

Abstract
While ribosomes must maintain translational reading frame in order to translate primary genetic information into polypeptides, cis‐acting signals located in mRNAs represent higher order information content that can be used to fine‐tune gene expression. Classes of signals have been identified that direct a fraction of elongating ribosomes to shift reading frame by one base in the 5′ (−1) or 3′ (+1) direction. This is called programmed ribosomal frameshifting (PRF). Although mechanisms of PRF differ, a common feature is induction of ribosome pausing, which alters kinetic partitioning rates between in‐frame and out‐of‐frame codons at specific ‘slippery’ sequences. Many viruses use PRF to ensure synthesis of the correct ratios of virus‐encoded proteins required for proper viral particle assembly and maturation, thus identifying PRF as an attractive target for antiviral therapeutics. In contrast, recent studies indicate that PRF signals may primarily function as mRNA destabilizing elements in cellular mRNAs. These studies suggest that PRF may be used to fine‐tune gene expression through mRNA decay pathways. The possible regulation of PRF by noncoding RNAs is also discussed. WIREs RNA 2012 doi: 10.1002/wrna.1126
This article is categorized under:
-
1
RNA Structure and Dynamics > Influence of RNA Structure in Biological Systems
-
2
RNA Evolution and Genomics > Computational Analyses of RNA
-
3
Translation > Translation Regulation
INTRODUCTION
The Players: mRNAs, tRNAs, and the Ribosome
The primary genetic information contained in messenger RNA (mRNA) is bundled into packets of three nucleotides termed codons (reviewed in Ref 1). Each codon is recognized by the anticodon loop of a specific transfer RNA (tRNA), the 3′ ends of which are charged with specific amino acids. Thus, each codon encodes a specific amino acid (or information instructing the translational apparatus to terminate translation). The ribosome is an ancient molecular machine that brings mRNAs and tRNAs together to synthesize the proteins encoded by the mRNAs using the amino acids supplied by the tRNAs (reviewed in Ref 2). The ribosome is composed of multiple RNAs (called ribosomal RNAs, rRNAs) and proteins divided into two separate subunits (SU), termed large (LSU) and small (SSU). mRNAs are threaded through the SSU of the ribosome, and the stepwise, codon by codon, progression of the ribosome along the mRNA from the 5′ to 3′ direction is a process called translocation. The direct interaction between mRNA and tRNAs occurs on the surface of the SSU in a region called the decoding center: here the codons of mRNAs form stable base pairing interactions with the anticodon loops of tRNAs. A second functional center located in the LSU is called the peptidyltransferase center (PTC). Here, the 3′ ends of tRNAs are brought into close proximity with one another in an entirely RNA‐based environment that catalyzes formation of peptide bonds (reviewed in Ref 3). A third major functional center is the elongation and termination factor binding site. This is composed of a complex surface area formed by both subunits where trans‐acting GTPases bind to the ribosome to either (1) deliver aminoacyl‐tRNAs (aa‐tRNAs) to the ribosome or (2) deliver a structurally similar translocase or termination factors. Interactions between these factors both enhance the intrinsic rate of peptide synthesis and ensure the directionality of ribosome movement along the mRNA. Given the triplet nature of codons, each mRNA contains three potential translational reading frames. Thus, one of the central questions in molecular and structural biology is to understand how the ribosome initially chooses the ‘correct’ reading frame, and how it manages to coordinate the activities of multiple functional centers so as to maintain reading frame throughout the course of translating mRNAs.
TRANSLATIONAL READING FRAME: ESTABLISHMENT AND MAINTENANCE
The issue of translational reading frame establishment also appears to have ancient origins: the so‐called ‘universal’ start signal for protein translation is specified by a single codon, AUG, encoding methionine. In eukaryotes, the correct reading frame of an mRNA is generally defined by the first AUG (as read from the 5′ to 3′ direction) which denotes where translation is to begin, while in polycistronic prokaryotic mRNAs containing multiple open reading frames (ORFs), reinitiation of translation is a bit more complicated (reviewed in Ref 4). While translation initiation at non‐AUG codons, and ribosomal bypassing of the first (or even subsequent) AUG codons have been well documented, these tend to be exceptions to the general rule, and in fact have served as useful tools to further our understanding of this general rule that the first AUG defines translational reading frame (reviewed in Ref 5). Indeed, data emerging from ribosome profiling experiments suggests that a significant fraction of translation initiation events occur at a small subset of non‐AUG codons.6
Establishment of Translational Reading Frame
To understand how translational reading frame is established, it is important to understand how the structure of the ribosome affects its function. The SSU contains a single rRNA species in all organisms, which ranges in size from 16S in bacteria and archae, to 18S in eukaryotes, and contains a minimum of 21 proteins, which expands to up to 33 in higher organisms (reviewed in Ref 7). As discussed above, base pairing interactions between mRNA codons and tRNA anticodons occur in the decoding center of the SSU, which is located near the 3′ end of its rRNA. The LSU of bacteria and archae contain two rRNAs, 23S and 5S, and approximately 31 proteins. In eukaryotes, the proteinacious component has expanded to up to 49 proteins, and a small fragment of the 23S rRNA appears to have become detached and evolved into a separate rRNA, called 5.8S. Interestingly, while 5.8S rRNA is a distinct molecule, structural analyses reveals that it is physically located in the same place, along the ‘rear’ solvent accessible side of the LSU, as its corresponding sequence in bacterial and archaeal ribosomes.8, 9, 10 Also different is the expansion of the major LSU rRNA in eukaryotes, ranging from 25S rRNA in yeast to 28S rRNA in metazoans. The LSU interacts with the SSU, and contains three distinct pockets for binding of tRNAs: the A‐site specifically binds aa‐tRNAs, the P‐site binds initiator tRNAs and tRNAs linked to elongating polypeptides (peptidyl‐tRNAs), and the E‐site binds deacylated tRNAs. Nascent peptides are extruded from the PTC through a tunnel where they exit from the ‘back’ side of the LSU.10 In addition, trans‐acting factors involved in delivering aa‐tRNAs and involved in termination are recruited through interactions with both the SSU and LSU.11 While these functional centers are the features of the ribosome most pertinent to this chapter, it should be noted that the ribosome contains additional functional elements.
Returning to the question of reading frame establishment, translation initiation differs between bacteria/archae, and eukaryotes. In bacteria/archae, special sequences (the Shine–Dalgarno sequence, or SD) located near the 5′ ends of their mRNAs are complementary to sequence located near the 3′ end of the 16S rRNA (the anti‐SD sequence). This complementarity enables the SD on the mRNA to hybridize with the anti‐SD on the SSU, properly positioning the initiation codon in the P‐site of the decoding center in the SSU, and independent initiation events can be directed on polycistronic mRNAs by separate SD sequences 5′ of each initiation site.12 Furthermore, variability in the distance between the SD and the initiation codon means that the SD is not sufficient to determine reading frame by itself: this process also requires initiator tRNA and initiation factor 2 (IF2). In eukaryotes, correct positioning of the initiator AUG on the ribosome is a much more complex process involving many more steps and trans‐acting factors. This includes recruitment of the SSU in complex with initiator tRNA and other IFs to the 5′ end of the mRNA and scanning of this 43S preinitiation complex along the mRNA in the 3′ direction. Nonetheless, recruitment of the initiator‐Met‐tRNA to the correct AUG remains the central element in reading frame establishment.13 Similarly, initiator tRNAs are universally recruited to the ribosomal P‐site (again with a few exceptions that have been of great utility). Recruitment of tRNA to this site is unique to translation initiation: during the remaining course of mRNA translation, subsequent aa‐RNAs, and even protein factors that mediate translation termination are recruited to the A‐site. In bacteria and archae, the initiator‐Met‐tRNA is formylated (fMet‐tRNA), unlike elongator Met‐tRNAs. Eukaryotes encode two distinct species of Met‐tRNAs, one specializing in initiation, the other in elongation. While the mechanics of translation initiation differ greatly between bacteria/archae and eukaryotes, recruitment of initiator‐Met‐tRNAs to the P‐site of the SSU is the central feature of translation initiation in all organisms. In sum, translational reading frame is generally established by bringing a special species of tRNA to an AUG codon positioned at the ribosomal P‐site.
