

Abstract
Background
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is directly linked to mutations in proteins (e.g., RyR2R4496C) responsible for intracellular Ca2+ homeostasis in the heart. However, the mechanism of Ca2+ release dysfunction underlying CPVT has only been investigated in isolated cells but not in the in situ undisrupted myocardium.
Methods and Results
We investigated in situ myocyte Ca2+ dynamics in intact Langendorff perfused hearts (ex vivo) from wildtype (WT) and RyR2R4496C+/− mice using laser scanning confocal microscopy. We found that myocytes from both WT and RyR2R4496C+/− hearts displayed uniform, synchronized Ca2+ transients. Ca2+ transients from beat to beat were comparable in amplitude with identical activation and decay kinetics in WT and RyR2R4496C+/− hearts, suggesting that excitation-contraction (EC) coupling between the sarcolemmal Ca2+ channels and mutated RyR2R4496C+/− channels remains intact under baseline resting conditions. Upon adrenergic stimulation, RyR2R4496C+/− hearts exhibited a high degree of Ca2+ release variability (CRV). The varied pattern of Ca2+ release was absent in single isolated myocytes, independent of cell cycle length, synchronized among neighboring myocytes, and correlated with CPVT. A similar pattern of action potential variability, which was synchronized among neighboring myocytes, was also revealed under adrenergic stress in intact hearts but not in isolated myocytes.
Conclusions
Our studies using in situ confocal imaging approach suggest that mutated RyR2s are functionally normal at rest but display a high degree of CRV upon intense adrenergic stimulation. CRV is a Ca2+ release abnormality resulting from electrical defects rather than the failure of the Ca2+ release response to action potentials in mutated ventricular myocytes. Our data provide important insights into Ca2+ release and electrical dysfunction in an established model of CPVT.
Keywords: arrhythmia (mechanisms), calcium, catecholaminergic polymorphic ventricular tachycardia, sarcoplasmic reticulum, ryanodine receptors
Introduction
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a lethal genetic disease characterized by exercise/stress-induced malignant ventricular arrhythmias and sudden cardiac death in young individuals with structurally normal hearts.1 The genetic foundation of CPVT is linked to mutations within two important Ca2+ handling proteins: autosomal dominant mutations of intracellular Ca2+ release channels or type 2 ryanodine receptors (RyR2),2, 3 and autosomal recessive mutations of calsequestrin (CASQ), a Ca2+ binding protein in the sarcoplasmic reticulum (SR).4 Patients with CPVT mutations are typically healthy at rest but develop arrhythmias under emotional or physical stress. Electrocardiograms of CPVT patients reveal bidirectional ventricular tachycardia and polymorphic ventricular tachycardia.2
Both RyR2 and CASQ play important roles in controlling intracellular Ca2+ handling and homeostasis.5, 6 Ca2+ release dysfunction is believed to be the underlying mechanism for the above mentioned ventricular arrhythmias in human patients due to the critical importance of the two key proteins in Ca2+ regulation of cardiomyocytes.3, 7, 8 Indeed, many studies have identified Ca2+ handling defects using mutant channels in isolated lipid bilayers,9–11 cultured myocytes, HEK 293 cells stably transfected with these mutants,12–15 or in ventricular myocytes isolated from mouse models that carry mutant RyR2.16–19 Notably, a recent study by Fernandez-Velasco, et al., showed that, compared to wild type (WT) myocytes, untreated RyR2R4496C+/− myocytes have an enhancement of Ca2+ sensitivity and an increase in spontaneous Ca2+ release in diastole during electrical pacing, which is augmented by isoproterenol and increasing the pacing frequency.20 Their results were further supported by Kang, et al., who demonstrated spontaneous Ca2+ release events in both RyR2R4496C+/− ventricular and Purkinje cells.21 However, these in vitro studies are controversial because it is unclear if these mutant proteins behave abnormally under resting conditions (i.e., in the absence of catecholamine stimulation).22, 23 One unresolved issue, however, is that the resting defect in Ca2+ dynamics does not correspond to the clinical manifestations of CPVT patients with the same mutation, whose hearts are structurally normal and free of arrhythmias unless under stress.
A second question is related to the pathophysiology of CPVT RyR2 mutations, in particular how RyR2 mutations cause CPVT. Several previous studies demonstrated that ventricular myocytes harboring the RyR2 R4496C mutation are prone to spontaneous Ca2+ release and delayed after depolarizations (DADs).13, 16, 20, 21 One of the leading hypotheses is that the mutation induces SR calcium leak in ventricular myocytes during diastole, thereby generating DADs that, in turn, trigger fatal cardiac arrhythmias.24 Those previous results were obtained from isolated myocytes, and it remains unclear whether mutated RyR2s in physiologically coupled myocytes in intact heart behave differently from isolated myocytes during steady state beating. It is therefore important to study the nature of Ca2+ handling under physiological conditions in RyR2-mutated hearts.
In the present study, we aimed to identify the in situ feature of Ca2+ mis-handling in CPVT hearts and to enhance our understanding of Ca2+-dependent arrhythmogenesis using a RyR2R4496C+/− mouse model. We performed in situ, ex vivo Ca2+ / action potential (AP) imaging in Langendorff-perfused intact hearts under near physiological conditions, using laser scanning confocal microscopy at baseline and following catecholamine stimulation. We also mapped Ca2+ dynamics to simultaneously recorded ECGs. Our data demonstrate that myocyte EC coupling between the sarcolemmal Ca2+ channels and mutated RyR2R4496C+/− channels remains intact under baseline resting conditions. However, under intense adrenergic stress, we identified a previously unappreciated pattern of Ca2+ handling dysfunction in physiologically-coupled ventricular myocytes with the RyR2 R4496C mutation. Interestingly, highly variable SR Ca2+ release in RyR2R4496C+/− hearts was synchronized among neighboring myocytes and correlated with CPVT occurrence as measured by ECG. Similarly, with in situ AP imaging, we detected stress-induced beat-to-beat variability in AP, which was also synchronized among neighboring mutated myocytes.
