

Abstract
Metallo-β-lactamase (MBL) genes confer resistance to virtually all β-lactam antibiotics and are rapidly disseminated by mobile genetic elements in Gram-negative bacteria. MBLs belong to three different subgroups, B1, B2, and B3, with the mobile MBLs largely confined to subgroup B1. The B3 MBLs are a divergent subgroup of predominantly chromosomally encoded enzymes. AIM-1 (Adelaide IMipenmase 1) from Pseudomonas aeruginosa was the first B3 MBL to be identified on a readily mobile genetic element. Here we present the crystal structure of AIM-1 and use in silico docking and quantum mechanics and molecular mechanics (QM/MM) calculations, together with site-directed mutagenesis, to investigate its interaction with β-lactams. AIM-1 adopts the characteristic αβ/βα sandwich fold of MBLs but differs from other B3 enzymes in the conformation of an active site loop (residues 156 to 162) which is involved both in disulfide bond formation and, we suggest, interaction with substrates. The structure, together with docking and QM/MM calculations, indicates that the AIM-1 substrate binding site is narrower and more restricted than those of other B3 MBLs, possibly explaining its higher catalytic efficiency. The location of Gln157 adjacent to the AIM-1 zinc center suggests a role in drug binding that is supported by our in silico studies. However, replacement of this residue by either Asn or Ala resulted in only modest reductions in AIM-1 activity against the majority of β-lactam substrates, indicating that this function is nonessential. Our study reveals AIM-1 to be a subclass B3 MBL with novel structural and mechanistic features.
INTRODUCTION
β-Lactams are the most commonly used antibiotics globally (3, 58, 59), but the dissemination and increased prevalence of β-lactamases worldwide have seriously challenged their clinical effectiveness. β-Lactamases cleave the β-lactam ring by hydrolysis, inactivating the antibiotic. To date, more than 890 such enzymes have been discovered (6, 7). Metallo-β-lactamases (MBLs) are a distinct group of β-lactamases found in many clinically relevant Gram-negative bacteria such as Pseudomonas aeruginosa, Acinetobacter baumannii, and various Enterobacteriaceae (37). In addition, MBLs have been discovered in a range of environmental and clinically innocuous bacteria (2, 59, 61). The hydrolytic spectrum of MBLs includes all β-lactams, with the exception of monobactams (aztreonam) (59), and they are not inhibited by serine β-lactamase inhibitors, such as clavulanic acid, tazobactam, and sulbactam, in clinical use (19, 33, 45).
MBLs are divided into three subclasses (B1, B2, and B3) on the basis of sequence similarity and structural features (2). Prior to the identification of AIM-1 (Adelaide IMipenmase 1) in P. aeruginosa (63) and the recently published SMB-1 (57), other acquired or mobile MBLs (IMPs, VIMs, NDMs, SPM, GIM, SIM, DIM, TMB-1, and KHM-1) (58) were almost exclusively confined to the B1 subclass. AIM-1 belongs to subclass B3 and was the first such enzyme to be found on a mobile genetic element from a major human pathogen (P. aeruginosa) rather than from an environmental source (63). Compared to other B3 MBLs (e.g., Stenotrophomonas maltophilia L1, Janthinobacterium lividum THIN-B, Chryseobacterium meningosepticum GOB, Legionella gormanii FEZ-1, Caulobacter crescentus CAU-1, Bradyrhizobium japonicum BJP-1, and Erwinia caratovora CAR-1), AIM-1 is divergent in sequence, showing only 23% identity to FEZ-1, 29% to BJP-1, and 36% to L1.
Available three-dimensional (3D) structures for B3 subclass MBLs include those of L1 (Stenotrophomonas maltophilia) (53, 56) and, as inhibitor complexes (36, 43), FEZ-1 (Legionella gormanii) (23) and BJP-1 (Bradyrhizobium japonicum) (15). Although B3 enzymes share the characteristic αβ/βα fold of MBLs, compared to other subclasses they exhibit notable differences in their active site architecture. In B3 MBLs, the loop connecting helix α4 and strand β7, which generally spans residues 156 to 166, is important for interactions with substrates (25). In subclass B1 MBLs, two nonequivalent loops, the “flapping loop” between β2 and β3 and (so-called) loop2 between β11 and α5, perform this role (2, 3). In subclass B2, loop2 is replaced by an elongated α3 helix that is important for interactions with substrates (20). Furthermore, while both B1 and B3 MBLs generally bind two zinc (Zn) ions (Zn1 and Zn2) in the active site, the coordinating residues are different in the two subclasses. In the B1 enzymes, the Zn ions are coordinated by His116-His118-His196 and Asp120-Cys221-His263, referred to as the histidine (Zn1) site and cysteine (Zn2) sites, respectively. In contrast, while the histidine site of subclass B1 is also present in the great majority of B3 MBLs, in these enzymes the second Zn ion is coordinated by Asp120-His121-His263 and so lacks a cysteine ligand. In both subclasses, the two Zn sites are bridged by a water/hydroxide that is generally accepted to be the nucleophile in the hydrolytic reaction. Thus, Zn1 has a tetrahedral coordination, while in most cases an additional water molecule completes the Zn2 site, resulting in a trigonal bipyramidal or square pyramidal (5-fold) coordination. B3 MBLs differ from other subclasses in possessing intramolecular disulfide bridges (15, 23, 56) but, with the exception of L1 (tetrameric) (2), resemble all other MBLs in being monomeric in solution.
In this study, we have determined the three-dimensional structure of AIM-1 from P. aeruginosa in two crystal forms and of two engineered variants with mutations at position 157. AIM-1 is distinguished by three disulfide bridges, a unique residue, Gln157, at the active site, and an antibiotic binding site that differs from those described in other B3 subclass enzymes. On the basis of these data, we have used computational and biochemical approaches to explore the interactions of AIM-1 with β-lactam substrates and candidate inhibitors and the importance of Gln157 to this process. The Q157A and Q157N mutations showed clear but modest effects on enzymatic activity, supporting the proposal that this residue contributes to, but is not essential for, substrate binding. Our results show AIM-1 to be a distinctive member of the B3 MBL subclass and define intermolecular interactions that may be important to the design of inhibitors with reactivity across the different MBL subclasses.
MATERIALS AND METHODS
Cloning of blaAIM-1.
The blaAIM-1 gene, together with flanking regions on both sides of the gene, was amplified from Pseudomonas aeruginosa WCH2677 (63) by PCR using the forward primer 5′-TGAGAAATGGCTACGCACTG-3′ and the reverse primer 5′-GTACGGAAAACTCAGCACCC-3′, resulting in a PCR product of 1,241 bp. This PCR product was purified (QIAquick gel extraction kit; Qiagen, Germany) and used as a template for amplification of the blaAIM-1 gene performed using AIM-1-NdeI forward primer 5′-GGAATTCCATATGAAACGTCGCTTCACCCTGCTG-3′ and AIM-1-BamHI reverse primer 5′-CGCGGATCCTCAAGGCCGCGCGCCGCTGGA-3′ (the introduced NdeI and BamHI restriction sites are indicated in bold letters, and regions corresponding to the start and end of the blaAIM-1 open reading frame are underlined). The blaAIM-1 gene (913 bp) was purified from the agarose gel (QIAquick gel extraction kit), digested with NdeI and BamHI, and ligated into T7 expression vector pET-26b (Novagen Inc., Madison, WI), generating plasmid pET26b-AIM-1. pET26b-AIM-1 was transformed into Escherichia coli BL21(DE3) (Invitrogen) for recombinant protein expression.
Construction of AIM-I mutants by site-directed mutagenesis.
A QuikChange site-directed mutagenesis kit (Agilent Biosciences) was used to generate the Q157N and Q157A mutants of AIM-1 according to the manufacturer's protocol. For both mutants, the following primers were designed to introduce the desired mutations: AIM-1-Q157N-F (5′-GACCGCACCGACCCGAACTTCGAGGTGGCCGAAC-3′) and AIM-1-Q157N-R (5′-GTTCGGCCACCTCGAAGTTCGGGTCGGTGCGGTC-3′) for the Q157N mutant and AIM-1-Q157A-F (5′-CCGCACCGACCCGGCATTCGAGGTGGC-3′) and AIM-1-Q157A-R (5′ GCCACCTCGAATGCCGGGTCGGTGCGG-3′) for the Q157A mutant (the modified bases are underlined). Amplified DNA from the mutagenesis reaction was digested with DpnI to cleave the parental DNA (nonmutated), purified, and transformed into E. coli XL1-Blue supercompetent cells (Agilent) and subsequently into E. coli BL21(DE3) (Invitrogen). The mutations were verified by DNA sequencing.