Maintenance of Translational Reading Frame
Having established translational reading frame on an mRNA, the ribosome must maintain it throughout the remaining course of translation, the bulk of which is termed the elongation phase. The first step of elongation occurs when elongator tRNAs are brought to the ribosome in association with a trans‐acting factor, called EF‐Tu in bacteria/archae, and eEF1A in eukaryotes, and GTP, forming the ternary complex (TC). The anticodon loop of elongator aa‐tRNA is delivered to the decoding center in the A‐site of the SSU, where a correct match between the mRNA codon and aa‐tRNA anticodon results in formation of a mini‐helix that is recognized and stabilized by additional interactions between both SSU rRNA bases and proteins.14 This in turn results in a structural rearrangement of the SSU and aa‐tRNA, which transduces information that causes EF‐Tu/eEF1A to hydrolyze GTP, releasing the tRNA from the elongation factor (reviewed in Ref 13). The aminoacylated 3′ end of the aa‐tRNA then moves in a process called accommodation, from outside of the LSU through a structural element (the accommodation corridor), and into the A‐site side of the PTC. Simultaneously, EF‐Tu/eEF1A + GDP is released from the ribosome to be recharged with GTP and aa‐tRNAs.15 Catalysis, i.e., peptidyltransfer, occurs in the PTC, where the methionine is transferred from the initiator‐tRNA to the elongator tRNA in a process that involves both steric positioning and active catalysis through a transesterification reaction by the ribosome. After this process, the 3′ end of the deacylated tRNA moves to the E‐site on the LSU, the 3′ end of the dipeptidyl‐tRNA moves to the P‐site of the LSU, while the anticodon loops of both tRNAs remain bound to the P‐ and A‐sites of the SSU respectively. This conformation is called the ‘hybrid state’ because the tRNAs occupy one site on the LSU and another on the SSU.16 It is also at this step that the ribosome reorients itself from the ‘classical’ or ‘unrotated’ state to the ‘ratcheted’ or ‘rotated’ state, a process that involves a complex spatial repositioning of the two subunits relative to one another (reviewed in Refs 17, 18). The next step in the process is where reading frame maintenance comes into play: translocation. Here, a second trans‐acting factor, EF‐G/eEF2, is recruited to the ribosome. Hydrolysis of GTP by this protein leads to a transition state for translocation where the ribosome disengages from the tRNA–mRNA complexes to allow movement of the anticodons of the P and A‐site tRNAs. This movement, coupled with release of the elongation factor, results in full tRNA occupation of the E and P‐sites by the deacylated tRNA and dipeptidyl‐tRNA, respectively, leaving an empty A‐site ready for the next aa‐tRNA to decode the next codon.15 It is thought that the two subunit nature of the ribosome separates movement of the body of the tRNA on the LSU from that of the mRNA/tRNA complexes on the SSU, enabling it to faithfully maintain translational reading frame (reviewed in Ref 17). More recently, high resolution methods have revealed numerous structural features that are thought to work in concert to assure that tRNAs remain correctly positioned in the ribosome, and that translocation is precisely limited to three nucleotides.18 Subsequent rounds of elongation reiterate this cycle until the ribosome encounters a termination codon (UUA, UGA, or UAG). Termination codons are specifically recognized by release factors (RF1 and RF2 in bacteria/archae, eF1‐eRF3 complex in eukaryotes) that are structural mimics of the TC, which specifically recognize termination codons.19 The lack of an amino donor site by the RFs enables a water molecule to enter the PTC, promoting hydrolysis of the C‐terminus of the nascent polypeptide chain from the peptidyl‐tRNA, resulting in peptide release.20
MECHANISMS OF PROGRAMMED TRANSLATIONAL FRAMESHIFTING
While it is obvious that the translational apparatus needs to faithfully maintain reading frame, altering translational fidelity could be advantageous in special circumstances. Indeed, many viruses employ numerous molecular mechanisms, generically termed translational recoding.21 These include but are not limited to directing elongating ribosomes to shift into an alternate reading frame, directing ribosomes to utilize alternative start sites, and bypassing or recoding termination codons.5 This is particularly relevant when genomic space is physically constrained, e.g., in viruses, where genome size is limited by the volume of the viral particle. Here, expanding the information content of a viral mRNA by enabling it to encode multiple proteins may confer a selective advantage. Another idea is that the ability for a single RNA to encode multiple proteins without having to alter its sequence (e.g., through splicing), may have conferred a selective advantage in the prebiotic RNA world.22 Additionally, the ability to recode mRNAs provides yet another level at which gene expression can be controlled. Notably, these mechanisms are all ‘programmed’ to occur at specific sequences by cis‐acting elements present on mRNAs, and at rates that are two or more orders of magnitude more frequent than nonprogrammed events.
Having identified the players and defined the contexts during which PRF events occur, we will focus on molecular mechanisms that program elongating ribosomes to shift translational reading frame at specific sites along mRNAs, and discuss their physiological relevance.
−1 Programmed Ribosomal Frameshifting
Our understanding of −1 PRF originates from studies of RNA viruses, many of which use this molecular mechanism to expand the information content of their mRNAs. The relatively large number of viral −1 PRF signals has enabled definition of some of the parameters constituting a −1 PRF signal. The most well‐defined −1 PRF phenomena are directed by an mRNA sequence motif composed of three important elements: a ‘slippery site’ composed of seven nucleotides where the translational shift in reading frame actually takes place; a short spacer sequence of usually less than 12 nucleotides; and a downstream stimulatory structure (usually an mRNA pseudoknot). A ‘typical’ −1 PRF signal is shown in Figure 1(a). In eukaryotic viruses, the slippery site has the heptameric motif N NNW WWH (IUPAC notation), where the incoming reading frame is indicated by spaces.23 In general, it has been accepted that the downstream structure causes elongating ribosomes to pause with tRNAs positioned at the slippery site. The nature of the slippery sequence enables repairing of the nonwobble bases of both the aa and peptidyl‐tRNAs with the −1 frame codons.24 While it is generally accepted that mRNA pseudoknots are the most common type of downstream stimulatory elements, other mRNA structures are capable of filling this role as well.25, 26, 27 Generally, it is thought that the essential function of the stimulatory structure is to provide an energetic barrier to an elongating ribosome, and to position it over the slippery site. However, the thermodynamic stability of the downstream barrier is not the sole determinant of frameshift efficiency: additional parameters, both known and unknown influence this parameter.28
Figure 1.

−1 programmed ribosomal frameshifting (−1 PRF). (a) From 5′ to 3′, a typical −1 PRF signal contains a heptameric slippery site, a short spacer, and a complex tertiary mRNA structure, typically an H‐type pseudoknot. The original translational reading frame at the slippery site is indicated by spaces. The 22 functional slippery sites are shown. (b) The many paths to −1 PRF. As described in the text, −1 PRF can occur at three different times during translation at the frameshift signal. The pseudoknot can direct a two nucleotide translocation event either as the ribosome enters (left, boxed 1) or exits (right, boxed 3) the slippery site. Alternatively, accommodation of the aminoacyl tRNA (aa‐tRNA) into the slippery site pulls the downstream mRNA into the ribosome by 9Å, creating tension between the slippery site and pseudoknot (center, boxed 2). The tension is relieved by decoupling tRNAs from the mRNA, with the mRNA slipping backward by one base.
