

Abstract
Host mitogen-activated protein kinases (MAPKs) are deregulated by herpes simplex virus 1 (HSV-1). Unlike p38 MAPK and Jun N-terminal protein kinase (JNK), which require ICP27 for their activation early in infection, extracellular signal-regulated kinase (ERK) activity is suppressed by an unknown mechanism. Here, we establish that HSV-1-induced suppression of ERK activity requires viral gene expression, occurs with delayed-early kinetics, and requires the functional virus-encoded Us3 Ser/Thr protein kinase. Finally, Us3 expression in uninfected cells was necessary and sufficient to suppress ERK activity in the absence of any other virus-encoded gene products. This demonstrates that inhibition of ERK activity in HSV-1-infected cells is an intrinsic Us3 function and defines a new role for this alphaherpesvirus Us3 kinase in regulating MAPK activation in infected cells.
INTRODUCTION
At the molecular level, numerous interactions between virus-encoded functions and host cell components regulate cellular signaling pathways. In many cases this represents a key means through which viruses effectively create permissive environments for their productive replication by capturing and controlling critical host functions and incapacitating innate host antiviral defenses. Mitogen-activated protein kinases (MAPKs), in particular, are important for integrating a diverse array of stimuli into defined cellular responses (20, 27, 35, 44, 49). Many DNA and RNA viruses subvert host MAPK signaling pathways to stimulate their productive replication, control cell proliferation, suppress apoptosis, or regulate latency (17, 25, 39, 50–52, 55, 56). MAPK activation is also associated with virus-induced cytokine production and inflammation (39, 41). Importantly, in cells infected with herpes simplex virus 1 (HSV-1), all three major MAPK pathways are deregulated in response to virus infection (14, 28, 51, 57).
Jun N-terminal kinases (JNK) and p38 MAPK pathways, referred to as stress-activated protein kinases, are activated by environmental and genotoxic stresses and play important roles in inflammation, proliferation, and cell survival (reviewed in reference 49). Both JNK and p38 are activated in HSV-1-infected cultured cells (28, 57). While the precise mechanism underlying JNK activation is incompletely understood, it is dependent upon the ICP27 regulatory protein, and some JNK activation was observed upon induction of ICP27 expression in an uninfected, established cell line. Under these conditions, ICP27 expression was sufficient to robustly activate p38, raising the possibility that additional viral factors may contribute to JNK activation in infected cells (12). p38, in turn, is required to activate the eukaryotic initiation factor 4E (eIF4E) kinase Mnk, which stimulates cap-dependent translation of viral mRNAs (51).
The extracellular signal-regulated kinases (ERKs) are activated by mitogens and are often activated in human cancers (reviewed in references 27 and 44). In contrast to p38 and JNK, ERK activity is suppressed in HSV-1-infected cells (28, 51). With this in mind, we set out to define the requirements for ERK inactivation in HSV-1-infected cells and to identify the viral gene product(s) required to control ERK signaling.
MATERIALS AND METHODS
Cell culture, viruses, and chemicals.
All cells were propagated in 5% CO2 with Dulbecco's modified Eagle's medium (DMEM) supplemented with 2 mM glutamine, 50 U of penicillin per milliliter, and 50 μg of streptomycin per milliliter, plus serum as follows: primary normal human dermal fibroblasts (NHDFs; Clonetics) were maintained in 5% fetal bovine serum (FBS) and serum deprived in 0.2% FBS as described earlier (51), U2OS cells (American Type Culture Collection [ATCC]) were propagated in 10% FBS, and Vero (ATCC) cells were propagated in 5% calf serum. F-strain wild-type (WT) and ΔUs3 (VRR1202), K220A Us3 mutant (VRR1204), and ΔUs3-Repair (VRR1202repair) HSV-1 were described earlier (45); UL13-deficient virus (d 13 lac z) was a gift from S. Rice (University of Minnesota, Minneapolis, MN [26]); the ICP4-deficient virus was a gift from N. DeLuca (University of Pittsburgh School of Medicine, Pittsburgh, PA [9]). UV-inactivated virus was prepared as described earlier (51). Antibodies Akt (no. 9272), Akt-pSer473 (no. 9271), TSC2 (no. 3611), p70 S6K T389, and total (no. 9430) were from Cell Signaling; and anti-FlagM2 monoclonal antibody was from Sigma. The ICP6 antibody was a kind gift from V. Preston (MRC Institute of Virology, Glasgow, United Kingdom [40]). All other antibodies were described earlier (51). PD-325901 was from Sigma, and rapamycin was from Calbiochem. PP242 was generously provided by K. Shokat (UCSF, San Francisco, CA) and was described earlier (11). TSC2 small interfering RNA (siRNA) was described earlier (7).
Immunoblotting, infections, transfections, and siRNA depletion.
Immunoblotting, infections, transfections, and small siRNA depletion procedures were performed as described in reference 7.
