

Abstract
The gp120 subunit of the HIV Env glycoprotein is responsible for receptor interactions leading to viral entry and is a primary target for neutralizing antibodies. Most structural studies have focused on the heavily truncated, deglycosylated gp120 core, leaving fundamental aspects of the glycoprotein that are responsible for immune evasion and receptor-induced activation unresolved. Here we investigate full-length, glycosylated HIV gp120 in unliganded and CD4-bound forms by using small-angle X-ray scattering to visualize global structural reorganization and hydrogen/deuterium exchange to track changes in local conformational dynamics. The studies revealed unliganded full-length gp120 to be considerably more dynamic, particularly at the CD4 binding site, than suggested by previous studies of the subunit core alone. The large V1/V2 loops, previously unmapped, are positioned to mask the coreceptor binding site in an orientation that recapitulates that observed in the Env trimer. CD4 binding shifts V1/V2 to unmask the coreceptor binding site and triggers profound dynamic changes in gp120 spanning from the binding site to the gp41-interactive face of gp120. These findings provide further insights on the structural basis of Env antigenicity and immunogenicity and of allosteric effects upon receptor binding.
INTRODUCTION
The Env glycoprotein complex is the sole viral antigen on the surface of HIV. This trimeric complex of gp120/gp41 heterodimers mediates delivery of the viral nucleocapsid to host cells through a series of receptor-induced conformational changes that drive fusion of the viral and host membranes. The gp120 subunit is primarily responsible for interactions with the CD4 receptor and chemokine coreceptors (CCR5 or CXCR4) and bears the majority of epitopes that are targeted by the humoral immune response. Early vaccination studies with monomeric or trimeric Env constructs elicited responses that were largely isolate specific and minimally effective against most primary HIV-1 isolates (3, 4, 20, 21, 38). However, the modest efficacy observed in the RV144 trial (52) has renewed interest in soluble monomeric gp120 as a vaccine immunogen.
Env, and in particular the gp120 subunit, evades antibody recognition of conserved epitopes by several means, including decoration with a dense glycan shield, hypervariable loops that mask conserved features, and high intrinsic conformational dynamics that render it a poorly defined antigen (45). These characteristics have also hindered structure determination, and significant gaps remain in our understanding of the overall topography and dynamics of the complete glycoprotein. Cryo-electron microscopy has been used to image the general architecture of the Env spike on HIV and simian immunodeficiency virus (SIV) virions (35, 73, 78, 82) and in solubilized trimeric forms (19, 75). Crystal structures are available for the gp120 core (6, 10, 22, 28, 30, 44), with most variable loops, flexible terminal extensions, and glycans truncated. These studies have revealed the architecture of the gp120 core in its CD4-bound conformation to be composed of inner and outer domains along with a 4-stranded β-sheet called the bridging sheet (Fig. 1).
Fig 1.
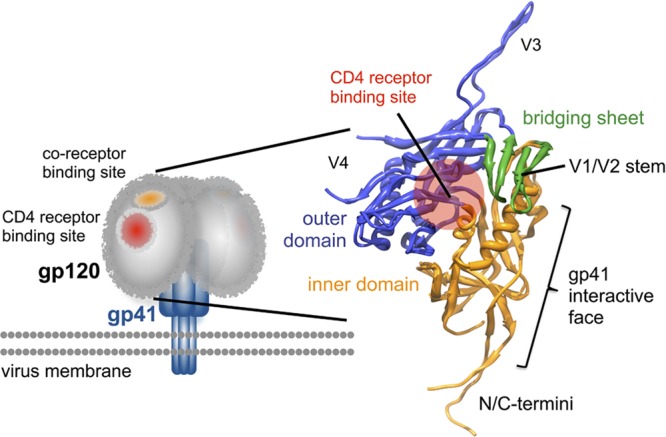
gp120 core structure. Crystal structure of gp120 from the gp120-sCD4-48D Fab complex (PDB 3JWD) superimposed with the gp120 core containing an intact V3 loop (gp120-sCD4-X5; PDB 2B4C) reveals the organization of gp120 in terms of inner (orange) and outer (blue) domains. The 4-stranded β-sheet subdomain termed the bridging sheet (green) is composed of two strands from the outer domain and the V1/V2 stem from the inner domain. The position of the CD4 binding site on gp120 is indicated by the red circle. gp120 is believed to interact with gp41 primarily through interactions involving the inner domain and N-/C-terminal extensions.
The variable loops, V1/V2 and V3, have been inferred to mask conserved epitopes from antibody recognition (77); however, the structural basis for this has not yet been determined. A recent study implicated the variable loops in maintaining the gp120 subunit in a pre-CD4-bound conformation, because the unliganded core, with V1/V2 and V3 truncated, adopts essentially the same structure as the CD4-bound state (28). The structure of the full-length gp120 subunit in its unliganded state remains undetermined. Such data would elucidate the disposition of the V1/V2 and V3 subdomains relative to the core and key epitopes, such as the CD4 and coreceptor binding sites.
CD4 binds at one of the few highly conserved sites on gp120, a complex, discontinuous epitope composed of facets of the inner and outer domains. Thermodynamic studies have shown CD4 binding to have a large stabilizing effect on gp120 (29, 43). The CD4 interaction induces changes in gp120 that are believed to generate and unmask the coreceptor binding surface, including the formation of the bridging sheet (30, 53, 63, 77). Hydrogen/deuterium exchange coupled with mass spectrometry (HDX-MS) of deglycosylated gp120 core has probed some of these CD4-induced effects (26). However, in light of the observation that truncations modulate the conformational equilibrium of the subunit as a whole, a study of full-length, glycosylated gp120 is expected to reveal important differences (28).
Here, we combined two solution-phase methods, small-angle X-ray scattering (SAXS) and HDX-MS, to examine full-length glycosylated gp120. SAXS provides a moderate-resolution (∼20 Å) 3-dimensional (3-D) shape reconstruction for the complete gp120 glycoprotein (50). HDX-MS provides a sensitive and detailed measure of conformational dynamics by monitoring the kinetics of backbone amide solvent exchange, revealing regions of order and disorder throughout the protein (41). Together, the combined approach reveals the morphology of the complete gp120 subunit structure in solution and directly identifies the local structural ordering and global changes induced by CD4 binding.
MATERIALS AND METHODS
Protein and complex production and purification for small-angle X-ray scattering.