Expression and purification.
Wild-type AIM-1 and the mutants Q157N and Q157A were expressed and purified similarly to previous descriptions (4, 49). In brief, recombinant bacteria were grown in Terrific broth (Sigma) with kanamycin (Sigma) (50 mg/liter) at 37°C and expression was induced in mid-log phase using 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG; Sigma, St. Louis, MO). The cells were harvested by centrifugation (10,000 × g, 15 min) 4 h after induction. AIM-1 (wild type and mutants) was purified from the periplasm followed by ion-exchange chromatography (Q-Sepharose HP column; GE Healthcare, United Kingdom) and gel-filtration chromatography (Superdex 200 column; GE Healthcare) as described for other MBLs (4, 49). The buffer system used for the purification consisted of 50 mM Tris (pH 7.2) and 100 μM ZnCl2, with or without NaCl. Partly purified fractions of AIM-1 mutants were further purified using a hydrophobic column (HiTrap Phenyl FF; GE Healthcare, United Kingdom) starting with the proteins in 1 M ammonium sulfate, followed by washing with 1.5 M ammonium sulfate, 50 mM Tris (pH 7.2), and 100 μM ZnCl2 and eluting with only 50 mM Tris (pH 7.2) and 100 μM ZnCl2. Purified enzymes were concentrated by ultrafiltration (Amicon) to a final concentration of 15 to 17 mg/ml protein with 95% purity as judged by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis. The purified protein was subjected to tandem mass spectroscopy (MS-MS) analysis for identification. A typical yield from 1 liter of cell culture was about 5 mg of protein.
AIM-1 crystallization.
Crystallization trials of wild-type AIM-1 were performed using commercial sparse matrix screening kits (Molecular Dimensions; Qiagen) and a Phoenix RE nanoliter crystallization robot (Rigaku), setting up 200 nl of protein and 200 nl of reservoir solution in a sitting-drop method in 96-well MRC 2 plates with a 60-μl reservoir volume. Optimization was done using 24-well pregreased plates (Hampton Research) by the hanging-drop method with 500 μl of reservoir volume and drops containing 1 μl of reservoir and 1 μl of protein solution. Diffraction-quality crystals were obtained after about 2 weeks of incubation at room temperature with 19% polyethylene glycol monomethyl ethers (PEG MME) 2000–0.1 M calcium acetate–0.1 M sodium cacodylate at pH 5.0. Crystals of the AIM-1 mutants Q157A and Q157N (both at 15 mg/ml) were also grown as hanging drops with reservoir solutions containing 17% to 18% polyethylene glycol monomethyl ethers (PEG MME) 2000, 0.10 or 0.15 M calcium acetate, and 0.1 M sodium cacodylate at pH 5.0.
Data collection, structure determination, and refinement.
All crystals were cryoprotected in 5% glycerol–30% PEG MME 2000–0.1 M calcium acetate–0.1 M sodium cacodylate at pH 5.0 and then flash frozen in liquid nitrogen. Diffraction data sets were collected from two different wild-type crystal forms (AIM-1 and AIM-1-3mol) at beamline ID29 of the European Synchrotron Radiation Facility (ESRF), Grenoble, Switzerland. For the AIM-1 crystal form, 75° of data (250 images with 0.3° oscillation per image) were collected at 10% transmission, with exposure for 0.3 s per image for a total exposure time of 75 s. For the AIM-1-3mol crystal form, 79° of data (395 images with 0.2° oscillation per image) were collected at 5% transmission, with exposure for 0.36 s per image for a total exposure time of 142 s. For the AIM-1-Q157A and AIM-1-Q157N mutants, data were collected at a less intense beamline at I911-3 (MAX II laboratory, Lund with full beam, 40 s per image), where the AIM-1-Q157A data set includes 40° of data (80 images with 0.5° oscillation) and the AIM-1-Q157N data are from 60° of data (120 images with 0.5° oscillation). All data sets were integrated and scaled with the program XDS (31), and structure factors were obtained using TRUNCATE (11).
The AIM-1 structure was solved by molecular replacement, using the structure of S. maltophilia L1 (Protein Data Bank [PDB] 1SML) (56) as a search model in the program PHASER (38). However, with this model, no clear rotation or translation solution was found, probably due to the low sequence identity (39%) between AIM-1 and L1. To improve the phases, a homology model of AIM-1 was built which included the correct AIM-1 sequence starting from residue Ala23, thus including the protruding N terminus involved in tetramerization of L1 (2). Upon rerunning PHASER with the homology model and accepting all solutions, including those with overlapping atoms, the top solution with the highest log likelihood gain (LLG) was correct. From this solution, ARP/wARP (10) was able to build 259 of a total of 270 residues into the resulting 2Fo-Fc electron density map, all of which had been correctly docked to the sequence. The remaining loops were built manually using WinCoot (18). The AIM-1-3mol, AIM-1-Q157A, and AIM-1-Q157N structures were then solved by molecular replacement using PHASER with the refined AIM-1 structure as a search model. All structures were refined with REFMAC5 (42), with water molecules added with the ARP_WATERS routine within REFMAC5 and manually while rebuilding the models in WinCoot (18). Anisotropic B-factors for all atoms were employed in the final stages of AIM-1 (1.6 Å), AIM-1-Q157A (1.73 Å), and AIM-1-Q157N (1.4 Å) refinements, as these gave a substantial (>4%) drop in Rfree values and significant improvements to the electron density maps and overall model statistics.
QM/MM calculations of the AIM-1:cefoxitin complex and docking of d-captopril.
The Maestro tool (Schrodinger Inc., Portland, OR) was used to build and optimize the geometry of hydrolyzed cefoxitin. Hydrolyzed cefoxitin was docked into AIM-1 by use of the docking program GOLD (29). The docked conformation of hydrolyzed cefoxitin was further optimized using a quantum mechanics/molecular mechanics (QM/MM) approach with the Q-Site program (2010; Schrödinger, LLC, New York, NY). The quantum part consists of hydrolyzed cefoxitin, the two Zn atoms, the bridging water molecule, and the side chains (Cβ and subsequent atoms) of all residues coordinating the two Zn atoms (His116, His118, His196, Asp120, His121, and His263). The geometry was optimized with the LACVP** basis set (17, 27) for the two Zn atoms, and the 6-31** basis set was used for the remaining atoms treated quantum mechanically. Density functional theory (DFT) with the B3LYP functional was used for the QM part, while the OPLS-2005 force field (30) was used to model the MM part. Link atoms (1) were used for the junction between the QM (Cβ atom) and the MM (Cα atom) parts of the amino acids coordinating with the two Zn atoms. The inhibitor d-captopril was docked into AIM-1 by the use of Glide software (Schrodinger Inc., Portland, OR) starting with the FEZ-1:d-captopril structure (PDB 1JT1) (23) and replacing the bridging water with a sulfate ion as previously observed (23).
Protein structure accession numbers.
Coordinates and structure factors for AIM-1, AIM-1-3mol, AIM-1-Q157A, and AIM-1-Q157N have been deposited in the PDB with accession codes 4AWY, 4AWZ, 4AX0, and 4AX1.
RESULTS AND DISCUSSION
Structure determination.
Recombinant AIM-1 was readily overexpressed in E. coli BL21(DE3) and purified from the periplasm, yielding the mature polypeptide without the periplasmic export sequence. Two crystal forms of the wild type, AIM-1 (space group P6122; one molecule in the asymmetric unit) and AIM-1-3mol (space group P61; three molecules in the asymmetric unit), were obtained, diffracting to 1.60 and 1.80 Å resolutions, respectively (Tables 1 and 2).
Table 1.
X-ray data collection statisticsa
| X-ray statistic | Value for indicated PDB entry |
|||
|---|---|---|---|---|
| 4AWY | 4AWZ | 4AX0 | 4AX1 | |
| Beamline | ID29 | ID29 | I911-3 | I911-3 |
| Space group | P6122 | P61 | P6122 | P6122 |
| Unit cell (Å) | a = b = 77.83; c = 239.02 | a = b = 79.47; c = 229.27 | a = b = 78.04; c = 239.48 | a = b = 77.99; c = 239.77 |
| Resolution (Å) (highest bin) | 28-1.60 (1.68-1.60) | 30-1.80 (1.90-1.80) | 30-1.83 (1.83-1.73) | 30-1.40 (1.40-1.48) |
| Wavelength (Å) | 1.23985 | 1.10481 | 1.0000 | 1.0000 |
| No. of unique reflections | 56,470 | 74,731 | 43,793 | 82,502 |
| Multiplicity | 5.5 (3.9) | 5.0 (4.8) | 4.4 (2.3) | 6.4 (3.4) |
| Completeness (%) | 97.8 (87.0) | 98.9 (99.6) | 96.2 (76.4) | 96.3 (76.7) |
| Mean (<I>/<σI>) | 20.3 (6.6) | 17.5 (4.0) | 9.3 (2.0) | 10.5 (2.1) |
| Rmerge (%)b | 4.6 (13.8) | 7.3 (37.8) | 8.0 (45.7) | 8.6 (46.0) |
| Wilson B-factor (Å2) | 14.57 | 12.84 | 17.06 | 15.21 |
Numbers in parentheses represent values for the highest of 10 resolution shells except where indicated.