RESULTS AND DISCUSSION
While herpesviruses induce a variety of changes to cell signaling pathways in infected cells, not all instances of virus-induced activation or inhibition of cell signaling require viral entry into host cells or virus gene expression, as these changes can be triggered simply by virions engaging cell surface receptors or deposition of tegument proteins into the cytoplasm. To investigate if viral gene expression was required to inhibit ERK activation, growth-arrested NHDFs were infected at a high multiplicity of infection (MOI) with untreated WT HSV-1 or UV-inactivated virus. At 13 h postinfection (hpi), total protein was harvested, fractionated by SDS-PAGE, and analyzed by immunoblotting using antibodies directed against total ERK (p42ERK2 and p44ERK1) and phosphospecific ERK antibodies that recognize activated ERK1/2 (Fig. 1A). Whereas Erk1/2 activation was suppressed in cells infected with untreated, WT virus, activated ERK levels detected in cells infected with UV-inactivated virus were similar to those observed in mock-infected cells.
Fig 1.
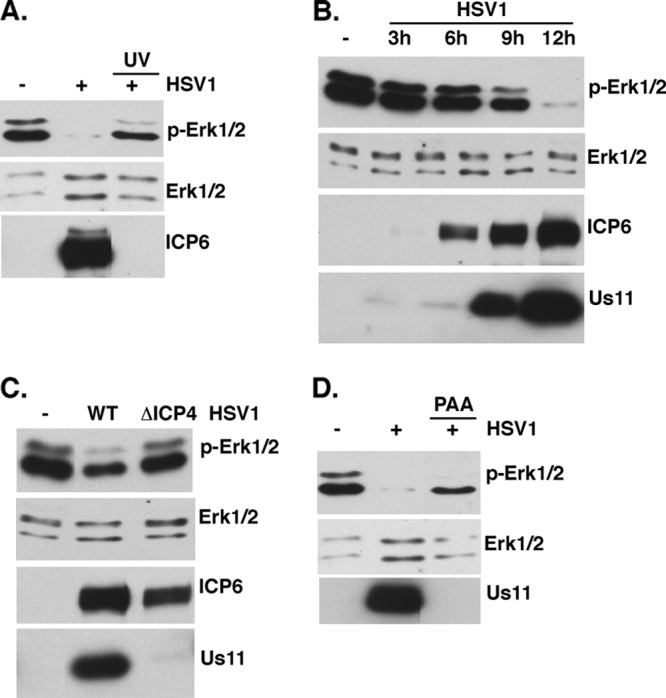
Requirements for suppressing ERK activation in HSV-1-infected cells. (A) Serum-deprived NHDFs were mock infected or infected with HSV-1 or UV-inactivated HSV-1 at a MOI of 5. Total protein was collected at 13 hpi, separated by SDS-PAGE, and analyzed by immunoblotting with the indicated antisera. (B) Same as for panel A, except that total protein was collected at the indicated times postinfection. (C) Same as for panel A, except that cells were mock infected or infected with WT KOS or the ICP4-deficient N12 mutant (MOI = 5). Total protein was collected at 9 hpi and analyzed as described for panel A. (D) Same as for panel A, except that cells were pretreated with ±300 μg/ml phosphonoacetic acid (PAA) for 30 min and then mock infected or infected with HSV-1 (MOI = 5) in the presence or absence of PAA. At 13 hpi, total protein was collected and analyzed as described for panel A.
To determine when in the HSV-1 productive replication cycle ERK activity is suppressed, ERK activation in infected NHDFs was evaluated at different times postinfection. Following high-MOI infection of growth-arrested NHDFs with WT HSV-1, total protein was isolated and fractionated by SDS-PAGE, and ERK activation was analyzed by immunoblotting. Figure 1B demonstrates that a significant decline in steady-state levels of activated ERK is evident between 6 and 9 hpi. By 12 hpi, ERK activation is no longer detectable. Relative to the well-characterized lytic gene expression profile, accumulation of the virus-encoded early protein ICP6 is observed at 6 hpi without any detectable reduction in ERK activation. This suggests that ERK inactivation occurs at a point in the viral replication cycle that lies downstream of ICP6 expression. While the ICP6 gene is classified as an early gene, it is unique in its expression kinetics in that it is responsive to the immediate early (IE) ICP0 transactivator (8, 9, 13, 46). By 12 hpi, ERK activation was markedly suppressed and abundant levels of the “true-late” Us11 gene product were readily observed. Together, these data crudely position the suppression of ERK activation in the viral life cycle somewhere between expression of the early ICP6 gene and a true-late gene whose expression is stringently dependent upon DNA replication.
To precisely delineate the viral gene expression requirements needed to suppress ERK activity, a genetic and pharmacological approach was employed. By utilizing a virus deficient in the essential IE regulator ICP4, the viral life cycle is arrested at the IE stage and only a limited set of viral proteins accumulate. While ERK activation was suppressed in cells infected with WT HSV-1, NHDFs infected with the well-characterized N12 (9) ICP4-deficient virus displayed levels of activated ERK similar to those of mock-infected cells (Fig. 1C). Accumulation of ICP6 in cells infected with either WT or N12 virus validates that these cells were infected and expressed ICP0, which activates ICP6 expression. However, accumulation of Us11, a product of a true-late gene whose expression depends upon ICP4 and subsequent DNA synthesis, was detected only in cells infected with WT HSV-1, validating that cells infected with the N12 mutant did in fact arrest their life cycle without initiating DNA synthesis. This suggested that the virus-encoded function required to suppress ERK activity was dependent upon ICP4 and therefore upon either ICP4 itself, a virus-borne early gene(s), a delayed-early or leaky-late gene(s) (γ1), or a true-late (γ2) gene(s).