HIV Env gp120 (isolate SF162) was expressed in Chinese hamster ovary (CHO) cells or human embryonic kidney (HEK) 293E cells and purified as described previously (55, 56). Monomeric and dimeric gp120s were resolved by size exclusion chromatography (HiLoad Superdex 200; GE Healthsciences, Piscataway, NJ) equilibrated with phosphate-buffered saline (PBS; 10 mM NaPO4, 150 mM NaCl, 0.02% NaN3; pH 7.4), concentrated by centrifugal filtration (Amicon Ultra 10-kDa cutoff; Millipore, Billerica, MA) to ∼2 mg/ml, and analyzed by blue native (BN)-PAGE (see Fig. S1 in the supplemental material).
Soluble two-domain (D1D2) CD4 (sCD4; Progenics) was purified by gel filtration chromatography (in PBS) to remove higher-molecular-weight (MW) contaminants. To form the gp120-sCD4 complex, purified gp120 monomer from HEK 293E cells was incubated with a 4-fold excess of sCD4 overnight at 4°C, and the complex was isolated by size exclusion chromatography using a Superdex 200 16/60 column, followed by concentration to 2 mg/ml using an Amicon Ultra centrifugal filter with 10-kDa cutoff (see Fig. S1C). An additional sCD4 complex was prepared with gp120 expressed in CHO cells. Due to limitations in sample quantities, a 3:1 molar ratio of sCD4:gp120 was incubated overnight at room temperature and concentrated using an Amicon Ultra centrifugal filter (30-kDa cutoff; Millipore), with 4 wash steps to remove excess sCD4. Gel densitometry and dynamic light scattering were used to verify that excess sCD4 was removed and only the stable complex was isolated.
17b Fab was isolated from IgG by using the Pierce Fab preparation kit (Thermo Fisher Scientific, Rockford, IL). To generate the gp120-sCD4-17b Fab complex, gp120 monomer (HEK 293E expressed) was incubated with a 4-fold excess of sCD4 and 2-fold excess of 17b Fab overnight at 4°C. The ternary complex was isolated by size exclusion chromatography (see Fig. S1C in the supplemental material) and concentrated with an Amicon Ultra spin filter with 10-kDa cutoff.
Static and dynamic light scattering were used to verify sample molecular weights and monodispersity. Ten acquisitions of 5 s at 20°C on a Dynapro Nanostar (Wyatt Technology, Santa Barbara, CA) were carried out for each sample. All data were fit using the Wyatt Dynamics analysis software, assuming a spherical model, with errors reported as standard deviations.
Small-angle X-ray scattering.
SAXS measurements were conducted on beam line 4-2 at the Stanford Synchrotron Radiation Laboratory (62). The focused 11-keV X-ray beam irradiated a thin-wall quartz capillary cell, which contained a sample aliquot, placed at 1.7 m upstream of the Rayonix MX 225HE detector (Evanston, IL). The detector pixel numbers were converted to the momentum transfer (Q) based on the following equation: Q = 4π × sinθ/λ, where 2 × θ is the scattering angle and λ the X-ray wavelength of 1.127 Å with a silver behenate powder standard placed at the capillary position. A set of 16 consecutive 1-s X-ray exposures were made on each sample aliquot (30 μl injected) with 15-μl oscillations between exposures to minimize radiation damage to the specimen. Protein scattering data were processed by using MarParse, scaled for the transmitted beam intensity integrated for each exposure, and azimuthally averaged (62). Exposures were averaged with rejection criteria of 1.3 standard deviations relative to the initial exposure to omit frames exhibiting radiation damage or other artifacts. Buffer scattering data were processed in the same way and subtracted from corresponding protein scattering data.
For each sample, concentration series ranging from 0.4 to 2.0 mg/ml were gathered. Molecular weights were calculated using both scattering intensity at the zero angle relative to water (42) and the program SAXS MoW (15). The partial specific volume of gp120 used for MW determination was calculated to be 0.694 cm3/g by using a weighted average of carbohydrate and protein components as described previously (32).
Ab initio shape reconstruction.
The 1-D SAXS curves were processed first using autoGNOM, which excludes higher-angle data, to determine approximate radius of gyration (Rg) and Dmax values for the P(r) plots, the pairwise distance distribution functions (48). A final P(r) plot was generated with GNOM v4.5, including Q values from 0.0105 to 0.472 (65, 66). The output was the basis for ab initio shape reconstruction using DAMMIN v5.3 running in jagged or slow mode, to generate a set of 16 to 24 models (67). Individual bead models are shown in Fig. S2, S3, and S10 in the supplemental material. The bead models were aligned using SUPCOMB13 with enantiomers considered (27) and then averaged using DAMAVER (69). Positions of low bead occupancy in the averaged model were filtered out using DAMFILT with an excluded volume set to the Porod volume, determined using Autoporod (47). The resulting model was then converted to a volume envelope by using the program pdb2vol within the SITUS2.2 package (74).
Atomic coordinates for the gp120 core, including the gp41-interactive region (PDB 3JWD) (44) and a complete V3 loop (PDB 2B4C) (22), were first superimposed in Chimera (49), and this composite PDB model was docked into the SAXS envelope by using the automated fit-in-map algorithm in Chimera (Fig. 2C) with repeated trials, starting with several distinct initial orientations. Because enantiomeric structures give rise to identical SAXS patterns, we examined both enantiomeric variants of the averaged SAXS models, with one form clearly showing a better agreement with the shape of the composite gp120 core PDB model. In the case of the CD4-bound state, the CD4 coordinates from the PDB 3JWD coordinate set were kept in the identical position relative to the gp120 core as found in the crystal structure, and the pair was docked into the SAXS envelope. The models shown correspond to the orientations of the PDB models that produced the optimal fits to the SAXS envelope when the envelope was contoured to enclose the Porod excluded volume, obtained from Autoporod (47).
Fig 2.

Solution small-angle X-ray scattering structures for full-length gp120 monomer. (A) SAXS pattern for glycosylated SF162 clade B gp120 monomer. Solid black line represents the calculated SAXS pattern for a typical DAMMIN reconstruction. Inset shows the Guinier fit. (B) Corresponding P(r) pairwise distance distribution plot. Black lines represent the calculated SAXS patterns for the modeled structures. (C) “Averaged” SAXS structures for full-length, glycosylated gp120 monomer, shown as an envelope drawn over the averaged, filtered DAMMIN structure (see Fig. S2 in the supplemental material). Structures for gp120 core with N-/C-terminal extensions and intact V3 loop (PDB 3JWD and 2B4C; blue) are fitted into the SAXS density. The CD4 binding site contact residues are shown in orange. The position of the V1/V2 stem (cyan) in the CD4-bound conformation reflected in the CD4-bound gp120 core crystal structures does not fit within the SAXS envelope for unliganded full-length gp120. (D) The V1/V2 loop structure from the crystallographically determined complex with bNAb PG9 (PDB 3U4E; cyan spheres) occupies approximately the same volume of the crest as suggested by the SAXS shape reconstructions. See also Movie S1 in the supplemental material.