Rmerge = [ΣhΣi | Ii (h) − <I(h)>|)/(ΣhΣI I(h)], where Ii(h) is the i-th measurement of reflection h and <I(h)> is the weighted mean of all measurements of h.
Table 2.
Refinement statistics for crystal structures
| Refinement statistic | Value for: |
|||
|---|---|---|---|---|
| AIM-1 | AIM-1-3mol | AIM-1-Q157A | AIM-1-Q157N | |
| Resolution (Å) | 20-1.60 | 20-1.80 | 25-1.73 | 25-1.4 |
| R-factor (all reflections) (%)a | 12.89 | 13.89 | 12.12 | 12.97 |
| R-free (%)a | 15.80 | 17.27 | 15.86 | 15.02 |
| No. of protein atoms | 2,160 | 6,159 | 2,160 | 2,163 |
| No. of water molecules | 385 | 799 | 365 | 410 |
| No. of other molecules | 3 Zn2+, 1 Ca2+, 4 Mg2+ | 6 Zn2+, 3 Ca2+, 3 Mg2+ | 2 Zn2+, 2 Ca2+, 1 acetate | 2 Zn2+, 2 Ca2+, 2 Mg2+, 1 acetate |
| RMSD bond lengths (Å) | 0.0161 | 0.0141 | 0.0142 | 0.0155 |
| RMSD bond angles (°) | 1.619 | 1.517 | 1.578 | 1.753 |
| Average B-factor (Å2) | ||||
| All atoms | 17.1 | 13.7 | 22.7 | 19.2 |
| Protein (A/B/C) | 13.7 | 11.3/12.2/13.9 | 20.1 | 16.2 |
| Water molecules | 35.8 | 23.0 | 37.9 | 34.9 |
| Zn2+/Ca2+/Mg2+ ions | 13.2/11.9/49.8 | 11.5/12.1/11.1 | 15.9/19.5/- | 11.9/15.8/40.9 |
Σh ||Fobs| − |Fcalc||/(Σh |Fobs|), where |Fobs| and |Fcalc| are observed and calculated structure factor amplitudes for all reflections (R-factor) and the reflections applied in the test R-free set (reflections not used in the structure refinement), respectively.
The final AIM-1 structure (Fig. 1a) consists of 270 protein residues, Ala28 to Ser312, according to the BBL numbering scheme (21) which is used throughout this paper. The structure was well defined in the electron density maps, although high B-factors were observed for both the N and C termini and multiple main- and side-chain conformations for residues Thr87 to Gly92 were modeled. In total, there were 20 residues with alternative conformations and some atoms with occupancy reduced or set to zero to fit to the observed electron density.
Fig 1.

Overall ribbon structures of (a) AIM-1, (b) BJP-1 (PDB 3LVZ), (c) FEZ-1 (PDB 1K07), and (d) L1-Mox (PDB 2AIO), with the Zn coordinating residues and Zn ions (orange) outlined. Residues 156 to 162 and 223 to 230 adjacent to the active site are shown in green. The figure was made with PyMol (http://www.pymol.org).
The AIM-1-3mol model consisted of 269 (chains A and C) or 267 (chain B) residues. Poorly defined electron density was observed for residues Cys208 to Cys213 (chains A, B, and C), and a total of 22 alternative conformations were included. Noncrystallographic symmetry was not used in refinement. Overall, both structures were well defined, with low R-factors (<13.5%) and low R-free values (<16%) and with satisfactory geometries (Table 2). In the Ramachandran plots for both AIM-1 and AIM-1-3mol, Asp84 was an outlier (disallowed region), as observed in most structures of subclass B1 and B3 MBLs (see, e.g., reference 4). In addition, AIM-1 has one residue (Pro89) and AIM-1-3mol one residue (Ala281 chain B) in the disallowed region.
The root mean square deviation (RMSD) between the AIM-1 and AIM-1-3mol structures was 0.21 to 0.39 Å between the CA atoms, indicating no large conformational differences. The 3 molecules in AIM-1-3mol had CA RMSDs of 0.39 to 0.49 Å, where chains B and C were the most divergent (0.49 Å) and chains A and B were most similar (0.39 Å). Upon superposition of the three chains in AIM-1-3mol, it was clear that most of the loops and both termini exhibit different conformations due to different crystal packing interactions.
Overall structure and active site.
The overall fold of AIM-1 consists of an αβ/βα sandwich, with the α helices exposed to the solvent and a central compact core formed by two β-sheets composed of 5 and 7 β-strands, respectively (Fig. 1a). The structure contains six α helices, five 310 helices (η1 to η5; Fig. 2), and a dinuclear Zn-binding site that is situated in the shallow groove located at the interface of the two αβ domains. This active site groove is largely defined by two loops, residues 156 to 162, connecting helix α4 and strand β7, and residues 223 to 230, connecting strand β11 and helix α5.
Fig 2.

Sequence alignment of AIM-1, BJP-1, FEZ-1, and L1, with the secondary-structure elements of AIM-1 (top) and L1 (bottom; PDB 1SML) also included. The zinc binding residues (His116, His118, Asp120, His121, His196, and His263) are indicated by triangles (▲). The loops important for the substrate specificity are outlined. The figure was made with ESPript (26).
The AIM-1 active site (Fig. 3a to d) contains two well-defined zinc ions: Zn1, coordinated by His116, His118, and His196, and Zn2, bound to Asp120, His121, and His263. Details of metal coordination in the active site are given in Table 3. In addition to the protein ligands, a water molecule or hydroxyl ion (W1) bridges the two zinc ions and is the most likely candidate to perform the nucleophilic attack on the β-lactam carbonyl carbon during β-lactam hydrolysis (13, 32, 56, 60). W1 is also closely bound to OD2 of Asp120. Zn1 is tetrahedrally coordinated, while the Zn2 site holds an additional water molecule (W2) and is thus 5-fold coordinated, with geometry that has been described as trigonal, or square, pyramidal (15). The zinc ions were refined with full occupancy, resulting in B-factors of 9.0 Å2 (Zn1) and 11.0 Å2 (Zn2) for AIM-1 and 8.2 to 9.4 Å2 (Zn1) and 11.6 to 14.1 Å2 (Zn2) for AIM-1-3mol. These data are consistent with the presence in AIM-1 of two zinc sites of similar affinities. In the various AIM-1 structures, slight differences in the interaction distances (Table 3) were observed for Asp120 OD2-W1 (2.94 Å in AIM-1 versus 3.03 Å in AIM-1-3mol), Zn1-W1 (1.94 Å versus 2.02 Å), and Zn2 W1 (1.98 Å versus 2.06 Å) interactions.
Fig 3.

(a to d) The active sites in (a) AIM-1 and (b) AIM-1-3mol and the adjacent additional metal binding sites found in (c) AIM-1 and (d) AIM-1-3mol (chain A). (e to g) Disulfide bonds in AIM-1 with reduced or full occupancy for (e) Cys32-Cys66, (f) Cys208-Cys213, and (g) Cys256-Cys290. All 2Fo-Fc electron density maps are displayed at 1.6 σ.
Table 3.