The original simultaneous‐slippage model24 of −1 PRF suggested that peptidyl and aa‐tRNAs simultaneously slip by one base in the 5′ direction to base pair with the −1 frame codons in the slippery site. From this general conceptual framework, the precise mechanistic details of −1 PRF have been debated, and three apparently competing models were proposed for the mechanism of −1 PRF. In each of these, physical slippage of the ribosome is coupled to the energetic input of GTP hydrolysis. The ‘integrated model’ model of −1 PRF posited that the shift occurs after delivery of the aa‐tRNA to the A‐site, but before peptidyltransfer.23 This corresponds to Box 2 in Figure 1(b). A refinement of this model, called the ‘9Å solution’ proposed that the downstream stimulatory element plays an active role in −1 PRF by resisting 5′ movement of the mRNA subsequent to aa‐tRNA accommodation (enabled by eEF1A hydrolysis of GTP). This creates tension along the mRNA that can be relieved by disengaging the tRNAs from the mRNA, thus allowing the mRNA to shift forward by one base relative to the tRNA/ribosome complex.29 A second model proposed that −1 PRF occurs during translocation, where the energy driving slippage is supplied by eEF2 hydrolysis of GTP, and the downstream stimulatory element resists forward movement of the ribosome, resulting in translocation by only two nucleotides. Importantly, this cotranslocation model can occur through two discrete kinetic pathways. One of these pathways occurs after peptidyltransfer, with the two tRNAs moving to P/E and A/P states, followed by an incomplete, two‐base translocation event.30, 31 This is denoted by Box 3 in Figure 1(b). The second cotranslocational model proposed that incomplete translocation stimulated by the downstream element occurs one elongation cycle earlier: here, the E and P‐site tRNAs slip so that the new A‐site codon is in the −1 frame32 as indicated by Box 1 in Figure 1(b).
Importantly, there is strong experimental evidence supporting all three models, suggesting that rather than explaining −1 PRF through a single molecular mechanism, −1 PRF should be conceived as a problem of kinetic partitioning occurring within the context of the translation elongation cycle. With this in mind, the ‘many pathways model’ of −1 PRF unified all three models and revealed the major steps in the translation elongation cycle that affect −1 PRF.33 This is shown in Figure 1(b). Importantly, this model provides a mathematical framework within which estimates can be calculated regarding the relative contributions of each of the elements to rates of −1 PRF, how often ribosomes will partition between the 0 and −1 frames, and how this partitioning will distribute at each of the three possible stages of the elongation cycle. In sum, the ‘many pathways model’ of −1 PRF presents a unified theory of −1 PRF that also provides a toolbox for quantitative prediction of −1 PRF rates.
+1 Programmed Ribosomal Frameshifting
In contrast to −1 PRF where the translational reading frame is recoded by one nucleotide toward the 5′ direction of the mRNA, the elongating ribosome is induced to bypass one nucleotide toward 3′ direction in +1 PRF. +1 PRF has been observed in Escherichia coli in the translation of prfB to produce RF2.34 In the yeast Saccharomyces cerevisiae two retrotransposable elements, Ty1 and Ty3,35, 36 and three genes, ABP140,37 EST3,38 and OAZ1 39 use +1 PRF. The expression of the mammalian equivalent of yeast OAZ1, ornithine decarboxylase antizyme (i.e., OAZ), has also been shown to involve +1 PRF.40
Unlike −1 PRF, where there is only one generally well‐understood type of frameshift signal, +1 PRF signals appear case specific. However, it is clear that +1 PRF is also driven by cis‐acting elements that cause elongating ribosomes to kinetically partition into the +1 frame, and that slippage of P‐site tRNA appear to be the most important parameter. However, the precise mechanisms are different for different +1 PRF signals. In the bacterial cases such as the E. coli prfB mRNA, the U CUU UGA slippery site contains the in frame UGA termination codon which is recognized by RF2.41 While translation termination is efficient when RF2 levels are high, low RF2 levels result in inefficient recognition of the UGA codon. This causes the ribosome to pause. An SD‐like sequence located in the prfB mRNA immediately 5′ of the slippery site interacts with the anti‐SD sequence on the 16S rRNA so as to reposition the ribosome in the +1 frame. Thus, RF2 production is autoregulated through +1 PRF.42 More recently, mathematical modeling of the prfB +1 PRF signal revealed that this mechanism is influenced by three distinct kinetic parameters: (1) destabilization of deacylated tRNA in the E‐site, (2) rearrangement of peptidyl‐tRNA in the P‐site, and (3) the availability of cognate aa‐tRNA corresponding to the A‐site.43 While all three function synergistically to promote efficient +1 PRF, a rate constant of ≈︁1.9 s−1 for slippage of the P‐site tRNA from CUU to UUU is the driving force behind this mechanism. The +1 PRF is also enhanced by the presence of a ‘hungry codon’ in the A‐site (i.e., low abundance of RF2), and destabilization of tRNA–mRNA interactions in the E‐site.
Eukaryotic translation does not utilize mechanisms analogous to the SD/anti‐SD interactions that direct prokaryotic initiation and +1 PRF. Thus, the +1 PRF kinetic partitioning must be driven by other mechanisms. In OAZ mRNA +1 PRF, the primary kinetic trap appears to be the presence of a strong secondary mRNA structure 3′ of the slippery site. However, the element that stimulates OAZ +1 PRF has undergone a significant amount of evolutionary divergence. For example, while almost all vertebrate OAZ +1 PRF signals involve mRNA pseudoknots, fewer protostome OAZ sequences contain predicted pseudoknots, most nematodes lack the ability to form this type of structure, and no pseudoknots can be calculated in any yeast/fungi or insect OAZ +1 PRF signals.39 Similarly, the slippery sites of OAZ have diverged. The metazoan OAZ slippery site is UCC UGA U,44 but has degenerated in fungi and arthropods.45 Importantly, similar to prfB, the OAZ +1 frameshift is stimulated by a 0‐frame A‐site UGA codon, and is also primarily dependent on tRNA–mRNA interactions in the ribosomal P‐site. For example, mutation of the rat OAZ P‐site sequence from UCC to CCC inhibited frameshifting in S. cerevisiae.39 Also similar to pfrB, the E‐site of the OAZ +1 PRF signal also modulates frameshifting efficiency, although this is less well understood.39 OAZ +1 PRF is also autoregulated: it is stimulated by polyamines.46 Neutralization of negative charge repulsion by positively charged polyamines may facilitate the formation of mRNA–rRNA interactions that enhance tRNA slippage in the P‐site while the ribosome is paused at the 0‐frame UGA termination codon. Importantly, OAZ +1 PRF is autoregulated. Ornithine decarboxylase (ODC) catalyzes the first step in polyamine biosynthesis, while OAZ downregulates polyamine synthesis by stimulating ubiquitin‐independent degradation of ODC by the proteasome. Thus, increased levels of polyamines negatively feedback on polyamine synthesis by stimulating +1 PRF, and hence the synthesis of OAZ.