We noted that the resulting averaged SAXS model did not vary significantly, whether only the 10 models in best agreement (as judged by their computed normalized spatial discrepancy [NSD] values) or the full set was used. This suggests that the DAMMIN models are reasonably convergent, and the averaged model represents a conservative envelope of spatial occupancy for the macromolecular density.
All-atom modeling.
The sequence of gp120 (SF162) was threaded onto available crystal structures of b12-bound and sCD4/48D-bound gp120 (PDB 2NY7 and 3JWD), using MODELLER (11). As there are currently no tools for generating large ensembles of homology models with glycans, a new methodology was developed within MODELLER to include attached glycans as flexible residues (unpublished data). The simulated annealing step was modified to include carbohydrate parameters from the CHARMM36 force field (16, 17). Monosialylated biantennary complex glycans with core fucosylation (positions 87, 135, 154, 186, 195, 274, 299, 352, 392, 398, 401, and 454) and Man8 high-mannose glycans (positions 232, 229, 260, 293, 329, 336, 382, 388, and 438) were based on the predominant glycoform observed in the MS data (see Fig. S11 in the supplemental material). Glycans in regions outside peptic coverage were inferred from glycosylation at similar positions reported in previous studies (31). Additional restraints in the calculations were included to introduce disulfide bonds absent from the reference structures. Models threaded to 2NY7 (V1/V2 “up”) lacked coordinates for the N-terminal extension/layer 1 (up to residue 81); therefore, initial coordinates were obtained from the same region in 3JWD and held in place with generous (16 Å, upper distance) restraints to the inner domain. The ensembles of 100 glycosylated models from MODELLER were fit to the raw SAXS patterns by using FoXS (59). Minimal ensemble searches with up to 3 conformations were attempted to minimize χ2 values (46). Models of gp120-sCD4 and gp120-sCD4-17bFab were calculated similarly (PDB 3JWD and 1GC1). In the latter case, the N-/C-terminal residues of 17b Fab, absent in the crystal structures, were included in the models. Models of free 17b Fab were generated with a template (PDB 1RZ8) (23), excluding restraints for the linker between the V1 and C1 domains. Figures were created using PyMOL (7) and Chimera (49).
Hydrogen/deuterium exchange.
Deuterium exchange was initiated by dilution of stock gp120 (1 to 2 mg/ml) into PBS to a final deuterium level of 85% and incubation at 22°C. Reactions were quenched at 15 s, 1 min, 5 min, 30 min, and 4 h with a 1:1 dilution into ice-cold 0.1% formic acid (FA), 1 M guanidine-HCl (GndHCl), 500 mM Tris-carboxy-ethane-phosphate (TCEP) for a final pH of 2.5. Pepsin (Worthington Labs, Lakewood, NJ) was immediately added to a final ratio of 1:1 (by mass), and the mixture was incubated on ice for 5 min. Samples were frozen in liquid nitrogen and stored at −80°C for up to several weeks. A zero time point to correct for deuterium uptake during digestion was obtained by dilution of the stock protein into prequenched deuterated PBS, followed by identical pepsin digestion. A fully deuterated sample was prepared by incubating denatured gp120 (50 mM Tris [pH 8.0], 4 M GndHCl, 50 mM dithiothreitol treated at 60°C for 30 min) in deuterium (85%) overnight at 40°C. The CD4 complex was prepared by incubating a 3:1 molar ratio of sCD4-gp120 (3 mg/ml total protein) overnight at 4°C prior to deuterium exchange. Deuterium oxide (D2O; 99.96%) was purchased from Cambridge Isotope Labs (Andover, MA).
Mass shifts were determined using HX-Express (71). The percent deuteration at each time point was calculated using the following equation: percent D = 100 × [(m – m0%)/(m100% − m0%)], where m is the mass centroid of the fragment at a given time point, m0% is the zero time point centroid, and m100% is the fully deuterated centroid (34). Centroids for the most abundant glycoforms of the same peptide were averaged, because no significant differences were observed within these populations. Several mass envelopes displayed heavy broadening due to CD4 binding, and bimodal fitting was applied for deconvolution of these data (60, 72) (see Fig. S14 and the methods section in the supplemental material). Protection factors and conformational stabilities were determined by fitting exchange rates of peptic fragments and comparing them to intrinsic rates, as described previously (2, 61, 79).
Mass spectrometry.
A combination of tandem mass spectrometry (MS/MS) and exact mass data were used to manually identify peptic fragments of gp120. Fragmentation data of glycopeptides were insufficient for assignment, but glycan mass and type could be obtained (40). Peptic digests of gp120 (10 μg) were neutralized (100 mM Tris [pH 8.0]) and treated with 2 mU PNGase F (Prozyme, Hayworth, CA) overnight to remove all N-linked glycans. MS/MS analysis of these deglycosylated fragments allowed for unambiguous assignments of glycopeptides.
Deuterium uptake was analyzed by liquid chromatography-MS (LC-MS) using a Waters Acquity UPLC integrated with a Synapt HDMS instrument (Waters, Milford, MA). Peptides were resolved on a Zorbax 300SB C18 column (1 by 50 mm, 3 μm; Agilent Technologies, Santa Clara, CA) with a gradient of 5 to 50% acetonitrile over 12 min at 50 μl/min with 0.05% trifluoroacetic acid in both mobile phases. The column injection loop and lines were kept at 0°C on melting ice to minimize back-exchange. The loop and syringe were washed repeatedly with 0.1% formic acid and methanol between injections to minimize sample carryover. Source and desolvation temperatures of 85°C and 175°C were used to minimize back-exchange (41).
RESULTS
Small-angle X-ray scattering characterization of gp120 in solution.
Small-angle X-ray scattering was used to obtain shape information for the full-length, glycosylated monomeric gp120 in its unliganded state, bound to sCD4, and in complex with both sCD4 and 17b Fab. No sign of significant aggregation, interparticle interactions, or other concentration-dependent effects were observed within the SAXS data over a range of concentrations from 0.4 to 2.0 mg/ml at 15°C, as reflected by linearity of the Guinier regions (QRg, <1.3). Furthermore, the Rg from the Guinier analysis was in excellent agreement with that derived from the P(r) pairwise distance distribution plots (Table 1) (65). Molecular mass results obtained from SAXS and static light scattering (SLS) showed the expected mass for monomeric gp120 along with the expected increase for each complex (Table 1; see also Table S1 in the supplemental material). Dynamic light scattering (DLS) verified the monodispersity of samples at concentrations used for SAXS measurements. A comparison of SAXS patterns at 37°C and 15°C did not reveal significant differences in scattering.