Details from the Zinc ligand interactions in the active site of AIM-1 and AIM-1-3mol
| Ligand | Interaction partner | Interaction distance (Å) for: |
|||
|---|---|---|---|---|---|
| AIM-1 | AIM-1-3mol |
||||
| Chain A | Chain B | Chain C | |||
| Zn1 | His116 NE2 | 2.07 | 2.11 | 2.03 | 2.01 |
| His118 ND1 | 2.10 | 2.07 | 2.04 | 2.15 | |
| His196 NE2 | 2.02 | 2.09 | 2.13 | 2.14 | |
| W1 | 1.94 | 2.02 | 1.96 | 1.93 | |
| Zn2 | Asp120 OD2 | 2.12 | 2.19 | 2.22 | 2.08 |
| His121 NE2 | 2.09 | 2.06 | 2.07 | 2.00 | |
| His263 NE2 | 2.03 | 2.08 | 2.08 | 2.09 | |
| W1 | 2.27 | 2.26 | 2.25 | 2.30 | |
| W2 | 1.98 | 2.02 | 2.20 | 2.04 | |
| Asp120 OD1 | W1 | 2.74 | 2.70 | 2.61 | 2.74 |
| Zn1 | Zn2 | 3.48 | 3.58 | 3.45 | 3.40 |
In addition to the two active site Zn ions, other bound metal ions were observed in both the AIM-1 and AIM-1-3mol structures. One metal ion, present in all AIM-1 molecules (AIM-1, AIM-1-3mol chains A, B, and C), was bound to Ser221 OH and His263 N and two water molecules. From the electron density measurements, this was interpreted as a calcium (Ca) ion with 85% occupancy and a B-factor of 11.9 Å2 in AIM-1. Refinement with a magnesium (Mg) ion gave a positive difference in electron density at 6σ, indicating a heavier metal at this position. The three Ca ions with 85% occupancy in the AIM-1-3mol model also gave B-factors for the metals that resembled those of their surroundings. The Ca ion is only 3.8 to 4.0 Å away from the conserved residue Tyr293 in the four AIM-1 chains. In the L1-moxalactam complex and BJP-1 structures, this position is occupied by a water molecule with the same four interacting partners, whereas in FEZ-1, the side chain of Met266 occupies this position (15, 23, 53).
Disulfide bridges.
Intramolecular disulfide bonds play an important role in protein folding. The AIM-1 structure features three intramolecular disulfide bonds: Cys256-Cys290, as also found in FEZ-1 and L1, and the additional Cys32-Cys66 and Cys208-Cys213 disulfide bonds. Some of these covalent bonds were clearly broken, as shown by negative difference density in the experimental electron density maps, and were therefore refined in two conformations. Thus, in the AIM-1 structure, the Cys32-Cys66, Cys208-Cys213, and Cys256-Cys296 disulfide bonds were found to have only 70%, 70%, and 50% occupancies, respectively (Fig. 3e to g). In the AIM-1-3mol structure, the Cys32-Cys66 bond was observed with reduced occupancy for chain A (70%) and chain C (50%) but not chain B. The Cys208-Cys213 bond was intact, whereas the Cys256-Cys296 bond had only 50% occupancy for all 3 chains (A, B, and C), and the alternative side-chain conformations for the Cys residues that are rotated resulted in a displacement of the SG atom of 1.0 to 1.5 Å. All three of these disulfide bridges are likely to contribute to the stability of the AIM-1 structure. The Cys256-Cys290 bond links the α6 helix to the α5 helix, Cys32-Cys66 positions the N-terminal portion of AIM-1 by connecting strands β1 and β3, and Cys208-Cys213 stabilizes the positions of β10 and β11.
As X-ray diffraction data for the two wild-type structures were collected at the intense insertion-device beamline ID29 at ESRF, these broken disulfide bonds are most probably caused by radiation damage occurring despite the use of attenuation. Disulfide bond breakage is a known specific form of damage accruing at intense synchrotrons (34, 46, 47). Although particularly associated with high dose rates (short exposure time and high intensity) (35), the total absorbed X-ray dose is the most important (24) factor in the accumulation of radiation damage such as that observed here.
Comparison with other B3 MBLs.
Crystal structures for three other MBLs of subclass B3 are available (14, 15, 23, 53). A comparison of the structure of AIM-1 with those previously determined structures is shown in Fig. 1. Superpositions show that the overall fold is reasonably well conserved between the four proteins, with overall CA RMSD values for AIM-1 compared to L1 (PDB 2QJS) (15) of 1.40 Å, L1 in complex with hydrolyzed moxalactam (L1-mox; PDB 2AIO) (53) of 1.48 Å, FEZ-1 (PDB 1JT1) (23) of 1.61 Å, and BJP-1 (PDB 2GMN) (15) of 1.53 Å. However, there are some significant structural differences, involving in particular the N termini which form loops whose conformations and orientations differ. For AIM-1, the N terminus, starting at Ala28, forms a long loop which folds into a hairpin before continuing into β1 (Leu45 onwards). The first residue that can be structurally aligned between AIM-1, BJP-1, and L1 is Trp38 (conserved in these structures), while residue 39 (Asn39 in AIM-1; Pro39 in FEZ-1) overlaps structurally with a CA-CA distance of 2.1 Å in the superimposed AIM-1 and FEZ-1 structures. By way of contrast, the C termini of all four structures are formed by long α helices of comparable lengths and positions, with low RMSD values in this region. A number of loops connecting secondary-structure elements also differ in AIM-1 from their equivalents in the other B3 enzymes. First, the loop (residues 277 to 281) connecting helices β12 and α6 is shorter in AIM-1. Further, the conformations of two of the loops that enclose the AIM-1 active site (residues 156 to 162 and 223 to 230) are substantially different.
In general, two loops define the active site of MBLs from all subclasses. For example, in the subclass B1 enzymes, the so-called “flapping loop” between β2 and β3 (BBL residues 60 to 66) performs an important role in recognition of, and interactions with, the substrate (2). In the B3 subgroup, this loop is much shorter, due to shortened β2 and β3 strands in these enzymes, and so does not contribute to substrate binding. However, studies of L1 suggest that a second loop (residues 156 to 166 between α4 and β7) may play a similar role in the B3 subgroup (8, 28, 41). In AIM-1, this loop adopts a notably different conformation compared to other B3 subclass MBLs (Fig. 1) and differs substantially from them in sequence (Fig. 2). In particular, the conformation of this loop brings Gln157, a residue unique to AIM-1 among known B3 structures, into the vicinity of the Zn1 site to interact with a W3 water molecule (Fig. 3a and b) that is in turn hydrogen bonded to the W1 “bridging” water molecule. In addition, the loop connecting β11 to α5 (residues 223 to 230) is longer by two (FEZ-1, BJP-1) or three (L1) amino acids than its equivalents in other B3 MBL structures and projects across the AIM-1 active site groove.
Zinc binding.
In AIM-1, as in other subclass B3 MBLs, the HXHXDH sequence motif that defines MBL superfamily proteins forms the core of the dinuclear zinc site. Coordination of the Zn ions in the AIM-1 and AIM-1-3mol active sites closely resembles that observed in the structures of the other B3 enzymes. The Zn-Zn distances of AIM-1 (3.48Å) and AIM-1-3mol (3.40 to 3.58 Å) are comparable with those observed in the BJP-1 (3.45 Å; PDB 3LVZ) (15), FEZ-1 (3.66Å; PDB 1K07) (23), L1 (3.46Å; PDB 1SML) (56), and L1-Mox (3.68Å; PDB 2AIO) (53) structures. Among other active site residues of potential functional importance, Ser221 (conserved across B3 MBLs) adopts the conformation (pointing into the active site) observed in structures of the L1 enzyme, rather than pointing toward residue 233, as is observed in the captopril structures of BJP-1 (15) and FEZ-1 (PDB 1JT1) (23).
Comparison of drug binding sites (R1/R2): docking of hydrolyzed cefoxitin and d-captopril into AIM-1.
The B3 MBLs show variable substrate preferences (Table 4) which might be explained by structural comparisons. Both L1 and FEZ-1 are broad-spectrum enzymes, hydrolyzing a wide range of β-lactam substrates (9, 19, 39, 52). In the L1 substrate binding site, as defined in the complex with hydrolyzed moxalactam (53), the moxalactam R1 group (C7 substituent) occupies a hydrophobic pocket formed by Tyr32, Trp38, Phe156, and Ile162, and the C3 carboxylate interacts with Ser221 and Ser223 (43, 53). Similar interactions are involved in ligand binding in other L1-inhibitor complexes (43). In FEZ-1, structurally equivalent residues (Phe119, Tyr36, Tyr156, and Thr163) define a similar R1 pocket, with the additional possibility of interactions between Asp160 and the charged R1 substituents of some substrates. In BJP-1, although some of these hydrophobic residues (Trp38, Tyr151, and Leu162) are conserved, additional bulky amino acids (Phe31 and Leu226) restrict the R1 pocket and affinity for many substrates is reduced. In the complex of BJP-1 with the inhibitor 4-nitrobenzenesulfonamide (NBSA; PDB 3M8T) (15), inhibitor binding in the R1 site requires displacement of Phe31 and of the extended N terminus that also occludes the unliganded active site (15). While less structural information is available regarding potential interactions of B3 MBLs with the R2 substituents of bound β-lactams, in the crystal structure of FEZ-1 complexed with d-captopril (PDB 1JT1) (23), the d-proline ring of the inhibitor occupies a pocket defined by the side chains of Met266 and Tyr293 and the aliphatic portion of Lys297. Although Tyr293 is conserved across the majority of B3 MBLs of known structure (Fig. 2), the sizes and charges of the residues defining the likely R2 site differ among the four compared enzymes.
