Holden DW, Paton JC. 2001. Immunization with components of two iron uptake ABC transporters protects mice against systemic Streptococcus pneumoniae infection. Infect. Immun. 69:6702–6706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cognet I, et al. 2003. Expression of recombinant proteins in a lipid A mutant of Escherichia coli BL21 with a strongly reduced capacity to induce dendritic cell activation and maturation. J. Immunol. Methods 272:199–210 [DOI] [PubMed] [Google Scholar]
- 10. Giefing C, et al. 2008. Discovery of a novel class of highly conserved vaccine antigens using genomic scale antigenic fingerprinting of pneumococcus with human antibodies. J. Exp. Med. 205:117–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harvey RM, Ogunniyi AD, Chen AY, Paton JC. 2011. Pneumolysin with low hemolytic activity confers an early growth advantage to Streptococcus pneumoniae in the blood. Infect. Immun. 79:4122–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hava DL, Camilli A. 2002. Large-scale identification of serotype 4 Streptococcus pneumoniae virulence factors. Mol. Microbiol. 45:1389–1406 [PMC free article] [PubMed] [Google Scholar]
- 13. Hava DL, LeMieux J, Camilli A. 2003. From nose to lung: the regulation behind Streptococcus pneumoniae virulence factors. Mol. Microbiol. 50:1103–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Horton RM, et al. 1993. Gene splicing by overlap extension. Methods Enzymol. 217:270–279 [DOI] [PubMed] [Google Scholar]
- 15. Hoskins J, et al. 2001. Genome of the bacterium Streptococcus pneumoniae strain R6. J. Bacteriol. 183:5709–5717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jomaa M, et al. 2005. Antibodies to the iron uptake ABC transporter lipoproteins PiaA and PiuA promote opsonophagocytosis of Streptococcus pneumoniae. Infect. Immun. 73:6852–6859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kerr AR, et al. 2004. The Ami-AliA/AliB permease of Streptococcus pneumoniae is involved in nasopharyngeal colonization but not in invasive disease. Infect. Immun. 72:3902–3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lau GW, et al. 2001. A functional genomic analysis of type 3 Streptococcus pneumoniae virulence. Mol. Microbiol. 40:555–571 [DOI] [PubMed] [Google Scholar]
- 19. LeMessurier KS, Ogunniyi AD, Paton JC. 2006. Differential expression of key pneumococcal virulence genes in vivo. Microbiology 152:305–311 [DOI] [PubMed] [Google Scholar]
- 20. Lipsitch M, et al. 2000. Competition among Streptococcus pneumoniae for intranasal colonization in a mouse model. Vaccine 18:2895–2901 [DOI] [PubMed] [Google Scholar]
- 21. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 22. Mahdi LK, Ogunniyi AD, LeMessurier KS, Paton JC. 2008. Pneumococcal virulence gene expression and host cytokine profiles during pathogenesis of invasive disease. Infect. Immun. 76:646–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marra A, et al. 2002. Differential fluorescence induction analysis of Streptococcus pneumoniae identifies genes involved in pathogenesis. Infect. Immun. 70:1422–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McAllister LJ, Ogunniyi AD, Stroeher UH, Leach AJ, Paton JC. 2011. Contribution of serotype and genetic background to virulence of serotype 3 and serogroup 11 pneumococcal isolates. Infect. Immun. 79:4839–4849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McCluskey J, Hinds J, Husain S, Witney A, Mitchell TJ. 2004. A two-component system that controls the expression of pneumococcal surface antigen A (PsaA) and regulates virulence and resistance to oxidative stress in Streptococcus pneumoniae. Mol. Microbiol. 51:1661–1675 [DOI] [PubMed] [Google Scholar]
- 26. Meinke A, Henics T, Hanner M, Minh DB, Nagy E. 2005. Antigenome technology: a novel approach for the selection of bacterial vaccine candidate antigens. Vaccine 23:2035–2041 [DOI] [PubMed] [Google Scholar]
- 27. Meng JP, et al. 2008. Identification of Streptococcus pneumoniae genes specifically induced in mouse lung tissues. Can. J. Microbiol. 54:58–65 [DOI] [PubMed] [Google Scholar]
- 28. Moffitt KL, et al. 2011. TH17-based vaccine design for prevention of Streptococcus pneumoniae colonization. Cell Host Microbe 9:158–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Molzen TE, et al. 2011. Genome-wide identification of Streptococcus pneumoniae genes essential for bacterial replication during experimental meningitis. Infect. Immun. 79:288–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. O'Brien KL, et al. 2009. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet 374:893–902 [DOI] [PubMed] [Google Scholar]