Table 1.
Macromolecular properties derived from SAXS and light-scattering measurements
| Sample |
Rg (Å) |
Dmaxa (Å) | Porod vol (Å3)b | Rh (DLS)c (Å) | Mass (kDa) |
||
|---|---|---|---|---|---|---|---|
| Guinier | GNOMa | Measuredd | Expectede | ||||
| gp120 | 37.1 ± 0.03 | 37.6 ± 0.1 | 130 | 230,374 | 46.5 ± 0.4 | 98.1 ± 13.0 | 93 |
| gp120−sCD4 | 37.0 ± 0.05 | 38.0 ± 0.1 | 135 | 246,073 | 48.3 ± 0.4 | 106.8 ± 6.0 | 113.4 |
| gp120−sCD4-17b | 42.8 ± 0.06 | 43.5 ± 0.1 | 150 | 299,816 | 54.2 ± 0.8 | 149.7 ± 16.5 | 162.1 |
| gp120 dimer | 47.0 ± 0.05 | 47.9 ± 0.1 | 166 | 449,665 | 52.7 ± 0.9 | 168.1 ± 33.4 | 186 |
| sCD4 (D1D2) | 20.9 ± 0.05 | 21.7 ± 0.2 | 73 | 25,216 | 23.8 ± 0.2 | 18.9 ± 5.8 | 20.3 |
| 17b Fab | 26.7 ± 0.03 | 26.5 ± 0.2 | 90 | 63,701 | 31.5 ± 0.2 | 49.1 ± 6.2 | 48.7 |
Real space Rg and Dmax values from GNOM (65).
Porod were volumes determined using Autoporod (47).
Hydrodynamic radii (Rh) were obtained from DLS measurements.
Value is the mean molecular mass determined from SLS and by two SAXS methods; individual values are presented in Table S1 of the supplemental material.
The expected MW was calculated based on the most predominant glycoforms observed during HDX-MS analysis.
We and others have observed that gp120 “monomer” preparations from a variety of sources often contain nearly equal proportions of monomeric and disulfide-bonded dimeric gp120 (13), which can be resolved by gel filtration (see Fig. S1A in the supplemental material). As expected, dimeric gp120 exhibited a much larger Rg, 47.7 versus 37.6 Å. The dimensions we observed for gp120 monomer were markedly smaller than those reported previously for gp120 (also isolate SF162; Rg of 42.4 Å [1]). This difference was likely due to the presence of dimeric material in the previously studied samples, as an equal mixture of monomer and dimer would yield an intermediate Rg value in this range. We note that our measurements for isolated sCD4 were in excellent agreement with values reported by Ashish et al. (21.5 versus 22.3 Å) (1); thus, methodological differences are not likely to be a major factor in the discrepancies.
Ab initio shape reconstruction of unliganded gp120 subunit generated structures with three main features: a ventral stalk, a central bulky core region, and a crest situated on the dorsal surface of the core (Fig. 2C). The reconstruction runs resulted in models whose SAXS patterns were in good agreement with the experimentally measured patterns, as reflected by the goodness of fit between model and experimental SAXS patterns (χ2, 1.319 ± 0.004 [mean ± standard deviation]) among 16 DAMMIN models (see Fig. S2 in the supplemental material), where χ2 is the discrepancy between DAMMIN model and experimentally measured SAXS patterns (67); perfect agreement would yield a value of 0, while values around 1 indicate good agreement. The models exhibited good structural convergence, as reflected by the low normalized spatial discrepancy (NSD) values determined following alignment (0.708 ± 0.014) (27); perfect superposition would yield an NSD of 0, and values less than 1 are generally in good agreement.
A composite PDB structure for the SF162 gp120 core was composed using crystal structures of the gp120 core, including N-/C-terminal extensions and an intact V3 (PDB 3JWD and 2B4C, respectively [22, 44]). The core regions of these gp120 structures could be aligned (root mean square deviation [RMSD], 0.924Å), providing the relative positions of the N-/C-terminal extensions and the V3 loop. This structure was docked into the SAXS envelope by using the automated fit-in-map procedure in Chimera (49). The N-/C-terminal extensions dock into the ventral stalk, and the core region fits neatly into the central bulk of density (Fig. 2C; see also Movie S1 in the supplemental material). Based on the crystal structure with an intact V3 loop (PDB 2B4C; albeit with sCD4 and the X5 antibody bound [22]), V3 would sit within the crest of density in the unliganded gp120 SAXS envelope. In contrast, the hairpin stem for truncated V1/V2 loops in the CD4-bound crystal structures, with a bridging sheet formed, unambiguously fall outside the SAXS-determined envelope for unliganded gp120. As the bridging sheet is expected to be absent in unliganded gp120, the b12-complexed core crystal structure (PDB 2NY7 [81]), lacking this structural motif, was similarly modeled into the SAXS density. The orientation of the V1/V2 stem in this structure was more consistent with that in the unliganded SAXS model, projecting into the crest region (see Fig. 5A).
Fig 5.

Comparison of SAXS models of free and CD4-bound gp120. (A) Structures of sCD4 (red) bound to gp120 core (blue; 3JWD and 2B4C) are shown fit into the SAXS envelopes for unbound (gray) and bound with sCD4 (tan), showing the shift of the density corresponding to V1/V2 upon CD4 binding. Positions of the V1/V2 stem are shown in white for the unbound form (based on the b12-liganded gp120 core structure [PDB 2NY7]), and cyan shows the stem in the sCD4 bound form (PDB 3JWD; after bridging sheet formation). (B) SAXS model of unliganded gp120, showing the crest of density atop the core compared with the expected position of 17b (ribbons, dark purple heavy chain, and light purple light chain), based upon superposition of the gp120 core structures in 1GC1 (gp120 core-sCD4-17b Fab) and 3JWD structures. The same 17b/coreceptor binding surface on gp120 is exposed in the CD4-bound gp120 complex as determined by SAXS. The ternary gp120-sCD4-17b Fab complex determined by SAXS showed good agreement with the available crystal structures (1GC1 superimposed with 3JWD).
Based on the proximity of the V1/V2 stem, we attribute the majority of the crest density to the V1/V2 and V3 loops. This region accounts for 20 to 25% of the volume (and number of assigned DAMMIN beads) in the unliganded gp120 SAXS models, consistent with glycosylated V1/V2 and V3 loops, comprising ∼20% of gp120. The crest also accommodates the approximate volume expected for V1/V2, based on the recently reported structure for V1/V2 in complex with PG9 antibody (39) (Fig. 2D). Overall, the unliganded gp120 monomer in solution closely resembled the primary lobes of density observed by cryo-electron tomography of Env on the surface of virus as well as in soluble trimeric form (Fig. 3) (19, 35, 73).