Functions encoded by delayed-early (or leaky-late [γ1]) or “true-late” (γ2) genes can be distinguished by their sensitivity to phosphonoacetic acid (PAA), an inhibitor of viral DNA synthesis. Whereas ICP4-dependent early gene functions are typically resistant to PAA treatment, transcription of γ2 genes is blocked and transcription of γ1 genes is reduced by PAA treatment. To determine if HSV-1-induced ERK suppression was sensitive or resistant to PAA, ERK activation in NHDFs mock infected or infected with WT HSV-1 in the presence and absence of PAA was evaluated. While WT HSV-1 infection effectively suppressed ERK activation in infected cells, PAA treatment reduced activated ERK to levels below those observed in mock-infected cells but clearly above those detected in cultures that were not treated with PAA (Fig. 1D). In contrast, the γ2 Us11 protein was detected only in untreated, infected cultures, establishing the efficacy of the PAA block (Fig. 1D). The intermediate suppression of ERK activation in response to PAA is consistent with the involvement of a viral function whose expression is reduced but not completely eliminated by PAA. This suggests that an early or leaky-late (γ1) virus-borne gene product is required to suppress ERK activation in infected cells.
MAPK signaling is intricately regulated by protein phosphorylation. Thus, we considered the possibility that a virus-encoded kinase might play a role in deregulating ERK activity in infected cells. The Us3 and UL13 genes, which encode two Ser/Thr kinases and are, respectively, expressed with early and γ1 kinetics in the productive growth cycle, were chosen as candidate genes, and their contributions to MAPK activation in HSV-1-infected cells were investigated. NHDFs were either mock infected or infected with WT virus, a UL13-deficient virus (ΔUL13), or a Us3-deficient virus (ΔUs3), and MAPK activation in cell lysates was evaluated by immunoblotting with phosphospecific antibodies. While wild-type levels of p38 activation and ERK suppression were observed in cells infected with ΔUL13, only p38 activation was observed to proceed normally in cells infected with ΔUs3 (Fig. 2A and B). Remarkably, ERK suppression was detected only in cells infected with WT virus, whereas normal levels of activated ERK persisted in cells infected with ΔUs3 (Fig. 2A). All virus-infected cultures expressed the true-late, HSV-1 Us11 protein (Fig. 2C). Even though less Us11 accumulated in cultures infected with ΔUL13, the extent of ERK inactivation and p38 activation was similar to that observed in cultures infected with ΔUL13-Repair. Reduced protein accumulation in cells infected with UL13-deficient viruses could reflect reduced transcription from the viral genome (26) or altered protein synthesis (37), both of which have been reported.
Fig 2.
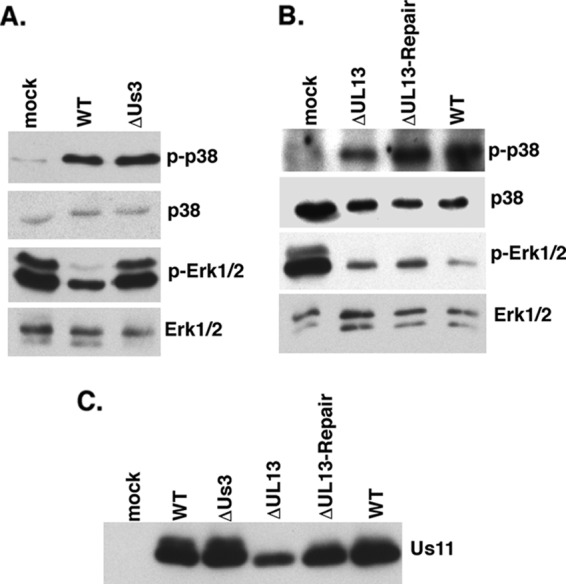
Suppression of ERK activation in HSV-1-infected cells is dependent upon the Us3 Ser/Thr kinase. (A) Serum-deprived NHDFs were mock infected or infected with WT HSV-1 or a Us3-deficient mutant (ΔUs3) at a MOI of 5. Total protein was harvested at 15 hpi and analyzed by immunoblotting using the indicated antisera. (B) Same as for panel A, except that cells were infected with a UL13-deficient virus (ΔUL13), a UL13-deficient virus in which the mutation was repaired (ΔUL13-Repair), or WT virus. (C) Samples as described for panels A and B were analyzed by immunoblotting using anti-Us11.
