

Abstract
Growth factors have been shown to be potent mediators of osteogenesis. However, their use in tissue-engineered scaffolds not only can be costly but also can induce undesired responses in surrounding tissues. Thus, the ability to specifically induce osteogenic differentiation in the absence of exogenous growth factors through manipulation of scaffold material properties would be desirable for bone regeneration. Previous research indicates that addition of inorganic or hydrophobic components to organic, hydrophilic scaffolds can enhance multipotent stem cell (MSC) osteogenesis. However, the combined impact of scaffold inorganic content and hydrophobicity on MSC behavior has not been systematically explored, particularly in three-dimensional (3D) culture systems. The aim of the present study was therefore to examine the effects of simultaneous increases in scaffold hydrophobicity and inorganic content on MSC osteogenic fate decisions in a 3D culture environment toward the development of intrinsically osteoinductive scaffolds. Mouse 10T½ MSCs were encapsulated in a series of novel scaffolds composed of varying levels of hydrophobic, inorganic poly(dimethylsiloxane) (PDMS) and hydrophilic, organic poly(ethylene glycol) (PEG). After 21 days of culture, increased levels of osteoblast markers, runx2 and osteocalcin, were observed in scaffolds with increased PDMS content. Bone extracellular matrix (ECM) molecules, collagen I and calcium phosphate, were also elevated in formulations with higher PDMS:PEG ratios. Importantly, this osteogenic response appeared to be specific in that markers for chondrocytic, smooth muscle cell, and adipocytic lineages were not similarly affected by variations in scaffold PDMS content. As anticipated, the increase in scaffold hydrophobicity accompanying increasing PDMS levels was associated with elevated scaffold serum protein adsorption. Thus, scaffold inorganic content combined with alterations in adsorbed serum proteins may underlie the observed cell behavior.
Introduction
Multipotent stem cell (MSC) differentiation is known to be influenced by a range of environmental stimuli, among the strongest of which are growth factors. However, the use of growth factors in tissue-engineering scaffolds not only can be costly but also can induce undesired responses in surrounding tissues. Thus, MSC-based bone regeneration strategies would be benefited by the identification of scaffold material properties that intrinsically promote osteoblast lineage progression in the absence of exogenous growth factors.
A number of studies have shown that the addition of inorganic components to organic scaffolds can enhance both their osteoconductivity and osteoinductivity.1–10 These inorganic additives have included not only hydroxyapatite, but also a range of silicon-containing compounds, such as silica, silane, and siloxane.3–5,7–10 In addition, scaffold hydrophobicity has been demonstrated to influence osteogenic differentiation.11–13 However, the combined effects of scaffold inorganic content and hydrophobicity have not been systematically explored. Furthermore, most of the aforementioned studies were performed in two-dimensional (2D) culture, and, thus, may not be indicative of the effects of the same scaffold variables in more biomimetic, three-dimensional (3D) culture systems. The aim of the present study was therefore to examine the impact of simultaneous increases in scaffold hydrophobicity and inorganic content on MSC osteogenic lineage progression in a 3D culture environment.
Toward this goal, we developed a novel series of scaffolds comprised of varying levels of hydrophobic, inorganic poly(dimethylsiloxane) (PDMS) and hydrophilic, organic poly(ethylene glycol) (PEG). PEG was selected for the hydrophilic, organic component due to its established biocompatibility and its previous use in bone regeneration applications.6,11,14,15 Similarly, PDMS was chosen as the inorganic component over silica, silane, and other polysiloxanes due to its processability and its intermediate level of hydrophobicity. In addition, PDMS is used extensively in biomedical applications and is considered biocompatible.16–19 For the present study, PDMS and PEG molecular weights (Mn) and concentrations were selected to yield scaffolds with moduli within the osteogenic range identified in the 2D human MSC studies of Engler et al.20 and the 3D human and mouse MSC studies of Huebsch et al.21
In analyzing the lineage progression of encapsulated mouse 10T½ MSCs, quantitative immunoassays were performed for mid-to-late term markers associated with osteoblast, chondrocytic, smooth muscle cell (SMC), and adipocytic fates. Furthermore, biochemical and histological analyses were conducted to assess the deposition of extracellular matrix (ECM) components associated with mature bone (e.g., collagen I and calcium phosphate)22 as well as ECM molecules, which would be considered undesirable for bone regeneration (e.g., elastin and collagen type III).
Materials and Methods
Preparation of diacrylate-terminated PEG
Diacrylate-terminated PEG (PEG-DA, Fig. 1) was prepared in accordance to known methods.23 Briefly, PEG (Mn=3.4 kDa; Sigma) was dissolved in dry dichloromethane (20 mL/mmol PEG) in a 250-mL round bottom flask purged with Ar. Triethylamine (2:1 molar ratio; Sigma) was added slowly to the solution, followed by the dropwise addition of acryloyl chloride (4:1 molar ratio; Sigma). The reaction mixture was allowed to stir at room temperature overnight. Removal of HCl was accomplished by washing the mixture twice with 2M K2CO3 and separating into aqueous and organic phases. The organic phase was then dried with anhydrous MgSO4, and the volatiles were removed under reduced pressure. The resulting crude oil was precipitated in diethyl ether in an ice bath, filtered, and dried under vacuum at room temperature overnight. The extent of diacrylation was confirmed to be ∼85% by 1H NMR.
FIG. 1.

Schematic of the PEG-DA and PDMSstar-MA components of PEG-PDMSstar hydrogels. PDMS, poly(dimethylsiloxane); PEG-DA, diacrylate-terminated poly(ethylene glycol); PDMSstar-MA, methacrylate-terminated star PDMS.
Synthesis of methacrylate-terminated star PDMS
Methacrylate-terminated star PDMS (PDMSstar-MA, Mn=2 kDa) was prepared via a two-step synthetic strategy per a modification of the methodology in Grunlan et al.24 A star architecture of PDMS was selected over a linear form to reduce phase separation between the hydrophobic PDMS and hydrophilic PEG before hydrogel polymerization.25 In brief, silane (SiH)-terminated star PDMS (PDMSstar-SiH) was first prepared by the acid-catalyzed equilibration of octamethylcyclotetrasiloxane with tetrakis(dimethylsiloxane)silane (tetra-SiH).24 Octamethylcyclotetrasiloxane and tetra-SiH were combined in a 100-mL round bottom flask under N2 at a 4.3:1 molar ratio, with the ratio of octamethylcyclotetrasiloxane and tetra-SiH selected to yield a product of the desired Mn. Triflic acid (0.060 μL/mmol octamethylcyclotetrasiloxane) was then added, and the reaction was allowed to stir for 16 h at room temperature. After cooling, the mixture was neutralized by combining with excess hexamethyledisilazane and stirring for 2 h. The polymer mixture was precipitated three times in toluene/methanol and the isolated polymer dried under reduced pressure. The chemical structure and Mn of the resulting colorless product (PDMSstar-SiH) were confirmed by 1H NMR, 13C NMR, IR, and gel permeation chromatography.