The yeast Ty retrotransposable elements utilize +1 PRF to direct synthesis of Gag‐pol fusion proteins.47 The Ty1 slippery site is CUU AGG C.35 Like prfB and OAZ, the frameshift is primarily driven by slippage of the P‐site tRNA from CUU to UUA. Unlike the prior two examples however, this slippery site does not contain a 0‐frame termination codon. Rather, the kinetic trap is supplied by the rare A‐site AGG codon, which is decoded by the low abundance Arg‐tRNACCU tRNA. Overexpression of this tRNA caused a 50‐fold decrease in +1 PRF, while deleting it caused +1 PRF efficiency to approach 100%.48 The +1 frameshifts of Ty2 and Ty4, and other members of the copia family of retrotransposable elements are thought to utilize this mechanism of tRNA slippage, as well as the yeast ABP140 frameshift signal.37, 49 While, the genome organization of the Ty3 gypsy‐like yeast retrotransposon is similar to Ty1,50 its +1 PRF signal is different. The GCG AGU U slippery site disallows the possibility of the 0‐frame tRNA in the P‐site to base pair with the +1 frame.36 It is thought that Ty3‐directed +1 PRF involves skipping the first A of the 0‐frame P‐site codon followed by recognition of the +1 frame GUU codon. Further analysis demonstrated that +1 PRF depended on some special characteristic of the Ala‐tRNAUGC, and that this was also shared by four more tRNAs. A downstream stimulatory element is also been proposed to constitute the kinetic trap utilized in Ty3 +1 PRF. While the original hypothesis involving direct base pairing between this sequence and the 18S rRNA helix 1851 has been ruled out, a very stringent set of mutagenesis experiments suggest that the Ty3 stimulatory element may interact with rRNA and ribosomal proteins in the ribosomal entry tunnel, as well as unknown constituents of the solvent face of the 40S subunit.52 Interestingly, while the EST3 slippery site is identical to that of Ty1 and frameshifting is dependent on limiting quantities of cognate A‐site tRNA, its +1 PRF signal also contains a downstream stimulatory element.53 It has been speculated that interaction of this element with specific targets of the paused ribosome may limit A‐site access by tRNAs.
IMPLICATIONS OF PROGRAMMED FRAMESHIFTING IN VIROLOGY
Many RNA viruses utilize PRF to posttranscriptionally regulate expression of multiple genes encoded by their monocistronic mRNAs. The mRNAs of many such viruses, e.g., totiviruses, Ty elements, and most retroviruses, contain two or more overlapping ORFs in which the major viral nucleocapsid proteins (e.g., Gag) are encoded by a 5′ ORF, while sequences encoding proteins with enzymatic functions (typically Pro and Pol) are located 3′ of, and out‐of‐frame with, the Gag ORF. The enzymatic proteins are only translated as a result of PRF events that occur at frequencies of 1–40% depending on the specific virus and assay system employed.54 This ensures production of a greater ratio of structural nucleocapsid proteins to products having enzymatic/replicative activities. The importance of maintaining precise ratios of structural to enzymatic proteins on viral propagation has been demonstrated using two endogenous viruses of the yeast S. cerevisiae and two retroviruses. In the yeast dsRNA L‐A ‘killer’ virus, Gag‐pol dimerization nucleates formation of the viral particle.55 Small alterations in programmed frameshifting efficiencies promote rapid loss of the virus, and it is thought that increasing the amount of Gag‐pol protein synthesized may cause too many particles to initiate nonproductively while producing too little may prevent efficient dimerization.56 Similarly, increasing or decreasing the efficiency of the +1 ribosomal frameshift in the Ty1 retrotransposable element of yeast results in reduced retrotranspostion frequencies by inhibiting proteolytic processing of the TyA‐TyB polyprotein (Gag‐pol equivalent), thus blocking formation of the mature forms of RNase H, integrase and reverse transcriptase.48 Similarly, changing the ratio of Gag to Gag‐pol proteins in retroviruses like HIV or Moloney Murine Leukemia Virus interferes with virus‐particle formation.57, 58, 59, 60, 61 In these viruses, overexpression of the Gag‐pol protein also resulted in inefficient processing of the polyprotein and inhibition of virus production.
Coronaviruses also utilize −1 PRF to synthesize C‐terminally extended fusion proteins that are subsequently proteolytically processed.62 The genomic organization of coronaviruses is different in that the structural proteins are encoded in subgenomic mRNAs while the genes regulated by −1 PRF are involved in replicase/transcriptase function. A study examining the consequences of altering −1 PRF efficiencies in the SARS‐associated coronavirus (SARS‐CoV) demonstrated that, although the functional genesets involved are very different, they supported the general hypothesis that viral PRF efficiencies have been finely tuned to deliver a ‘golden mean’ of proteins required for optimal virus replication and viability63(Figure 2).
Figure 2.
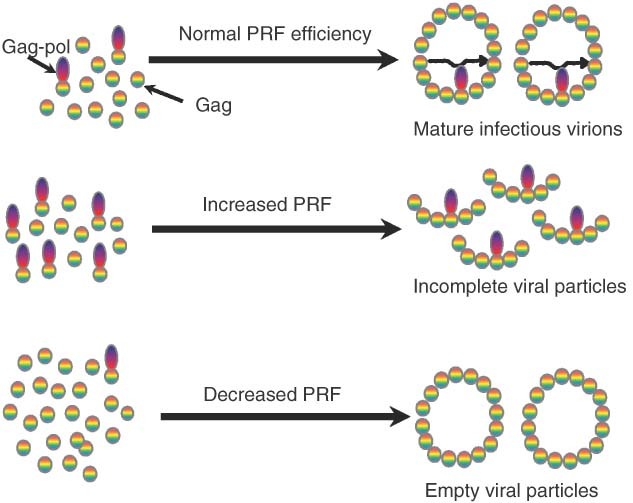
Programmed ribosomal frameshifting (PRF) efficiency is critical for viral particle assembly. Top panel: normal rates of PRF result in the correct stoichiometric ratios of viral structural (Gag) to enzymatic (Gag‐pol) proteins, enabling efficient viral particle assembly, viral genome packaging, and maturation. Middle panel: increased rates of PRF result in formation of incomplete viral particles. Bottom panel: decreased rates of PRF promote formation of empty viral particles.
The requirement of many RNA viruses for precise rates of −1 PRF suggested a target for antiviral therapeutics.64 The peptidyltransferase inhibitors anisomycin, sparsomycin, and preussin all affect −1 PRF efficiency and inhibit virus propagation in yeast,65, 66 and the eEF‐2 inhibitor sordarin alters +1 PRF and Ty1 retrotranspositon.66 Biochemical and computational screens have identified small compounds capable of binding the −1 PRF signals of HIV‐159, 67, 68 and SARS‐CoV.69 Synthetic oligonucleotide‐based compounds have also been shown to alter rates of −1 PRF.70, 71, 72, 73, 74, 75 The recent development of cell‐based dual‐fluorescence reporter systems provide inexpensive platforms for high throughput screens directed at viral PRF signals.76, 77 Genetic methods have been employed to identify numerous cellular gene products that affect both −1 and +1 PRF (reviewed in Ref 78). Importantly however, all of the mutants generated by the genetics approaches promote deleterious cellular phenotypes, suggesting that global dysregulation of PRF may interfere with expression of cellular genes (see next section). Indeed, the recent demonstration that defects in rRNA pseudouridylation promote increased rates of −1 PRF support this, and suggest that such defects may contribute to the pathologies associated with the human diseases X‐linked Dyskeratosis Congenita and Hoyeraal‐Hreidarsson syndrome.79
IMPLICATIONS OF PROGRAMMED FRAMESHIFTING IN CONTROL OF CELLULAR GENE EXPRESSION
As with most basic molecular mechanisms, although first described in viruses, it is now clear that PRF is much more widespread and is likely employed by organisms representing every branch in the tree of life (for reviews see Refs 21, 80, 81). While functional PRF signals in expressed eukaryotic genes have been identified, until recently these discoveries have been serendipitous.38, 40, 82, 83, 84 The past few years have seen the publication of several reports describing in silico identification of ‘recoding signals’ using a wide variety of computational approaches.85, 86, 87, 88, 89, 90, 91 While the methodologies of each study covered a broad range of bioinformatics techniques, the general goal of most was to try to first identify overlapping reading frames, and then to test sequences in the overlap regions for their ability to promote PRF. The strength of this approach is that it can identify new classes of PRF‐promoting elements. However, this strategy is based on the assumption that PRF outcomes should mimic those observed in viral genomes: thus, it cannot identify new functional outcomes of frameshifting.