To determine if the Us3 Ser/Thr kinase activity was required to suppress ERK activation, cells were either mock infected or infected with a virus expressing K220A kinase-deficient Us3 protein (K220A Us3), ΔUs3, a virus in which the Us3 deletion was repaired (ΔUs3-repair), or WT HSV1 (Fig. 3A). After 15 h, total protein was isolated, fractionated by SDS-PAGE, and analyzed by immunoblotting. Importantly, only cells infected with Us3-deficient viruses were unable to suppress ERK activity (Fig. 3A, compare lanes 3 and 4 to lanes 2 and 5). The suppression of ERK activity observed by repairing the ΔUs3 mutation (Fig. 3A, compare lane 3 to lane 5) demonstrates that the loss of Us3 function is indeed responsible for the failure to inactivate ERK in cells infected with ΔUs3. Furthermore, ERK activation in cells infected with K220A Us3 (Fig. 3A, compare lane 4 with lane 2 or 5) establishes that a single amino acid substitution that impairs Us3 kinase activity prevents the inhibition of ERK activation. This suggests that the Us3 Ser/Thr kinase activity is required to suppress ERK activation in infected cells. Similar levels of a viral late protein (Us11) accumulated in infected cells.
Fig 3.

The Us3 Ser/Thr kinase activity is required to suppress ERK activation. (A) Serum-deprived NHDFs were mock infected or infected (MOI = 5) with WT HSV-1, ΔUs3, a Us3-deficient virus in which the Us3 allele was restored (ΔUs3-Repair), or a virus expressing a kinase-deficient Us3 gene containing a K-to-A single amino acid substitution at residue 220 (K220A Us3). At 15 hpi, total protein was isolated and analyzed by immunoblotting with the indicated antisera. RhoGDI served as a loading control. (B) U2OS cells were mock infected or infected with WT HSV-1 or ΔU3 (MOI = 10), and at 9 hpi total protein was collected and analyzed as described for panel A. A modest reduction in Us11 accumulation was observed in cells infected with ΔUs3, as Us3-deficient viruses are unable to activate mTORC1 and display reduced viral protein synthesis (6, 7). (C) U2OS cells were transfected with the vector control plasmid, a K220A Us3-Flag-expressing plasmid, or a WT Us3-Flag-expressing plasmid. After 24 h, total protein was isolated and analyzed as described for panel A.
Having demonstrated that suppressing ERK activation in HSV-1-infected cells was Us3 dependent, we next asked if Us3 was sufficient to preclude ERK activation when transiently expressed in uninfected cells. The established U2OS cells were chosen for this purpose because they can express Us3 following transfection and exhibited Us3-dependent ERK suppression upon infection with HSV-1 (Fig. 3B). To determine if Us3 was necessary and sufficient to suppress ERK activation, U2OS cells were transfected with plasmids expressing WT FLAG-tagged Us3, FLAG-tagged K220A, or a vector-alone control. After 1 day, total protein was harvested and MAPK activation analyzed by immunoblotting. Whereas ERK activation in K220A Us3-expressing cells was similar to that observed in cells transfected with the vector alone, FLAG WT Us3 expression was sufficient to markedly reduce ERK activation, even though it was present at reduced levels relative to the K220A protein (Fig. 3C). The reduced mobility of WT FLAG-Us3 relative to the K220A variant has been observed previously (7) and reflects autophosphorylation resulting from catalytic activity intrinsic to the WT Us3 protein. This demonstrates that a catalytically active Us3 is necessary and sufficient to suppress ERK activation in the absence of any other virus-encoded proteins.
Us3 is unrelated to any one cellular Ser/Thr kinase but recently has been shown to phosphorylate several Akt substrates (TSC2, FOXO, and GSK3) in infected cells (6, 7). In particular, direct phosphorylation of TSC2 by Us3 is required for mTORC1 activation in HSV-1-infected cells. Several reports have suggested that cross talk between Ras-MAPK and phosphatidylinositol 3-kinase (PI3K)-AKT pathways exists, allowing each pathway to exert a regulatory effect on the other (5; reviewed in reference 30). In particular, the PI3K-AKT pathway inhibits RAF-MEK-ERK signaling (42), and preventing mTOR activation can reportedly result in ERK activation under certain conditions (4). To investigate if Us3-stimulated mTORC1 activity was required to suppress ERK activation, cultures were treated with dimethyl sulfoxide (DMSO) (control), the mTORC1-selective inhibitor rapamycin, or the active-site mTOR inhibitor PP242 and either mock infected or infected with WT HSV-1 (Fig. 4A). While none of the chemical treatments altered ERK activation significantly in uninfected cells, HSV-1 infection effectively suppressed ERK activation. Treatment of infected cultures with either the mTORC1-selective inhibitor rapamycin or the mTOR active-site inhibitor PP242 effectively abrogated hyperphosphorylation of the translational repressor 4E-BP1 as reported previously (7). However, only a modest recovery of ERK activation resulted from treatment with PP242, which unlike rapamycin, also prevents Akt activation by inhibiting S473 phosphorylation. Similarly, TSC2 depletion restored the ability of ΔUs3 to activate mTORC1, as measured by p70 S6K phosphorylation (Fig. 4B, compare lanes 1 and 2), but did not detectably suppress ERK activation (Fig. 4B, compare lanes 1 and 2). While Us3 shares with Akt the ability to suppress ERK activation, this likely proceeds through a substrate other than TSC2 and is not detectably affected by mTORC1 in HSV-1-infected cells.