The SiH terminal groups of the PDMSstar-SiH were converted into photosensitive methacrylate moieties by the subsequent hydrosilylation reaction with allyl methacrylate.26 Briefly, PDMSstar-SiH was dissolved in dry toluene using a 100-mL round bottom flask and purged with N2. The Karstedt's catalyst (Pt–divinyltetramethyldisiloxane complex in xylene, 2% Pt) was added at 33 μL/mmol, and the reaction mixture was heated to 90°C. Allyl methacrylate was then added over 15 min in a 4:1 molar ratio, after which the reaction was heated to 90°C and stirred overnight. Completion of the reaction was confirmed by the disappearance of the Si-H (∼2100 cm−1) absorbance in the IR spectrum. The reaction mixture was decolorized by refluxing with activated carbon for 12 h. After removal of volatiles under reduced pressure, the catalyst was removed from the residue via flash column chromotography on silica gel. The resulting product (PDMSstar-MA, Fig. 1) was confirmed by 1H-NMR.
Synthesis of acrylate-derivatized cell adhesion ligand
The cell adhesion peptide RGDS (American Peptide) was reacted with acryloyl-PEG-N-hydroxysuccinimide (ACRL-PEG-NHS, 3.4 kDa; Nektar) at a 1:1 molar ratio for 2 h in 50 mM sodium bicarbonate buffer, pH 8.5.27,28 The product (ACRL-PEG-RGDS) was purified by dialysis, lyophilized, and stored at −20°C until use.
Hydrogel preparation
PEG-PDMSstar hybrid hydrogels were prepared by the photopolymerization of aqueous mixtures of PEG-DA and PDMSstar-MA. Five distinct precursor solutions were prepared: (a) three with 10 wt% total polymer, but with a 100:0, 95:5, or 80:20 wt ratio of PEG-DA to PDMSstar-MA, and (b) two additional PEG-DA formulations containing 9.5wt% and 8.0 wt% total polymer, but no PDMS (referred to as 95:0 and 80:0 constructs, respectively). ACRL-PEG-RGDS was added to each solution so as to yield 1 mM RGDS in the swollen gels. A photoinitiator consisting of a 30 wt% solution of 2,2-dimethyl-2-phenyl-acetophenone in N-vinylpyrrolidone was added to the precursor solutions at 10 μL/mL. The resulting mixtures were vortexed and passed through 0.22-μm polyethersulfone (PES) filters, which served both to sterilize the solutions as well as to create fine, stable dispersions of hydrophobic PDMSstar in the aqueous PEG solutions. The filtered precursor solutions were immediately poured into 0.75-mm-thick transparent rectangular molds, and polymerized by a 6-min exposure to long-wave UV light (∼6 mW/cm2, 365 nm; Spectroline).
Evaluation of initial scaffold material properties
Dynamic light scattering
The average size of PDMSstar-MA particles in the PEG-PDMSstar hydrogels was estimated by dynamic light scattering (DLS) measurements of aqueous solutions of 0.5 wt% and 2 wt% PDMSstar-MA. After 0.22-μm filtration, DLS measurements were performed on each solution using a Brookhaven ZetaPALS instrument. A laser with a wavelength of 660 nm was used as the incident beam, and the scattered light was detected at a 90° scattering angle at 25°C. The light-scattering data were analyzed with BI-DLSW control software, and a non-negative-constrained least-squares algorithm was used to determine the size distributions.
Hydrogel mechanical properties
Bulk mechanical testing: After an 18-h immersion in phosphate-buffered saline (PBS), four 8-mm discs were cored from each hydrogel formulation and mechanically tested under unconstrained compression using an Instron 3342. After application of a 0.05-N preload, each hydrogel was subjected to compression at a rate of 1 mm/min. The compressive modulus of each hydrogel was extracted from the resulting stress–strain data over a 5%–25% strain range.
Atomic force microscopy (AFM): After 18 h of swelling in deionized water (dIH2O),29 PEG-PDMSstar hydrogels were locally, mechanically tested using a Bioscope System AFM (Veeco Instruments) equipped with a Nanoscope IIIa controller and mounted on a Zeiss Axiovert 100 TV inverted optical microscope. Each hydrogel was fixed to a 60-mm-diameter polystyrene Petri-dish with an RP30 instant adhesive (Adhesive Systems, Inc). The hydrogels were then submerged in 1.5 mL of dIH2O to reduce tip-adhesion effects and to maintain sample hydration during testing. The gel samples were indented with a silicon dioxide glass bead with a radius (R) of 300 nm attached to a silicon-nitride cantilever with a manufacturer-calibrated spring constant of 360 pN/nm (Novascan). Deflection sensitivity for the AFM probe was determined to be 54 nm/V. All indentations were performed at a frequency of 0.5 Hz with a z-scan size of 800 nm (speed of 0.8 μm/s). After recording typically 30 consecutive indentation force–depth curves at each location on the hydrogel, the tip was retracted to its original position. At least, ten distinct locations were analyzed per hydrogel sample.
The applied force f sphere (pN) during indentation was taken to be the product of the measured cantilever deflection d (V), cantilever spring constant k (pN/nm), and deflection sensitivity ds (nm/V). The indentation depth δ (nm) of the probe during an approach–retraction cycle was calculated as the difference between the piezo displacement z (nm) and effective cantilever deflection dsd (nm). An effective Young's modulus (E) was determined from AFM force curves based on the Hertzian equation.30–32 However, the strict use of this equation requires simplifying assumptions, such as a flat, homogeneous, semi-infinite elastic material, and a rigid probe.33 Thus, to accommodate potential nonlinearities, we calculated the pointwise apparent modulus 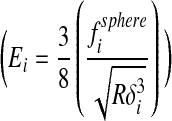 , or material stiffness, at each point (i) along all postcontact approach curves. All pointwise modulus values appeared to converge upon an asymptotic value at an indentation depth typically <30 nm. These asymptotic values were used.32,34
, or material stiffness, at each point (i) along all postcontact approach curves. All pointwise modulus values appeared to converge upon an asymptotic value at an indentation depth typically <30 nm. These asymptotic values were used.32,34
Hydrogel mesh size
The PEG-based hydrogel mesh structure cannot be readily visualized using techniques such as conventional scanning electron microscopy. Thus, a variety of methods to estimate the PEG-DA hydrogel mesh size have been developed, including correlations linking measurable quantities, such as equilibrium hydrogel swelling and PEG-DA Mn, to mesh size.35,36 Although these correlations yield reasonable average mesh size estimates for homopolymer hydrogels,35,36 they cannot readily be applied to PEG-PDMSstar hybrid hydrogels. Thus, in this study, the average hydrogel mesh size was characterized via dextran diffusion based on an adaptation of the methodology of Watkins et al.37 Briefly, four 8-mm discs were cored from each hydrogel formulation after 24-h swelling and immersed in 0.5 mL HBS–azide (HEPES-buffered saline plus 0.05 wt% azide) containing 50 μg/mL FITC-labeled dextran (10 kDa; Sigma). Dextran was then allowed to diffuse into the hydrogels for 24 h at 37°C, after which each disc was gently blotted and transferred to 0.5 mL fresh HBS–azide. After 24 h at 37°C, the fluorescence of the HBS–azide solution surrounding each disc was measured at ex/em 488/532. Each fluorescence measure was converted to micrograms dextran using dextran standard curves and then divided by gel weight to yield a quantitative indicator of hydrogel permissivity (C). These permissivity measures were used to estimate the mesh size (ξ) of each hydrogel (x) relative to the 100:0 PEG-PDMSstar formulation as follows: 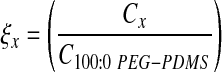 .
