

Abstract
Staphylococcus aureus pneumonia causes significant morbidity and mortality. Alpha-hemolysin (Hla), a pore-forming cytotoxin of S. aureus, has been identified through animal models of pneumonia as a critical virulence factor that induces lung injury. In spite of considerable molecular knowledge of how this cytotoxin injures the host, the precise host response to Hla in the context of infection remains poorly understood. We employed whole-genome expression profiling of infected lungs to define the host response to wild-type S. aureus compared with the response to an Hla-deficient isogenic mutant in experimental pneumonia. These data provide a complete expression profile at 4 and at 24 h postinfection, revealing a unique response to the toxin-expressing strain. Gene ontogeny analysis revealed significant differences in the extracellular matrix and cardiomyopathy pathways, both of which govern cellular interactions in the tissue microenvironment. Evaluation of individual transcript responses to Hla-secreting staphylococci was notable for upregulation of host cytokine and chemokine genes, including the p19 subunit of interleukin-23. Consistent with this observation, the cellular immune response to infection was characterized by a prominent Th17 response to the wild-type pathogen. These findings define specific host mRNA responses to Hla-producing S. aureus, coupling the pulmonary Th17 response to the secretion of this cytotoxin. Expression profiling to define the host response to a single virulence factor proved to be a valuable tool in identifying pathways for further investigation in S. aureus pneumonia. This approach may be broadly applicable to the study of bacterial toxins, defining host pathways that can be targeted to mitigate toxin-induced disease.
INTRODUCTION
Staphylococcus aureus is a Gram-positive human pathogen that causes a considerable burden of disease characterized by a spectrum of illness ranging from mild skin infections to life-threatening infections such as sepsis, pneumonia, endocarditis, and osteomyelitis (47). Pneumonia due to S. aureus is one of the most common invasive forms of disease, carrying an associated mortality rate up to 50% (1, 24, 34, 39). The combination of high incidence, high pathogenicity, and frequent resistance to multiple antibiotics results in S. aureus being one of the most significant human pathogens.
S. aureus harbors a number of virulence factors, including several cytolytic, pore-forming toxins (19, 21). Among the best studied of these toxins is alpha-hemolysin (Hla), a secreted toxin that assembles into a membrane-perforating homoheptamer upon binding to its eukaryotic cellular receptor, ADAM10 (5, 36, 67). Hla induces pulmonary hypertension and inflammation in several animal models (52, 66) and has been demonstrated to be essential in the pathogenesis of lethal pneumonia in a murine model, causing widespread alveolar injury and concomitant epithelial barrier disruption (9, 10). Several studies shed light on the mechanism by which Hla induces inflammation and lung damage. Hla induces platelet-activating factor production in endothelial cells (25, 69, 70) and release of nitric oxide and inflammatory mediators from pulmonary epithelium-derived cells (64). The toxin also promotes cytokine release from macrophages (56) and induces cell death and cytokine release from monocytes (4). Cell death is triggered through the NLRP3-dependent program of cellular necrosis, resulting in the release of endogenous proinflammatory molecules (15); indeed, NLRP3−/− mice display reduced interleukin-1β (IL-1β) production in response to Hla and demonstrate more mild clinicopathologic features of S. aureus pneumonia (37). Hla has been shown to injure alveolar epithelial cells by upregulation of the enzymatic activity of the toxin receptor ADAM10, leading to E cadherin cleavage and disruption of the intercellular adherens junction that is essential for intact epithelial barrier function (30, 75). Conversely, innate host protection from the effects of Hla is accomplished through activation of type I interferon signaling, recently shown to be mediated by phospholipid scramblase 1 (PLSCR1)-induced mitigation of cellular ATP loss (46). Together, these studies indicate that Hla targets critical host defenses provided by both innate immune cells and the tissue barrier and highlight the complexity of the host response to the toxin.
Neutralization of key virulence factors is one approach to find effective preventative and therapeutic agents to combat staphylococcal infection; indeed, immunization and small-molecule strategies targeting Hla afford a high degree of protection against lethal pneumonia (11, 61, 62). Similarly, antivirulence strategies associated with reduced expression of Hla protect against disease (59, 60, 73). In the setting of bacterial toxin-induced disease, a viable alternative approach may be modulation of the host response to minimize the deleterious effects of intoxication. This strategy circumvents the challenges associated with pathogen-acquired resistance, especially to small-molecule-based therapies. We hypothesized that whole-genome transcriptional analysis of lung tissue infected with wild-type (WT) or Hla-negative (Hla−) S. aureus would allow us to pinpoint host signature responses during the course of infection that were specifically linked to the presence of the toxin. Such studies were anticipated to enhance our understanding of the host-pathogen interaction in S. aureus pneumonia and suggest avenues for disease modification.
MATERIALS AND METHODS
Bacterial strains.
Staphylococcus aureus Newman WT and its isogenic toxin-deficient mutant, designated Hla−, have been previously described (9, 10).
Mouse model of lung infection.
Seven-week-old female C57BL/6J mice (Jackson Laboratories, Wilmington, MA) were inoculated via the intranasal route with either phosphate-buffered saline (PBS) or the S. aureus Newman or Hla− strain as described previously (9, 10). Following a 1:100 dilution of an overnight culture into fresh tryptic soy broth, the bacteria were grown with shaking at 37°C to an optical density of 0.5 at 660 nm. Culture aliquots (50 ml) were sedimented by centrifugation, and the bacteria were washed and resuspended in 1.0 to 1.5 ml of PBS. Animals utilized for microarray and transcription-based analyses received inocula ranging from 3 × 108 to 5 × 108 CFU, associated with clinical signs of severe lung disease. Animals utilized for T cell subset analyses received a sublethal inoculum of 1 × 108 CFU for initial infection, followed in 8 days by a second sublethal infection of 0.8 × 108 to 2 × 108 CFU and tissue harvesting at 24 h. Animals were anesthetized with ketamine and xylazine as previously described (49). After appropriate anesthesia was documented, 30 μl of the bacterial slurry was inoculated into the left nare and the animals were held upright for 1 min postinoculation. All animals were housed under standard conditions and provided food and water ad libitum. At time points of 4, 8, 12, or 24 h, animals underwent bronchoalveolar lavage fluid collection and were then sacrificed for removal of the lung tissue. All animal studies were performed in concordance with principles set forth by the Animal Welfare Act and the National Institutes of Health guidelines for the care and use of animals in biomedical research and were reviewed and approved by the University of Chicago Institutional Animal Care and Use Committee.
RNA isolation.