Fig 4.
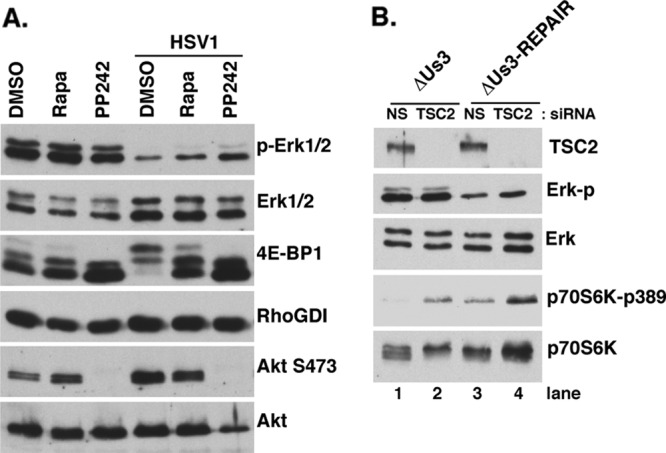
Us3-dependent suppression of ERK activity does not require Us3-mediated mTORC1 activation. (A) Serum-deprived NHDFs were pretreated with DMSO, 100 nM rapamycin, or 2.5 μM PP242 for 30 min and subsequently mock infected or infected with HSV-1 (MOI = 5) in the presence or absence of inhibitors. At 15 hpi, total protein was isolated and analyzed by immunoblotting with the indicated antisera. (B) NHDFs were transfected with control or nonsilencing (NS) or TSC2 siRNA for 48 h, serum deprived for 72 h, and infected with ΔUS3 or ΔUs3-Repair (MOI = 5) as described previously (7). At 14 hpi, total protein was harvested and analyzed as described for panel A.
Having defined the Us3-dependent suppression of ERK activation, the impact of preventing ERK activation on productive growth in normal cultured cells was investigated. Previous studies have shown that HSV-1 can replicate in a variety of established human cell lines with various degrees of MEK activation (47). However, the outputs of activated ERK signaling vary greatly depending upon their biological context and the strength of signaling, ranging from proapoptotic to proliferative (35, 58). It therefore was important to evaluate the consequences of Us3-dependent ERK suppression in normal human cells. While TSC2 depletion substantially restores the ability of Us3-deficient viruses to replicate in NHDFs (7), it is possible that suppressing ERK activation may also enhance Us3-deficient virus growth. NHDFs were infected at low MOI with ΔUs3 or Us3-Repair virus in the presence or absence of the MEK inhibitor PD-325901, and infectious virus production was quantified after 4 days by plaque assay on Vero cells. While the Us3-deficient virus replicated 20-fold less than viruses with WT Us3 genes, neither virus was detectably affected by a small-molecule MEK inhibitor that prevents ERK activation (Fig. 5A and B). Figure 5C shows that the inhibitor effectively prevented ERK activation through at least 72 h.
Fig 5.

Replication of a Us3-deficient virus in the presence of a MEK inhibitor. (A) Confluent serum-deprived NHDFs seeded in a 12-well dish (1 × 105 cells/well) were infected with 100 PFU/well an HSV-1 Us3 deletion mutant (ΔUs3) or a Us3-deficient virus in which the Us3 allele was restored (ΔUs3-Repair) in the presence of control DMSO or 100 nM PD-325901. After 4 days, cell-free lysates were prepared by freeze-thawing, and the amount of infectious virus produced was determined by plaque assay in Vero cells. (B) Cells as described for panel A were infected with ΔUs3 or ΔUs3-Repair virus (MOI = 5) as described above. At 15 hpi, total protein was harvested and analyzed by immunoblotting with the indicated antisera. (C) NHDFs as described for panel A were treated with DMSO or with 100 nM PD-325901. At the indicated times posttreatment, total protein was collected and analyzed as described for panel B.
While the precise contribution(s) that suppressing ERK activation makes to HSV-1 replication remains elusive, it is nevertheless important to realize that the Us3 Ser/Thr protein kinase has the intrinsic capacity to suppress ERK activation, and this potentially could account for a subset of the phenotypes associated with Us3-deficient viruses. While activating stress-activated kinases like p38 concomitant with the inhibition ERK has been observed in apoptotic cells, this can be antagonized by phosphatidylinositol 3-kinase signaling (3, 54). By suppressing ERK activation and acting in part like an Akt mimic, Us3 ensures that p38 can be safely activated without driving the cells into a proapoptotic program. Activation of p38 is required in part to activate Mnk, which in turn stimulates viral protein synthesis via eIF4E phosphorylation (51). Cytoskeletal rearrangements in alphaherpesvirus-infected cells require Us3-mediated activation of PAK (48), and PAK acts to transmit anchorage-dependent protein kinase A signaling to ERK (15). Since inhibiting ERK activity inhibits anoikis in cells deprived of anchorage (21), Us3 may suppress anoikis by inhibiting ERK activation as infected cells rearrange their cytoskeleton and lose contacts with the extracellular matrix. Finally, Us3 can block expression of gamma interferon-dependent genes (24), which is known to involve C/EBP-β phosphorylation by ERK1/2 (16, 43). Thus, our observation that Us3 suppresses ERK activity may explain how Us3 prevents gamma interferon-induced gene expression.