.
Serum protein adsorption
After 24 h of swelling, four 36-mm discs were harvested from each PEG-PDMSstar hydrogel formulation to evaluate the dependence of bulk serum protein adsorption on increasing PDMSstar content. Each disc sample was exposed to 10% fetal bovine serum (FBS) in PBS for 24 h at 37°C. Nonadsorbed proteins were then removed by immersing the specimens in dIH2O at 37°C for 60 min, with dIH2O changes every 20 min. Adsorbed proteins were subsequently stripped from the hydrogels by progressive extraction with a series of aqueous isopropanol solutions of increasing hydrophobicity.38 Specifically, each hydrogel sample was successively immersed in 10%, 30%, 50%, and 70% aqueous isopropanol solutions (I10, I30, I50, and I70, respectively) for 20 min per solution at 37°C. The isopropanol fractions for a particular hydrogel sample were then combined, evaporated under vacuum, and resuspended in 500 μL of PBS. The total amount of protein stripped from each hydrogel sample was quantified using the CBQCA total protein quantitation kit (Invitrogen). For the purpose of comparison, the average protein measures for each gel composition were normalized to the 100:0 PEG-PDMSstar formulation.
Relative bulk hydrophobicity
Contact angle measurements are widely used to analyze differences in the hydrophobic character of material surfaces. However, this method is limited to the assessment of surface hydrophobicity, and associated results cannot be readily extended to bulk behavior. We therefore developed a method to assess differences in bulk hydrogel hydrophobicity by comparing the extent of swelling in solvents of varying polarities.39 Specifically, solvents of higher polarity indices (P) interact preferentially with hydrophilic domains of the bulk material, whereas solvents of lower P indices show greater affinity for hydrophobic segments of the material. Thus, differences in the relative uptake of solvents of distinct polarities by a given hydrogel formulation can serve as an indicator of its bulk hydrophobicity. In brief, four discs from each hydrogel formulation were submerged in dIH2O (P=12.1) or I70 (P=10.0)40 for 24 h at room temperature, after which the swollen weight (Ws) of each disc was recorded. The samples were then dried under vacuum for 48 h and their corresponding dry weights (Wd) measured. The equilibrium mass swelling ratio (q) of each formulation in each solvent (x) was calculated as 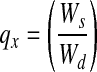 . The relative bulk hydrophobicity (H) of each hydrogel formulation was estimated as follows:
. The relative bulk hydrophobicity (H) of each hydrogel formulation was estimated as follows: 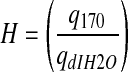 .
.
Cell culture
Cryopreserved 10T½ mouse MSCs (ATCC) were thawed and expanded in monolayer culture. Before encapsulation, cells were maintained at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium (DMEM; Hyclone) supplemented with 10% heat-inactivated FBS (Hyclone).
Fabrication and culture of cell-laden constructs
Precursor solutions for the 100:0, 95:5, and 80:20 PEG-PDMSstar gels and the 95:0 and 80:0 controls were prepared in PBS. ACRL-PEG-RGDS was then added to each precursor solution so as to yield an RGDS concentration of 1 mM in the swollen gels. After addition of a photoinitiator, the mixtures were vortexed and sterilized by 0.22-μm filtration. 10T½ cells were then resuspended in each precursor solution so as to yield a post-swelling cell density of ∼2×106 cells/g. The resulting suspensions were polymerized into hydrogel networks by 6-min exposure to long-wave UV light (∼6 mW/cm2, 365 nm), a process that has previously been demonstrated to be cytocompatible.41–43 Gels were immersed in the DMEM supplemented with 10% heat-inactivated FBS, 100 U/mL penicillin, and 100 mg/L streptomycin and maintained at 37°C and 5% CO2. After 24 h, samples (n=4 per formulation) were collected for bulk compressive analyses per the above procedure. Medium in the remaining samples was changed every 2 days for 21 days.
Endpoint construct analyses
After 21 days of culture, a series of samples (n=4 per formulation) were mechanically analyzed under bulk compression. Separate samples were collected from each hydrogel formulation for biochemical and histological analyses. All steps associated with the biochemical and histological procedures described below took place at room temperature unless otherwise noted.
Biochemical analyses
Competitive ELISAs for differentiation markers: Proteins extracted by sample homogenization in Trizol followed by protein isolation44 were evaluated for the housekeeping protein GAPDH as well as for mid-to-late term markers of osteogenesis, chondrogenesis, SMC progression, and adipogenesis via competitive ELISAs. In brief, appropriate primary antibodies and their corresponding peptide antigens were purchased from Santa Cruz Biotechnology (SCBT). High-binding EIA 96-well plates (Costar) were coated overnight at 4°C with 200 ng per well of peptide for adipocyte–fatty acid binding protein (A-FABP, C-15), PPARγ (I-18), myocardin (M-16), smooth muscle 22α (SM22α, P-15), runx2 (S-19), collagen I (Col1A1, D-13), osteocalcin (M-15), sox-9 (C-20), or collagen II (Col2A1, N-19) and with 100 ng per well of peptide for GAPDH (V-18). The coated wells were then blocked with bovine serum albumin (BSA). Peptide standards and isolated sample proteins were diluted in PBS containing 3% BSA and 0.05% Tween 20 and incubated with a primary antibody for 1 h, after which the mixtures were transferred to coated wells and incubated for an additional hour. The primary antibody that had bound to each coated well surface was then detected using an appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (Jackson ImmunoResearch Laboratories [JIRL]), followed by application of 2,2'-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) [ABTS; Sigma]. The resulting sample absorbance was read at 410 nm and translated to a concentration using the associated standard curve. Each target protein was analyzed in duplicate for each sample (n=3–6 per gel formulation) and normalized to GAPDH. For the purposes of comparison, the resulting protein concentrations for each formulation were further normalized to the 100:0 PEG-PDMSstar formulation.