Table 4.
Comparison of enzyme kinetics data for AIM-1 wild type, AIM-1-Q157A, and AIM-1-Q157N and the B3 subclass MBLs BJP-1 (15, 54), FEZ-1 (39), and L1 (9, 52)
| Substrate | AIM-1 |
AIM-1-Q157A |
AIM-1-Q157N |
BJP-1 |
FEZ-1 |
L1 |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Km (μM) | kcat (s−1) | kcat/Km (s−1/M) | Km (μM) | kcat (s−1) | kcat/Km (s−1/M) | Km (μM) | kcat (s−1) | kcat/Km (s−1/M) | Km (μM) | kcat (s−1) | kcat/Km (s−1/M) | Km (μM) | kcat (s−1) | kcat/Km (s−1/M) | Km (μM) | kcat (s−1) | kcat/Km (s−1/M) | |
| Penicillin G | 110 ± 21 | 590 ± 31 | 5.4 × 106 | 380 ± 92 | 560 ± 56 | 1.4 × 106 | 200 ± 65 | 440 ± 50 | 2.2 × 106 | 130 | 18 | 1.3 × 105 | 590 | 70 | 1.1 × 105 | 50 | 1,110 | 2.2 × 107 |
| Ampicillin | 24 ± 3 | 150 ± 5 | 6.3 × 106 | 50 ± 9 | 390 ± 17 | 7.8 × 106 | 87 ± 15 | 36 ± 17 | 4.1 × 106 | 670 | 13 | 1.3 × 104 | >5,000 | >5.5 | 1.1 × 104 | 40 | 175 | 4.4 × 106 |
| Cefoxitin | 22 ± 2 | 52 ± 1 | 2.4 × 105 | 110 ± 16 | 35 ± 2 | 3.2 × 104 | 100 ± 23 | 4.9 ± 0.5 | 4.9 × 103 | 140 | 10 | 7.1 × 104 | 11 | 3 | 2.7 × 105 | 2 | 1.1 | 5.5 × 105 |
| Cefuroxime | 35 ± 4 | 170 ± 5 | 4.8 × 106 | 140 ± 8 | 370 ± 7 | 2.7 × 105 | 110 ± 17 | 61 ± 3 | 5.5 × 105 | 115 | 58 | 5.0 × 105 | 50 | 320 | 6.4 × 105 | 30 | 80 | 2.7 × 106 |
| Ceftazidime | 730 ± 180 | 46 ± 7 | 6.3 × 104 | 710 ± 120 | 5.4 ± 1 | 7.6 × 103 | 400 ± 120 | 5.4 ± 1 | 1.3 × 104 | >700 | >3 | 4.3 × 103 | >1,000 | >4 | 4.0 × 103 | 145 | 27 | 0.2 × 106 |
| Cefepime | 440 ± 60 | 37 ± 1 | 8.4 × 103 | 390 ± 190 | 2.9 ± 1 | 7.4 × 103 | 180 ± 51 | 1.2 ± 0.2 | 6.7 × 102 | >400 | >0.08 | 2.0 × 102 | >1,000 | >6 | 6.0 × 103 | 130 | 0.33 | 2.5 × 104 |
| Imipenem | 410 ± 16 | 2,200 ± 50 | 5.4 × 106 | 610 ± 34 | 360 ± 12 | 5.9 × 105 | 1,600 ± 300 | 300 ± 40 | 1.9 × 105 | 260 | 15 | 6.0 × 104 | >1,000 | >200 | 2.0 × 105 | 90 | 65 | 7.3 × 105 |
| Meropenem | 41 ± 4 | 760 ± 16 | 1.8 × 107 | 130 ± 15 | 270 ± 11 | 2.0 × 106 | 93 ± 6 | 120 ± 3 | 1.3 × 106 | 190 | 156 | 8.3 × 105 | 85 | 45 | 5.0 × 105 | 13 | 77 | 5.9 × 106 |
| Ertapenem | 45 ± 4 | 340 ± 9 | 7.5 × 106 | 17 ± 1 | 94 ± 1 | 5.5 × 106 | 62 ± 7 | 33 ± 1 | 5.2 × 105 | NAa | NA | NA | NA | NA | NA | NA | NA | NA |
NA, not available.
Some, but not all, of the interactions described are likely to be replicated in the binding of substrates and inhibitors to AIM-1. For example, while Trp38 and Phe119 are present in the AIM-1 R1 pocket, the distinctive conformation of the α4-β7 loop ensures that residues such as L1 Phe156 and Ile162 do not have obvious structural equivalents. In order to explore these differences further, we have used docking and quantum mechanics/molecular mechanics (QM/MM) calculations to model the interactions of AIM-1 with a β-lactam. Hydrolyzed cefoxitin was docked into AIM-1 using the program GOLD, and the results were compared with the experimentally determined L1-Mox structure (53). The similarity between the modes of binding in the two complexes gave us confidence in the docking result, which was therefore used as a starting point for further modeling in which drug:protein interactions were optimized using accurate QM/MM calculations.
In the QM/MM-optimized complex, the orientation of the cefoxitin R1 substituent is defined by the side chains of residues Trp38, Phe119, Gln157, Val160, and Glu162 (Fig. 4a and b). The presence of a relatively bulky residue, Ile, at AIM-1 position 225, facing Trp38 across the active site groove, may also affect substrate binding by creating a narrower active site than is found in other B3 MBLs and defining the position and orientation of the cefoxitin 6-membered ring. The 7α-methoxy group of cefoxitin binds to Asp120 with a strong polar interaction. As described above, Trp38 and a hydrophobic residue at position 119 are conserved in B3 MBLs. However, other interactions correspond less well with previously observed structures, due in large part to the distinctive structures of the two loops (α4 to β7 [residues 156 to 166] and β11 to α5 [residues 223 to 230]) that surround the AIM-1 active site groove. Most strikingly, Gln157 contacts the cefoxitin C8 carboxylate (i.e., that formed on addition of water/hydroxide to the β-lactam carbonyl) and C10 carbonyl (R1 substituent). Gln157 is not conserved in other B3 MBL sequences (Fig. 2), although the side-chain NE2 atom occupies a position similar to that seen with ND1 of FEZ-1 Asn225.
Fig 4.

Interactions between hydrolyzed cefoxitin and AIM-1 in the optimized protein-drug complex after the QM/MM calculations. (a and b) Views of active site shown (a) as a schematic and (b) in the context of the structure. (c) Calculated electrostatic surface of (DelPhi) (48) AIM-1. (d) Ribbon diagram showing location of the active site. The color coding is red, white, and blue, representing the charges of −10, 0, and +10 KbT/e, and the docked hydrolyzed cefoxitin (green) is included in both panels. (e and f) Closeup views of cefoxitin (green) and d-captopril (magenta) docked into the AIM-1 active site as described here. R1 and R2 binding sites are shown with (e) and without (f) the electrostatic surface. Residues 156 to 162 and 223 to 230 are shown in green.
As cefoxitin does not contain a bulky C3 substituent, our model was less informative regarding possible interactions involving the AIM-1 R2 pocket. We used the program Glide to dock d-captopril (a small-molecule inhibitor whose interactions with numerous MBLs have been studied previously; see, e.g., references 22, 23, and 43) into the AIM-1 active site (Fig. 4e and f) to provide a comparison with the available structure of the FEZ-1:d-captopril complex and to identify residues that may be involved in interactions with the C3 substituents (R2 group) of cephalosporin or carbapenem substrates. Although this region is variable in sequence, the main chain conformation varies little between the four available structures of B3 MBLs. However, we note that, in AIM-1, the presence of bulky Thr and Ile side chains at positions 223 and 225, respectively, is likely to influence the orientation of some R2 substituents. In addition, Arg300 creates a more strongly positively charged R2 pocket in AIM-1 than in other B3 enzymes, where Asp or Lys is present at this position (Fig. 4e and f). In the docked AIM-1:d-captopril complex, Arg300 is hydrogen bonded to the C9 carboxyl group. The docking results show that the AIM-1 R2 pocket is narrower, better defined, and more positively charged than is the case with the other B3 structures.
Role of Gln157 and AIM-1 specificity.