Methods
Animal experiments were performed in accordance with the protocol approved by the Institutional Animal Care and Use Committee at the University of Iowa. In situ confocal Ca2+ imaging in intact hearts with / without ex vivo electrocardiogram, in situ confocal Aaction Potential (AP) imaging in intact hearts were adapted from published reports.25–27 Ca2+ imaging in adult single isolated ventricular myocytes and in primary cultured neonatal myocytes were performed as previously described.28, 29 Confocal Ca2+ / AP images were analyzed offline with custom routines composed with IDL image analysis software (ITT VIS Inc., Boulder, CO).30 Pseudo ECG data were processed offline with Clampfit 10. Data were expressed as mean ± SE, and median with interquartile range in boxplots. Multiple regression analysis was performed to determine the correlation coefficient and significance. Student’s t-tests were applied for pair-wise comparison. Bonferroni procedure following a global test based on linear mixed effects model was performed for multiple group comparisons (NCSS, Kaysville, Utah). A compound symmetry correlation structure was assumed for linear mixed effects model tests. A p value of <0.05 was considered statistically significant. Expanded methods are available in Supplementary Material.
Results
Normal EC coupling in in situ RyR2R4496C+/− myocytes at rest
RyR2R4496C+/− mutant mice are susceptible to CPVT under catecholamine stimulation.31 We performed in situ Ca2+ imaging of ventricular myocytes from intact hearts attached to an oxygenated Langendorff-perfusion system.25 We examined autonomous Ca2+ signals, initiated by sinus rhythm, in ventricular myocytes. In linescan mode, each myocyte from both WT and RyR2R4496C+/− hearts displayed uniform, synchronized Ca2+ transients. Ca2+ transients from beat-to-beat were comparable in amplitude with identical activation and decay kinetics between these groups (Figure 1), suggesting that EC coupling between the sarcolemmal Ca2+ channels and mutated RyR2R4496C+/− channels (e.g. calcium-induced calcium release) remains intact under baseline resting conditions. In addition, spontaneous Ca2+ sparks or waves were rarely observed at diastolic phase during steady state beating in both WT and RyR2R4496C+/− myocytes, indicating mutated RyR2s are not leaky under resting condition.
Figure 1.

In situ confocal Ca2+ imaging in WT and RyR2R4496C+/− hearts: normal Ca2+ transients under resting conditions. A–B, Autonomous, synchronized Ca2+ transients driven by sinus rhythm in (A) WT and (B) RyR2R4496C+/− hearts. Spontaneous Ca2+ waves or Ca2+ sparks were rarely detected during steady state beating under resting conditions. Bottom panels are spatial averages of Ca2+ signals from corresponding images. C, Boxplots of Ca2+ transients (amplitude, time to peak, and T50 relaxation), ◆ and error bars inside the box denotes mean±SE. N=5–7 hearts, n=54 or 82 frames of transients (most of frames include multiple myocytes) for WT and RyR2R4496C+/−, respectively. Green bars indicate edges of myocytes in this and subsequent figures.
High beat-to-beat variability of Ca2+ transients in RyR2R4496C+/− myocytes under adrenergic stress
CPVT occurs under physical or emotional stress in patients with the RyR2R4496C+/− mutation and are elicited exclusively by adrenergic stress.2 RyR2R4496C+/− mice also display similar ECG abnormalities under high catecholamine and caffeine stimulation.32 To further understand Ca2+ performance in RyR2R4496C+/− myocytes during high adrenergic stress, we next compared in situ Ca2+ dynamics of WT and RyR2R4496C+/− myocytes in intact hearts under sinus rhythm with perfusion of epinephrine (1 μM) and caffeine (0.6 mM), a condition that induces CPVT in RyR2R4496C+/− mice.16, 31, 32 In WT myocytes, epinephrine plus caffeine increased the amplitude of Ca2+ transients and accelerated both the activation and decay kinetics (Figure 2A, D–F). Epinephrine with caffeine also significantly accelerated the kinetics of Ca2+ transients (both activation and relaxation) in RyR2R4496C+/− myocytes (Figure 2D–F). Strikingly, RyR2R4496C+/− myocytes under high sympathetic stress exhibited high beat to beat Ca2+ release variability (CRV) in the amplitude of Ca2+ transients (Figure 2BC). While some Ca2+ transients with a higher amplitude were detected in mutant myocytes after adrenergic stimulation, many of the Ca2+ transients were markedly reduced, which resulted in a lower average amplitude as compared to WT myocytes (Figure 2D). It should be noted that these transients with reduced amplitude differed from asynchronous Ca2+ waves. Specifically, they were synchronized with fast rising kinetics (Tpeak), similar to that of WT myocytes (Figure 2E). CRV activity often followed an irregular pattern of amplitude fluctuation of Ca2+ transients. In RyR2R4496C+/− myocytes, CRV was reversible and disappeared upon washout of epinephrine and caffeine, suggesting that this CRV defect is induced by adrenergic stress. Variance analysis from thousands of Ca2+ transient samples (5–7 hearts in each group) further supports our observation that Ca2+ transients are highly varied in amplitude from beat-to-beat under high sympathetic stress conditions, but not at baseline (Figure 3AB). Importantly, this CRV pattern was not observed in single isolated RyR2R4496C+/− myocytes under field stimulation with the same adrenergic stimulation (Supporting Information, Figure S1). Our data suggest that adrenergic stress-induced CRV requires physiological cellular coupling in RyR2R4496C+/− hearts.
Figure 2.