Total collagen, sulfated glycosaminoglycan (sGAG), elastin, and collagen III assays: Based on the cell marker results, the 100:0, 95:5 and 80:20 gels were further analyzed for various ECM components. Sample proteins isolated by base hydrolysis45 were used to quantify total collagen, sGAG, elastin, and collagen III. For each of the following assays, the standards used were subjected to the same association with PEG-DA and PDMSstar-MA and the same digestion conditions as the samples.
sGAG production was measured using a modification of the Blyscan assay (Biocolor). In brief, 40 μL of protein from each sample (n=5–6 per formulation) was neutralized, mixed with 60 μL Blyscan dye reagent, and the absorbance immediately read at 525 nm relative to a chondroitin sulfate-B standard (Sigma). Similarly, levels of the amino acid hydroxyproline were quantified as an indirect measure of total collagen. Extracted proteins were first further hydrolyzed for 18 h at 110°C in 6 M HCl. The samples (n=5–6 per formulation) were then dried under vacuum followed by resuspension in dIH2O and reaction with chloramine T and p-dimethylbenzaldehyde reagents.46 The sample absorbance was read at 550 nm relative to an L-4-hydroxyproline standard (Sigma). Total collagen content was estimated from measured grams of hydroxyproline by dividing by 0.13.
For collagen III quantification, samples (n=4–6 per formulation) and standards were applied to a high-binding 96-well EIA plate for 3 h, after which the plate was blocked with 3 wt% BSA. After application of primary antibody for collagen III (Calbiochem), donkey anti-rabbit HRP secondary antibody (JIRL) and ABTS were applied. Absorbance was read at 410 nm, with human collagen III (Sigma) serving as a standard. To quantify elastin, the above procedure was followed, except that isolated sample proteins were first exposed to 0.25 M oxalic acid at 100°C overnight to convert elastin to α-elastin. Oxalic acid was then removed and exchanged for PBS using Microcon YM-3 centrifugal filters (Millipore). Resulting samples (n=5–6 per formulation) were then applied to a high-binding EIA 96-well plate for 3 h. Adsorbed elastin fragments were detected by applying an elastin primary antibody (BA-4; SCBT), with bovine α-elastin (Sigma) serving as a standard.
Measured total collagen, sGAG, collagen III, and elastin levels were normalized to the cell number as assessed by the PicoGreen assay (Invitrogen). For the purposes of comparison, the resulting concentrations for each formulation were further normalized to the 100:0 PEG-PDMSstar hydrogels.
Total calcium deposition: To assess construct calcium levels, hydrogel discs (n=3–4 per formulation) were homogenized in 640 μL lysis buffer (1% TritonX-100 and 0.5% SDS in calcium-free PBS) per 100 mg hydrogel. Total calcium was quantified using 10-μL aliquots of each sample homogenate via the Calcium CPC liquid color kit (Stanbio).
Histological analyses
Samples harvested for histological analyses were fixed with 10% formalin for 30 min, embedded in Tissue-Tek media, and cut into 35-μm sections using a cryomicrotome.
Immunostaining: Immunohistochemical staining was conducted using the same A-FABP, SM22α, osteocalcin, and collagen-II primary antibodies used for competitive ELISA analyses. After 10-min treatment with peroxidase (Biocare Medical), sections were blocked with Terminator (Biocare Medical) for 30 min followed by 1-h exposure to the primary antibody diluted in HBS. The bound primary antibody (SCBT) was detected by using either alkaline phosphatase (AP)- or HRP-conjugated secondary antibodies (JIRL) followed by application of an appropriate chromogen (LabVision). Three sections from each sample of each formulation were stained per antibody. Immunostained sections were imaged under brightfield using an Axiovert microscope (Zeiss).
von Kossa staining: To detect calcium deposits, three sections per sample of each formulation were stained using a standard von Kossa kit (American MasterTech Scientific). In brief, rehydrated sections were rinsed with dIH2O, after which a 5% silver nitrate solution was applied. Sections were then exposed to full-spectrum light in a humidified chamber for 45 min. After rinsing with dIH2O, sections were exposed to 5% sodium thiosulfate for 2.5 min, briefly rinsed, and mounted. Stained sections were imaged as previously described.
Statistical analyses
Data are reported as mean±standard deviation. Statistical differences among material property measures (e.g., modulus) were performed using ANOVA followed by a Tukey post hoc test, p<0.05. Statistical differences in biochemical or cell marker data were assessed via ANOVA followed by the Tamhane T2 post hoc test for samples with unequal variances, p<0.05 (SPSS software).
Results
Hydrogel material properties
To gain insight into the shifts in scaffold material properties underlying observed alterations in cell behavior across formulations, various hydrogel properties were characterized.
PDMSstar particle size
A previous study of PEG-PDMSstar hydrogels demonstrated that the phase separation between PDMSstar and PEG observed in aqueous solution resulted in PEG hydrogels embedded with spherical PDMSstar particles.47 To characterize the average diameter of the PDMSstar particles incorporated into the present PEG-PDMSstar hydrogels, DLS studies were performed. These assays revealed an average PDMSstar particle size of 190 nm with a polydispersity index of 1.4 for both the 0.5% and 2% PDMSstar aqueous solutions (representative of the PDMSstar concentrations in the 95:5 and 80:20 hydrogels, respectively). A representative DLS curve is provided in Supplementary Figure S1 (Supplementary Data are available online at www.liebertonline.com/tea). These DLS results indicate that the scaffold inorganic content to which encapsulated cells could potentially be exposed (i.e., the PDMSstar total particle surface area) increased monotonically from the 100:0 to the 80:20 gels.
Mechanical properties
As shown in Figure 2A, the hydrogel modulus under bulk compression was reduced ∼7% by the increase in PDMS content associated with the 100:0 and the 95:5 gels. In contrast, the bulk compressive modulus of the 80:20 PEG-PDMSstar hydrogels was ∼75% of that of the 95:5 formulation (p<0.001). These bulk modulus values were not significantly affected by inclusion of cells (at 2×106 cells/g) (Table 1 vs. Fig. 2A). In addition, no significant differences in gel modulus were noted between day 1 and 21 of culture (Table 1), indicating minimal scaffold degradation over the culture period, consistent with previous PEG-DA literature.48 This resistance to degradation implied that minimal alterations in bulk material properties occurred over the culture period and facilitated the correlation between initial scaffold material properties and observed cell behaviors.
FIG. 2.

Average mechanical property measures of PEG-PDMSstar hydrogels obtained from (A) bulk compression and (B) AFM-based local compression tests. *Significantly different from 100:0 gels; #significantly different from 95:5 gels; ζsignificantly different from 95:0 gels. Samples for bulk compressive tests: n=3–5 per formulation. Sample locations for AFM-based tests: n=10 per formulation. AFM, atomic force microscopy.
Table 1.
Moduli of Cell-Laden PEG-PDMSstar Hydrogels Under Bulk Compression
| PEG-PDMS ratio | Modulus at 24 ha | Modulus at 21 daysa |
|---|---|---|
| 100:0 | 148.5±5.1 | 149.6±16.0 |
| 95:5 | 140.0±5.2 | 140.4±10.4 |
| 80:20 | 108.8±10.0b,c | 111.7±14.2b,c,d |
| 95:0 | 134.7±27.8 | 144.1±11.7 |
| 80:0 | 100.9±33.0b,c,d | 102.3±31.6b,c,d |
n=4 per formulation per assay.