In contrast, while ‘outcome‐neutral’ approaches using mRNA motifs known to promote efficient PRF cannot identify new classes of frameshift signals, they can enable an expansion of our understanding of functional uses for PRF. With this in mind, rather than focusing on identifying two overlapping out‐of‐frame ORFs, our first computational search for eukaryotic −1 PRF signals aimed to identify −1 PRF promoting motifs that resembled well‐characterized examples of viral −1 PRF signals.87 This first study identified ∼260 putative −1 PRF signals in the annotated portion of the S. cerevisiae genome. However, it was limited by incomplete annotation of the yeast genome and relatively insufficient computational resources available at the time (ca. 1995–1998). Each new iteration of this approach has been more comprehensive and powerful, utilizing new informatics tools applied to faster and more robust computational platforms. The ‘second generation’ analysis utilizing pattern matching approaches coupled with a statistical feature based on RNA folding algorithms using the S. cerevisiae genome as the testbed demonstrated that (1) ∼10% of yeast genes contain at least one high probability −1 PRF signal and (2) >95% of all −1 PRF events would direct elongating ribosomes to encounter premature termination codons (PTCs).85 Expansion of this analysis to >20 genomes suggests that these two important findings may be a universal feature of eukaryotic transcriptomes 92. The predicted ribosomal frameshift database (PRFdB, http://prfdb.umd.edu) contains a searchable catalog putative eukaryotic −1 PRF signals.
As noted above, while viral −1 PRF events result in synthesis of fusion proteins with N‐terminal domains encoded by the original reading frame and C‐terminal extensions encoded by the ‐1 frame ORF, genomic frameshifting directs elongating ribosomes to PTCs. This engendered the hypothesis that −1 PRF is used by cells to control mRNA abundance and stability through the nonsense‐mediated mRNA decay (NMD) pathway. A proof‐of‐principle study demonstrated that (1) a well‐characterized viral −1 PRF signal can act as an mRNA destabilizing element in cis, (2) mRNA destabilization required an intact NMD pathway, and (3) the extent of mRNA destabilization was inversely proportional to −1 PRF efficiency.93 A follow‐up study using a series of −1 PRF signals isolated from four endogenous cellular mRNAs from S. cerevisiae showed that a subset of these also promoted mRNA degradation through the no‐go decay (NGD) pathway, presumably because frameshift‐promoting mRNA secondary structures also promote sufficiently long ribosome pausing to activate this pathway.94 These two mRNA destabilization pathways are shown in Figure 3. In that study, more detailed investigations revealed that the EST2 mRNA, encoding the catalytic subunit of telomerase, was destabilized by −1 PRF, and that ablation of −1 PRF signals in this mRNA promoted its stabilization. Unpublished work in our laboratory has identified functional −1 PRF signals in mRNAs encoding additional subunits in yeast telomerase, suggesting that PRF may play a role in telomere length homeostasis by controlling the abundance and relative ratios of telomerase‐associated factors.
Figure 3.
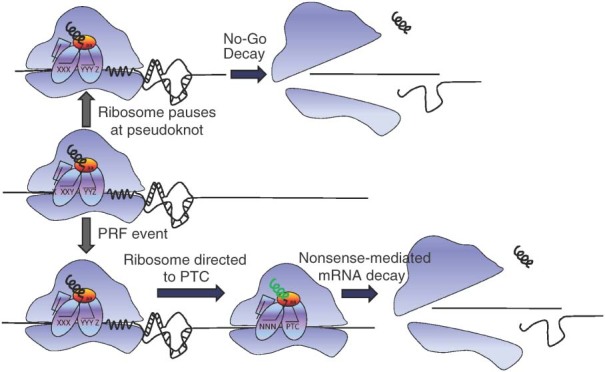
−1 programmed ribosomal frameshifting (−1 PRF) signals promote mRNA destabilization through the nonsense‐mediated decay (NMD) and the no‐go decay (NGD) pathways. Middle: an elongating ribosome encounters a −1 PRF signal. Top: the mRNA pseudoknot induced ribosome pause results in activation of the NGD pathway, releasing the ribosome and promoting degradation of the mRNA. Bottom: a −1 PRF event directs an elongating ribosome to a premature termination codon (PTC), activating the NMD pathway, resulting in ribosome release and mRNA degradation.
If PRF is used to control cellular gene expression, then it is reasonable to hypothesize that it is regulated. As described above, regulation would have to involve mechanisms that would target specific signals rather than promoting global changes in PRF. Autoregulation as described for OAZ and prfB +1 PRF provide one such PRF signal‐specific mechanism. However, in these cases the products encoded by frameshift events directly feedback on the PRF signals. This would not apply to most other cellular −1 PRF signals because they do not encode any new protein products. Thus, regulation should involve the use of sequence‐specific trans‐acting factors. In addition, the ability of −1 PRF signals to direct mRNA destabilization in a manner that is inversely proportional to −1 PRF rates93,97 would suggest that, rather than turning −1 PRF completely on or off, trans‐acting regulatory factors may function to increase or decrease rates of −1 PRF, thus providing for nuanced effects on gene expression that could be fine‐tuned to specific circumstances. As discussed above, the ability of synthetic oligonucleotide‐based compounds to alter rates of −1 PRF suggests that naturally occurring oligonucleotides, e.g., small noncoding RNAs, may hold the key to regulation of −1 PRF. Base pairing interactions would provide the requisite sequence specificity, and their potential ability to either disrupt or stabilize −1 PRF promoting mRNA tertiary structures would provide them with the capacity to promote increased or decreased rates of −1 PRF. These two possibilities are shown in Figure 4. Indeed, preliminary studies from our laboratory have identified at least two endogenous micro‐RNAs (miRNAs) capable of stimulating ‐1 PRF in a human mRNA. As a final thought, if PRF is an important regulator of cellular gene expression, then mutations in −1 PRF signals might impact PRF rates, and hence mRNA stability and protein abundance. We suggest that this may explain why some single nucleotide polymorphisms have been shown to promote discernable phenotypes, e.g., inherited diseases, despite the fact that they are silent with regard to the amino acids that they encode.
Figure 4.
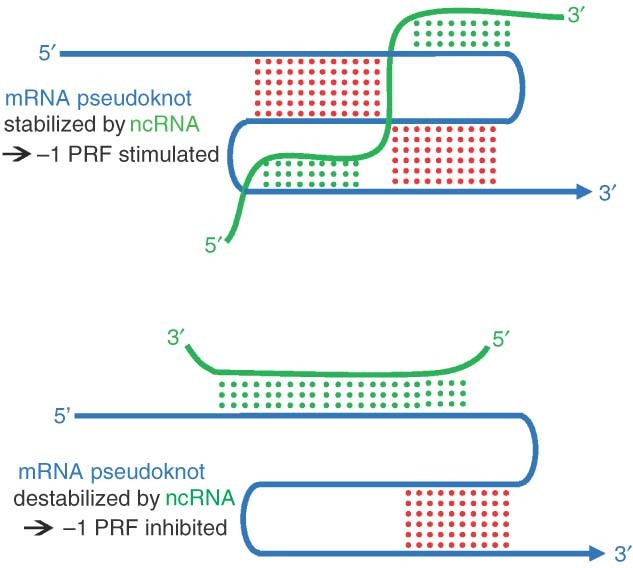
Possible mechanisms through which ncRNAs could be used to regulate −1 programmed ribosomal frameshifting (−1 PRF). Top panel: stimulation of −1 PRF through interaction of an ncRNA (gray) that further stabilizes a −1 PRF promoting mRNA pseudoknot (black). Bottom panel: inhibition of −1 PRF by an ncRNA that competes for mRNA sequence elements required for pseudoknot formation.