High degree of Ca2+ release variability in RyR2R4496C+/− myocytes from intact heart as demonstrated by in situ confocal imaging after adrenergic stimulation. A, Autonomous Ca2+ transients in myocytes from WT hearts under epinephrine (1 μM) and caffeine (0.6 mM) perfusion. Beat-to-beat Ca2+ transients were stable in WT myocytes even under adrenergic stress challenge. B–C, Ca2+ transients from RyR2R4496C+/− myocytes of intact hearts, exhibiting high degree of variability in Ca2+ transient amplitude. These Ca2+ transients with varied amplitude were action potential-triggered events with a quick rising and decay phase, which is distinct from asynchronous propagated Ca2+ wave events. D–F, Boxplots of Ca2+ transients (amplitude, time to peak - Tpeak, and decay rate T50). ◆ and error bars inside the box denotes mean±SE. The averaged amplitude of Ca2+ transients of RyR2R4496C+/− myocytes under epinephrine plus caffeine perfusion was less than that of control. N=5–7 hearts for each group, n=107 or 74 frames of transients for WT and RyR2R4496C+/− myocytes, respectively. * p<0.05 and ** p<0.01 vs WT or RyR2R4496C+/− without epinephrine and caffeine (control), ## p<0.01 vs WT under the same condition (epinephrine and caffeine). Global test: p=0.00001, 0.005, 0.02 among the 4 groups, for D, E, F, respectively.
Figure 3.
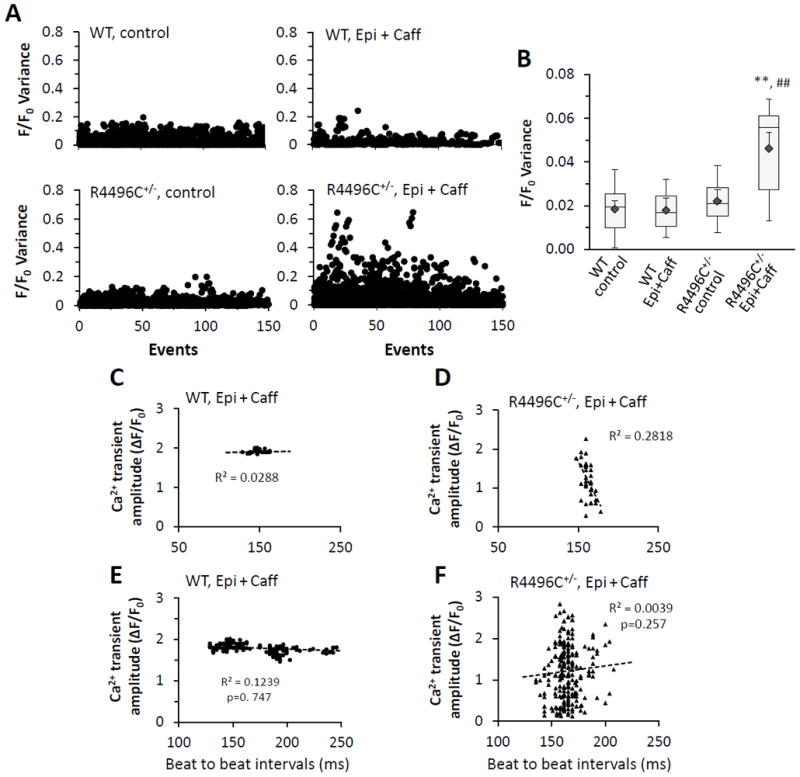
Ca2+ release variability in RyR2R4496C+/− myocytes from intact hearts is independent of the beating interval preceding each transient. A–B, Variance analysis of CRV. A, Variance was calculated as the square of variation (sample value minus mean value) of each transient amplitude. Scatter plots of F/F0 variance from WT and RyR2R4496C+/− myocytes under resting (control) and stress (epinephrine plus caffeine) conditions, respectively, are shown. B, Boxplot of F/F0 variance from each group. RyR2R4496C+/− myocytes exhibited significant higher variance comparing to other groups, ◆ and error bars inside the box denotes mean±SE. n=5–7 hearts, n=1947, 1502, 2405, or 2449, respectively, for each group. **p<0.01 vs RyR2R4496C+/− under control condition, ## p<0.01 vs WT under the same condition (epinephrine and caffeine). p=0.008 among the 4 groups (global test). C–F, No Correlation between Ca2+ transient amplitude (F/F0) and the beating interval preceding each transient in WT (C) and mutant (D) hearts. C–D, Examples displaying Ca2+ transient amplitude – time interval correlation from a representative Ca2+ image of WT (C) and RyR2R4496C+/− (D) heart, respectively, under epinephrine and caffeine stimulation. E–F, Multiple regression analysis on data from 3–4 hearts of WT and RyR2R4496C+/−(n=163, 227 events, respectively), indicating no correlation between Ca2+ transient amplitude and time interval preceding each transient in both WTs and mutant hearts.
High CRV is not due to beating interval variability
We postulated that the high CRV observed in mutated myocytes from intact hearts is due to variable beating intervals preceding each Ca2+ transient, which would cause varied SR Ca2+ loading under ex vivo conditions. To test this hypothesis, we measured the time intervals preceding each Ca2+ transient and correlated it with the amplitude of the corresponding transient from WT and RyR2 mutant hearts. As shown in Figure 3C–F, the range of amplitudes of Ca2+ transients from WT myocytes (after epinephrine + caffeine) was narrow, while the amplitudes of Ca2+ transients from mutated myocytes were dispersed over a much wider range. More importantly, there was no correlation between the beating intervals and Ca2+ transient amplitudes in either WT or mutated myocytes under stress. This analysis clearly indicates that the high level of CRV observed in RyR2R4496C+/− myocytes under ex vivo conditions and with high sympathetic stimulation is not caused by beating interval variability and is independent of stimulation frequency.