Statistical difference with the 100:0 formulation, p<0.05.
Statistical difference with the 95:5 formulation, p<0.05.
Statistical difference with the 95:0 formulation, p<0.05.
PEG, poly(ethylene glycol); PDMS, poly(dimethylsiloxane).
To assess the degree to which the decrease in PEG between the 100:0 and 80:20 gels was contributing to the observed differences in mechanical properties, control hydrogels containing 9.5 wt% PEG-DA and 8.0 wt% PEG-DA, but no PDMSstar were prepared. These control gels are referred to as 95:0 and 80:0 PEG-PDMSstar hydrogels, respectively. Comparison of the bulk modulus values of the 95:5 and 95:0 gels as well as those of the 80:20 and 80:0 hydrogels (Fig. 2A) suggests that the PEG-containing phase was the primary determinant of the observed bulk compressive modulus values.
To evaluate spatial variations in the modulus associated with PEG-PDMSstar phase separation, microscale AFM-based compressive measurements were performed. In contrast to the bulk compression results, the average AFM modulus values indicated that PDMSstar incorporation substantially altered hydrogel modulus at the microscale (Fig. 2B). This was reflected in the increased standard deviations associated with the average local moduli of the PDMSstar-containing gels relative to the pure PEG control gels. The difference in the magnitude of the bulk compression and AFM-based modulus values can be attributed to differences in mechanical testing method and conditions. In particular, the AFM assessments were conducted over a much lower strain range than the bulk compressive tests.
Average mesh size and relative bulk hydrophobicity
The bulk average mesh size data reflected the AFM-modulus assessments. Namely, the average mesh size increased significantly as the ratio of PEG to PDMSstar decreased (p<0.001, Table 2). In addition, the average mesh sizes of the 95:0 and the 80:0 control gels were significantly greater than that of the 95:5 (p=0.001) and 80:20 (p=0.001) hydrogels, respectively. These results reflect the inclusion of densely cross-linked nanoparticles of 2-kDa PDMSstar within a more loosely cross-linked 3.4-kDa PEG hydrogel network.
Table 2.
Relative Mesh Size, Bulk Hydrophobicity, and Serum Protein Adsorption
| PEG-PDMS ratio | Relative mesh sizea | HqI70/qdIH2Oa | Relative adsorbed proteina |
|---|---|---|---|
| 100:0 | 1.00±0.02 | 1.04±0.01 | 1.00±0.15 |
| 95:5 | 1.05±0.03 | 1.10±0.02b,d | 1.97±0.29b |
| 80:20 | 1.37±0.04b,c,d,e | 1.17±0.03b,c,d,e | 5.06±0.48b,c,d |
| 95:0 | 1.10±0.02b | 1.04±0.02 | 1.12±0.02 |
| 80:0 | 1.52±0.02b,c,d | 1.03±0.03c | 2.76±0.33b,d |
n=4 per formulation per assay.
Statistical difference with the 100:0 formulation, p<0.05.
Statistical difference with the 95:5 formulation, p<0.05.
Statistical difference with the 95:0 formulation, p<0.05.
Statistical difference with the 80:0 formulation, p<0.05.
dIH2O, deionized water.
In terms of relative bulk hydrophobicity, hydrogel H-values were significantly higher in the 80:20 and 95:5 hydrogels than in the 100:0 gels (p<0.001 and p=0.003, respectively; Table 2). However, the H-values for the 80:0 and 95:0 control gels could not be distinguished from those of the 100:0 hydrogels. Thus, the present data suggest that the increase in relative bulk hydrophobicity observed between the 100:0 and 80:20 hydrogels can be attributed to the increased incorporation of PDMSstar rather than to decreasing PEG-DA levels.
Serum protein adsorption
A primary mechanism by which scaffold hydrophobicity impacts cell behavior is through modulation of the levels, identities, and orientations of absorbed proteins.1,3,49,50 We therefore measured the amount of serum proteins adsorbed by the various hydrogel formulations. The levels of serum protein adsorbed onto the 95:5 and 80:20 hydrogels were approximately two- and five-fold greater than the 100:0 gels (p=0.021 and p<0.001, respectively; Table 2). Importantly, this increase in adsorbed serum proteins could not be attributed solely to the associated decrease in PEG levels from the 100:0 to the 80:20 gels. Namely, the serum protein adsorption by the 80:20 gels was 80% greater than that of the 80:0 hydrogels (p<0.001). Thus, the presence of hydrophobic PDMSstar particles within the PEG-PDMSstar hydrogels altered bulk serum protein adsorption.
Cell phenotypic markers and ECM production
To investigate the potential influence of alterations in PEG-PDMSstar hydrogel composition on 10T½ fate decisions, markers associated with chondrocyte-, osteoblast-, SMC-, and adipocyte-like fates were examined via competitive ELISA. Specifically, isolated sample proteins were evaluated for mid-to-late term markers of osteogenesis (runx2 and osteocalcin), chondrogenesis (sox-9 and collagen II), SMC progression (myocardin and SM22α), and adipogenesis (PPARγ and A-FABP).
ELISA analyses revealed that expression of transcription factors sox-9, myocardin, and PPARγ did not vary significantly across formulations (Fig. 3A). In agreement with the sox-9 data, collagen II production was also not significantly altered with increasing PDMSstar content (Fig. 3B). Similarly, although A-FABP and SM22α levels appeared to increase slightly in the 80:20 hydrogels relative to the 100:0 hydrogels, these differences fell below statistical significance. However, levels of the osteogenic transcription factor runx2 increased approximately four-fold as PDMS levels increased from the 100:0 gels to the 80:20 gels (p<0.001). In addition, osteocalcin deposition was approximately 4 times greater in the 80:20 hydrogels than in the 100:0 hydrogels (p<0.001). Immunostaining results for collagen II, osteocalcin, SM22α, and A-FABP were consistent with these ELISA results. A subset of these immunostaining images are shown in Figure 4.
FIG. 3.

Analyses of cell differentiation markers. (A) Levels of runx2, PPARγ, sox-9, and myocardin per PEG-PDMSstar formulation, and (B) levels of osteocalcin, A-FABP, collagen II, and SM22α per PEG-PDMSstar gel type. Results were normalized to the 100:0 PEG-PDMSstar formulation for the purpose of comparison. *Significantly different from 100:0 gels; #significantly different from 95:5 gels; ζsignificantly different from 95:0 gels; ξsignificantly different from 80:0 gels. Samples per assay: n=3–6 per formulation.
FIG. 4.