Lung tissue was snap-frozen in liquid nitrogen and stored at −80°C. At the time of processing, total mouse lung RNA was isolated by directly solubilizing lung tissue in TRIzol LS reagent (Life Technologies) using a Brinkman Polytron tissue disrupter. RNA was then purified using an RNeasy purification kit according to the manufacturer's protocol (Qiagen). Approximately 10 μg of purified, total RNA was used for analyses.
Microarray data analysis.
The microarray data analysis was performed using an Affymetrix GeneChip platform and Expression Analysis Manual protocols (Affymetrix Inc., Santa Clara, CA). Three mice were included for each condition. The signal intensity fluorescent images produced during Affymetrix GeneChip Mouse 430_2 array hybridizations were scanned using a Hewlett-Packard GeneArray G2500A scanner. We evaluated chip quality using GeneChip operating software (GCOS), dChip (43), and the affy package (6) in Bioconductor software. All RNA samples and chips used in this study passed established quality criteria (data not shown). The expression level of each probe set was summarized by the gcrma package in Bioconductor with GC robust multichip average (GCRMA) normalization (76). To identify differentially expressed genes, pairwise comparisons were conducted using the samr package (71) in Bioconductor. Only probe sets present (determined by the mas5calls function in the affy package in Bioconductor) in three replicates of at least one group were retained. The heat map, gene ontology biological process terms, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were generated using criteria of a false discovery rate (FDR) of <1% and a minimum of a 3-fold change between examined groups. The summary of the number of genes differentially regulated and tables of specific genes differing between S. aureus WT and Hla− infections, including the genes identified in the extracellular matrix (ECM) and cardiomyopathy pathways, were prepared using an FDR of <5% and a minimum of a 2-fold change. For probe sets with the same Entrez Gene or UniGene identifier, only the probe set with the lowest FDR or the highest fold changes in the gene list were included. We searched for any enriched KEGG (35) physiological pathways among the differential genes using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) (18, 29). A corrected P value of <0.05 (Fisher's exact test) after the Benjamini-Hochberg procedure (3) was used as the cutoff.
qRT-PCR analysis.
Quantative reverse transcription-PCR (qRT-PCR) was performed with the two-step reaction protocol using SsoFast EvaGreen supermix (Bio-Rad Laboratories, Hercules, CA). First-strand cDNAs were synthesized using an iScript Select cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA) at 42°C for 70 min, followed by 85°C for 5 min and a hold at 4°C. Actin was used as the endogenous control. Primer sets used for the quantitative PCR analysis included IL1r2 (interleukin 1 receptor type II) forward (For; 5′-GTTTCTGCTTTCACCACTCCA-3′) and reverse (Rev; 5′-GAGTCCAATTTACTCCAGGTCAG-3′), Ccl11 (chemokine [C-C motif] ligand 11) For (5′-GAATCACCAACAACAGATGCAC-3′) and Rev (5′-ATCCTGGACCCACTTCTTCTT-3′), Sdc4 (syndecan 4) For (5′-GTCCCCGGAGAGTCGATTC-3′) and Rev (5′-GCACCAAGGGCTCAATCACTT-3′), Figf (c-fos-induced growth factor) For (5′-TCACGCTCAGCATCCCATC-3′) and Rev (5′-ACTTCTACGCATGTCTCTCTAGG-3′), Col5a2 (procollagen type V alpha 2) For (5′-ACAGGTGAAGTGGGATTCTCA-3′) and Rev (5′-CCATAGCACCCATTGGACCA-3′), Tgfb3 (transforming growth factor beta 3) For (5′-CAGGCCAGGGCAGTCAGAG-3′) and Rev (5′-ATTTCCAGCCTAGATCCTGCC-3′), IL-17A (interleukin-17A) For (5′-CTCCAGAAGGCCCTCAGACTAC-3′) and Rev (5′-AGCTTTCCCTCCGCATTGACACAG-3′), and Actin For (5′-TGGCATTGTTACCAACTGGGACG-3′) and Rev (5′-GCTTCTCTTTGATGTCACGCACG-3′). All qRT-PCR assays were carried out using primer sets from Integrated DNA Technologies. qRT-PCR parameters used were as follows: 95°C for 30 s and then 95°C for 5 s and 55°C for 5 s for 40 cycles. A melting curve was constructed for verification of the specificity of PCR products by increasing the temperature from 60 to 95°C in 0.5°C-step increments. PCR was performed with SsoFast EvaGreen supermix, using a CFX96 optical reaction module and C1000 thermal cycler (Bio-Rad Laboratories, Hercules, CA). The relative fold change in gene expression was calculated following actin normalization using the comparative threshold cycle (CT) method as described previously (45, 65). The mean value for the control mice was used to calculate the fold change for each experimental mouse.
Lung processing for flow cytometry.
Mice were anesthetized and lungs were perfused with 6 ml PBS with 50 units/ml heparin injected through the right ventricle. Then, the lung was surgically excised and minced with sterile glass coverslips. The pieces were incubated in a shaker at 37°C for 30 min in complete Dulbecco modified Eagle medium (DMEM; Cellgro, Manassas, VA), 5% fetal bovine serum, 0.05 mM β-mercaptoethanol, 10 mM HEPES, 100 U/ml penicillin, and 100 μg/ml streptomycin with 146 U/ml collagenase I (Invitrogen, Carlsbad, CA). Samples were pipetted up and down 50 times to break up larger tissue sections, any remaining pieces of tissue were removed by straining the solution through a Nytex filter, and the cells in suspension were centrifuged and resuspended in complete DMEM. Cells were then resuspended in 44% Percoll (Sigma, St. Louis, MO) and spun in 44% and 66% Percoll gradients. Cells were collected at the interface of the two solutions. Live cell count was determined via trypan blue exclusion, and then the cells were divided for antibody staining and in vitro stimulation studies. For stimulation, cells were incubated overnight at 37°C in complete DMEM with IL-2 (20 U/ml) and then stimulated with phorbol myristate acetate (PMA; 20 ng/ml) plus ionomycin (400 ng/ml) for 6 h at 37°C, with brefeldin A (25 μg/ml) added for the final 2 h.
Flow cytometry.
Prior to staining with antibodies, cells were washed and nonspecific antibody binding was blocked with anti-FcγR (2.4G2) antibodies. Surface staining antibodies included anti-CD3 allophycocyanin (APC) (clone 145-2c11; eBioscience, San Diego, CA) and anti-GR1 fluorescein isothiocyanate (clone 1A8; BD Biosciences Pharmingen, San Diego, CA). For intracellular staining, cells were first surface stained for 30 to 60 min and then fixed with 4% paraformaldehyde for 15 min and washed in permeabilization buffer (0.25% saponin in fluorescence-activated cell sorter buffer [0.2% bovine serum albumin and 0.012% sodium azide in PBS]), followed by intracellular staining for 60 min at 4°C. Intracellular antibodies were anti-IL-17A phycoerythrin (PE) antibody (clone eBio17B7) or isotype control antibody rat IgG2a PE (eBioscience, San Diego, CA). The samples were then washed and flow cytometric analysis was performed with an LSRII apparatus (BD Biosciences, Mountain View, CA). Data were analyzed with FlowJo software (Tree Star, Ashland, OR).