CRV is synchronized among neighboring myocytes with mutant RyR2
An advantage of in situ confocal imaging is that it allows examination of the Ca2+ dynamics of multiple, physiologically interconnected myocytes simultaneously. The data in Figure 2B&C revealed a coordinated pattern of CRV between two neighboring cells from RyR2R4496C+/− hearts. To further confirm this phenomenon, we evaluated the pattern of CRV among multiple myocytes from RyR2R4496C+/− hearts. Surprisingly, we found that CRV is indeed present in a coordinated pattern among many neighboring myocytes (Figure 4). This novel pattern of Ca2+ release dysfunction was consistently observed in RyR2R4496C+/− hearts during adrenergic stress but not in WT hearts. Interestingly, CRV was not observed in RyR2R4496C+/− cultured neonatal myocytes or among neighboring cells under both control and adrenergic stimulation conditions (Supporting Information, Figure S2). Collectively, these data strongly suggest that CRV is an integrated, tissue level response to adrenergic stress, and that the source of CRV is likely not ventricular myocytes themselves, but from an abnormality in the upstream electrical signal.
Figure 4.

Ca2+ release variability in RyR2R4496C+/− myocytes under sympathetic stress: synchrony among neighboring myocytes. A, A typical Ca2+ image of 5 myocytes from a RyR2R4496C+/− heart. B, Spatial profile of Ca2+ signals from each myocyte showing that stress-induced Ca2+ release variability was coordinated among many neighboring mutated myocytes.
CRV is associated with CPVT
In order to test if increased CRV was related to CPVT, we performed simultaneous recordings of in situ confocal linescan Ca2+ images and real time ex vivo ECG measurements in Langendorff perfused intact hearts. WT hearts exhibited normal ECGs and regular, stable Ca2+ transients from beat-to-beat among different neighboring myocytes (Figure 5A) in both the absence and presence of epinephrine and caffeine. RyR2R4496C+/− hearts also displayed normal ECG and Ca2+ dynamics at baseline. However, under high sympathetic stimulation, CRV occurred in RyR2R4496C+/− hearts simultaneously with CPVT (Figure 5B). These data suggest that CRV, a tissue level measure of SR Ca2+ release abnormality in RyR2R4496C+/− hearts, is associated with electrical abnormalities, including CPVT.
Figure 5.

Ca2+ release variability and CPVT in RyR2R4496C+/− hearts. Confocal Ca2+ imaging was performed with simultaneous recordings of ex vivo ECG in hearts with continuous Langendorff perfusion. A, A WT heart exhibited normal ex vivo ECG and stable Ca2+ transients from beat-to-beat under stress (Note: p wave is visible but has very small magnitude). B, RyR2R4496C+/− hearts concurrently displayed CRV and CPVT.
Physiologically coupled RyR2R4496C+/− myocytes have abnormal APs
To investigate the underlying mechanism of CRV, we then measured the in situ APs under the same recording conditions as for in situ Ca2+ imaging,27 except that a fast voltage-sensitive dye, ANNINE-6plus, was loaded into the intact hearts through Langendorff perfusion to track the dynamic changes of transmembrane potential. We applied this imaging technique to examine the in situ electrical properties of RyR2R4496C+/− myocytes in intact hearts under adrenergic stress. Surprisingly, we found that RyR2R4496C+/− myocytes also exhibited high heterogeneity in AP morphology under adrenergic stress in comparison to WT myocytes (Figure 6AB). On average, RyR2R4496C+/− myocytes had similar ratio of fluorescence change (indicative of AP amplitude) and shorter AP duration than those of WT myocytes (Figure 6CD). Variance analysis showed that mutant myocytes had a greater variability in both AP amplitude and duration (Figure 6EF). More importantly, AP variability was also synchronized among neighboring myocytes from the RyR2R4496C+/− hearts (Figure 6B & Supporting Information, Figure S3). By contrast, AP studies in single isolated myocytes using conventional current clamp recordings showed no difference in variance in these parameters between WT and RyR2R4496C+/− hearts under adrenergic stress (Supporting Information, Figure S4), suggesting that in situ electrical abnormalities in RyR2R4496C+/− hearts are non-ventricular in origin. Taken together, our data support the notion that electrical abnormalities of non-ventricular origin underlie the mechanism of CRV in physiologically coupled ventricular myocytes.
Figure 6.
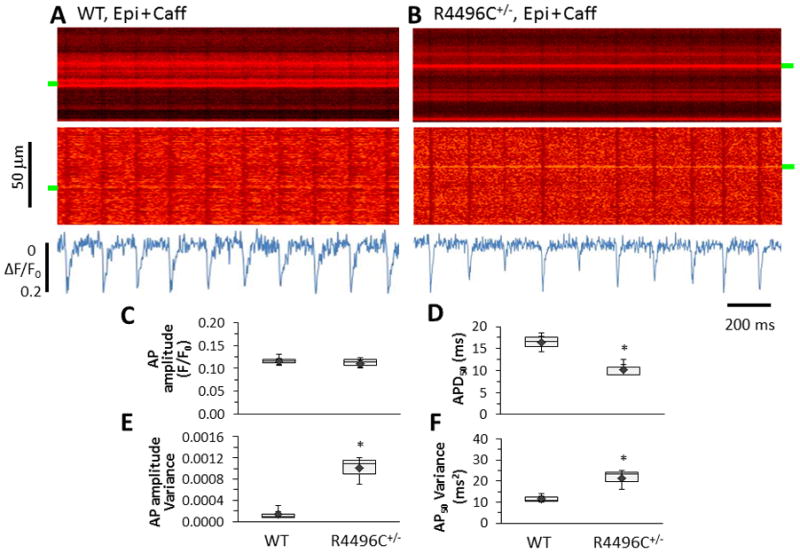
In situ imaging of myocyte APs in intact hearts. A–B, Representative examples of ANNINE-6plus fluorescence from WT and RyR2R4496C+/− hearts perfused with epinephrine plus caffeine. Top to bottom: raw images, images after normalization, and spatial average of ANNINE-6plus fluorescence. RyR2R4496C+/− myocytes displayed beat-to-beat variation in AP morphology. C–D, Summary of AP amplitude (ΔF/F0) and APD50 in boxplots. E–F, Boxplots of variance of AP amplitude and APD50. * p<0.05, n=4 and 5 for WT and RyR2R4496C+/− hearts, and 326 or 213 AP events for WT or RyR2R4496C+/− hearts, respectively.