CONCLUSION
The history of modern molecular biology is replete with examples in which basic biological regulatory mechanisms were first discovered in viruses. This is not because viruses are special, but rather because their small genomes help to increase signal‐to‐noise ratios, thus facilitating the scientific discovery process. Indeed, as obligate intracellular parasites, viruses are subject to the same rules and regulations that govern their host cells. Thus, while PRF was first thought to be a virus‐specific mechanism, it is becoming clear that cellular mRNAs employ this mechanism as well. The study of PRF continues to illuminate our understanding of how ribosomes normally maintain reading frame. In particular, viewed from a kinetic standpoint, we have come to understand that −1 PRF represents an endpoint resulting from changes in kinetic partitioning at different steps along a reaction pathway rather than a single mechanism. We hope that this kinetic view of −1 PRF will be useful in identifying specific targets for antiviral therapeutics. Another recurring motif in virology is that viruses tend to re‐purpose molecular mechanisms that were originally taken from host cells. The finding that viruses use PRF to make C‐terminally extended fusion proteins differs from most of their cellular counterparts, which appear to use −1 PRF to regulate gene expression through mRNA stability. Indeed, this observation may help to explain why global changes in −1 PRF efficiency are detrimental to cell viability, and may even help to elucidate one of the underlying causes of human ribosomopathies. This would also explain why −1 PRF should be regulated by sequence‐specific mechanisms, e.g., by ncRNAs as proposed here. Corollary to this may be the explanation for why viruses that utilize −1 PRF are able to successfully replicate in their host cells because permissive cells would presumably not express ncRNAs capable of interacting with the viral −1 PRF signals. It is our hope that these observations and suggestions will spur new investigators to enter this new and expanding field.
Acknowledgements
I acknowledge the many postdoctoral fellows, graduate and undergraduate students with whom I have had the pleasure to work with during these past 15 or so years. They keep me young. In particular, I thank a few critical people. I have had the good fortune and pleasure of working with Dr. Arturas Meskauskas over the past decade: he has been my second brain. During his tenure as a graduate student in my laboratory, Dr. Jason Harger developed a seemingly simple, yet powerful assay that continues to this day to be used by many laboratories to investigate translational fidelity. Dr. Ewan Plant provided critical insight and expertise that both solidified our view of PRF and virology, as well as uncovering mechanism through which −1 PRF may be regulated. Dr. Jonathan Jacobs was the first computer wizard who cracked the problem of cellular −1 PRF: the PRFdB is built on his original source code. Most recently, Ashton Trey Belew has expanded and improved upon this project, making the PRFdB a much more powerful and user friendly tool, and providing heroic service to the quest to understand how cellular gene expression is regulated by −1 PRF. This work was supported by a grant to J.D.D. by the National Institutes of Health (R01 GM058859).
REFERENCES
- 1. Nirenberg M. Historical review: deciphering the genetic code–a personal account. Trends Biochem Sci 2004, 29:46–54. [DOI] [PubMed] [Google Scholar]
- 2. Schmeing TM, Ramakrishnan V. What recent ribosome structures have revealed about the mechanism of translation. Nature 2009, 461:1234–1242. [DOI] [PubMed] [Google Scholar]
- 3. Leung EK, Suslov N, Tuttle N, Sengupta R, Piccirilli JA. The mechanism of peptidyl transfer catalysis by the ribosome. Annu Rev Biochem 2011, 80:527–555. [DOI] [PubMed] [Google Scholar]
- 4. Kozak M. The scanning model for translation: an update. J Cell Biol 1989, 108:229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Atkins JF, Gesteland RF. Recoding: Expansion of Decoding RulesEnriches Gene Expression. New York: Springer; 2010.. [Google Scholar]
- 6. Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 2011, 147:789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taylor DJ, Devkota B, Huang AD, Topf M, Narayanan E, Sali A, Harvey SC, Frank J. Comprehensive molecular structure of the eukaryotic ribosome. Structure 2009, 17:1591–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ben Shem A, Jenner L, Yusupova G, Yusupov M. Crystal structure of the eukaryotic ribosome. Science 2010, 330:1203–1209. [DOI] [PubMed] [Google Scholar]
- 9. Klinge S, Voigts‐Hoffmann F, Leibundgut M, Arpagaus S, Ban N. Crystal structure of the eukaryotic 60s ribosomal subunit in complex with initiation factor 6. Science 2011, 334:941–948. [DOI] [PubMed] [Google Scholar]
- 10. Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science 2000, 289:905–920. [DOI] [PubMed] [Google Scholar]
- 11. Agirrezabala X, Frank J. Elongation in translation as a dynamic interaction among the ribosome, tRNA, and elongation factors EF‐G and EF‐Tu. Q Rev Biophys 2009, 42:159–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jenner L, Rees B, Yusupov M, Yusupova G. Messenger RNA conformations in the ribosomal E site revealed by X‐ray crystallography. EMBO Rep 2007, 8:846–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 2009, 136:731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ogle JM, Brodersen DE, Clemons WM Jr, Tarry MJ, Carter AP, Ramakrishnan V. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science 2001, 292:897–902. [DOI] [PubMed] [Google Scholar]
- 15. Rodnina MV, Gromadski KB, Kothe U, Wieden HJ. Recognition and selection of tRNA in translation. FEBS Lett 2005, 579:938–942. [DOI] [PubMed] [Google Scholar]
- 16. Moazed D, Noller HF. Intermediate states in the movement of transfer RNA in the ribosome. Nature 1989, 342:142–148. [DOI] [PubMed] [Google Scholar]
- 17. Dunkle JA, Cate JH. Ribosome structure and dynamics during translocation and termination. Annu Rev Biophys 2010, 39:227–244. [DOI] [PubMed] [Google Scholar]
- 18. Pisarev AV, Kolupaeva VG, Yusupov MM, Hellen CU, Pestova TV. Ribosomal position and contacts of mRNA in eukaryotic translation initiation complexes. EMBO J 2008, 27:1609–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nakamura Y, Ito K. tRNA mimicry in translation termination and beyond. Wiley Interdiscip Rev RNA 2011, 2:647–668. [DOI] [PubMed] [Google Scholar]
- 20. Brunelle JL, Shaw JJ, Youngman EM, Green R. Peptide release on the ribosome depends critically on the 2' OH of the peptidyl‐tRNA substrate. RNA 2008, 14:1526–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baranov PV, Gesteland RF, Atkins JF. Recoding: translational bifurcations in gene expression. Gene 2002, 286:187–201. [DOI] [PubMed] [Google Scholar]
- 22. Dinman JD, Icho T, Wickner RB. A‐1 ribosomal frameshift in a double‐stranded RNA virus forms a Gag‐pol fusion protein. Proc Natl Acad Sci USA 1991, 88:174–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harger JW, Meskauskas A, Dinman JD. An ‘integrated model’ of programmed ribosomal frameshifting and post‐transcriptional surveillance. TIBS 2002, 27:448–454. [DOI] [PubMed] [Google Scholar]
- 24. Jacks T, Varmus HE. Expression of the Rous Sarcoma Virus pol gene by ribosomal frameshifting. Science 1985, 230:1237–1242. [DOI] [PubMed] [Google Scholar]
- 25. Kollmus H, Hentze MW, Hauser H. Regulated ribosomal frameshifting by an RNA‐protein interaction. RNA 1996, 2:316–323. [PMC free article] [PubMed] [Google Scholar]