Data analysis and statistics.
Data analysis and compilation were performed using Prism (version 5.0) software (GraphPad Software, San Diego, CA). Statistical analysis was performed using Student's t test, where a P value of <0.05 indicates a significant result.
Microarry data accession number.
The microarray data obtained in this study have been deposited in the NCBI Gene Expression Omnibus (GEO) site (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE38053.
RESULTS
S. aureus Hla induces unique features of the host response to lung infection.
To investigate the host response to Hla, we utilized an established mouse model of pneumonia in which 7-week-old C57BL/6J mice were infected intranasally with S. aureus Newman (9). In this model, lung injury is induced within the first 24 h postinfection, manifest as alveolar destruction, infiltration of large numbers of immune cells, especially neutrophils, and proliferation of S. aureus within the lung tissue. Mice infected with an Hla− variant of WT S. aureus demonstrate less severe lung inflammation, recovering from infection (9, 10). To first define the host transcriptional response to S. aureus pneumonia, we examined the gene expression profile in infected lung tissue using RNA prepared following infection with WT S. aureus and compared it to that in lung tissue following control treatment with PBS. Using the Affymetrix platform (Mouse Genome 430 [version 2.0] array) and a false discovery rate of <5% and requiring a minimum of a 2-fold change in gene expression, we identified 2,459 genes differentially expressed between WT-infected animals and the PBS controls (Table 1, top). A more pronounced effect was noted at the 24-h time point postinfection, wherein 7,306 genes were differentially expressed between WT-infected animals and control mice (Table 1, bottom). A total of 2,878 transcripts were upregulated, while 4,428 were downregulated at 24 h. Additional controls for these studies are shown in Table S1 in the supplemental material. Both WT S. aureus and the toxin-deficient strain induced a brisk host response to infection; at 4 h postinfection, the number of host genes with altered regulation was quantitatively similar (Table 1, top).
Table 1.
Comparison of gene expression for infection with WT S. aureus, Hla− isogenic mutant strain, or PBS control
| Time (h) | Treatment |
Gene expressiona |
|||
|---|---|---|---|---|---|
| Group 1 | Group 2 | Total | Upregulated | Downregulated | |
| 4 | WT | PBS | 2,459 | 1,430 | 1,029 |
| 4 | Hla− | PBS | 2,738 | 1,465 | 1,274 |
| 4 | WT | Hla− | 0 | 0 | 0 |
| 24 | WT | PBS | 7,306 | 2,878 | 4,428 |
| 24 | Hla− | PBS | 2,704 | 1,241 | 1,463 |
| 24 | WT | Hla− | 1,281 | 540 | 741 |
FDR < 5%; fold change ≥ 2; total, upregulated or downregulated for group 1.
To understand the host cellular pathways impacted by S. aureus infection, gene ontology analysis was performed by comparison of the genes differentially expressed between WT- and Hla−-infected mice and PBS control-treated mice (FDR < 1% and fold change > 3). Prominent biological processes altered by infection with either strain included the immune response, defense response, inflammatory response, response to wounding, and chemotaxis (see Fig. S1 in the supplemental material), ontologies very consistent with those described in published studies of infectious lung damage in rodent models (42, 53, 58). Consistent with this finding, no specific host pathways were identified within 4 h of infection to be uniquely affected by the expression of Hla on the basis of gene ontogeny analysis (see Fig. S2 in the supplemental material).
Microarray analysis demonstrated 2,704 differentially regulated genes at 24 h postinfection by comparison of mice infected with the S. aureus Hla− strain and PBS control-treated mice (1,241 upregulated and 1,463 downregulated genes; Table 1, bottom), a number smaller than the 7,306 differentially regulated genes seen for WT. In contrast to the analysis at 4 h, a comparison of the host response to infection with WT S. aureus and the Hla− strain at 24 h revealed a large number of differentially regulated genes (Table 1, bottom), with the WT strain inducing upregulation of 540 genes and downregulation of 741 genes compared to the gene induction by the Hla− strain. Both quantitative and qualitative differences in gene regulation induced by these infecting strains are appreciated by heat map analysis (Fig. 1). Gene ontogeny analysis revealed three pathways that differ significantly between WT infection and Hla− infection at 24 h: dilated cardiomyopathy, ECM receptor interaction, and hypertrophic cardiomyopathy (Fig. 2). The ECM is made of macromolecules such as collagens and elastin, as well as proteoglycans such as hyaluronan, providing a scaffold that supports cells and tissue structure. The pathway is also very important for the response to injury, modulating activities such as cell proliferation, differentiation, migration, and adhesion (8, 33). As seen in Table S2 in the supplemental material, collagens, integrins, and syndecans are among the genes differentially regulated in the presence of the toxin.
Fig 1.

Heat map of gene expression levels for differentially regulated genes in the mouse lung obtained by pairwise comparison between PBS control and WT infection at 24 h postinoculation. Criteria for analysis: FDR of <1% and a minimum of a 3-fold change. Red represents increased gene expression; blue represents downregulation.
Fig 2.

Top 10 significant KEGG pathways enriched by the differentially regulated genes in the mouse lung in response to WT S. aureus infection at 24 h postinoculation compared to PBS control. The corresponding results for the comparison between Hla− infection and PBS control are also listed. The differentially expressed genes with an FDR of <1% and a minimum of a 3-fold change were included in this analysis. The vertical dashed line indicates the cutoff of significance (P < 0.05). P values were corrected by the Benjamini-Hochberg procedure (3).
A more detailed and comprehensive analysis of specific genes from many pathways, not only the three mentioned above, that exhibit differential regulation in the presence of Hla was performed, revealing genes that were upregulated (Table 2) or downregulated (Table 3) by greater than 6-fold in WT infection compared to Hla− infection. qRT-PCR analysis for a subset of genes was performed on lung RNA prepared from mice infected with either WT or Hla− S. aureus, validating the findings of the microarray (see Fig. S3 in the supplemental material).
Table 2.