Discussion
Ca2+ handling abnormalities play an important role in the pathophysiology of heart disease, including heart failure, arrhythmias, and sudden cardiac death.33 Patients with a specific mutation (R4496C) in the cardiac Ca2+ release channel, RyR2, suffer from exercise/stress induced CPVT and sudden cardiac death. The genetically modified mouse model bearing the same mutation provides an excellent model for studying the mechanisms of Ca2+-dependent arrhythmogenesis and the pathophysiology of CPVT.32 In this study, we applied the in situ confocal Ca2+ imaging techniques to study Ca2+ handling in intact hearts.25 This approach allows investigation under near physiological conditions. To our surprise, we obtained distinctly different results as compared to results in ventricular myocytes isolated from these same mice.20, 21, 34 We found that 1) mutated RyR2s are functionally normal during non-stressed, resting condition; 3) we identified a new pattern of Ca2+ release dysfunction, CRV, in RyR2R4496C+/− but not WT myocytes under adrenergic stress; 4) CRV is an integrated, tissue level response of mutated myocytes to adrenergic stress and is observed in intact hearts but not in single isolated myocytes; 5) CRV is synchronized among neighboring myocytes, independent of beating interval or stimulation frequency; 6) CRV is tightly associated with CPVT; and 7) AP variability in intact hearts (but not in single isolated myocytes) is the likely cause of CRV.
Normal CICR function in ventricular myocytes with RyR2R4496C+/− mutation at rest
Whether mutant RyR2 channels are functionally normal at rest is a controversial yet fundamentally important question. Particularly for patients with this mutation, it is critical to determine if the dysfunctional Ca2+ handling is constantly present and is exacerbated with stress, or if the mutated channels are normal at resting state and become dysfunctional only with adrenergic stimulation. The answer to this issue may provide important insights in developing therapeutic strategies for CPVT patients. Marks and colleagues demonstrated using single channel recording in lipid-bilayers that RyRs harboring human mutations (including R4496C) show similar gating features as WT RyRs at baseline conditions but are much more leaky upon high protein kinase A stimulation (10-fold increase in channel open probability).9, 10 George and Lai also showed unaltered baseline Ca2+ activity in HL-1 myocytes transfected with mutated RyR2s.35 However, these results were challenged by other groups who reported an increase in abnormal Ca2+ release and higher sensitivity of mutant RyR2s compared to WT at resting conditions. Jiang et al. reported that CPVT mutations enhance the sensitivity of the RyR2 channel to luminal Ca2+ activation and lower the threshold for spontaneous Ca2+ release in the setting of Ca2+ overload.11, 13 Recently, Fernandez-Velasco et al. showed an increased incidence of Ca2+ sparks and Ca2+ waves at baseline in RyR2R4496C+/− myocytes, which is further enhanced by either isoproterenol or high pacing rates.20 Liu et al. reported similar results of increased spark frequency at baseline and after isoproterenol challenge in isolated RyR2R4496C+/− myocytes.19 However, when we examined Ca2+ signals from intact WT and mutant hearts using the in situ imaging method, we found RyR2 mutated myocytes exhibit no abnormal Ca2+ release, e.g., Ca2+ waves, at resting conditions (Figure 1). These discrepancies may be explained by differences in experimental conditions that resulted in exposure of RyR2 to different intra-luminal Ca2+ levels. Enzymatic and mechanical dissociation of heart tissue exerts a significant stress to the myocytes, causing myocyte Ca2+ overload and spontaneous Ca2+ release.34, 36, 37 Instead, under conditions which did not exist or cause SR Ca2+ overloading such as in lipid bilayers, 9, 10 cultured HL-1 myocytes 35 and intact hearts (of the present study), findings were surprisingly consistent, that is, RyR2s carrying R4496C mutation are not leaky at baseline. It has been shown that CPVT-linked RyR2 mutations, including R4496C, increase the sensitivity of the RyR2 channel to SR Ca2+ overload.13 In the absence of SR Ca2+ overload (i.e., in situ under resting conditions or studies with lipid bilayers 9, 10 or cultured HL-1 myocytes 35), the R4496C mutation-linked Ca2+ release defect would not be apparent. Importantly, our in situ findings are also consistent with the clinical characteristics of CPVT patients whose hearts are structurally normal throughout their lives but only develop fatal arrhythmias under emotional or physical stress. If mutated RyR2 channels expressed in myocytes are continuously leaky at resting conditions, this persistent abnormality of intracellular Ca2+ signaling is predicted to lead to alterations in myocyte gene transcription that could eventually promote cardiac pathophysiology (e.g., cardiomyopathy).