(A) Calcium measures per hydrogel formulation (n=3–4 per formulation). *Significantly different from 100:0 gels; #significantly different from 95:5 gels. (B) Representative images of PEG-PDMSstar hydrogel sections stained for osteocalcin (blue), SM22α (red), and calcium deposits (brown–black), n=3 sections per sample per gel type. The scale bar in the upper right-hand image equals 40 μm and applies to all images. Color images available online at www.liebertpub.com/tea
Based on the above differentiation marker results, the 100:0, 95:5, and 80:20 gels were further analyzed for ECM molecules associated with osteogenesis (collagen I and calcium deposits), chondrogenesis (sGAG), adipogenesis (elastin and collagen III), and SMC-lineage progression (collagen III and elastin). As shown in Figure 5, collagen III deposition did not vary significantly with increasing PDMSstar content. Similarly, elastin and sGAG concentrations could not be distinguished across hydrogel formulations (Fig. 5). However, collagen-I levels were significantly higher in the 80:20 hydrogels relative to remaining formulations (p<0.016). These collagen-I results were reflected in the total collagen data (p<0.010 for all pairwise comparisons). Furthermore, quantitative cresolphthalein complexone (CPC) assays revealed a marked increase in calcium deposits in the 80:20 gels relative to the 100:0 and 95:5 gels (p<0.002, Fig. 4A). Qualitative von Kossa staining supported these quantitative CPC results (Fig. 4B). Cumulatively, the cell marker and ECM data indicated increased MSC osteogenic differentiation with increasing scaffold PDMSstar content.
FIG. 5.

Levels of total collagen, collagen I, collagen III, sGAG, and elastin in each PEG-PDMSstar formulation. Results were normalized to the 100:0 PEG-PDMSstar formulation for the purpose of comparison. *Significantly different from 100:0 gels; #significantly different from 95:5 gels. Samples per assay: n=4–6 per formulation. sGAG, sulfated glycosaminoglycan.
Discussion
In the present study, mouse 10T½ MSCs were encapsulated in a series of PEG-PDMSstar hydrogels of varying inorganic content and hydrophobicity. Quantitative analyses indicated marked increases in a range of osteogenic markers with increasing PDMS levels. Specifically, runx2, osteocalcin, and calcium deposits were each three to four times greater in the 80:20 formulation versus the 100:0 control gels. Expression of collagen I, the primary collagen type in bone, was also elevated in hydrogels with increased PDMS content. In addition, the agreement in the collagen I and total collagen profiles suggests that collagen I was the dominant collagen type synthesized by the encapsulated cells.
In contrast, markers for adipogenesis (PPARγ and A-FABP), chondrogenesis (sox-9 and collagen II), and SMC lineage progression (myocardin and SM22α) were not altered in scaffolds with increased PDMS content, indicating that the observed osteogenic response was specific. To further confirm this, the levels of ECM molecules collagen III, elastin, and sGAG were quantified. Specifically, elastin- and collagen III-rich matrices are generally associated with smooth muscle51 and loose connective tissues,52,53 whereas increased production of sGAG is generally associated with chondrogenesis.54 Levels of these ECM moieties were similar across gel formulations, supporting the specificity of the associated osteogenic response.
Since the introduction of PDMSstar-MA into the PEG-DA hydrogel network could potentially alter the bulk hydrogel modulus and mesh structure, the observed cell responses could not be attributed directly to the hydrophobic–inorganic character of PDMS without further material property assessments. Based on the collected AFM modulus data, the decrease in average modulus from ∼35 kPa in the 100:0 hydrogels to ∼22 kPa in the 80:20 gels did not appear to play a significant role in the observed osteogenic response. Specifically, although osteogenic markers runx2 and osteocalcin were significantly greater in the 80:20 gels than in the 80:0 control gels, the average modulus values (both bulk and AFM-based) could not be distinguished between these gels. The apparent independence of cell responses on the average gel modulus is consistent with literature, in that each of the analyzed PEG-PDMSstar scaffolds fell within the osteogenic modulus range identified by Huebsch et al.21 for both mouse and human MSCs in 3D culture. By a similar argument, the variations in the mesh size across hydrogel formulations are an unlikely source of the osteogenic response associated with the 80:20 gels.
Thus, the hydrophobic–inorganic character of PDMS itself appears to promote the observed cellular responses. This would be consistent with prior studies linking increased scaffold silane7 or siloxane10 content to increased osteogenic responses. It is likely that both the inorganic nature of PDMS as well as its hydrophobicity played critical roles in the present results. In particular, silica, which is inorganic, but hydrophilic, has been associated with increased osteogenesis.3,4 Similarly, the introduction of hydrophobic, but noninorganic species within scaffolds has been linked to bone formation.4
Several limitations of the present study merit comment. First, the potential influence of the nanotopography introduced by the PDMS nanoparticles on observed cell responses requires further examination. In addition, the sample number per formulation and the range of PDMS levels examined were limited. More extensive in vitro studies followed by in vivo work would be required to definitely demonstrate that increased PDMS incorporation intrinsically promotes osteogenesis.
Conclusions
We have employed a novel class of hydrogels formed from the photocrosslink of PEG-DA and PDMSstar-MA to examine the influence of simultaneous increases in scaffold hydrophobicity and inorganic content on the osteogenic responses of encapsulated MSCs. Based on ECM and cell phenotypic data, elevated scaffold inorganic content and hydrophobicity were indeed correlated with increased osteogenic differentiation, although the potential influence of variations in scaffold nanotopography could not be ruled out. That said, the present results indicate that PEG-PDMSstar hydrogels may be promising scaffolds for bone regeneration, warranting further investigation of the primary mechanisms through which they alter MSC behavior.
The current work represents the first MSC study performed with these hybrid hydrogels. Future studies will probe the impact of a broader range of PDMSstar levels. In addition, various solvents will be employed to modulate the presence of PDMS nanoparticles within the hydrogel matrix, so that the impact of scaffold hydrophobicity and inorganic content can be more clearly parsed from the influence of nanotopography.
Supplementary Material
Acknowledgments
The authors acknowledge an NIH R21 for funding. We also thank Rebecca McMahon, Ph.D., and Allen Bulick, Ph.D., for their help with immunostaining and Viviana Guiza, B.S., for careful reading of the article.
Disclosure Statement
No competing financial interests exist.