Genes upregulated during WT S. aureus infection compared to expression during Hla− infection at 24 ha
| Symbol | Gene name | Fold change |
|---|---|---|
| Ccl20 | Chemokine (C-C motif) ligand 20 | 56 |
| Cyp27b1 | Cytochrome P450, family 27, subfamily b, polypeptide 1 | 25 |
| Ccl11 | Chemokine (C-C motif) ligand 11 | 23 |
| Tmem45b | Transmembrane protein 45b | 23 |
| Aldh1a3 | Aldehyde dehydrogenase family 1, subfamily A3 | 22 |
| Cldn4 | Claudin 4 | 16 |
| Slc13a3 | Solute carrier family 13 (sodium-dependent dicarboxylate transporter), member 3 | 14 |
| Has1 | Hyaluronan synthase 1 | 11 |
| Ngp | Neutrophilic granule protein | 11 |
| Pitpnm2 | Phosphatidylinositol transfer protein, membrane associated 2 | 11 |
| Ceacam10 | Carcinoembryonic antigen-related cell adhesion molecule 10 | 10 |
| Csf3 | Colony-stimulating factor 3 (granulocyte) | 10 |
| Il18rap | Interleukin-18 receptor accessory protein | 9.8 |
| Kcnk5 | Potassium channel, subfamily K, member 5 | 9.5 |
| Fosl1 | Fos-like antigen 1 | 9.3 |
| Mctp2 | Multiple C2 domains, transmembrane 2 | 9.0 |
| Nipal1 | NIPA-like domain containing 1 | 8.9 |
| Sprr1a | Small proline-rich protein 1A | 8.8 |
| Ereg | Epiregulin | 8.5 |
| Il6 | Interleukin-6 | 8.4 |
| Dbn1 | Drebrin 1 | 7.9 |
| Lif | Leukemia inhibitory factor | 7.8 |
| Hamp | Hepcidin antimicrobial peptide | 7.7 |
| Cxcl1 | Chemokine (C-X-C motif) ligand 1 | 7.5 |
| Pcp4 | Purkinje cell protein 4 | 7.2 |
| Selp | Selectin, platelet | 7.1 |
| Zfp783 | Zinc finger protein 783 | 6.9 |
| Ptx3 | Pentraxin-related gene | 6.9 |
| Plekha7 | Pleckstrin homology domain containing, family A, member 7 | 6.8 |
| Ffar2 | Free fatty acid receptor 2 | 6.8 |
| Cd80 | CD80 antigen | 6.7 |
| Mbp | Myelin basic protein | 6.7 |
| Il23a | Interleukin-23, alpha subunit p19 | 6.7 |
| Artn | Artemin | 6.5 |
| Cldn1 | Claudin 1 | 6.3 |
FDR < 5%.
Table 3.
Genes downregulated during WT S. aureus infection compared to expression during Hla− infection at 24 ha
| Symbol | Gene name | Fold change |
|---|---|---|
| Adh1 | Alcohol dehydrogenase 1 (class I) | 0.040 |
| Aspn | Asporin | 0.055 |
| Ptprd | Protein tyrosine phosphatase, receptor type D | 0.058 |
| Lrrn4 | Leucine rich repeat neuronal 4 | 0.066 |
| Figf | c-Fos-induced growth factor | 0.068 |
| Lphn3 | Latrophilin 3 | 0.074 |
| Col8a1 | Collagen, type viii, alpha 1 | 0.076 |
| Hhip | Hedgehog-interacting protein | 0.092 |
| Prkg2 | Protein kinase, cyclic GMP dependent, type II | 0.10 |
| Aard | Alanine- and arginine-rich domain-containing protein | 0.10 |
| Timp4 | Tissue inhibitor of metalloproteinase 4 | 0.11 |
| Kcnj13 | Potassium inwardly rectifying channel, subfamily J, member 13 | 0.11 |
| Mfap5 | Microfibrillum-associated protein 5 | 0.11 |
| Fzd2 | Frizzled homolog 2 (Drosophila) | 0.11 |
| Prrx1 | Paired related homeobox 1 | 0.11 |
| Gria3 | Glutamate receptor, ionotropic, AMPA3 (alpha 3) | 0.11 |
| Stmn2 | Stathmin-like 2 | 0.11 |
| Angpt1 | Angiopoietin 1 | 0.11 |
| Sema3a | Sema domain, immunoglobulin domain (Ig), short basic domain, secreted (semaphorin) 3A | 0.12 |
| Nox4 | NADPH oxidase 4 | 0.12 |
| Speg | SPEG complex locus | 0.12 |
| Sfrp1 | Secreted frizzled-related protein 1 | 0.12 |
| Agr2 | Anterior gradient 2 (Xenopus laevis) | 0.12 |
| Cckar | Cholecystokinin A receptor | 0.12 |
| Wisp2 | WNT1-inducible signaling pathway protein 2 | 0.12 |
| Synm | Synemin, intermediate filament protein | 0.13 |
| Fam38b | Family with sequence similarity 38, member B | 0.13 |
| Osbpl6 | Oxysterol binding protein-like 6 | 0.13 |
| Gas1 | Growth arrest specific 1 | 0.14 |
| Pcdh12 | Protocadherin 12 | 0.14 |
| Vgll3 | Vestigial like 3 (Drosophila) | 0.14 |
| Gulp1 | GULP, engulfment adaptor PTB domain containing 1 | 0.14 |
| Tppp | Tubulin polymerization-promoting protein | 0.14 |
| Rspo1 | R-spondin homolog (Xenopus laevis) | 0.14 |
| Decr1 | 1,2-Dienoyl coenzyme A reductase 1, mitochondrial | 0.15 |
FDR < 5%.
Patterning of T cell response to S. aureus pneumonia by Hla.
To appreciate the biological significance of microarray-based findings of differential host responses to Hla-expressing S. aureus, we chose to further examine the upregulated expression of IL-23 p19 observed in WT infection compared to Hla− infection (6.7-fold increase; Table 2). Production of IL-23 in the context of infection and autoimmunity has been linked to the differentiation of IL-17-producing CD4+ T helper cells, or a Th17 response (7). The secretion of IL-17 in the context of infection promotes the mobilization of neutrophils, augments cytokine production by epithelial cells, and maintains the integrity of the tight junction. Together, these responses enhance protective immunity to extracellular pathogens (16, 38, 51, 74). IL-17 is produced both by Th17 cells and by innate immune cells as part of the immediate host defense response to bacteria. These responses are of particular relevance at epithelial surfaces, key sites of human infection with S. aureus. Quantitative changes in the IL-23 p19 transcript were of considerable interest to us, given our prior studies of vaccine-mediated immunity to Hla and our ongoing efforts to define novel strategies by which to protect the host against toxin-mediated injury (11, 30). Th17 responses have been demonstrated to be important in the context of S. aureus skin infection (14, 55) and influenza virus coinfection (40). Th17-dominant responses have been suggested to play an important role in adaptive immunity to S. aureus (44) and have recently been highlighted to be a potential means by which to elicit vaccine-mediated immunity (68).
qRT-PCR for IL-23 p19 was performed on RNA harvested from lungs of mice infected with WT or Hla− S. aureus (or treated with PBS control) at four different time points. IL-23 p19 was upregulated more than 60-fold following infection with both WT and Hla− strains as early as 4 h after inoculation (Fig. 3). At 8, 12, and 24 h, however, WT infection was characterized by amplification and persistence of the IL-23 p19 transcriptional response greater than those obtained with Hla− infection (Fig. 3). Commensurate with these findings, the IL-17A transcriptional response was also greater in WT-infected mice than Hla−-infected mice at 24 h (see Fig. S4 in the supplemental material). These data indicate that the expression of IL-23 p19 is associated in part with the presence of Hla and suggest that the toxin may shape the nature of the host T cell response.