Ca2+ release variability and electrical abnormalities in physiologically coupled myocytes with RyR2R4496C+/− mutation
In this study, we identified a new pattern of Ca2+ release dysfunction - a high degree of variation in Ca2+ transient amplitude in RyR2R4496C+/− myocytes. This pattern of Ca2+ release abnormality was present in mutated myocytes under adrenergic stimulation; it was not observed in WT cells, in RyR2 mutated myocytes under baseline conditions, or in mutated neonatal myocytes in culture. This result was only detectable by in situ imaging methods. Ca2+ release variability is distinct in nature from rapid pacing-induced Ca2+ alternans,25 in that 1) CRV is independent of the cycle length; 2) the change in Ca2+ transient amplitude is predominantly very irregular; 3) CRV is not inducible in isolated myocytes (either WT or mutated myocytes); and 4) rapid high-rate pacing-induced Ca2+ alternans may not be coordinated or synchronized among different myocytes from beat to beat.25 These features suggest that proarrhythmic defects in RyR2R4496C+/− hearts are not limited to ventricular myocytes. Recently, two elegant studies using different approaches (optical mapping and confocal imaging) both concluded that the RyR2R4496C mutation associated CPVT originated from abnormalities in Purkinje cells. Cerrone et al. studied the mechanisms and origin of CPVT arrhythmias in Langendorff-perfused RyR2R4496C+/− mouse hearts using optical mapping.38 Their data provide compelling evidence that CPVT arrhythmias originate from the specialized conduction system. Very recently, Fishman and colleagues examined the frequency and severity of spontaneous Ca2+ activities in Purkinje cells in comparison to ventricular myocytes with the aid of a novel fluorescent reporter in the cardiac conduction system (including the distal Purkinje fiber network). These studies revealed that Ca2+ handling defects in Purkinje cells are more pronounced and frequent than those of ventricular myocytes with the same mutation.21 Our data are consistent with these studies by demonstrating that CRV occurs in intact hearts but not in single isolated myocytes with RyR2 mutation, and that CRV is synchronized among different neighboring myocytes.
Our data suggest that CRV may originate from an abnormality in electrical activity in other regions, such as Purkinje fibers or ectopic ventricular foci. Providing additional support for this conclusion, our in situ AP optical recordings and single cell AP studies indicate that this defect is in electrical conduction system rather than ventricular in origin. Therefore, it is postulated that under stress conditions, defective RyR2-mediated abnormal Ca2+ release in Purkinje cells leads to aberrant electrical activities, thereby triggering variability in Ca2+ release in ventricular myocytes. We speculate that the mechanism is as follows: Under stress condition, spontaneous Ca2+ releases in Purkinje fibers with RyR2R4496C+/− mutation cause DADs, which conduct and produce abnormal APs in ventricular myocytes, as we have observed using in situ AP imaging technique. These abnormal APs, specifically, high variability of AP amplitude and duration but synchronized among neighboring ventricular myocytes, may lead to a varied magnitude of Ca2+ influx, causing coordinated Ca2+ release variability among mutated ventricular myocytes. Also contributing to the mechanism, it is very possible that upon stress the Purkinje fibers initiate the arrhythmia, which is further sustained by ventricular myocytes carrying defective mutant of RyR2s.
Limitations
In human patients with this or other similar CPVT-linked mutations, intense emotional or physical stress may trigger the appearance of symptoms, such as ventricular arrhythmias, syncope, and sudden cardiac death. However, in RyR2R4496C+/− mice, the combination of caffeine and epinephrine, instead of epinephrine alone, is necessary to induce CPVT. This may suggest that humans with the same mutation are more susceptible to Ca2+ disorder-induced CPVT than mice. This difference does not appear to be related to basal catecholamine levels since β-adrenergic blockade failed to completely prevent sudden cardiac death in patients with CPVT mutations.39 Future studies towards understanding the mechanisms underlying this difference are warranted.
Summary
Our study using the in situ confocal imaging approach provides compelling evidence that the RyR2R4496C+/− mutants are functionally normal in situ under resting conditions, but display high degree of CRV upon intense adrenergic stimulation. CRV is an integrated, tissue level response of mutated myocytes to adrenergic stress and is closely correlated with CPVT. Our data reveal that CRV results from electrical defects rather than the failure of Ca2+ release response to action potentials in mutated ventricular myocytes. This study provides important insights into Ca2+ release and electrical dysfunction in an established model of CPVT and has important implications in understanding the mechanism of CPVT in patients.
Supplementary Material
Ca2+ handling abnormalities play important roles in the pathophysiology of heart failure, arrhythmias, and sudden cardiac death. Patients with a specific mutation (R4496C) in the cardiac Ca2+ release channel, RyR2, suffer from exercise/stress-induced catecholaminergic polymorphic ventricular tachycardia (CPVT) and sudden cardiac death, but are typically healthy at rest. The reason that the resting defect in Ca2+ dynamics in vitro in isolated myocytes does not correspond to the clinical manifestations of CPVT patients with the same mutation is unexplained. In a genetically modified mouse model of CPVT (RyR2R4496C+/−), we applied in situ confocal Ca2+ imaging techniques to study Ca2+ handling in undisrupted myocardium of intact hearts. This approach allows investigation under near physiological conditions. The RyR2R4496C+/− mutants were found to be functionally normal in situ under resting conditions, but had a high degree of Ca2+ release variability upon intense adrenergic stimulation. This new pattern of Ca2+ handling abnormality is an integrated, tissue level response of mutated myocytes to adrenergic stress, closely correlated with CPVT. Our data reveal that Ca2+ release variability results from electrical defects, likely originating from Purkinje fibers, rather than the failure of Ca2+ release response to action potentials in mutated ventricular myocytes. This study provides insights into Ca2+ release and electrical dysfunction in CPVT.
Acknowledgments
We sincerely thank Dr. Kai Wang (a biostatistician at the University of Iowa, College of Public Health) for kind help in statistical analysis.
Funding Sources: This work was supported by NIH R01 HL090905 (L.S.S.) and American Heart Association Scientific Development Grant 0635056N (L.S.S.); the research grants from the Canadian Institutes of Health Research and the National Institutes of Health (2R01HL075210) to S.R.W.C.; NIH R01 HL079031, HL62494, HL70250 and HL113001 (M.E.A.) and a grant from the Fondation Leducq for the Alliance for CaMKII Signaling (M.E.A.)