- 26. Baril M, Dulude D, Steinberg SV, Brakier‐Gingras L. The frameshift stimulatory signal of human immunodeficiency virus type 1 group O is a pseudoknot. J Mol Biol 2003, 331:571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yu CH, Noteborn MH, Pleij CW, Olsthoorn RC. Stem‐loop structures can effectively substitute for an RNA pseudoknot in ‐1 ribosomal frameshifting. Nucleic Acids Res 2011, 39:8952–8959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kontos H, Napthine S, Brierley I. Ribosomal pausing at a frameshifter RNA pseudoknot is sensitive to reading phase but shows little correlation with frameshift efficiency. Mol Cell Biol 2001, 21:8657–8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Plant EP, Jacobs KLM, Harger JW, Meskauskas A, Jacobs JL, Baxter JL, Petrov AN, Dinman JD. The 9‐angstrom solution: how mRNA pseudoknots promote efficient programmed ‐1 ribosomal frameshifting. RNA 2003, 9:168–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weiss RB, Dunn DM, Shuh M, Atkins JF, Gesteland RF. E. coli ribosomes re‐phase on retroviral frameshift signals at rates ranging from 2 to 50 percent. New Biol 1989, 1:159–169. [PubMed] [Google Scholar]
- 31. Namy O, Moran SJ, Stuart DI, Gilbert RJ, Brierley I. A mechanical explanation of RNA pseudoknot function in programmed ribosomal frameshifting. Nature 2006, 441:244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leger M, Dulude D, Steinberg SV, Brakier‐Gingras L. The three transfer RNAs occupying the A, P and E sites on the ribosome are involved in viral programmed ‐1 ribosomal frameshift. Nucleic Acids Res 2007, 35:5581–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liao PY, Choi YS, Dinman JD, Lee KH. The many paths to frameshifting: kinetic modeling and analysis of the effects of different elongation steps on programmed −1 ribosomal frameshifting. Nucleic Acids Res 2011, 39:300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Craigen WJ, Caskey CT. Expression of peptide chain release factor 2 requires high‐ efficiency frameshift. Nature 1986, 322:273–275. [DOI] [PubMed] [Google Scholar]
- 35. Belcourt MF, Farabaugh PJ. Ribosomal frameshifting in the yeast retrotransposon Ty: tRNAs induce slippage on a 7 nucleotide minimal site. Cell 1990, 62:339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Farabaugh PJ, Zhao H, Vimaladithan A. A novel programmed frameshift expresses the Pol3 gene of retrotransposon‐Ty3 of yeast—frameshifting without transfer‐RNA slippage. Cell 1993, 74:93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Asakura T, Sasaka I, Nagano F, Satoh A, Obaishi H, Nishioka H, Imamura H, Hotta K, Tanaka K, Nakanishi H, et al. Isolation and characterization of a novel actin filament‐binding protein from Saccharomyces cerevisiae. Oncogene 1998, 16:121–130. [DOI] [PubMed] [Google Scholar]
- 38. Lundblad V, Morris DK. Programmed translational frameshifting in a gene required for yeast telomere replication. Curr Biol 1997, 7:969–976. [DOI] [PubMed] [Google Scholar]
- 39. Ivanov IP, Anderson CB, Gesteland RF, Atkins JF. Identification of a new antizyme mRNA +1 frameshifting stimulatory pseudoknot in a subset of diverse invertebrates and its apparent absence in intermediate species. J Mol Biol 2004, 339:495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Matsufuji S, Matsufuji T, Wills NM, Gesteland RF, Atkins JF. Reading two bases twice: mammalian antizyme frameshifting in yeast. EMBO J 1996, 15: 1360–1370. [PMC free article] [PubMed] [Google Scholar]
- 41. Craigen WJ, Cook RG, Tate WP, Caskey CT. Bacterial peptide chain release factors: conserved primary structure and possible frameshift regulation of release factor 2. Proc Natl Acad Sci U S A 1985, 82:3616–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Adamski FM, Donly BC, Tate WP. Competition between frameshifting, termination and suppression at the frameshift site in the Escherichia coli release factor‐2 mRNA. Nucleic Acids Res 1993, 21:5074–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liao PY, Gupta P, Petrov AN, Dinman JD, Lee KH. A new kinetic model reveals the synergistic effect of E‐, P‐ and A‐sites on +1 ribosomal frameshifting. Nucleic Acids Res 2008, 36:2619–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ivanov IP, Gesteland RF, Atkins JF. Survey and summary: antizyme expression: a subversion of triplet decoding, which is remarkably conserved by evolution, is a sensor for an autoregulatory circuit. Nucleic Acids Res 2000, 28:3185–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Palanimurugan R, Scheel H, Hofmann K, Dohmen RJ. Polyamines regulate their synthesis by inducing expression and blocking degradation of ODC antizyme. EMBO J 2004, 23:4857–4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rom E, Kahana C. Polyamines regulate the expression of orinithine decarboxylase antizyme in vitro by inducing ribosomal frameshifting. Proc Natl Acad Sci U S A 1994, 91:3959–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Farabaugh PJ. Post‐transcriptional regulation of transposition by Ty retrotransposons of Saccharomyces cerevisiae. J Biol Chem 1995, 270:10361–10364. [DOI] [PubMed] [Google Scholar]
- 48. Kawakami K, Paned S, Faioa B, Moore DP, Boeke JD, Farabaugh PJ, Strathern JN, Nakamura Y, Garfinkel DJ. A rare tRNA‐Arg(CCU) that regulates Ty1 element ribosomal frameshifting is essential for Ty1 retrotransposition in Saccharomyces cerevisiae. Genetics 1993, 135:309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Farabaugh PJ, Kramer E, Vallabhaneni H, Raman A. Evolution of +1 programmed frameshifting signals and frameshift‐regulating tRNAs in the order Saccharomycetales. J Mol Evol 2006, 63:545–561. [DOI] [PubMed] [Google Scholar]
- 50. Hansen LJ, Chalker DL, Sandmeyer SB. Ty3, a yeast retrotransposon associated with tRNA genes, has homology to animal retroviruses. Mol Cell Biol 1988, 8:5245–5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li Z, Stahl G, Farabaugh PJ. Programmed +1 frameshifting stimulated by complementarity between a downstream mRNA sequence and an error‐correcting region of rRNA. RNA 2001, 7:275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Guarraia C, Norris L, Raman A, Farabaugh PJ. Saturation mutagenesis of a +1 programmed frameshift‐inducing mRNA sequence derived from a yeast retrotransposon. RNA 2007, 13:1940–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Taliaferro D, Farabaugh PJ. An mRNA sequence derived from the yeast EST3 gene stimulates programmed +1 translational frameshifting. RNA 2007, 13:606–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Plant EP, Dinman JD. Comparative study of the effects of heptameric slippry site composition on ‐1 frameshifting among different translational assay systems. RNA 2006, 12:666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Caston JR, Trus BL, Booy FP, Dinman JD, Wickner RB, Steven AC. Structural aspects of the capsid of L‐A dsRNA virus of yeast. In: Givosen U, Hendrix R, Russel M. Proceedings of the XIV InternationalPhage/Virus Assembly Conference. 1995.. [Google Scholar]
- 56. Dinman JD, Wickner RB. Ribosomal frameshifting efficiency and Gag/Gag‐pol ratio are critical for yeast M1 double‐stranded RNA virus propagation. J Virol 1992, 66:3669–3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Biswas P, Jiang X, Pacchia AL, Dougherty JP, Peltz SW. The human immunodeficiency virus type 1 ribosomal frameshifting site is an invariant sequence determinant and an important target for antiviral therapy. J Virol 2004, 78:2082–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Felsenstein KM, Goff SP. Expression of the gag‐pol fusion protein of moloney murine leukemia virus without gag protein does not induce viron formation or proteolytic processing. J Virol 1988, 62:2179–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hung M, Patel P, Davis S, Green SR. Importance of ribosomal frameshifting for human immumodeficiency virus type 1 assembly and replication. J Virol 1998, 72: 4819–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Karacostas V, Wolffe EJ, Nagashima K, Gonda MA, Moss B. Overexpression of the HIV‐1 gag‐pol polyprotein results in intracellular activation of HIV‐1 protease and inhibition of assembly and budding of virus‐like particles. Virology 1993, 193:661–671. [DOI] [PubMed] [Google Scholar]