Fig 3.
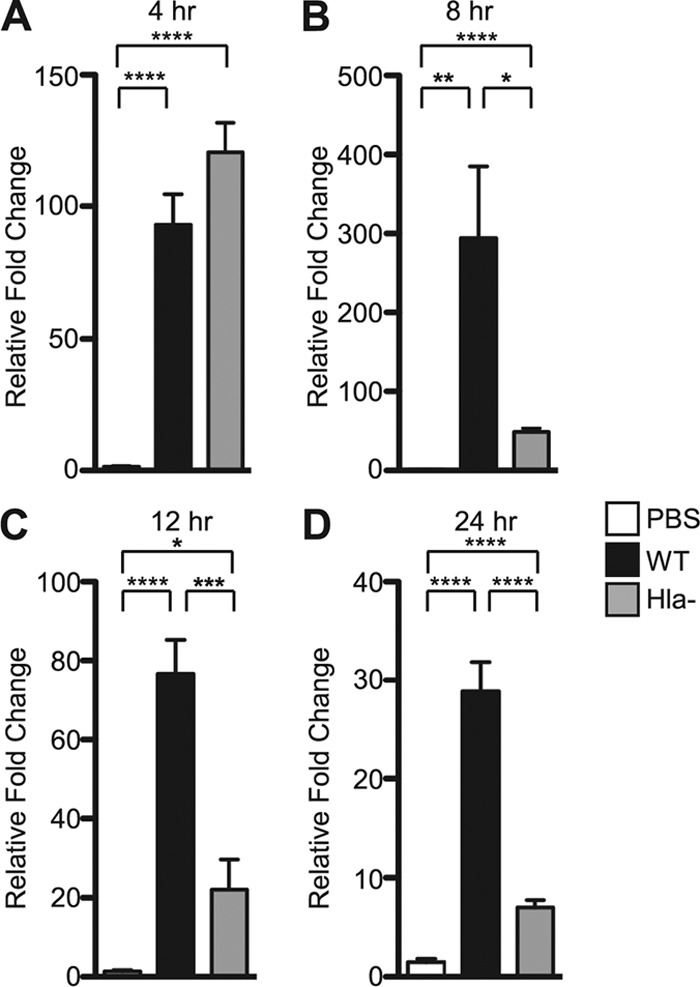
Sustained IL-23 p19 gene expression in S. aureus pneumonia depends on bacterial expression of Hla. qRT-PCR analysis of IL-23 p19 gene expression in the mouse lung in response to infection with WT or Hla− S. aureus or PBS control at 4 h (A), 8 h (B), 12 h (C), and 24 h (D) postinoculation. Bars represent average ± SEM. P values are indicated: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
To examine the adaptive T cell immune response to S. aureus pneumonia, mice were infected with a sublethal dose of WT or Hla− S. aureus and allowed to recover. Eight days later, mice were reinfected with the same staphylococcal strain. The mice were then sacrificed at 24 h postinfection and lungs were harvested for cellular analysis. A fraction of the recovered cells was subjected to flow cytometric analysis of CD3 and GR1 expression to quantify T cell and neutrophil infiltration into the lung, respectively. Mice infected with WT S. aureus demonstrated an increased overall cell count in the lung compared to that for mice infected with the Hla− strain, consistent with our prior observations (Fig. 4A) (30). The absolute number of T cells and neutrophils was significantly higher in the WT infection than the Hla− infection (Fig. 4B and C). Given the initial finding that the IL-23 p19 transcript was markedly increased in mice infected with WT S. aureus compared to those infected with the Hla− strain, we examined the proportion of IL-17-positive (IL-17+) T cells as a subset of the CD3+ population. To facilitate these studies, harvested cells were plated overnight in the presence of IL-2, stimulated with PMA and ionomycin, and then analyzed for IL-17 expression. The absolute number of IL-17+ cells was higher in WT-infected mice (Fig. 5A to D), consistent with an overall increase in cell recovery. More importantly, the percentage of CD3+/IL-17+ cells was significantly higher in mice infected with WT S. aureus (Fig. 5E to H), suggesting that Hla expression in the context of staphylococcal pneumonia induces a qualitatively distinct T cell response, enhancing the generation of the Th17 population. Specificity of staining was confirmed by staining of lung cells from WT-infected mice with the isotype control antibodies (data not shown). Collectively, these data demonstrate that Hla is a strong inducer of Th17 cells during staphylococcal pneumonia.
Fig 4.
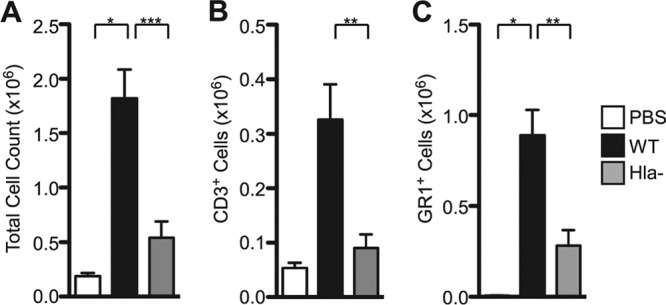
Infection with WT S. aureus is associated with increased immune cell recovery from lung tissue. Histogram plots of total cell count (A), CD3+ T cells (B), and GR1-positive (GR1+) neutrophils (C) recovered from the lungs of mice infected with WT S. aureus, Hla− S. aureus, or PBS control. Bars represent average ± SEM. Results are derived from 4 PBS-treated mice, 21 WT-infected mice, and 14 Hla−-infected mice. P values are indicated: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Fig 5.