Fig 3.
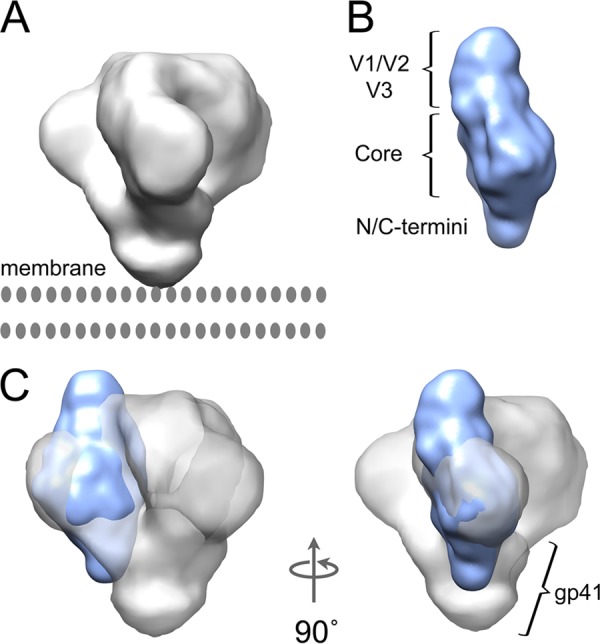
Agreement of unliganded gp120 monomer with cryo-electron tomography (cryo-ET) reconstructions for Env trimers on the surface of virions. The gp120 monomer structure fits into the density determined by electron cryo-tomography for the ectodomain portion of trimeric Env spikes (electron microscopy density map EMD-5019). (A) Cryo-ET map for unliganded HIV-1 Env. (B) SAXS envelope for unliganded full-length HIV-1 gp120; in this view, the CD4 binding site would be on the face on the right-hand side. (C) Docking of the unliganded gp120 SAXS model (blue) into the cryo-ET density map (gray) shows good overall agreement of core, variable loop crest, and termini placement. In the context of the trimer, the termini and variable loops are expected to adjust to some extent, as they are believed to participate in intersubunit contacts.
To test the dependence on the algorithm used to obtain the shape reconstruction, a different shape reconstruction program, GASBOR (68), was also used. Unlike DAMMIN, which restricts model beads to a regular lattice, GASBOR attempts to produce a 3-dimensional model composed of a chain-like ensemble of dummy residues. GASBOR resulted in reconstructions with similar overall shapes for unliganded gp120 monomer (see Fig. S4 and Movie S2 in the supplemental material). The χ2 value of 1.366 ± 0.032 and NSD value of 1.518 ± 0.031 showed slightly more model-to-model variation than among DAMMIN models; this is generally observed with GASBOR, in large part due to the greater spatial degrees of freedom afforded the dummy residues in GASBOR. Nevertheless, the average GASBOR model in all essential features recapitulated the findings with the DAMMIN model, namely, the V1/V2 stem in the CD4-bound conformation falls outside the density, and a prominent lobe is positioned atop the core.
While glycans contribute to X-ray scattering and also to the assigned bead positions in the ab initio structures, they were not resolved as distinct recognizable features in the low-resolution models in the present study. They likely contribute additional bulk in the individual bead models, and what discrete knobs of density are present in those models are largely averaged out in the process of aligning, averaging, and filtering to produce the final conservative SAXS envelopes.
Structure of the gp120 monomer-sCD4 complex.
A similar modeling process was applied to SAXS measurements for the gp120-sCD4 complex. Again, the DAMMIN reconstruction method produced a set of models that agreed well with experimentally measured data (χ2, 1.362 ± 0.003), and the models exhibited reasonable convergence of structures, with an average NSD of 0.739 ± 0.051 (Fig. 4; see also Fig. S3 in the supplemental material). When the composite PDB model (3JWD + 2B4C), including CD4 coordinates from 3JWD, was docked into the SAXS envelope for full-length gp120-sCD4, additional density for sCD4 was observed in a position consistent with the crystal structure (Fig. 4C; see also Movie S3 in the supplemental material). In addition, comparisons of SAXS structures for gp120 with and without sCD4 indicated that major shifts in mass beyond the core take place upon receptor binding. Whereas unliganded gp120 exhibited a lobe of density in the dorsal crest position, sCD4-bound gp120 exhibited a lobe in a lateral position (relative to the core), where it was proximal to the CD4 binding site (Fig. 4C and 5A). This shift exposes the dorsal surface of the core and was interpreted to reflect the movement of the V1/V2 loops into their CD4-bound conformation. Indeed, the CD4-bound conformation of the V1/V2 stem observed in CD4-gp120 core crystal structures fit entirely into the gp120-sCD4 SAXS envelope, unlike in the unliganded case (Fig. 2C). The density alongside CD4 in the SAXS envelope also accommodates the recently reported V1/V2 structure (39) (Fig. 4D), and it is roughly consistent with the position of the loops following CD4 binding suggested by cryo-electron tomography studies for Env trimers (19, 35, 73). The feature in the SAXS models is not quite as prominent as Subramaniam and coworkers observed in subtomogram averaged trimers from some isolates; however, they noted that the size of this feature was largely isolate dependent (see Fig. S9D in the supplemental material) (19). For example, compared with JRFL, KNH1144 gp140 SOSIP trimers exhibited a much smaller feature for V1/V2 in the CD4 bound state. It is also conceivable that in the context of the trimer, persistent intersubunit interactions even after CD4 binding may influence the positioning of the variable loops, leading to subtle differences in their presentation versus what we observed in gp120 monomers.
Fig 4.

Solution small-angle X-ray scattering structures for full-length gp120 monomer in complex with D1D2 sCD4. (A) SAXS pattern for the full-length, glycosylated gp120-sCD4 complex. Solid black line represents the calculated SAXS pattern for a typical DAMMIN reconstruction. Inset shows the Guinier fit. (B) Corresponding P(r) pairwise distance distribution plot. (C) “Averaged” SAXS structures for the gp120-sCD4 complex, shown as an envelope drawn over the averaged, filtered DAMMIN structure (see Fig. S3 in the supplemental materia). Structures for gp120 core with N-/C-terminal extensions and an intact V3 loop (PDB 3JWD and 2B4C; blue) were fit into the SAXS density. D1D2 sCD4, which was cocrystallized with gp120 core, is shown in orange. (D) The V1/V2 loop structure from the complex with bNAb PG9 (PDB 3U4E; cyan spheres) occupies approximately the same volume as suggested by the SAXS shape reconstructions, although the specific conformation may be different. See also Movie S2 in the supplemental material.