The mechanism of β-lactam hydrolysis by MBLs is proposed to involve the hydroxide ion (W1) that bridges the two Zn ions (Zn1 and Zn2) (13, 56, 60). This ion performs a nucleophilic attack on the carbonyl carbon atom (C7/C8) of the β-lactam ring (Fig. 5). In serine β-lactamases, addition of the nucleophile to the β-lactam carbonyl is aided by the presence of an oxyanion hole that helps to polarize the β-lactam carbonyl bond and stabilizes the negative charge that develops on the tetrahedral intermediate during catalysis (13). Oxyanion holes are also present in serine proteases such as trypsin (formed by two main-chain amines; see, e.g., reference 50) and in the subtilisin family (formed by a conserved asparagine; see, e.g., reference 51). However, while similar factors might be expected to contribute to β-lactam hydrolysis by MBLs, there is no experimental evidence for populated tetrahedral intermediates in any studied enzyme. Consistent with this conclusion, investigations of the contributions of several elements of the MBL active site to β-lactam hydrolysis have not yielded strong support for the idea of the presence of an oxyanion hole. In the B1 enzymes, investigations of structures of inhibitor complexes (12, 62) have led to proposals that Asn233 forms part of an oxyanion hole, but these are only partially supported by mutagenesis experiments, which show that hydrolysis of some substrates is little affected by substitution at Asn233 in the IMP-1 enzyme (5, 16). Furthermore, Asn233 is not uniformly conserved in B1 MBLs. In the B3 enzymes, Tyr228 (L1) and Asn225 (FEZ-1) have been variously proposed to stabilize oxyanionic species (22, 56) but the effects of substitutions at these positions are also substrate dependent (9, 40). Taken together, these data indicate that, although Zn1 is believed to polarize the β-lactam carbonyl for addition of W1 (44), involvement of other elements of the MBL active site in this process, or in stabilizing more transiently populated species, has not been demonstrated.
Fig 5.

Possible mechanism of cephalosporin hydrolysis catalyzed by AIM-1. The proposed role of the NE2 Gln157 in interactions with the C10 carbonyl (all panels) and with the C8 carbonyl of the intact substrate (panel a), the oxyanion (panel b), and the C8 carboxylate of hydrolyzed β-lactam (panels c and d) is highlighted (red lines).
Our structures show that AIM-1 does not contain residues at position 225 (Ile) or 228 (Tyr) that are compatible with a role in oxyanion hole formation (Fig. 4), and Asp233 is located too far from the active site to fulfil this function. However, structural comparisons and docking and QM/MM experiments are all consistent with the possibility of catalytically significant interactions between bound substrate and the NE2 nitrogen of AIM-1 Gln157. Our structures show Gln157 to overlap in space with FEZ-1 Asn225, while our models show that Gln157 NE2 interacts both with one oxygen of the C8 carboxylate group (derived from addition of W1 to the β-lactam carbonyl) and with the R1 (C10) carbonyl oxygen of hydrolyzed cefoxitin. Thus, the model implies that the NH2 group of the Gln157 side chain might assist with substrate binding, through interaction with the C10 carbonyl oxygen, and hydrolysis, by making stabilizing interactions with transiently populated species formed upon addition of W1 to C8 (Fig. 5b and c). A comparison of the kinetic parameters for B3 MBLs (Table 4) shows that, with the exception of ceftazidime, AIM-1 in general hydrolyzes many β-lactam substrates more efficiently than BJP-1, FEZ-1, and L1. We speculated that the presence of Gln157 may contribute to enhanced β-lactam hydrolysis by AIM-1 compared to other B3 MBLs. To investigate the role of Gln157 in the reactivity and specificity of AIM-1, the kinetic properties of the wild-type enzyme and two Gln157 mutants (Gln157Ala and Gln157Asn) were investigated and the crystal structures of both mutants were determined (Table 1 and Table 2).
Wild-type AIM-1 is a broad-spectrum enzyme that efficiently hydrolyzes penicillin (penicillin G, ampicillin) and carbapenem (imipenem, meropenem, ertapenem) substrates (Table 4). Turnover of cephalosporins is more variable, with activity significantly reduced for cefepime and ceftazidime, which possess cyclic positively charged R2 groups, compared to cefuroxime or cefoxitin (Fig. 6). The neutral-to-positive charge of the AIM-1 R2 site, which may be due to the presence of Arg300, may explain the high Km values for these substrates (Fig. 4e and Table 4). The effect of mutations at position 157 was dependent upon both the nature of the substitution and the substrate assayed. However, replacement of Gln157 by Asn or Ala generally led to modest (less than 10-fold) increases in Km values and similar (less than 10-fold) reductions in kcat, giving overall reductions in catalytic efficiency of up to 2 log orders of magnitude. Surprisingly, the effects on kcat were more pronounced for the Asn than the Ala mutant, with the greatest reductions observed for the cephalosporins and for ertapenem. For the Gln157Ala mutation, kcat increased for hydrolysis of ampicillin and cefuroxime. Effects on Km were more consistent in comparisons of the two mutants, although enhanced binding (i.e., reductions in Km) was observed for hydrolysis of ertapenem by the Ala and ceftazidime and cefepime by the Asn mutants.
Fig 6.
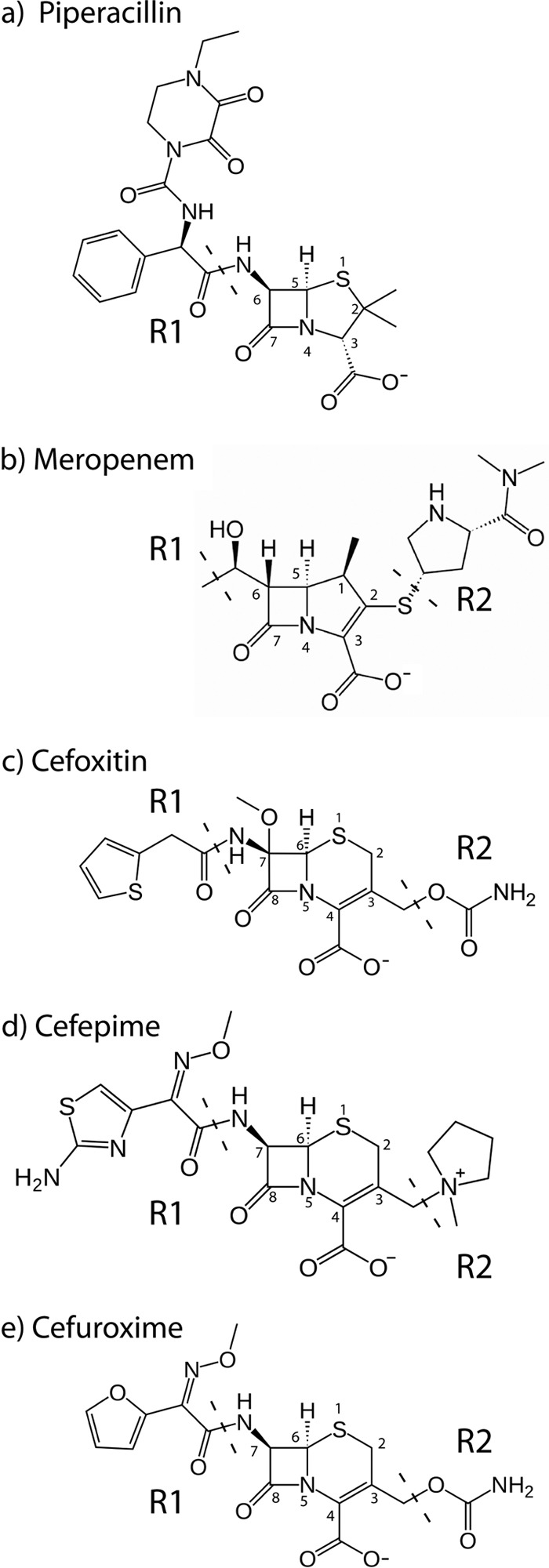
Structures of the β-lactam drugs (a) piperacillin, (b) meropenem, (c) cefoxitin, (d) cefepime, and (e) cefuroxime.
Crystal structure data demonstrate that neither of the two mutations results in gross distortion of the overall fold of the protein. Superposition of the complete AIM-1 and AIM-1-Q157N structures (RMSD = 0.09Å for 270 CA atoms) revealed that the side-chain nitrogens of Gln157 (NE2 in AIM-1) and Asn157 (ND2 in AIM-1-Q157N) occupy comparable positions (see Fig. S1 in the supplemental material). This is achieved through local rearrangement of the main chain of Asn157 in the mutant enzyme. Comparison of the AIM-1 Q157A mutant crystal structure with that of the wild type (RMSD = 0.10 Å for 270 CA atoms) shows that the position of the main chain is little affected by the mutation, thus removing the possibility of any interaction with the bound substrate at this position.