Hla expression is required for induction of a pulmonary Th17 response to S. aureus infection. Flow cytometric analysis of cells stimulated with PMA and ionomycin and then stained with anti-CD3 allophycocyanin (APC) and anti-IL-17 PE. (A to C) Live cell gate from mouse lung following infection with PBS (A), WT S. aureus (B), or Hla− S. aureus (C). (D) Histogram quantifying IL-17+ cells recovered from the lungs of mice. (E to G) CD3+ lymphocyte gate from mouse lung following infection with PBS (E), WT S. aureus (F), or Hla− S. aureus (G). Representative plots are shown. The percentage of IL-17+ cells is indicated. (H) Histogram of the percentage CD3+ cells that are IL-17+. Bars represent the average ± SEM. Results are derived from 4 PBS-treated mice, 21 WT-infected mice, and 14 Hla−-infected mice. P values are indicated: *, P < 0.05; ***, P < 0.001; ****, P < 0.0001.
DISCUSSION
We have used a whole-transcriptome approach as a tool to discern the impact of a single bacterial virulence factor on the host response to infection. S. aureus is unique among bacterial pathogens, given the multitude of organ systems and tissues that can be targeted during infection and the pleiotropic manifestations of disease apparent in different individuals. The wide array of virulence factors elaborated by S. aureus clearly patterns these diverse disease states. The pathological features of S. aureus pneumonia observed in animal model systems are intimately linked with the expression of Hla, providing a window of opportunity to dissect the host response to toxin exposure.
We observed that the general host response to both wild-type and toxin-deficient S. aureus is characterized by alterations in genes that regulate the biological processes of inflammation, defense, the immune response, wounding, and chemotaxis, extending earlier findings (2, 58). Specific alterations observed following expression with the Hla-expressing strain, however, serve to highlight the unique role of the toxin in host injury. Gene ontogeny analysis revealed that the extracellular matrix and cardiomyopathy pathways have significantly different regulation in the presence of Hla. These findings are consistent with several prior observations, namely, that staphylococcal isolates involved in necrotizing pneumonia display increased adhesion to extracellular matrix molecules (17) and that the Hla−–ADAM10 complex is associated with disruption of focal adhesions and intercellular adherens junction formation (30, 75). Future studies of the roles of differentially regulated genes in the ECM and cardiomyopathy pathways will likely lead to a more comprehensive understanding of the mechanisms of toxin-induced lung damage, perhaps also affording insight into the nascent mechanisms that underlie the host reparative response to injury.
The relevance of IL-17 to pulmonary infection has previously been established for Gram-negative bacteria (27, 28, 41, 77), and we have now examined IL-17 in the context of pneumonia due to a Gram-positive organism. Our focus on the Th17 response as a specific biological response pathway to intoxication was predicated on knowledge of the increased susceptibility of humans and mice lacking in this response to infection with S. aureus (48, 63), the association of Th17 cells with the host response to S. aureus (14, 23, 26, 31, 40), and the more recent findings from Niebuhr and colleagues that S. aureus Hla induced IL-17A in peripheral blood mononuclear cells and Th17 cells in the setting of atopic dermatitis (55).
Host recognition of staphylococcal cell wall components and bacterial lipopeptides leads to activation of Toll-like receptor (TLR) and Nod-like receptor (NLR) signaling on antigen-presenting cells such as monocytes and dendritic cells (12, 22, 23, 54, 72). These innate cellular responses pattern cytokine and chemokine production, immune cell recruitment, and the induction of the adaptive immune response. Collectively, these host responses clearly establish the balance between favorable immunologic clearance of the pathogen and deleterious immunopathology that contributes to host death in the setting of lung infection (2, 11, 15, 37, 50, 57, 72). Relevant to the current study, these early signaling events result in the production of IL-23, templating induction of the Th17 response (13, 20, 32, 51, 72).
The link between expression of Hla and the Th17 response observed in our studies may be accounted for by one of several molecular mechanisms. First, we have demonstrated previously that S. aureus recovery from the lungs of infected mice is reduced when the cellular action of Hla is antagonized during infection (11, 61, 62). In this context, the tissue bacterial load may emerge as a critical regulator of the host response. Infection with an Hla− S. aureus strain would be predicted to provide a lesser degree of innate immune stimulation by peptidoglycan and lipopeptides, dampening the magnitude or duration of the IL-23/IL-17 response. Indeed, we observed that the induction of IL-23 by WT and Hla− S. aureus was indiscernible at 4 h postinfection and then rapidly waned in response to the Hla− strain. Interestingly, a considerable number of staphylococci (≥5 × 106/lung) are still recovered at 24 h postinfection in the setting of Hla antagonism (11, 61, 62), suggesting that the nature of the host response may be determined within a relatively narrow window predicated on both the absolute burden of S. aureus in the tissue and the length of time over which the host is exposed.
As a second plausible mechanism, the direct actions of Hla on antigen-presenting cells may shape the host adaptive response. This may be accomplished by (i) stimulation of the NLRP3 inflammasome by the toxin (15, 37), (ii) enhanced inflammatory cytokine and/or chemokine production in response to lung epithelial injury caused by Hla (2, 11), or (iii) toxin-mediated cytolytic injury to innate antigen-presenting cells (4, 56). Deciphering between these possibilities will likely shed additional light on both the nature of the antistaphylococcal response and the role of Hla in patterning this response.
We have now extended previous studies by connecting the presence of a secreted cytotoxin to the pulmonary Th17 response in staphylococcal pneumonia. This emphasizes the importance of Hla in disease pathogenesis, most specifically, in contemplating the nature of the host immunologic response. Through these studies, we have demonstrated microarray technology and gene ontogeny analysis to be a powerful tool in the selective identification of toxin-responsive pathways, highlighting a number of toxin-induced cellular signatures for ongoing study. As Hla has emerged as a strategic target for vaccine and passive immunotherapy approaches to S. aureus disease, understanding how this toxin primes the host immune system is anticipated to inform the rational design of strategies capable of specifically enhancing the generation of adaptive immunity.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Departments of Pathology, Pediatrics, and Microbiology at the University of Chicago and a grant from the National Institute of Allergy and Infectious Diseases to J.B.W. (grant AI097434). J.B.W. also acknowledges membership in and support from the Region V Great Lakes RCE (NIH award 2-U54-AI-057153).
We thank Jesse Williams and Anne Sperling for providing protocols and advice for flow cytometry experiments, Ting Wang for advice on qRT-PCR studies, Ryan Duggan and David Leclerc for flow cytometry expertise, and Georgia Sampedro for assistance with the preparation of figures.