Other alphaherpesviruses use different strategies to manipulate ERK. Pseudorabies virus (PRV) Us2 tethers ERK to intracellular membranes and selectively inhibits activation of ERK nuclear targets, restricting activated ERK to the cytoplasm (18). Equine herpesvirus US2 has similar membrane binding properties and may also function similarly (29). However, varicella-zoster virus (VZV) open reading frame 12 (ORF12) protein triggers ERK activation in established, transformed cell lines, whereas the HSV-1 ORF12 homolog encoded by the UL46 ORF did not (25). In addition, HSV-2 infection reportedly activates ERK in an ICP10-dependent manner, while this was not observed in HSV-1-infected cells (38).
Notably, only alphaherpesviruses encode Us3 homologs, and these viruses are all neurotrophic, raising the possibility that the biological consequences of suppressing ERK activation in HSV-1-infected cells are discernible only in neurons, in animal models of pathogenesis, or in the natural human host. Conflicting results regarding the ability of Us3-deficient viruses to prevent apoptosis in neurons or neuron-like cell lines have been reported (1, 31, 32). Finally, the possibility that HSV-1 Us3-dependent ERK suppression is of little consequence for the virus cannot be excluded. While the complete list of cellular substrates for Us3 remains to be defined, it potentially has a rather broad specificity encompassing Akt and PKA substrates, even though it lacks significant primary sequence homology to any of these kinases (2, 6, 7, 10, 19, 22, 23, 33, 34, 36, 45, 53). One possibility is that a certain degree of promiscuity in substrate selection is tolerated as long as the targets phosphorylated by Us3 either stimulate virus replication or do not negatively impact the viral replication program. Such a reservoir of bona fide substrates, whose modification does not result in measurable biological outcomes (i.e., they do not stimulate or inhibit virus replication), may not be limited to virus-encoded kinases and may also include substrates of cellular kinases. The resulting pool of substrates without associated biological functions could serve as an evolutionary breeding ground from which selected functional regulatory links or attributes appear or are remodeled over time. Alternatively, suppression of ERK activation may just be an unavoidable consequence that coincidentally results when a kinase with the required substrate specificity intrinsic to Us3 is selected. While it is difficult to prove the hypothesis that phosphorylation of certain genuine substrates is not associated with a measurable response, sifting through the potential biological noise associated with a genuine substrate that is not associated with a detectable biological outcome(s) will further complicate our understanding of alphaherpesvirus Us3 kinases, and perhaps cellular kinases as well.
ACKNOWLEDGMENTS
We thank N. DeLuca and S. Rice for viruses, V. Preston for the ICP6 antisera, and D. Walsh for critically reading the manuscript.
This work was supported by grants from the NIH to I.M. (GM056927 and AI073898) and R.R. (AI41478). U.C. was supported in part by an NIH training grant (T32 AI07180).
Footnotes
Published ahead of print 16 May 2012
REFERENCES
- 1. Aiamkitsumrit B, et al. 2007. Herpes simplex virus type 1 ICP4 deletion mutant virus d120 infection failed to induce apoptosis in nerve growth factor-differentiated PC12 cells. J. Neurovirol. 13:305–314 [DOI] [PubMed] [Google Scholar]
- 2. Benetti L, Roizman B. 2004. Herpes simplex virus protein kinase US3 activates and functionally overlaps protein kinase A to block apoptosis. Proc. Natl. Acad. Sci. U. S. A. 101:9411–9416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berra E, Diaz-Meco MT, Moscat J. 1998. The activation of p38 and apoptosis by the inhibition of ERK is antagonized by the phosphoinositide 3-kinase/Akt pathway. J. Biol. Chem. 273:10792–10797 [DOI] [PubMed] [Google Scholar]
- 4. Carracedo A, et al. 2008. Inhibition of mTORC1 leads to MAPK path-way activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Invest. 118:3065–3074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carracedo A, Pandolfi PP. 2008. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene 27:5527–5541 [DOI] [PubMed] [Google Scholar]
- 6. Chuluunbaatar U, Mohr I. 2011. A herpesvirus kinase that masquerades as Akt: you don't have to look like Akt to act like it. Cell Cycle 10:2064–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chuluunbaatar U, et al. 2010. Constitutive mTORC1 activation by a herpesvirus Akt surrogate stimulates mRNA translation and viral replication. Genes Dev. 24:2627–2639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DeLuca N, McCarthy AM, Schaffer P. 1985. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J. Virol. 56:558–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DeLuca N, Schaffer P. 1988. Physical and functional domains of the herpes simplex virus. J. Virol. 62:732–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Erazo A, Yee MB, Banfield BW, Kinchington PR. 2011. The alpha-herpesvirus US3/ORF66 protein kinases direct phosphorylation of the nuclear matrix protein matrin 3. J. Virol. 