- 61. Park J, Morrow CD. Overexpression of the gag‐pol precursor from human immunodeficiency virus type‐I proviral genomes results in efficient proteolytic processing in the absence of virion production. J Virol 1991, 65: 5111–5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Masters PS. The molecular biology of coronaviruses. Adv Virus Res 2006, 66:193–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Plant EP, Rakauskaite R, Taylor DR, Dinman JD. Achieving a golden mean: mechanisms by which coronaviruses ensure synthesis of the correct stoichiometric ratios of viral proteins. J Virol 2010, 84:4330–4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dinman JD, Ruiz‐Echevarria MJ, Peltz SW. Translating old drugs into new treatments: Identifying compounds that modulate programmed ‐1 ribosomal frameshifting and function as potential antiviral agents. Trends Biotech 1998, 16:190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dinman JD, Ruiz‐Echevarria MJ, Czaplinski K, Peltz SW. Peptidyl transferase inhibitors have antiviral properties by altering programmed ‐1 ribosomal frameshifting efficiencies: development of model systems. Proc Natl Acad Sci U S A 1997, 94:6606–6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kinzy TG, Harger JW, Carr‐Schmid A, Kwon J, Shastry M, Justice MC, Dinman JD. New targets for antivirals: the ribosomal A‐site and the factors that interact with it. Virology 2002, 300:60–70. [DOI] [PubMed] [Google Scholar]
- 67. Staple DW, Venditti V, Niccolai N, Elson‐Schwab L, Tor Y, Butcher SE. Guanidinoneomycin B recognition of an HIV‐1 RNA helix. Chembiochem 2008, 9:93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Palde PB, Ofori LO, Gareiss PC, Lerea J, Miller BL. Strategies for recognition of stem‐loop RNA structures by synthetic ligands: application to the HIV‐1 frameshift stimulatory sequence. J Med Chem 2010, 53: 6018–6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Park SJ, Kim YG, Park HJ. Identification of RNA pseudoknot‐binding ligand that inhibits the ‐1 ribosomal frameshifting of SARS‐coronavirus by structure‐based virtual screening. J Am Chem Soc 2011, 133: 10094–10100. [DOI] [PubMed] [Google Scholar]
- 70. Vickers TA, Ecker DJ. Enhancement of ribosomal frameshifting by oligonucleotides targeted to the HIV gag‐pol region. Nucleic Acids Res 1992, 20: 3945–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Aupeix‐Scheidler K, Chabas S, Bidou L, Rousset JP, Leng M, Toulme JJ. Inhibition of in vitro and ex vivo translation by a transplatin‐modified oligo(2'‐O‐methylribonucleotide) directed against the HIV‐1 gag‐pol frameshift signal. Nucleic Acids Res 2000, 28: 438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Howard MT, Gesteland RF, Atkins JF. Efficient stimulation of site‐specific ribosome frameshifting by antisense oligonucleotides. RNA 2004, 10:1653–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Henderson CM, Anderson CB, Howard MT. Antisense‐induced ribosomal frameshifting. Nucleic Acids Res 2006, 34:4302–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yu CH, Noteborn MH, Olsthoorn RC. Stimulation of ribosomal frameshifting by antisense LNA. Nucleic Acids Res 2010, 38:8277–8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ahn DB, Lee W, Choi JK, Kim SJ, Plant EP, Almazan F, Taylor DR, Enjuanes L, Oh JW. Interference of ribosomal frameshifting by antisense peptide nucleic acids suppresses SARS coronavirus replication. Antiviral Res 2011, 91:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cardno TS, Poole ES, Mathew SF, Graves R, Tate WP. A homogeneous cell‐based bicistronic fluorescence assay for high‐throughput identification of drugs that perturb viral gene recoding and read‐through of nonsense stop codons. RNA 2009, 15:1614–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rakauskaite R, Liao PY, Rhodin MH, Lee K, Dinman JD. A rapid, inexpensive yeast‐based dual‐fluorescence assay of programmed–1 ribosomal frameshifting for high‐throughput screening. Nucleic Acids Res 2011, 39:e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dinman JD, O'Connor M. Mutants that affect recoding In: Atkins JF, Gesteland RF, eds. Recoding: Expansion of Decoding Rules Enriches Gene Expression. New York, Dordrecht, London: Springer; 2010, 321–344. [Google Scholar]
- 79. Jack K, Bellodi C, Landry DM, Niederer R, Meskauskas A, Musalgoankar S, Kopmar N, Krasnykh O, Dean AM, Thompson SR, et al. rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells. Mol Cell 2011, 44:660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cobucci‐Ponzano B, Rossi M, Moracci M. Recoding in archaea. Mol Microbiol 2005, 55:339–348. [DOI] [PubMed] [Google Scholar]
- 81. Dinman JD. Programmed Ribosomal Frameshifting Goes Beyond Viruses: Organisms from all three kingdoms use frameshifting to regulate gene expression, perhaps signaling a paradigm shift. Microbe Washington DC 2006, 1:521–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Shigemoto K, Brennan J, Walls E, Watson CJ, Stott D, Rigby PWJ, Erith AD. Identification and characterisation of a developmentally regulated mammalian gene that utilises—1 programmed ribosomal frameshifting. Nucleic Acids Res 2001, 29:4079–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Manktelow E, Shigemoto K, Brierley I. Characterization of the frameshift signal of Edr, a mammalian example of programmed ‐1 ribosomal frameshifting. Nucleic Acids Res 2005, 33:1553–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wills NM, Moore B, Hammer A, Gesteland RF, Atkins JF. A functional ‐1 ribosomal frameshift signal in the human paraneoplastic Ma3 gene. J Biol Chem 2006, 281:7082–7088. [DOI] [PubMed] [Google Scholar]
- 85. Jacobs JL, Belew AT, Rakauskaite R, Dinman JD. Identification of functional, endogenous programmed ‐1 ribosomal frameshift signals in the genome of Saccharomyces cerevisiae. Nucleic Acids Res 2007, 35: 165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Namy O, Rousset JP, Napthine S, Brierley I. Reprogrammed genetic decoding in cellular gene expression. Mol Cell 2004, 13:157–168. [DOI] [PubMed] [Google Scholar]
- 87. Hammell AB, Taylor RL, Peltz SW, Dinman JD. Identification of putative programmed ‐1 ribosomal frameshift signals in large DNA databases. Genome Res 1999, 9: 417–427. [PMC free article] [PubMed] [Google Scholar]
- 88. Shah AA, Giddings MC, Parvaz JB, Gesteland RF, Atkins JF, Ivanov IP. Computational identification of putative programmed translational frameshift sites. Bioinformatics 2002, 18:1046–1053. [DOI] [PubMed] [Google Scholar]
- 89. Gao X, Havecker ER, Baranov PV, Atkins JF, Voytas DF. Translational recoding signals between gag and pol in diverse LTR retrotransposons. RNA 2003, 9:1422–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gurvich OL, Baranov PV, Zhou J, Hammer AW, Gesteland RF, Atkins JF. Sequences that direct significant levels of frameshifting are frequent in coding regions of Escherichia coli. EMBO J 2003, 22:5941–5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Moon S, Byun Y, Kim HJ, Jeong S, Han K. Predicting genes expressed via ‐1 and +1 frameshifts. Nucleic Acids Res 2004, 32:4884–4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Belew AT, Hepler NL, Jacobs JL, Dinman JD. PRFdb: a database of computationally predicted eukaryotic programmed ‐1 ribosomal frameshift signals. BMC Genomics 2008, 9:339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Plant EP, Wang P, Jacobs JL, Dinman JD. A programmed ‐1 ribosomal frameshift signal can function as a cis‐acting mRNA destabilizing element. Nucleic Acids Res 2004, 32:784–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Belew AT, Advani VM, Dinman JD. Endogenous ribosomal frameshift signals operate as mRNA destabilizing elements through at least two molecular pathways in yeast. Nucleic Acids Res 2010, 39:2799–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]