Footnotes
Published ahead of print 25 June 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Athanassa Z, Siempos, Falagas ME. 2008. Impact of methicillin resistance on mortality in Staphylococcus aureus VAP: a systematic review. Eur. Respir. J. 31:625–632 [DOI] [PubMed] [Google Scholar]
- 2. Bartlett AH, Foster TJ, Hayashida A, Park PW. 2008. Alpha-toxin facilitates the generation of CXC chemokine gradients and stimulates neutrophil homing in Staphylococcus aureus pneumonia. J. Infect. Dis. 198:1529–1535 [DOI] [PubMed] [Google Scholar]
- 3. Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. B 57:289–300 [Google Scholar]
- 4. Bhakdi S, Muhly M, Korom S, Hugo F. 1989. Release of interleukin-1 beta associated with potent cytocidal action of staphylococcal alpha-toxin on human monocytes. Infect. Immun. 57:3512–3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bhakdi S, Tranum-Jensen J. 1991. Alpha-toxin of Staphylococcus aureus. Microbiol. Rev. 55:733–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bolstad BM, Irizarry RA, Astrand M, Speed TP. 2003. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19:185–193 [DOI] [PubMed] [Google Scholar]
- 7. Boniface K, Blom B, Liu YJ, de Waal Malefyt R. 2008. From interleukin-23 to T-helper 17 cells: human T-helper cell differentiation revisited. Immunol. Rev. 226:132–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bowers SL, Banerjee I, Baudino TA. 2010. The extracellular matrix: at the center of it all. J. Mol. Cell. Cardiol. 48:474–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bubeck Wardenburg J, Bae T, Otto M, Deleo FR, Schneewind O. 2007. Poring over pores: alpha-hemolysin and Panton-Valentine leukocidin in Staphylococcus aureus pneumonia. Nat. Med. 13:1405–1406 [DOI] [PubMed] [Google Scholar]
- 10. Bubeck Wardenburg J, Patel RJ, Schneewind O. 2007. Surface proteins and exotoxins are required for the pathogenesis of Staphylococcus aureus pneumonia. Infect. Immun. 75:1040–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bubeck Wardenburg J, Schneewind O. 2008. Vaccine protection against Staphylococcus aureus pneumonia. J. Exp. Med. 205:287–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bubeck Wardenburg J, Williams WA, Missiakas D. 2006. Host defenses against Staphylococcus aureus infection require recognition of bacterial lipoproteins. Proc. Natl. Acad. Sci. U. S. A. 103:13831–13836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen K, et al. 2011. Th17 cells mediate clade-specific, serotype-independent mucosal immunity. Immunity 35:997–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cho JS, et al. 2010. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J. Clin. Invest. 120:1762–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Craven RR, et al. 2009. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS One 4:e7446 doi:10.1371/journal.pone.0007446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cua DJ, Tato CM. 2010. Innate IL-17-producing cells: the sentinels of the immune system. Nat. Rev. Immunol. 10:479–489 [DOI] [PubMed] [Google Scholar]
- 17. de Bentzmann S, et al. 2004. Staphylococcus aureus isolates associated with necrotizing pneumonia bind to basement membrane type I and IV collagens and laminin. J. Infect. Dis. 190:1506–1515 [DOI] [PubMed] [Google Scholar]
- 18. Dennis G, Jr, et al. 2003. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4:P3 doi:10.1186/gb-2003-4-5-p3 [PubMed] [Google Scholar]
- 19. Diep BA, Otto M. 2008. The role of virulence determinants in community-associated MRSA pathogenesis. Trends Microbiol. 16:361–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dubin PJ, et al. 2012. Interleukin-23-mediated inflammation in Pseudomonas aeruginosa pulmonary infection. Infect. Immun. 80:398–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Engelmann S, Hecker M. 2009. A proteomics view of virulence factors of Staphylococcus aureus. Genome Dyn. 6:187–197 [DOI] [PubMed] [Google Scholar]
- 22. Fournier B, Philpott DJ. 2005. Recognition of Staphylococcus aureus by the innate immune system. Clin. Microbiol. Rev. 18:521–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Frodermann V, et al. 2011. A modulatory interleukin-10 response to staphylococcal peptidoglycan prevents Th1/Th17 adaptive immunity to Staphylococcus aureus. J. Infect. Dis. 204:253–262 [DOI] [PubMed] [Google Scholar]
- 24. Gillet Y, et al. 2007. Factors predicting mortality in necrotizing community-acquired pneumonia caused by Staphylococcus aureus containing Panton-Valentine leukocidin. Clin. Infect. Dis. 45:315–321 [DOI] [PubMed] [Google Scholar]
- 25. Grimminger F, et al. 1997. Human endothelial cell activation and mediator release in response to the bacterial exotoxins Escherichia coli hemolysin and staphylococcal alpha-toxin. J. Immunol. 159:1909–1916 [PubMed] [Google Scholar]
- 26. Henningsson L, et al. 2010. Interleukin-17A during local and systemic Staphylococcus aureus-induced arthritis in mice. Infect. Immun. 78:3783–3790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hoshino H, et al. 2000. Increased elastase and myeloperoxidase activity associated with neutrophil recruitment by IL-17 in airways in vivo. J. Allergy Clin. Immunol. 105:143–149 [DOI] [PubMed] [Google Scholar]
- 28. Hoshino H, Lotvall J, Skoogh BE, Linden A. 1999. Neutrophil recruitment by interleukin-17 into rat airways in vivo. Role of tachykinins. Am. J. Respir. Crit. Care Med. 159:1423–1428 [DOI] [PubMed] [Google Scholar]
- 29. Huang da W, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4:44–57 [DOI] [PubMed] [Google Scholar]
- 30. Inoshima I, et al. 2011. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med. 17:1310–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Islander U, et al. 2010. Superantigenic Staphylococcus aureus stimulates production of interleukin-17 from memory but not naive T cells. Infect. Immun. 78:381–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ivanov S, et al. 2007. Functional relevance of the IL-23-IL-17 axis in lungs in vivo. Am. J. Respir. Cell Mol. Biol. 36:442–451 [DOI] [PubMed] [Google Scholar]
- 33. Jarvelainen H, Sainio A, Koulu M, Wight TN, Penttinen R. 2009. Extracellular matrix molecules: potential targets in pharmacotherapy. Pharmacol. Rev. 61:198–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kallen AJ, et al. 2010. Staphylococcus aureus community-onset pneumonia in patients admitted to children's hospitals during autumn and winter of 2006–2007. Epidemiol. Infect. 138:666–672 [DOI] [PubMed] [Google Scholar]
- 35. Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. 2004. The KEGG resource for deciphering the genome. Nucleic Acids Res. 32:D277–D280 doi:10.1093/nar/gkh063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kawate T, Gouaux E. 2003. Arresting and releasing staphylococcal alpha-hemolysin at intermediate stages of pore formation by engineered disulfide bonds. Protein Sci. 12:997–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kebaier C, et al. 2012. Staphylococcus aureus alpha-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the NLRP3 inflammasome. J. Infect. Dis. 205:807–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Khader SA, Gaffen SL, Kolls JK. 2009. Th17 cells at the crossroads of innate and adaptive immunity against infectious diseases at the mucosa. Mucosal Immunol. 2:403–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Klevens RM, et al. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771 [DOI] [PubMed] [Google Scholar]