Taken together, the results of our mutagenesis studies do not support the proposal of an essential role for an interaction between Gln157 NE2 and substrate (carboxylate at C8 or C10 carbonyl) in β-lactam hydrolysis by AIM-1. This conclusion arises from the overall similarity of the effects of the Asn substitution, where the structure suggests that such an interaction could be retained, to those of the Ala substitution, where it is expected to be lost, and the fact that the kinetic consequences of both mutations are relatively modest. On this basis, we conclude that Gln157 does not form part of a catalytically essential oxyanion hole. This is consistent with the results of recent investigations into the role of Asn233 in β-lactam hydrolysis by the IMP-1 MBL (5) and with much current thinking on the MBL mechanism, which holds that anionic species in which the β-lactam ring has already been opened (Fig. 5c), rather than tetrahedral oxyanionic structures (Fig. 5b), are the mechanistically significant intermediates in the MBL reaction (55) and are selectively stabilized by the enzyme. Thus, mutations at Gln157 may not affect kcat if the rate-determining step for the reaction occurs after cleavage of the β-lactam ring. However, our modeling results suggest that, as shown in Fig. 5, Gln157 may be involved in interactions with both intact (substrate) and hydrolyzed (transient high-energy species, intermediates, and products) β-lactams. This is not inconsistent with our kinetic data, since Km is derived from multiple microscopic rate constants and may not be significantly affected if mutation affects the affinities of different states to similar degrees or if changes cancel one another out.
Conclusion.
The structural data presented here show AIM-1, the first B3 MBL to be found on a mobile genetic element in a significant human pathogen, to have several distinctive properties compared to previously characterized members of this subclass (BJP-1, FEZ-1, and L1). Although AIM-1, despite low sequence identity (<36%), retains a overall fold (αβ/βα) similar to those of these enzymes, AIM-1 contains three, rather than one, disulfide bridges and differs notably from these enzymes in the composition and orientation of the loop (156 to 166) positioned in adjacence to the active site. The structure, and subsequent modeling experiments, indicates that the AIM-1 substrate binding site contains an R1 pocket that is narrower and better defined, and an R2 pocket that is more positively charged, than in other B3 MBLs. Furthermore, the residue Gln157, which does not have a direct equivalent in other MBLs, is positioned to interact with the C10 carbonyl and the C8 carboxylate (formed on addition of the hydroxide nucleophile to the β-lactam C8 [carbonyl] carbon) of the bound substrate. Thus, Gln157 seems important for substrate binding.
Determination of an accurate high-resolution 3D structure is a critical step in the process of structure-based drug design. Better knowledge of the biochemical properties of MBLs, of which structure determination is an important part, would also aid in development of MBL inhibitors. The accelerating dissemination of MBLs is increasingly limiting treatment possibilities for health care-associated infections by Gram-negative bacteria. Identification of clinically effective inhibitors of these enzymes is thus a clinical problem of growing urgency. MBL inhibitors may also be important to development of improved diagnostic methods that are important for infection control and to limit the spread of MBL genes among bacterial pathogens. Knowledge of the structure of AIM-1 now permits its inclusion in these efforts and will aid in the development of MBL inhibitors effective against all subclasses of these diverse and challenging drug targets.
Supplementary Material
ACKNOWLEDGMENTS
The Norwegian Structural Biology Centre (NorStruct) is supported by the national Functional Genomics Programme (FUGE) of the Research Council of Norway. Ø.S. is supported by a grant from the Northern Norway Regional Health Authority. J.S. is funded by the United Kingdom Medical Research Council (G1100135) and the U.S. National Institutes of Health (1R01AI100560-01).
Provision of beam time at ID29 at the European Radiation Facility (ESRF), Grenoble, France, and at I911-3, Max lab II, Lund, Sweden, is gratefully acknowledged. Assistance with data collection by K. A. Johnson and R. Helland and with docking by R. Ahmad are very much appreciated.
Footnotes
Published ahead of print 4 June 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Amara P, Field MJ. 2003. Evaluation of an ab initio quantum mechanical/molecular mechanical hybrid-potential link-atom method. Theor. Chem. Acc. 109:43–52 [Google Scholar]
- 2. Bebrone C. 2007. Metallo-β-lactamases (classification, activity, genetic organization, structure, zinc coordination) and their superfamily. Biochem. Pharmacol. 74:1686–1701 [DOI] [PubMed] [Google Scholar]
- 3. Bebrone C, et al. 2010. Current challenges in antimicrobial chemotherapy: focus on β-lactamase inhibition. Drugs 70:651–679 [DOI] [PubMed] [Google Scholar]
- 4. Borra PS, et al. 2011. Structural and computational investigations of VIM-7: insights into the substrate specificity of VIM metallo-β-lactamases. J. Mol. Biol. 411:174–189 [DOI] [PubMed] [Google Scholar]
- 5. Brown NG, Horton LB, Huang W, Vongpunsawad S, Palzkill T. 2011. Analysis of the functional contributions of Asn233 in metallo-β-lactamase IMP-1. Antimicrob. Agents Chemother. 55:5696–5702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bush K. 2010. Bench-to-bedside review: the role of β-lactamases in antibiotic-resistant Gram-negative infections. Crit. Care 14:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bush K, Fisher JF. 2011. Epidemiological expansion, structural studies and clinical challenges of new β-lactamases from Gram-negative bacteria. Annu. Rev. Microbiol. 65:455–478 [DOI] [PubMed] [Google Scholar]
- 8. Buynak JD, et al. 2004. Penicillin-derived inhibitors that simultaneously target both metallo- and serine-β-lactamases. Bioorg. Med. Chem. Lett. 14:1299–1304 [DOI] [PubMed] [Google Scholar]
- 9. Carenbauer AL, Garrity JD, Periyannan G, Yates RB, Crowder MW. 2002. Probing substrate binding to metallo-β-lactamase L1 from Stenotrophomonas maltophilia by using site-directed mutagenesis. BMC Biochem. 3:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cohen SX, et al. 2008. ARP/wARP and molecular replacement: the next generation. Acta Crystallogr. D Biol. Crystallogr. 64:49–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Collaborative Computational Project-4 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760–763 [DOI] [PubMed] [Google Scholar]
- 12. Concha NO, et al. 2000. Crystal structure of the IMP-1 metallo β-lactamase from Pseudomonas aeruginosa and its complex with a mercaptocarboxylate inhibitor: binding determinants of a potent, broad-spectrum inhibitor. Biochemistry 39:4288–4298 [DOI] [PubMed] [Google Scholar]
- 13. Concha NO, Rasmussen BA, Bush K, Herzberg O. 1996. Crystal structure of the wide-spectrum binuclear zinc β-lactamase from Bacteroides fragilis. Structure 4:823–836 [DOI] [PubMed] [Google Scholar]
- 14. Crisp J, et al. 2007. Structural basis for the role of Asp-120 in metallo-β-lactamases. Biochemistry 46:10664–10674 [DOI] [PubMed] [Google Scholar]
- 15. Docquier JD, et al. 2010. High-resolution crystal structure of the subclass B3 metallo-β-lactamase BJP-1: rational basis for substrate specificity and interaction with sulfonamides. Antimicrob. Agents Chemother. 54:4343–4351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Docquier JD, et al. 2003. On functional and structural heterogeneity of VIM-type metallo-β-lactamases. J. Antimicrob. Chemother. 51:257–266 [DOI] [PubMed] [Google Scholar]
- 17. Dunietz BD, et al. 2000. Large scale ab initio quantum chemical calculation of the intermediates in the soluble methane monooxygenase catalytic cycle. J. Am. Chem. Soc. 122:2828–2839 [Google Scholar]
- 18. Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 19. Felici A, et al. 1993. An overview of the kinetic parameters of class B β-lactamases. Biochem. J. 291(Pt. 1):151–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garau G, et al. 2005. A metallo-β-lactamase enzyme in action: crystal structures of the monozinc carbapenemase CphA and its complex with biapenem. J. Mol. Biol. 345:785–795 [DOI] [PubMed] [Google Scholar]