Interestingly, the V3 loop position reflected in the 2B4C crystal structure no longer falls within the SAXS envelope (22). It is unclear whether this loop gains steric freedom and hence is poorly resolved once V1/V2 shift, or if it becomes more closely associated with the core.
Shape reconstruction with GASBOR produced models that resembled the DAMMIN models and exhibited good model-to-experimental data fits, with χ2 values of 1.448 ± 0.029 and reasonable model convergence, although not as tight as for the DAMMIN models, with an NSD of 1.414 ± 0.044 (see Fig. S5 and Movie S4 in the supplemental material). Overall features, including the gp120 core, D1D2 sCD4, and presence of additional mass or density extending from the V1/V2 stem alongside the bound CD4, are consistently observed in shape reconstructions from the SAXS data regardless of the reconstruction approach.
Validation of the SAXS approach.
In order to compare our data against well-documented samples for which complete or nearly complete high-resolution structures are available, we gathered SAXS data for sCD4, 17b Fab, and the ternary complex of gp120-sCD4-17b Fab, and we processed the data using the same methodology described above for gp120 and gp120-sCD4. In addition, to test the dependence of the resulting SAXS models on the expression system, we examined unliganded gp120 monomers and gp120-sCD4 complexes using gp120 material produced from CHO cells.
All specimens were monodisperse as judged by dynamic light scattering and showed good agreement for Rg values obtained from analysis of the Guinier range and from real-space P(r) plots (Table 1). The sCD4 and 17b Fab SAXS data sets also served as a methodology control for ab initio reconstructions, showing excellent agreement with the crystallographically determined structures (see Fig. S6 in the supplemental material). DAMMIN model fits to experimental data and model convergence were excellent for both sCD4 (χ2, 1.409 ± 0.004; NSD, 0.938 ± 0.020) and 17b Fab (χ2, 1.338 ± 0.001; NSD, 0.617 ± 0.058).
CHO cell-expressed gp120 monomer produced a model for unliganded gp120 in excellent agreement with that observed for 293E cell-expressed gp120 monomers (see Fig. S7 in the supplemental material). The models exhibited SAXS patterns in good agreement with the experimentally measured patterns, as reflected by the goodness of fit between model and experimental SAXS patterns (χ2, 1.411 ± 0.007). Model convergence was also reasonable, as reflected by the low NSD values determined following alignment 0.805 ± 0.032) (27). DAMMIN reconstruction of the sCD4 complex with CHO cell-expressed gp120 produced a set of models that closely resembled the shape envelopes determined for 293E cell-expressed gp120-sCD4 and that agreed well with experimentally measured data (χ2, 1.487 ± 0.004). The models exhibited good convergence of structures, with an average NSD of 0.722 ± 0.035 (see Fig. S8 in the supplemental material).
The ternary complex of gp120-sCD4-17b Fab likewise showed excellent agreement with the available crystal structures for gp120 core with sCD4 and 17b Fab bound (30) (Fig. 5B; see also Fig. S9 and S10 in the supplemental material). The DAMMIN model's SAXS patterns and experimental measurements were in good agreement (χ2, 1.622 ± 0.002), and convergence among models was likewise reasonable (NSD, 0.870 ± 0.009).
All-atom modeling of gp120.
As an alternative approach to interpreting SAXS data, all-atom models were constructed to compare theoretical and experimentally measured scattering patterns. All-atom models of unliganded gp120, gp120-sCD4 and gp120-sCD4-17bFab were generated using available gp120 core structural data, with missing loops and glycans modeled using MODELLER (11). The coordinates for b12 (2NY7) and sCD4-48D Fab (3JWD) complexes with gp120 core served as templates for the pre- and postbridging sheet formed states, respectively, i.e., the V1/V2 stem “up” and “down.” A total of 100 structures of unliganded gp120 were generated for both templates, with random conformations for V1/V2, V3, and the N/C termini. High-mannose and complex-type N-linked glycan chains (21 in total) were added at the initial modeling step and treated as flexible residues during annealing (see Materials and Methods). The top 5 models obtained for the prebridging sheet conformation (V1/V2 “up”) fit the SAXS profile of unliganded gp120, with an average χ2 of 3.09 and a top score of 2.50 (Fig. 6A and D). In contrast, the models with the postbridging sheet template (V1/V2 “down”) were less consistent with these same SAXS data, with a top 5 average χ2 of 4.78 and a best fit of 4.61. Since a single model structure may not encapsulate gp120's conformation(s) in solution, we attempted minimal ensemble searches (MES) to fit a combination of different models against the SAXS data. Fits using 3 models from the prebridging sheet ensemble showed a considerably better fit (χ2, 1.91), with 4 or more models showing negligible improvement.
Fig 6.

All-atom modeling of gp120 structures. The top five all-atom models most consistent with SAXS data are shown in the same orientation for unbound gp120 (A), sCD4 complex (B), and sCD4-17b Fab complex (C). The gp120 core is shown surface rendered in blue with N-/C-terminal extensions (blue spheres), V1/2 (cyan), V3 (green), 21 N-linked glycans (beige), and sCD4 (red surface). Although sCD4 is absent in the unliganded gp120 models, the position of sCD4 is indicated by red spheres. Fits of SAXS data (blue) to the best all-atom model (red) are shown for unbound gp120 (D), the sCD4 complex (E), and the sCD4-17b Fab complex (F). Models in top of panels A to C are in an orientation similar to that shown in Fig. 5B.
While we primarily view this all-atom approach as a means for testing the general occupancy of large structural features, such as the approximate position of V1/V2 loops, we did examine the effect of including additional structural information for V1/V2 and V3 based upon available fragmentary structural information. Additional ensembles were built by incorporating structures of V3 (22, 70) and/or V1/V2 from the PG9-bound complex (39). Incorporation of these structures led to poorer fits to the SAXS data, with the exception of V3 from the 2B4C crystal structure, in which the extended conformation of V3 was similar to that obtained for most random orientations (see Table S2 in the supplemental material).
The same modeling approach was also used to generate an ensemble for the gp120-sCD4 and gp120-sCD4-17bFab complexes. The best-ranking models for both complexes were in excellent agreement with the SAXS data (Fig. 6E and F). In both cases, MES fitting using up to 3 models yielded a marginal improvement in the fit (see Table S2). Results of a comparison of the best fit models of each state of gp120 were consistent with sCD4 binding leading to spatial rearrangement of the V1/V2 loops relative to the core causing exposure of the 17b binding site (Fig. 6A to C).