85:568–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Feldman ME, et al. 2009. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 7:e38 doi:10.1371/journal.pbio.1000038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gillis PA, Okagaki LH, Rice SA. 2009. Herpes simplex virus 1 ICP27 induces p38 mitogen-activated protein kinase signaling and apoptosis in HeLa cells. J. Virol. 83:1767–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goldstein DJ, Weller SK. 1988. Herpes simplex virus type-1 ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: isolation and characterization of an ICP6 lacZ insertion mutant. J. Virol. 62:196–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hargett D, McLean T, Bachenheimer SL. 2005. Herpes simplex virus ICP27 activation of stress kinases JNK and p38. J. Virol. 79:8348–8360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Howe AK, Juliano RL. 2000. Regulation of anchorage-dependent signal transduction by protein kinase A and p21-activated kinase. Nat. Cell Biol. 2:593–600 [DOI] [PubMed] [Google Scholar]
- 16. Hu J, et al. 2001. ERK1 and ERK2 activate CCAAAT/enhancer-binding protein-beta-dependent gene transcription in response to interferon-gamma. J. Biol. Chem. 276:287–297 [DOI] [PubMed] [Google Scholar]
- 17. Izumi KM. 2004. Epstein-Barr virus signal transduction and B-lymphocyte growth transformation. Prog. Mol. Subcell. Biol. 36:269–288 [DOI] [PubMed] [Google Scholar]
- 18. Kang MH, Banfield BW. 2010. Pseudorabies virus tegument protein Us2 recruits the mitogen-activated protein kinase extracellular-regulated kinase (ERK) to membranes through interaction with the ERK common docking domain. J. Virol. 84:8398–8408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kato A, et al. 2005. Identification of proteins phosphorylated directly by the Us3 protein kinase encoded by herpes simplex virus 1. J. Virol. 79:9325–9331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kolch W. 2005. Coordinating ERK/MAPK signaling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 6:827–837 [DOI] [PubMed] [Google Scholar]
- 21. Lassus P, et al. 2000. Extinction of Rac1 and Cdc42Hs signaling defines a novel p53 dependent apoptotic pathway. Oncogene 19:2377–2385 [DOI] [PubMed] [Google Scholar]
- 22. Leach N, et al. 2007. Emerin is hyperphosphorylated and redistributed in herpes simplex virus type 1-infected cells in a manner dependent on both UL34 and Us3. J. Virol. 81:10792–10803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leader DP, Deana AD, Marchiori F, Purves FC, Pinna LA. 1991. Further definition of the substrate specificity of the α-herpesvirus protein kinase and comparison with protein kinases A and C. Biochim. Biophys. Acta 1091:426–431 [DOI] [PubMed] [Google Scholar]
- 24. Liang L, Roizman B. 2008. Expression of gamma interferon-dependent genes is blocked independently by virion host shutoff RNase and by the Us3 protein kinase. J. Virol. 82:4688–4696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu X, Li Q, Dowdell K, Fischer ER, Cohen JL. 2012. Varicella-zoster virus ORF12 protein triggers phosphorylation of ERK1/2 and inhibits apoptosis. J. Virol. 86:3143–3151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Long MC, Leong V, Schaffer PA, Spencer CA, Rice SA. 1999. ICP22 and the UL13 protein kinase are both required for herpes simplex virus-induced modification of the large subunit of RNA polymerase II. J. Virol. 73:5593–5604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McKay MM, Morrison DK. 2007. Integrating signals from RTKs to ERK/MAPK. Oncogene 26:3113–3121 [DOI] [PubMed] [Google Scholar]
- 28. McLean TI, Bachenheimer SL. 1999. Activation of cJun N-terminal kinase by herpes simplex virus type 1 enhances viral replication. J. Virol. 73:8415–8426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meindl A, Osterrieder N. 1999. The equine herpesvirus 1 Us2 homolog encodes a non-essential membrane associated virion component. J. Virol. 73:3430–3437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mendoza MC, Emrah Er E, Blenis J. 2011. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem. Sci. 36:320–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moria I, et al. 2003. The US3 protein kinase of herpes simplex virus attenuates the activation of the c-Jun N-terminal protein kinase signal transduction pathway in infected piriform cortex neurons of C57BL/6 mice. Neurosci. Lett. 351:201–205 [DOI] [PubMed] [Google Scholar]
- 32. Moria I, et al. 2006. Herpes simplex virus Us3 protein kinase regulates virus-induced apoptosis in olfactory and vomeronasal chemo-sensory neurons in vivo. Microbes Infect. 8:1806–1812 [DOI] [PubMed] [Google Scholar]
- 33. Mou F, Forest T, Baines JD. 2007. Us3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J. Virol. 81:6459–6470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Munger J, Roizman B. 2001. The US3 protein kinase of herpes simplex virus 1 mediates the posttranslational modification of BAD and prevents BAD-induced programmed cell death in the absence of other viral proteins. Proc. Natl. Acad. Sci. U. S. A. 98:10410–10415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Murphy LO, Blenis J. 2006. MAPK signal specificity: the right place at the right time. Trends Biochem. Sci. 31:268–275 [DOI] [PubMed] [Google Scholar]