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation. 1995;91:1512–1519. doi: 10.1161/01.cir.91.5.1512. [DOI] [PubMed] [Google Scholar]
- 2.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino VV, Danieli GA. Mutations in the cardiac ryanodine receptor gene (hryr2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 3.Priori SG, Napolitano C. Cardiac and skeletal muscle disorders caused by mutations in the intracellular Ca2+ release channels. J Clin Invest. 2005;115:2033–2038. doi: 10.1172/JCI25664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lahat H, Eldar M, Levy-Nissenbaum E, Bahan T, Friedman E, Khoury A, Lorber A, Kastner DL, Goldman B, Pras E. Autosomal recessive catecholamine- or exercise-induced polymorphic ventricular tachycardia: Clinical features and assignment of the disease gene to chromosome 1p13–21. Circulation. 2001;103:2822–2827. doi: 10.1161/01.cir.103.23.2822. [DOI] [PubMed] [Google Scholar]
- 5.Bers DM. Excitation-contraction coupling and cardiac contractile force. Boston: Kluwer Academic Publishers; 2001. [Google Scholar]
- 6.Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C, Pfeifer K. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116:2510–2520. doi: 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu N, Priori SG. Disruption of calcium homeostasis and arrhythmogenesis induced by mutations in the cardiac ryanodine receptor and calsequestrin. Cardiovasc Res. 2008;77:293–301. doi: 10.1093/cvr/cvm004. [DOI] [PubMed] [Google Scholar]
- 8.Wang SQ, Song LS, Lakatta EG, Cheng H. Ca2+ signalling between single l-type Ca2+ channels and ryanodine receptors in heart cells. Nature. 2001;410:592–596. doi: 10.1038/35069083. [DOI] [PubMed] [Google Scholar]
- 9.Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N, D’Armiento JM, Napolitano C, Memmi M, Priori SG, Lederer WJ, Marks AR. Fkbp12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829–840. doi: 10.1016/s0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 10.Lehnart SE, Wehrens XH, Laitinen PJ, Reiken SR, Deng SX, Cheng Z, Landry DW, Kontula K, Swan H, Marks AR. Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation. 2004;109:3208–3214. doi: 10.1161/01.CIR.0000132472.98675.EC. [DOI] [PubMed] [Google Scholar]
- 11.Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, Cheng H, Chen SR. Ryr2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (soicr) Proc Natl Acad Sci USA. 2004;101:13062–13067. doi: 10.1073/pnas.0402388101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang D, Xiao B, Zhang L, Chen SR. Enhanced basal activity of a cardiac Ca2+ release channel (ryanodine receptor) mutant associated with ventricular tachycardia and sudden death. Circ Res. 2002;91:218–225. doi: 10.1161/01.res.0000028455.36940.5e. [DOI] [PubMed] [Google Scholar]
- 13.Jiang D, Wang R, Xiao B, Kong H, Hunt DJ, Choi P, Zhang L, Chen SR. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of ryr2 mutations linked to ventricular tachycardia and sudden death. Circ Res. 2005;97:1173–1181. doi: 10.1161/01.RES.0000192146.85173.4b. [DOI] [PubMed] [Google Scholar]
- 14.George CH, Jundi H, Walters N, Thomas NL, West RR, Lai FA. Arrhythmogenic mutation-linked defects in ryanodine receptor autoregulation reveal a novel mechanism of Ca2+ release channel dysfunction. Circ Res. 2006;98:88–97. doi: 10.1161/01.RES.0000199296.70534.7c. [DOI] [PubMed] [Google Scholar]
- 15.Terentyev D, Nori A, Santoro M, Viatchenko-Karpinski S, Kubalova Z, Gyorke I, Terentyeva R, Vedamoorthyrao S, Blom NA, Valle G, Napolitano C, Williams SC, Volpe P, Priori SG, Gyorke S. Abnormal interactions of calsequestrin with the ryanodine receptor calcium release channel complex linked to exercise-induced sudden cardiac death. Circ Res. 2006;98:1151–1158. doi: 10.1161/01.RES.0000220647.93982.08. [DOI] [PubMed] [Google Scholar]
- 16.Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, Imbriani M, Napolitano C, Lai FA, Priori SG. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: Insights from a ryr2 r4496c knock-in mouse model. Circ Res. 2006;99:292–298. doi: 10.1161/01.RES.0000235869.50747.e1. [DOI] [PubMed] [Google Scholar]
- 17.Uchinoumi H, Yano M, Suetomi T, Ono M, Xu X, Tateishi H, Oda T, Okuda S, Doi M, Kobayashi S, Yamamoto T, Ikeda Y, Ohkusa T, Ikemoto N, Matsuzaki M. Catecholaminergic polymorphic ventricular tachycardia is caused by mutation-linked defective conformational regulation of the ryanodine receptor. Circ Res. 2010;106:1413–1424. doi: 10.1161/CIRCRESAHA.109.209312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rizzi N, Liu N, Napolitano C, Nori A, Turcato F, Colombi B, Bicciato S, Arcelli D, Spedito A, Scelsi M, Villani L, Esposito G, Boncompagni S, Protasi F, Volpe P, Priori SG. Unexpected structural and functional consequences of the r33q homozygous mutation in cardiac calsequestrin: A complex arrhythmogenic cascade in a knock in mouse model. Circ Res. 2008;103:298–306. doi: 10.1161/CIRCRESAHA.108.171660. [DOI] [PubMed] [Google Scholar]