Mapping local conformational order/disorder by HDX-MS.
In order to probe the solution behavior of gp120 and map CD4-induced conformational changes in greater detail, HDX-MS was used to measure local solvent exchange kinetics of the full-length, glycosylated SF162 gp120. Pepsin digestion yielded 57 observable (unique) peptides of sufficient signal to monitor 82% of the sequence of gp120. These included multiple glycoforms of 13 peptic segments containing N-linked glycan modifications (see Fig. S11 in the supplemental material). Although glycan microheterogeneity was evident at all sites, in each case, a predominant type of glycosylation could be assigned based on modification mass and tandem MS/MS data. Glycans at positions 232, 239, 260, 329, 336, and 438 were found to be predominantly high-mannose glycans, while sites 87, 274, 352, and 454 were mainly sialic acid-decorated biantennary glycans (numbering based on the complete SF162 Env). One limitation with these HDX-MS studies was relatively poor sequence coverage of the most highly glycosylated regions (i.e., V1 and V4). From an examination of fragments that were deglycosylated after peptic digestion, it appears that although these fragments are generated by pepsinization, they are either not retained by the C18 stationary phase used for LC-MS, or extensive glycan heterogeneity results in insufficient signal for any individual glycoform.
A C-terminal gp120 fragment was identified that bore O-linked glycosylation, with structures NeuAc1Hex1NAcHex1 and NeuAc2HexNAc1Hex1 at position T490, likely corresponding to mono- and disialylated type 1 core glycans. The nonglycosylated form of this fragment was also observed; thus, both decorated and undecorated forms are present in gp120 populations. O-linked glycosylation of Env has been reported, and T490 is predicted to have a high likelihood of glycosylation (18, 51), but to our knowledge this is the first direct, site-specific identification of an O-linked glycan on gp120. This finding may be notable, given that this specific segment coincides with the epitope of recently identified broadly neutralizing antibodies (24).
Exchange profile of unliganded, full-length glycosylated gp120 monomer.
Deuterium exchange kinetics were analyzed for all observable peptides. As hydrogen bonding protects amide protons from deuterium exchange, the exchange rates can be used to estimate the extent of stable secondary structure or hydrogen-bonded backbone present at each fragment. Regions that are highly disordered or unstructured will display no protection from solvent and become deuterated rapidly (within seconds). Regions involved in secondary structure are protected, but transient unfolding events will expose these amides over time and therefore the exchange kinetics can be used to analyze the relative dynamics of the structured regions.
Several fragments from both the inner and outer domain showed slowed exchange consistent with the presence of stable secondary structure, as expected from the crystallography-based model threaded with the clade B SF162 sequence (see Materials and Methods). Fragment 282–286, in a β-strand within the outer domain, showed the strongest protection, taking up no detectable amount of deuterium even after 4 h of incubation. Other fragments (257–263, 347–357, and 436–443) in the outer domain also showed protection for up to hours, suggesting that these structural elements are well ordered in unbound gp120 (Fig. 7B and D). Fragment 221–224, forming one of the sheets in the β-sandwich, and helices at positions 104 to 110 and 458 to 474 within the inner domain also showed moderate protection, indicating that portions of the inner domain are also reasonably ordered.
Fig 7.

Hydrogen/deuterium exchange of gp120 and the sCD4-gp120 complex. (A) Deuteration kinetics of fragments 52–82, 358–369, 111–125, and 407–435 in unbound gp120 (black) and sCD4-bound gp120 (gray). (B) Butterfly plot showing the deuterium exchange kinetics of unliganded (upper) and CD4-bound (lower) monomeric gp120. Percent deuteration is shown for each fragment at the midpoint of its position in the primary sequence. Time points of deuterium exchange are 15 s (yellow), 1 min (orange), 5 min (cyan), 30 min (blue), and 4 h (black). Gray bars indicate the positions of the variable loops. Much of the V1/V2 and V4 regions did not yield detectable peptic fragments and were invisible to this analysis. Raw data for each fragment are presented in Fig. S13 and S14 of the supplemental material. (C) Differences in the percent exchange for free and CD4-bound gp120 are plotted for each time point. The position of the CD4 binding loop, layer 1 of the inner domain, and the segments of the bridging sheet are highlighted. (D) Structure of the gp120 core with selected fragments observed to have relatively fast (red) or slow (blue) deuterium exchange kinetics in unliganded full-length gp120.
On the other hand several regions were exchanged rapidly, indicating a lack of stable secondary structure, including segments from V2, V3, and the N-/C-terminal extensions. Fragments 34–51 and 52–82 showed almost no protection, indicating that layer 1 of the inner domain is highly disordered (Fig. 7A). Interestingly, two segments (111–125 and 407–435), which together include 3 of the 4 β-strands of the bridging sheet, were also in rapid exchange, indicating that this motif is disordered within unliganded gp120 (Fig. 7A). The fragment corresponding to the CD4 binding loop (358–369) was only marginally protected, suggesting that the helical turn in this region is also relatively disordered (Fig. 7A).
No significant differences in exchange kinetics were observed between the distinct glycoforms of each glycopeptide. Similarly, the fragment 475–500, bearing an O-linked glycan, exhibited no significant difference in exchange compared to the nonglycosylated form.
Changes in gp120 exchange induced by CD4 binding.
Dramatic increases in protection from solvent exchange were observed throughout gp120 upon CD4 binding (Fig. 8). Several regions that showed moderate protection within the inner domain (residues 95–110 and 458–474) of unbound gp120 became even more protected, indicating structural rigidification upon sCD4 binding (Fig. 7C and D). Both halves of the bridging sheet, previously in rapid exchange, showed significant protection upon CD4 binding (Fig. 7A). The same change was also observed for the CD4 binding loop (residues 358–369), showing protection even up to 4 h. A slight increase in protection at V5, located proximal to the CD4 binding site, was also observed upon CD4 binding. Layer 1, which is in rapid exchange in unliganded gp120, showed a significant increase in protection, indicating that CD4 also induces the formation of this motif. Fragments in V2 and V3 were still in rapid exchange and thus showed no detectable change upon CD4 binding. Taken together, the differences in HDX profiles showed how CD4 binding induces the formation of the bridging sheet and locks down the conformation of the CD4 binding site and several structural elements within the inner domain.
Fig 8.