- 21. Garau G, et al. 2004. Update of the standard numbering scheme for class B β-lactamases. Antimicrob. Agents Chemother. 48:2347–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. García-Saez I, et al. 2003. The 1.5-Å structure of Chryseobacterium meningosepticum zinc β-lactamase in complex with the inhibitor, D-captopril. J. Biol. Chem. 278:23868–23873 [DOI] [PubMed] [Google Scholar]
- 23. García-Sáez I, et al. 2003. Three-dimensional structure of FEZ-1, a monomeric subclass B3 metallo-β-lactamase from Fluoribacter gormanii, in native form and in complex with D-captopril. J. Mol. Biol. 325:651–660 [DOI] [PubMed] [Google Scholar]
- 24. Garman EF. 2010. Radiation damage in macromolecular crystallography: what is it and why should we care? Acta Crystallogr. D Biol. Crystallogr. 66:339–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garrity JD, Pauff JM, Crowder MW. 2004. Probing the dynamics of a mobile loop above the active site of L1, a metallo-β-lactamase from Stenotrophomonas maltophilia, via site-directed mutagenesis and stopped-flow fluorescence spectroscopy. J. Biol. Chem. 279:39663–39670 [DOI] [PubMed] [Google Scholar]
- 26. Gouet P, Courcelle E, Stuart DI, Metoz F. 1999. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15:305–308 [DOI] [PubMed] [Google Scholar]
- 27. Hay PJ, Wadt WR. 1985. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 82:299–310 [Google Scholar]
- 28. Huntley JJ, Fast W, Benkovic SJ, Wright PE, Dyson HJ. 2003. Role of a solvent-exposed tryptophan in the recognition and binding of antibiotic substrates for a metallo-β-lactamase. Protein Sci. 12:1368–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jones G, Willett P, Glen RC. 1995. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 245:43–53 [DOI] [PubMed] [Google Scholar]
- 30. Jorgensen WL, Maxwell DS, Tirado-Rives J. 1996. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118:11225–11236 [Google Scholar]
- 31. Kabsch W. 1993. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallog. 24:795–800 [Google Scholar]
- 32. Kaminskaia NV, Spingler B, Lippard SJ. 2001. Intermediate in β-lactam hydrolysis catalyzed by a dinuclear zinc(II) complex: relevance to the mechanism of metallo-β-lactamase. J. Am. Chem. Soc. 123:6555–6563 [DOI] [PubMed] [Google Scholar]
- 33. Lassaux P, et al. 2010. Mercaptophosphonate compounds as broad-spectrum inhibitors of the metallo-β-lactamases. J. Med. Chem. 53:4862–4876 [DOI] [PubMed] [Google Scholar]
- 34. Leiros H-KS, McSweeney SM, Smalås AO. 2001. Atomic resolution structures of trypsin provide insight into structural radiation damage. Acta Crystallogr. D 57:488–497 [DOI] [PubMed] [Google Scholar]
- 35. Leiros H-KS, Timmins J, Ravelli RB, McSweeney SM. 2006. Is radiation damage dependent on the dose rate used during macromolecular crystallography data collection? Acta Crystallogr. D Biol. Crystallogr. 62:125–132 [DOI] [PubMed] [Google Scholar]
- 36. Liénard BM, et al. 2008. Structural basis for the broad-spectrum inhibition of metallo-β-lactamases by thiols. Org. Biomol. Chem. 6:2282–2294 [DOI] [PubMed] [Google Scholar]
- 37. Maltezou HC. 2009. Metallo-β-lactamases in Gram-negative bacteria: introducing the era of pan-resistance? Int. J. Antimicrob. Agents 33:405.e1–405.e7 [DOI] [PubMed] [Google Scholar]
- 38. McCoy AJ, et al. 2007. Phaser crystallographic software. J. Appl. Crystallogr. 40:658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mercuri PS, et al. 2001. Biochemical characterization of the FEZ-1 metallo-β-lactamase of Legionella gormanii ATCC 33297T produced in Escherichia coli. Antimicrob. Agents Chemother. 45:1254–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mercuri PS, et al. 2004. Probing the specificity of the subclass B3 FEZ-1 metallo-β-lactamase by site-directed mutagenesis. J. Biol. Chem. 279:33630–33638 [DOI] [PubMed] [Google Scholar]
- 41. Moali C, et al. 2003. Analysis of the importance of the metallo-β-lactamase active site loop in substrate binding and catalysis. Chem. Biol. 10:319–329 [DOI] [PubMed] [Google Scholar]
- 42. Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. 1999. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr. D Biol. Crystallogr. 55:247–255 [DOI] [PubMed] [Google Scholar]
- 43. Nauton L, Kahn R, Garau G, Hernandez JF, Dideberg O. 2008. Structural insights into the design of inhibitors for the L1 metallo-β-lactamase from Stenotrophomonas maltophilia. J. Mol. Biol. 375:257–269 [DOI] [PubMed] [Google Scholar]
- 44. Page MI, Badarau A. 2008. The mechanisms of catalysis by metallo β-lactamases. Bioinorg. Chem. Appl. 2008:576297 doi:10.1155/2008/576297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rasmussen BA, Bush K. 1997. Carbapenem-hydrolyzing β-lactamases. Antimicrob. Agents Chemother. 41:223–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ravelli RB, Leiros H-KS, Pan B, Caffrey M, McSweeney S. 2003. Specific radiation damage can be used to solve macromolecular crystal structures. Structure 11:217–224 [DOI] [PubMed] [Google Scholar]
- 47. Ravelli RB, McSweeney SM. 2000. The ‘fingerprint’ that X-rays can leave on structures. Structure 8:315–328 [DOI] [PubMed] [Google Scholar]
- 48. Rocchia W, et al. 2002. Rapid grid-based construction of the molecular surface and the use of induced surface charge to calculate reaction field energies: applications to the molecular systems and geometric objects. J. Comput. Chem. 23:128–137 [DOI] [PubMed] [Google Scholar]
- 49. Samuelsen Ø, Castanheira M, Walsh TR, Spencer J. 2008. Kinetic characterization of VIM-7, a divergent member of the VIM metallo-β-lactamase family. Antimicrob. Agents Chemother. 52:2905–2908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schmidt A, Jelsch C, Østergaard P, Rypniewski W, Lamzin VS. 2003. Trypsin revisited: crystallography AT (SUB) atomic resolution and quantum chemistry revealing details of catalysis. J. Biol. Chem. 278:43357–43362 [DOI] [PubMed] [Google Scholar]
- 51. Siezen RJ, Leunissen JA. 1997. Subtilases: the superfamily of subtilisin-like serine proteases. Protein Sci. 6:501–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Simm AM, et al. 2002. Characterization of monomeric L1 metallo-β-lactamase and the role of the N-terminal extension in negative cooperativity and antibiotic hydrolysis. J. Biol. Chem. 277:24744–24752 [DOI] [PubMed] [Google Scholar]
- 53. Spencer J, et al. 2005. Antibiotic recognition by binuclear metallo-β-lactamases revealed by X-ray crystallography. J. Am. Chem. Soc. 127:14439–14444 [DOI] [PubMed] [Google Scholar]
- 54. Stoczko M, Frere JM, Rossolini GM, Docquier JD. 2006. Postgenomic scan of metallo-β-lactamase homologues in rhizobacteria: identification and characterization of BJP-1, a subclass B3 ortholog from Bradyrhizobium japonicum. Antimicrob. Agents Chemother. 50:1973–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tioni MF, et al. 2008. Trapping and characterization of a reaction intermediate in carbapenem hydrolysis by B. cereus metallo-β-lactamase. J. Am. Chem. Soc. 130:15852–15863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ullah JH, et al. 1998. The crystal structure of the L1 metallo-β-lactamase from Stenotrophomonas maltophilia at 1.7 Å resolution. J. Mol. Biol. 284:125–136 [DOI] [PubMed] [Google Scholar]
- 57. Wachino J, et al. 2011. SMB-1, a novel subclass B3 metallo-β-lactamase, associated with ISCR1 and a class 1 integron, from a carbapenem-resistant Serratia marcescens clinical isolate. Antimicrob. Agents Chemother. 55:5143–5149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Walsh TR. 2010. Emerging carbapenemases: a global perspective. Int. J. Antimicrob. Agents 36S. 3:S8–S14 [DOI] [PubMed] [Google Scholar]
- 59. Walsh TR, Toleman MA, Poirel L, Nordmann P. 2005. Metallo-β-lactamases: the quiet before the storm? Clin. Microbiol. Rev. 18:306–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang Z, Fast W, Benkovic SJ. 1999. On the mechanism of the metallo-β-lactamase from Bacteroides fragilis. Biochemistry 38:10013–10023 [DOI] [PubMed] [Google Scholar]
- 61. Wright GD. 2007. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat. Rev. Microbiol. 5:175–186 [DOI] [PubMed] [Google Scholar]
- 62. Yamaguchi Y, et al. 2007. Crystallographic investigation of the inhibition mode of a VIM-2 metallo-β-lactamase from Pseudomonas aeruginosa by a mercaptocarboxylate inhibitor. J. Med. Chem. 50:6647–6653 [DOI] [PubMed] [Google Scholar]
- 63. Yong D, et al. A novel sub-group metallo-β-lactamase (MBL), AIM-1 emerges in Pseudomonas aeruginosa from Australia. Antimicrob. Agents Chemother., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.