- 40. Kudva A, et al. 2011. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J. Immunol. 186:1666–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Laan M, et al. 1999. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J. Immunol. 162:2347–2352 [PubMed] [Google Scholar]
- 42. Lewis CC, et al. 2008. Disease-specific gene expression profiling in multiple models of lung disease. Am. J. Respir. Crit. Care Med. 177:376–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li C, Hung Wong W. 2001. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol. 2:RESEARCH0032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lin L, et al. 2009. Th1-Th17 cells mediate protective adaptive immunity against Staphylococcus aureus and Candida albicans infection in mice. PLoS Pathog. 5:e1000703 doi:10.1371/journal.ppat.1000703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2-deltadeltaCT method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 46. Lizak M, Yarovinsky TO. 2012. Phospholipid scramblase 1 mediates type I interferon-induced protection against staphylococcal alpha-toxin. Cell Host Microbe 11:70–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lowy FD. 1998. Staphylococcus aureus infections. N. Engl. J. Med. 339:520–532 [DOI] [PubMed] [Google Scholar]
- 48. Ma CS, et al. 2008. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J. Exp. Med. 205:1551–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marketon MM, DePaolo RW, DeBord KL, Jabri B, Schneewind O. 2005. Plague bacteria target immune cells during infection. Science 309:1739–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Martin FJ, Parker D, Harfenist BS, Soong G, Prince A. 2011. Participation of CD11c(+) leukocytes in methicillin-resistant Staphylococcus aureus clearance from the lung. Infect. Immun. 79:1898–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McAleer JP, Kolls JK. 2011. Mechanisms controlling Th17 cytokine expression and host defense. J. Leukoc. Biol. 90:263–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McElroy MC, et al. 1999. Alpha-toxin damages the air-blood barrier of the lung in a rat model of Staphylococcus aureus-induced pneumonia. Infect. Immun. 67:5541–5544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Montgomery CP, Daum RS. 2009. Transcription of inflammatory genes in the lung after infection with community-associated methicillin-resistant Staphylococcus aureus: a role for Panton-Valentine leukocidin? Infect. Immun. 77:2159–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Muller-Anstett MA, et al. 2010. Staphylococcal peptidoglycan co-localizes with Nod2 and TLR2 and activates innate immune response via both receptors in primary murine keratinocytes. PLoS One 5:e13153 doi:10.1371/journal.pone.0013153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Niebuhr M, et al. 2011. Staphylococcal alpha-toxin is a strong inducer of interleukin-17 in humans. Infect. Immun. 79:1615–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Onogawa T. 2002. Staphylococcal alpha-toxin synergistically enhances inflammation caused by bacterial components. FEMS Immunol. Med. Microbiol. 33:15–21 [DOI] [PubMed] [Google Scholar]
- 57. Parker D, Prince A. 2012. Immunopathogenesis of Staphylococcus aureus pulmonary infection. Semin. Immunopathol. 34:281–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pennings JL, Kimman TG, Janssen R. 2008. Identification of a common gene expression response in different lung inflammatory diseases in rodents and macaques. PLoS One 3:e2596 doi:10.1371/journal.pone.0002596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Qiu J, et al. 2011. Isoalantolactone protects against Staphylococcus aureus pneumonia. FEMS Microbiol. Lett. 324:147–155 [DOI] [PubMed] [Google Scholar]
- 60. Qiu J, et al. 2012. Capsaicin protects mice from community-associated methicillin-resistant Staphylococcus aureus pneumonia. PLoS One 7:e33032 doi:10.1371/journal.pone.0033032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ragle BE, Bubeck Wardenburg J. 2009. Anti-alpha-hemolysin monoclonal antibodies mediate protection against Staphylococcus aureus pneumonia. Infect. Immun. 77:2712–2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ragle BE, Karginov VA, Bubeck Wardenburg J. 2010. Prevention and treatment of Staphylococcus aureus pneumonia with a beta-cyclodextrin derivative. Antimicrob. Agents Chemother. 54:298–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Renner ED, et al. 2008. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. J. Allergy Clin. Immunol. 122:181–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rose F, et al. 2002. Mediator generation and signaling events in alveolar epithelial cells attacked by S. aureus alpha-toxin. Am. J. Physiol. Lung Cell. Mol. Physiol. 282:L207–L214 [DOI] [PubMed] [Google Scholar]
- 65. Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3:1101–1108 [DOI] [PubMed] [Google Scholar]
- 66. Seeger W, et al. 1990. Staphylococcal alpha-toxin-induced vascular leakage in isolated perfused rabbit lungs. Lab. Invest. 63:341–349 [PubMed] [Google Scholar]
- 67. Song L, et al. 1996. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 274:1859–1866 [DOI] [PubMed] [Google Scholar]
- 68. Spellberg B, Daum R. 2010. A new view on development of a Staphylococcus aureus vaccine: insights from mice and men. Hum. Vaccin. 6:857–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Suttorp N, Buerke M, Tannert-Otto S. 1992. Stimulation of PAF-synthesis in pulmonary artery endothelial cells by Staphylococcus aureus alpha-toxin. Thromb. Res. 67:243–252 [DOI] [PubMed] [Google Scholar]
- 70. Suttorp N, Fuhrmann M, Tannert-Otto S, Grimminger F, Bhadki S. 1993. Pore-forming bacterial toxins potently induce release of nitric oxide in porcine endothelial cells. J. Exp. Med. 178:337–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tusher VG, Tibshirani R, Chu G. 2001. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U. S. A. 98:5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Volz T, et al. 2010. Natural Staphylococcus aureus-derived peptidoglycan fragments activate NOD2 and act as potent costimulators of the innate immune system exclusively in the presence of TLR signals. FASEB J. 24:4089–4102 [DOI] [PubMed] [Google Scholar]
- 73. Wang J, et al. 2011. Chrysin protects mice from Staphylococcus aureus pneumonia. J. Appl. Microbiol. 111:1551–1558 [DOI] [PubMed] [Google Scholar]
- 74. Wilke CM, Bishop K, Fox D, Zou W. 2011. Deciphering the role of Th17 cells in human disease. Trends Immunol. 32:603–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wilke GA, Bubeck Wardenburg J. 2010. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc. Natl. Acad. Sci. U. S. A. 107:13473–13478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wu Z, Irizarry RA, Gentleman R, Martinez-Murillo F, Spencer F. 2004. A model-based background adjustment for oligonucleotide expression arrays. J. Am. Statist. Assoc. 99:909–917 [Google Scholar]
- 77. Ye P, et al. 2001. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J. Exp. Med. 194:519–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.