- 19.Liu N, Ruan Y, Denegri M, Bachetti T, Li Y, Colombi B, Napolitano C, Coetzee WA, Priori SG. Calmodulin kinase ii inhibition prevents arrhythmias in ryr2(r4496c+/−) mice with catecholaminergic polymorphic ventricular tachycardia. J Mol Cell Cardiol. 2011;50:214–222. doi: 10.1016/j.yjmcc.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 20.Fernandez-Velasco M, Rueda A, Rizzi N, Benitah JP, Colombi B, Napolitano C, Priori SG, Richard S, Gomez AM. Increased Ca2+ sensitivity of the ryanodine receptor mutant ryr2r4496c underlies catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2009;104:201–209. doi: 10.1161/CIRCRESAHA.108.177493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang G, Giovannone SF, Liu N, Liu FY, Zhang J, Priori SG, Fishman GI. Purkinje cells from ryr2 mutant mice are highly arrhythmogenic but responsive to targeted therapy. Circ Res. 2010;107:512–519. doi: 10.1161/CIRCRESAHA.110.221481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Priori SG, Napolitano C. Intracellular calcium handling dysfunction and arrhythmogenesis: A new challenge for the electrophysiologist. Circ Res. 2005;97:1077–1079. doi: 10.1161/01.RES.0000194556.41865.e2. [DOI] [PubMed] [Google Scholar]
- 23.Kontula K, Laitinen PJ, Lehtonen A, Toivonen L, Viitasalo M, Swan H. Catecholaminergic polymorphic ventricular tachycardia: Recent mechanistic insights. Cardiovasc Res. 2005;67:379–387. doi: 10.1016/j.cardiores.2005.04.027. [DOI] [PubMed] [Google Scholar]
- 24.Marks AR, Priori S, Memmi M, Kontula K, Laitinen PJ. Involvement of the cardiac ryanodine receptor/calcium release channel in catecholaminergic polymorphic ventricular tachycardia. J Cell Physiol. 2002;190:1–6. doi: 10.1002/jcp.10031. [DOI] [PubMed] [Google Scholar]
- 25.Aistrup GL, Kelly JE, Kapur S, Kowalczyk M, Sysman-Wolpin I, Kadish AH, Wasserstrom JA. Pacing-induced heterogeneities in intracellular Ca2+ signaling, cardiac alternans, and ventricular arrhythmias in intact rat heart. Circ Res. 2006;99:e65–73. doi: 10.1161/01.RES.0000244087.36230.bf. [DOI] [PubMed] [Google Scholar]
- 26.Rubart M, Wang E, Dunn KW, Field LJ. Two-photon molecular excitation imaging of Ca2+ transients in langendorff-perfused mouse hearts. Am J Physiol Cell Physiol. 2003;284:C1654–C1668. doi: 10.1152/ajpcell.00469.2002. [DOI] [PubMed] [Google Scholar]
- 27.Bu G, Adams H, Berbari EJ, Rubart M. Uniform action potential repolarization within the sarcolemma of in situ ventricular cardiomyocytes. Biophys J. 2009;96:2532–2546. doi: 10.1016/j.bpj.2008.12.3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song LS, Guia A, Muth JN, Rubio M, Wang SQ, Xiao RP, Josephson IR, Lakatta EG, Schwartz A, Cheng H. Ca2+ signaling in cardiac myocytes overexpressing the α1 subunit of l-type Ca2+ channel. Circ Res. 2002;90:174–181. doi: 10.1161/hh0202.103230. [DOI] [PubMed] [Google Scholar]
- 29.Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci USA. 2006;103:4305–4310. doi: 10.1073/pnas.0509324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng H, Song LS, Shirokova N, Gonzalez A, Lakatta EG, Rios E, Stern MD. Amplitude distribution of calcium sparks in confocal images: Theory and studies with an automatic detection method. Biophys J. 1999;76:606–617. doi: 10.1016/S0006-3495(99)77229-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Q, Xiao J, Jiang D, Wang R, Vembaiyan K, Wang A, Smith CD, Xie C, Chen W, Zhang J, Tian X, Jones PP, Zhong X, Guo A, Chen H, Zhang L, Zhu W, Yang D, Li X, Chen J, Gillis AM, Duff HJ, Cheng H, Feldman AM, Song LS, Fill M, Back TG, Chen SR. Carvedilol and its new analogs suppress arrhythmogenic store overload-induced Ca2+ release. Nat Med. 2011;17:1003–1009. doi: 10.1038/nm.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cerrone M, Colombi B, Santoro M, di Barletta MR, Scelsi M, Villani L, Napolitano C, Priori SG. Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res. 2005;96:e77–82. doi: 10.1161/01.RES.0000169067.51055.72. [DOI] [PubMed] [Google Scholar]
- 33.Wehrens XH, Lehnart SE, Marks AR. Intracellular calcium release and cardiac disease. Annu Rev Physiol. 2005;67:69–98. doi: 10.1146/annurev.physiol.67.040403.114521. [DOI] [PubMed] [Google Scholar]
- 34.Herron TJ, Milstein ML, Anumonwo J, Priori SG, Jalife J. Purkinje cell calcium dysregulation is the cellular mechanism that underlies catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2010;7:1122–1128. doi: 10.1016/j.hrthm.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.George CH, Higgs GV, Lai FA. Ryanodine receptor mutations associated with stress-induced ventricular tachycardia mediate increased calcium release in stimulated cardiomyocytes. Circ Res. 2003;93:531–540. doi: 10.1161/01.RES.0000091335.07574.86. [DOI] [PubMed] [Google Scholar]
- 36.Powell T. The calcium paradox and isolated myocytes. Eur Heart J. 1983;4 (Suppl H):105–111. doi: 10.1093/eurheartj/4.suppl_h.105. [DOI] [PubMed] [Google Scholar]
- 37.Stern MD, Capogrossi MC, Lakatta EG. Spontaneous calcium release from the sarcoplasmic reticulum in myocardial cells: Mechanisms and consequences. Cell Calcium. 1988;9:247–256. doi: 10.1016/0143-4160(88)90005-x. [DOI] [PubMed] [Google Scholar]
- 38.Cerrone M, Noujaim SF, Tolkacheva EG, Talkachou A, O’Connell R, Berenfeld O, Anumonwo J, Pandit SV, Vikstrom K, Napolitano C, Priori SG, Jalife J. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2007;101:1039–1048. doi: 10.1161/CIRCRESAHA.107.148064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, DeSimone L, Coltorti F, Bloise R, Keegan R, Cruz Filho FE, Vignati G, Benatar A, DeLogu A. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69–74. doi: 10.1161/01.cir.0000020013.73106.d8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.