- 36. Ogg PD, McDonell PJ, Ryckman BJ, Knudson CM, Roller RJ. 2004. The HSV-1 Us3 protein kinase is sufficient to block apoptosis induced by over-expression of a variety of Bcl-2 family members. Virology 319:212–224 [DOI] [PubMed] [Google Scholar]
- 37. Overton H, McMillan D, Hope L, Wong-Kai-In P. 1994. Production of host shutoff-defective mutants of herpes simplex virus type 1 by inactivation of the UL13 gene. Virology 202:97–106 [DOI] [PubMed] [Google Scholar]
- 38. Perkins D, Pereira EF, Aurelian L. 2003. The herpes simplex virus type 2 R1 protein kinase (ICP10 PK) functions as a dominant regulator of apoptosis in hippocampal neurons involving activation of the ERK survival pathway and upregulation of the antiapoptotic protein Bag-1. J. Virol. 77:1292–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pleschka S. 2008. RNA viruses and the mitogenic Raf/MEK/ERK signal transduction cascade. Biol. Chem. 389:1273–1282 [DOI] [PubMed] [Google Scholar]
- 40. Preston VG, Palfreyman JW, Dutia BM. 1984. Identification of a herpes simplex virus type 1 polypeptide which is a component of the virus-induced ribonucleotide reductase. J. Gen. Virol. 65:1457–1466 [DOI] [PubMed] [Google Scholar]
- 41. Rajaiya J, Xiao J, Rajala RV, Chodosh J. 2008. Human adenovirus type 19 infection of corneal cells induces p38 MAPK-dependent interleukin-8 expression. Virol. J. 5:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rommel C, et al. 1999. Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science 286:1738–1741 [DOI] [PubMed] [Google Scholar]
- 43. Roy SK, Wachira SJ, Weihua X, Hu J, Kalvakolanu DV. 2000. CCAAT/enhancer-binding protein-beta regulates interferon-induced transcription through a novel element. J. Biol. Chem. 275:12626–12632 [DOI] [PubMed] [Google Scholar]
- 44. Rozengurt E. 2007. Mitogenic signaling pathways induced by G protein-coupled receptors. J. Cell. Physiol. 213:589–602 [DOI] [PubMed] [Google Scholar]
- 45. Ryckman BJ, Roller RJ. 2004. Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J. Virol. 78:399–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sacks WR, Schaffer PA. 1987. Deletion mutants in the gene encoding the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in cell culture. J. Virol. 61:829–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Smith KD, et al. 2006. Activated MEK suppresses activation of PKR and enables efficient replication and in vivo oncolysis by γ134.5 mutants of herpes simplex virus 1. J. Virol. 80:1110–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Van den Broekea C, et al. 2009. Alphaherpesvirus Us3-mediated reorganization of the actin cytoskeleton is mediated by group A p21-activated kinases. Proc. Natl. Acad. Sci. U. S. A. 106:8707–8712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wagner EK, Nebreda AR. 2009. Signal integration by JNK and p38 MAPK pathways in cancer and development. Nat. Rev. Cancer 9:537–540 [DOI] [PubMed] [Google Scholar]
- 50. Walsh D, et al. 2008. Eukaryotic translation initiation factor 4F architectural alterations accompany translation initiation factor redistribution in poxvirus-infected cells. Mol. Cell. Biol. 28:2648–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Walsh D, Mohr I. 2004. Phosphorylation of eIF4E by Mnk1 enhances HSV-1 translation and replication in quiescent cells. Genes Dev. 18:660–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Walsh D, Perez C, Notary J, Mohr I. 2005. Regulation of the translation initiation factor eIF4F by multiple mechanisms in human cytomegalovirus-infected cells. J. Virol. 79:8057–8064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Walters MS, Kinchington PR, Banfield BW, Silverstein SJ. 2010. Hyperphosphorylation of histone deacetylase 2 by alphaherpesvirus US3 kinases. J. Virol. 84:9666–9676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. 1995. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270:1326–1331 [DOI] [PubMed] [Google Scholar]
- 55. Yu F, et al. 2007. Systematic identification of cellular signals reactivating Kaposi sarcoma-associated herpesvirus. PLoS Pathog. 3(3):e44 doi:10.1371/journal.ppat.0030044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yu JH, Lin BY, Deng W, Broker TR, Chow LT. 2007. Mitogen-activated protein kinases activate the nuclear localization sequence of human papillomavirus type 11 E1 DNA helicase to promote efficient nuclear import. J. Virol. 81:5066–5078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zachos G, Clements B, Conner J. 1999. Herpes simplex virus type 1 infection stimulates p38/c-Jun N-terminal mitogen-activated protein kinase pathways and activates transcription factor AP-1. J. Biol. Chem. 274:5097–5103 [DOI] [PubMed] [Google Scholar]
- 58. Zugasti O, et al. 2001. Raf-MEK-Erk cascade in Anoikis is controlled by Rac1 and Cdc42 via Akt. Mol. Cell. Biol. 21:6706–6717 [DOI] [PMC free article] [PubMed] [Google Scholar]