References
- 1.Ducheyne P. Qiu Q. Bioactive ceramics: the effect of surface reactivity on bone formation and bone cell function. Biomaterials. 1999;20:2287. doi: 10.1016/s0142-9612(99)00181-7. [DOI] [PubMed] [Google Scholar]
- 2.Kretlow J.D. Mikos A.G. Review: mineralization of synthetic polymer scaffolds for bone tissue engineering. Tissue Eng. 2007;13:927. doi: 10.1089/ten.2006.0394. [DOI] [PubMed] [Google Scholar]
- 3.Garcia A.J. Ducheyne P. Boettiger D. Effect of surface reaction stage on fibronectin-mediated adhesion of osteoblast-like cells to bioactive glass. J Biomed Mater Res. 1998;40:48. doi: 10.1002/(sici)1097-4636(199804)40:1<48::aid-jbm6>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 4.Khan Y. Yaszemski M.J. Mikos A.G. Laurencin C.T. Tissue engineering of bone: Material and matrix considerations. J Bone Joint Surg Am. 2008;90:36. doi: 10.2106/JBJS.G.01260. [DOI] [PubMed] [Google Scholar]
- 5.Song J.-H. Yoon B.-H. Kim H.-E. Kim H.-W. Bioactive and degradable hybridized nanofibers of gelatin-siloxane for bone regeneration. J Biomed Mater Res Part A. 2008;84A:875. doi: 10.1002/jbm.a.31330. [DOI] [PubMed] [Google Scholar]
- 6.Benoit D.S.W. Schwartz M.P. Durney A.R. Anseth K.S. Small functional groups for controlled differentiation of hydrogel-encapsulated human mesenchymal stem cells. Nat Mater. 2008;7:816. doi: 10.1038/nmat2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curran J.M. Chen R. Hunt J.A. The guidance of human mesenchymal stem cell differentiation in vitro by controlled modifications to the cell substrate. Biomaterials. 2006;27:4783. doi: 10.1016/j.biomaterials.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 8.Ning C.Q. Mehta J. El-Ghannam A. Effects of silica on the bioactivity of calcium phosphate composites in vitro. J Mater Sci Mater Med. 2005;16:355. doi: 10.1007/s10856-005-0635-8. [DOI] [PubMed] [Google Scholar]
- 9.Gupta G. El-Ghannam A. Kirakodu S. Khraisheh M. Zbib H. Enhancement of osteoblast gene expression by mechanically compatible porous Si-rich nanocomposite. J Biomed Mater Res Part B Appl Biomater. 2007;81B:387. doi: 10.1002/jbm.b.30675. [DOI] [PubMed] [Google Scholar]
- 10.Ren L. Tsuru K. Hayakawa S. Osaka A. In vitro evaluation of osteoblast response to sol-gel derived gelatin-siloxane hybrids. J Sol Gel Sci Technol. 2003;26:1137. [Google Scholar]
- 11.Ayala R. Zhang C. Yang D. Hwang Y. Aung A. Shroff S.S., et al. Engineering the cell-material interface for controlling stem cell adhesion, migration, and differentiation. Biomaterials. 2011;32:3700. doi: 10.1016/j.biomaterials.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Fisher J.P. Lalani Z. Bossano C.M. Brey E.M. Demian N. Johnston C.M., et al. Effect of biomaterial properties on bone healing in a rabbit tooth extraction socket model. J Biomed Mater Res Part A. 2004;68A:428. doi: 10.1002/jbm.a.20073. [DOI] [PubMed] [Google Scholar]
- 13.Jansen E.J.P. Sladek R.E.J. Bahar H. Yaffe A. Gijbels M.J. Kuijer R., et al. Hydrophobicity as a design criterion for polymer scaffolds in bone tissue engineering. Biomaterials. 2005;26:4423. doi: 10.1016/j.biomaterials.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 14.Burdick J.A. Anseth K.S. Photoencapsulation of osteoblasts in injectable RGD-modified PEG hydrogels for bone tissue engineering. Biomaterials. 2002;23:4315. doi: 10.1016/s0142-9612(02)00176-x. [DOI] [PubMed] [Google Scholar]
- 15.Yang F. Williams C.G. Wang D.A. Lee H. Manson P.N. Elisseeff J. The effect of incorporating RGD adhesive peptide in polyethylene glycol diacrylate hydrogel on osteogenesis of bone marrow stromal cells. Biomaterials. 2005;26:5991. doi: 10.1016/j.biomaterials.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 16.Hron P. Hydrophilisation of silicone rubber for medical applications. Polym Int. 2003;52:1531. [Google Scholar]
- 17.Bradley S.G. Munson A.E. McCay J.A. Brown R.D. Musgrove D.L. Wilson S., et al. Subchronic 10 day immunotoxicity of polydimethylsiloxane (silicone) fluid, gel and elastomer and polyurethane disks in female B6C3F1 mice. Drug Chem Toxicol. 1994;17:175. doi: 10.3109/01480549409017860. [DOI] [PubMed] [Google Scholar]
- 18.Rowe V. Spencer H. Bass S. Toxicological studies on certain commercial silicones and hydrolyzable silane intermediates. J Ind Hyg Toxicol. 1948;30:332. [PubMed] [Google Scholar]
- 19.Curtis J. Colas A. Medical applications of silicones. In: Ratner B.D., editor; Hoffman A.S., editor; Schoen F.J., editor; Lemons J.E., editor. Biomaterials Science: An Introduction to Materials in Medicine. 2nd. San Diego: Elsevier Academic Press; 2004. pp. 697–707. [Google Scholar]
- 20.Engler A. Sen S. Sweeney H. Discher D. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 21.Huebsch N. Arany P.R. Mao A.S. Shvartsman D. Ali O.A. Bencherif S.A., et al. Harnessing traction-mediated manipulation of the cell/matrix interface to control stem-cell fate. Nat Mater. 2010;9:518. doi: 10.1038/nmat2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomopoulos S. Williams G.R. Gimbel J.A. Favata M. Soslowsky L.J. Variation of biomechanical, structural, and compositional properties along the tendon to bone insertion site. J Orthop Res. 2003;21:413. doi: 10.1016/S0736-0266(03)00057-3. [DOI] [PubMed] [Google Scholar]
- 23.Hahn M.S. McHale M.K. Wang E. Schmedlen R.H. West J.L. Physiologic pulsatile flow bioreactor conditioning of poly(ethylene glycol)-based tissue engineered vascular grafts. Ann Biomed Eng. 2007;35:190. doi: 10.1007/s10439-006-9099-3. [DOI] [PubMed] [Google Scholar]
- 24.Grunlan M.A. Lee N.S. Mansfeld F. Kus E. Finlay J.A. Callow J.A., et al. Minimally adhesive polymer surfaces (MAPS) prepared from star oligosiloxanes and star oligofluorosiloxanes. J Poly Sci Part A Polym Chem. 2006;44:2551. [Google Scholar]
- 25.Gong C. Fréchet J.M.J. End functionalization of hyperbranched poly(siloxysilane): novel crosslinking agents and hyperbranched-linear star block copolymers. J Polym Sci Part A Polym Chem. 2000;38:2970. [Google Scholar]
- 26.Boutevin B. Guida-Pietrasanta F. Ratsimihety J.A. Synthesis of photocrosslinkable fluorinated polydimethylsiloxanes: direct introduction of acrylic pendant groups via hydrosilylation. Polym Sci Part A Polym Chem. 2000;38:3722. [Google Scholar]