- 31. Oggioni MR, et al. 2006. Switch from planktonic to sessile life: a major event in pneumococcal pathogenesis. Mol. Microbiol. 61:1196–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ogunniyi AD, Folland RL, Briles DE, Hollingshead SK, Paton JC. 2000. Immunization of mice with combinations of pneumococcal virulence proteins elicits enhanced protection against challenge with Streptococcus pneumoniae. Infect. Immun. 68:3028–3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ogunniyi AD, Giammarinaro P, Paton JC. 2002. The genes encoding virulence-associated proteins and the capsule of Streptococcus pneumoniae are upregulated and differentially expressed in vivo. Microbiology 148:2045–2053 [DOI] [PubMed] [Google Scholar]
- 34. Ogunniyi AD, Grabowicz M, Briles DE, Cook J, Paton JC. 2007. Development of a vaccine against invasive pneumococcal disease based on combinations of virulence proteins of Streptococcus pneumoniae. Infect. Immun. 75:350–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ogunniyi AD, et al. 2009. Pneumococcal histidine triad proteins are regulated by the Zn2+-dependent repressor AdcR and inhibit complement deposition through the recruitment of complement factor H. FASEB J. 23:731–738 [DOI] [PubMed] [Google Scholar]
- 36. Ogunniyi AD, et al. 2010. Central role of manganese in regulation of stress responses, physiology, and metabolism in Streptococcus pneumoniae. J. Bacteriol. 192:4489–4497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ogunniyi AD, Woodrow MC, Poolman JT, Paton JC. 2001. Protection against Streptococcus pneumoniae elicited by immunization with pneumolysin and CbpA. Infect. Immun. 69:5997–6003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Orihuela CJ, Gao G, Francis KP, Yu J, Tuomanen EI. 2004. Tissue-specific contributions of pneumococcal virulence factors to pathogenesis. J. Infect. Dis. 190:1661–1669 [DOI] [PubMed] [Google Scholar]
- 39. Orihuela CJ, et al. 2003. Organ-specific models of Streptococcus pneumoniae disease. Scand. J. Infect. Dis. 35:647–652 [DOI] [PubMed] [Google Scholar]
- 40. Orihuela CJ, et al. 2004. Microarray analysis of pneumococcal gene expression during invasive disease. Infect. Immun. 72:5582–5596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Paton JC. 2004. New pneumococcal vaccines: basic science developments, p 382–402 In Tuomanen EI, Mitchell TJ, Morrison DA, Spratt BG. (ed), The pneumococcus. ASM Press, Washington, DC [Google Scholar]
- 42. Paton JC, Boslego JW. 2008. Protein vaccines, p 421–436 In Siber GR, Klugman KP, Makela PH. (ed), Pneumococcal vaccines: the impact of conjugate vaccines. ASM Press, Washington, DC [Google Scholar]
- 43. Paton JC, et al. 1991. Purification and immunogenicity of genetically obtained pneumolysin toxoids and their conjugation to Streptococcus pneumoniae type 19F polysaccharide. Infect. Immun. 59:2297–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Polissi A, et al. 1998. Large-scale identification of virulence genes from Streptococcus pneumoniae. Infect. Immun. 66:5620–5629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rappuoli R. 2000. Reverse vaccinology. Curr. Opin. Microbiol. 3:445–450 [DOI] [PubMed] [Google Scholar]
- 46. Smyth GK. 2004. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3:Article3 [DOI] [PubMed] [Google Scholar]
- 47. Smyth GK. 2005. Limma: linear models for microarray data, p 397–420 In Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W. (ed), Bioinformatics and computational biology solutions using R and Bioconductor. Springer, New York, NY [Google Scholar]
- 48. Smyth GK, Speed T. 2003. Normalization of cDNA microarray data. Methods 31:265–273 [DOI] [PubMed] [Google Scholar]
- 49. Tettelin H, et al. 2001. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 293:498–506 [DOI] [PubMed] [Google Scholar]
- 50. Weiser JN, Austrian R, Sreenivasan PK, Masure HR. 1994. Phase variation in pneumococcal opacity: relationship between colonial morphology and nasopharyngeal colonization. Infect. Immun. 62:2582–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Whalan RH, et al. 2005. PiuA and PiaA, iron uptake lipoproteins of Streptococcus pneumoniae, elicit serotype independent antibody responses following human pneumococcal septicaemia. FEMS Immunol. Med. Microbiol. 43:73–80 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.