- 27.Hahn M. Miller J. West J. Three dimensional biochemical and biomechanical patterning of hydrogels for guiding cell behavior. Adv Mater. 2006;18:2679. [Google Scholar]
- 28.Hahn M.S. Miller J.S. West J.L. Laser scanning lithography for surface micropatterning on hydrogels. Adv Mater. 2005;17:2939. [Google Scholar]
- 29.Nemir S. Hayenga H.N. West J.L. PEGDA hydrogels with patterned elasticity: novel tools for the study of cell response to substrate rigidity. Biotechnol Bioeng. 2010;105:636. doi: 10.1002/bit.22574. [DOI] [PubMed] [Google Scholar]
- 30.Engler A.J. Richert L. Wong J.Y. Picart C. Discher D.E. Surface probe measurements of the elasticity of sectioned tissue, thin gels and polyelectrolyte multilayer films: Correlations between substrate stiffness and cell adhesion. Surf Sci. 2004;570:142. [Google Scholar]
- 31.Costa K.D. Imaging and probing cell mechanical properties with the atomic force microscope. In: Taatjes D.J., editor; Mossman B.T., editor. Cell Imaging Techniques. Totowa, NJ: Humana Press; 2006. pp. 331–361. [DOI] [PubMed] [Google Scholar]
- 32.Costa K.D. Sim A.J. Yin F.C.-P. Non-Hertzian approach to analyzing mechanical properties of endothelial cells probed by atomic force microscopy. J Biomech Eng. 2006;128:176. doi: 10.1115/1.2165690. [DOI] [PubMed] [Google Scholar]
- 33.Johnson K. Contact Mechanics. Cambridge, United Kingdom: Cambridge University Press; 1985. [Google Scholar]
- 34.Engler A.J. Rehfeldt F. Sen S. Discher D.E. YuLi W. Dennis E.D. Microtissue elasticity: measurements by atomic force microscopy and its influence on cell differentiation. Methods Cell Biol. 2007;83:521. doi: 10.1016/S0091-679X(07)83022-6. [DOI] [PubMed] [Google Scholar]
- 35.Canal T. Peppas N.A. Correlation between mesh size and equilibrium degree of swelling of polymeric networks. J Biomed Mater Res. 1989;23:1183. doi: 10.1002/jbm.820231007. [DOI] [PubMed] [Google Scholar]
- 36.Mellott M.B. Searcy K. Pishko M.V. Release of protein from highly cross-linked hydrogels of poly(ethylene glycol) diacrylate fabricated by UV polymerization. Biomaterials. 2001;22:929. doi: 10.1016/s0142-9612(00)00258-1. [DOI] [PubMed] [Google Scholar]
- 37.Watkins A.W. Anseth K.S. Investigation of molecular transport and distributions in poly(ethylene glycol) hydrogels with confocal laser scanning microscopy. Macromolecules. 2005;38:1326. [Google Scholar]
- 38.Faucheux N. Haye B. Nagel M.D. Activation of the cyclic AMP pathway in cells adhering to biomaterials: regulation by vitronectin- and fibronectin-integrin binding. Biomaterials. 2000;21:1031. doi: 10.1016/s0142-9612(99)00277-x. [DOI] [PubMed] [Google Scholar]
- 39.Munoz-Pinto D. Hybrid polyethylene glycol hydrogels for tissue engineering applications [Ph.D. Chemical Engineering] Texas A&M University; College Station: 2011. [Google Scholar]
- 40.Freed B.K. Biesecker J. Middleton W.J. Spectral polarity index: a new method for determining the relative polarity of solvents [1] J Fluorine Chem. 1990;48:63. [Google Scholar]
- 41.Bryant S.J. Anseth K.S. Hydrogel properties influence ECM production by chondrocytes photoencapsulated in poly(ethylene glycol) hydrogels. J Biomed Mater Res. 2002;59:63. doi: 10.1002/jbm.1217. [DOI] [PubMed] [Google Scholar]
- 42.Bryant S.J. Nuttelman C.R. Anseth K.S. Cytocompatibility of UV and visible light photoinitiating systems on cultured NIH/3T3 fibroblasts in vitro. J Biomater Sci Polym Ed. 2000;11:439. doi: 10.1163/156856200743805. [DOI] [PubMed] [Google Scholar]
- 43.Williams C. Malik A. Kim T. Manson P. Elisseeff J. Variable cytocompatibility of six cell lines with photoinitiators used for polymerizing hydrogels and cell encapsulation. Biomaterials. 2005;26:1211. doi: 10.1016/j.biomaterials.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 44.Hummon A.B. Lim S.R. Difilippantonio M.J. Ried T. Isolation and solubilization of proteins after TRIzol extraction of RNA and DNA from patient material following prolonged storage. Biotechniques. 2007;42:467. doi: 10.2144/000112401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buxton A.N. Zhu J. Marchant R. West J.L. Yoo J.U. Johnstone B. Design and characterization of poly(ethylene glycol) photopolymerizable semi-interpenetrating networks for chondrogenesis of human mesenchymal stem cells. Tissue Eng. 2007;13:2549. doi: 10.1089/ten.2007.0075. [DOI] [PubMed] [Google Scholar]
- 46.Hahn M.S. Teply B.A. Stevens M.M. Zeitels S.M. Langer R. Collagen composite hydrogels for vocal fold lamina propria restoration. Biomaterials. 2006;27:1104. doi: 10.1016/j.biomaterials.2005.07.022. [DOI] [PubMed] [Google Scholar]
- 47.Hou Y. Schoener C.A. Regan K.R. Munoz-Pinto D. Hahn M.S. Grunlan M.A. Photo-crosslinked PDMSstar-PEG hydrogels: synthesis, characterization, and potential application for tissue engineering scaffolds. Biomacromolecules. 2010;11:648. doi: 10.1021/bm9012293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sawhney A.S. Pathak C.P. Hubbell J.A. Bioerodible hydrogels based on photopolymerized poly(ethylene glycol)-co-poly(a-hydroxy acid) diacrylate macromers. Macromolecules. 1993;26:581. [Google Scholar]
- 49.Lim J.Y. Shaughnessy M.C. Zhou Z. Noh H. Vogler E.A. Donahue H.J. Surface energy effects on osteoblast spatial growth and mineralization. Biomaterials. 2008;29:1776. doi: 10.1016/j.biomaterials.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 50.Anselme K. Osteoblast adhesion on biomaterials. Biomaterials. 2000;21:667. doi: 10.1016/s0142-9612(99)00242-2. [DOI] [PubMed] [Google Scholar]
- 51.Schmedlen R.H. Elbjeirami W.M. Gobin A.S. West J.L. Tissue engineered small-diameter vascular grafts. Clin Plast Surg. 2003;30:507. doi: 10.1016/s0094-1298(03)00069-5. [DOI] [PubMed] [Google Scholar]
- 52.Fischer G.M. Cox R.H. Detweiler D.K. Altered arterial connective tissue in racing greyhound dogs. Experientia. 1975;31:1426. doi: 10.1007/BF01923225. [DOI] [PubMed] [Google Scholar]
- 53.Sires B. Lemke B. Dortzbach R. Gonnering R. Characterization of human orbital fat and connective tissue. Ophthal Plast Reconstr Surg. 1998;14:403. doi: 10.1097/00002341-199811000-00004. [DOI] [PubMed] [Google Scholar]
- 54.Stevens M.M. Marini R.P. Martin I. Langer R. Shastri V.P. FGF-2 enhances TGF-beta 1-induced periosteal chondrogenesis. J Orthop Res. 2004;22:1114. doi: 10.1016/j.orthres.2003.12.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.