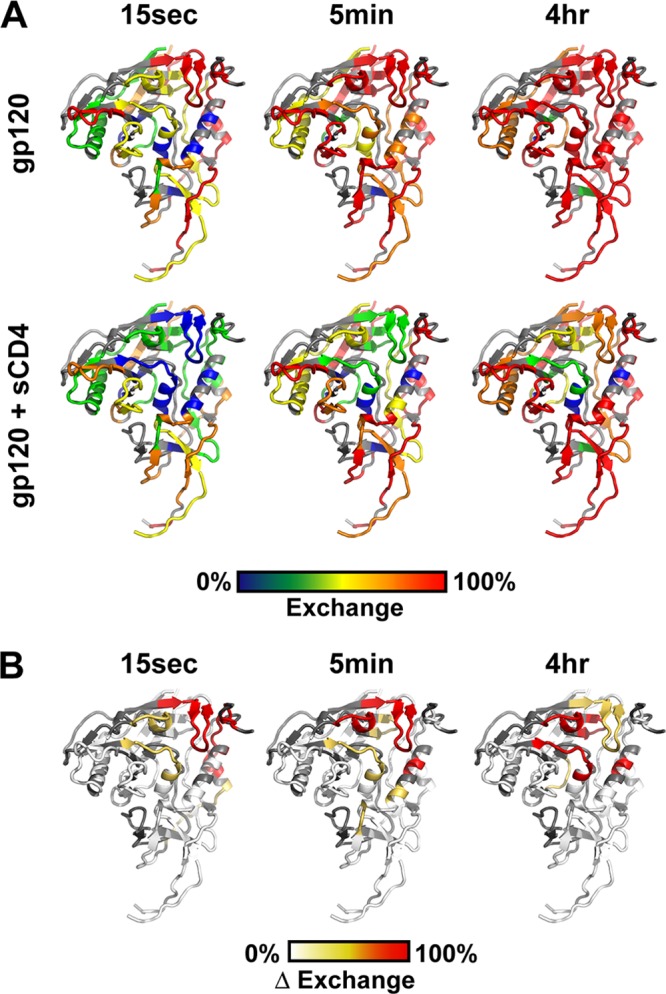
Exchange behavior of gp120. (A) Exchange data illustrated on the gp120 structure (modeled using PDB 3JWD and 2B4C) for free (top) and CD4-bound (bottom) forms. Coloring indicates the degree of deuterium uptake, from none (blue) to full (red). (B) Differences in deuterium exchange for free and CD4-bound gp120, plotted on the structure. The degrees of change between the two forms are colored from white (<20% difference) to orange (>20% difference) to red (>50% difference).
Comparison with HDX-MS study results with deglycosylated, minimal gp120 core.
Several observations of the current study provide notable differences from those of the single previous study of HDX-MS on the gp120 core, which were largely deglycosylated and lacked variable loops V1-V3 as well as the N-/C-terminal extensions (26). Although both studies indicated that the β-sandwich motif and helices within the inner domain were reasonably ordered in the unbound form, the CD4 binding loop and segments of the bridging sheet appeared considerably less protected (more disordered) in full-length glycosylated gp120. Increased protection upon CD4 binding was observed in both studies but, based on calculations of conformational stability, the stabilization in full-length gp120 was nearly double that observed for the core (see Table S3 in the supplemental material). This is consistent with thermodynamic study findings, which have reported a larger ΔG of CD4 binding to full-length versus core gp120 (43). As layer 1 (absent from the minimal core) became protected only upon CD4 binding, it is conceivable that the difference is partly due to the structuring of this region (Fig. 7).
Another significant difference between the full-length and core gp120 was seen in the loop near the inner-outer domain interface (residues 257–281). The two fragments from this region in full-length gp120 showed protection even at 4 h, with only a minor increase in protection with CD4 (ΔΔG, −0.9 and −0.5 kcal/mol). In contrast, this region in core gp120 exchanged quickly, with a drastic increase in protection upon CD4 binding (ΔΔG, −1.7 and −2.0 kcal/mol). This segment contained N-linked glycans at positions 260 and 274, both of which are highly conserved. Since glycans are thought to be relevant to the overall structure and antigenicity of Env (57), it is possible that this difference between core and full-length gp120 reflected structural stability imposed by the associated glycans. Alternatively, it is possible that some of the distinctions may be due to isolate-specific differences. Comparisons of gp120 from a variety of different isolates indicated that SF162 gp120 is in fact on the more-ordered end of the dynamic spectrum (T. M. Davenport, Guttman, Lee, unpublished data). Thus, other full-length gp120s are likely to exhibit even greater differences relative to the dynamics reported for the YU2 core.
Dimeric gp120 is linked by the V2 loop and N-terminal interactions.
Nonreducing SDS-PAGE revealed that dimeric gp120 is stabilized through disulfide bonds, as recently reported (13). HDX-MS of this dimeric form showed an exchange profile almost identical to that of monomeric gp120 (see Fig. S12 in the supplemental material). The only differences were increases in protection of fragments in the N-terminal extension and within the V2 loop. This result was consistent with results of deletion studies, which have shown that removal of the V2 loop or the extensions at the N/C termini decreased dimer formation (5, 13). From the current study, we specifically implicate residues 34–38 and 171–177 as being involved in the dimeric interface. Although these dimers are widely considered to be nonnative species, the exchange profile is very similar to monomeric gp120, indicating that apart from the dimer interface, both species are in similar conformations.
DISCUSSION
While a great deal of detailed information for the structure of HIV Env has been provided by X-ray crystallography (6, 10, 22, 30, 44), most commonly for the gp120 core in complex with stabilizing ligands, such as CD4 receptor or antibodies, the requirements for crystallization make it necessary to truncate and deglycosylate the glycoprotein, removing the very features that make each variant distinct and that can modulate structure and dynamics within the core itself. The combination of SAXS and HDX-MS allowed us to address two primary gaps in our understanding of gp120: (i) the disposition of the V1/V2 loops in the unliganded, full-length subunit and in the CD4-bound form and (ii) the local intrinsic dynamics within the glycoprotein and the changes upon ligand binding. These key aspects of gp120 profoundly influence how the glycoprotein interacts with receptors and antibodies, and thus an understanding of these features in detail is critical. This approach should also be applicable to other soluble Env constructs, such as gp140 ectodomain trimers.
Nature of the full-length, unliganded gp120 subunit.
The SAXS model for full-length, unliganded gp120 showed a crest of density on the top of the core region that was consistent with closely associated V1/V2 and V3 loops (Fig. 2 and 5). A significant body of evidence reported previously supports the hypothesis that the variable loops are associated and V3 is occluded by V1/V2 (37, 83). Whether this occlusion occurs within a single subunit or between neighboring gp120s remains controversial (36, 54). From the current study, it appears that V1/V2 contacts with V3 may already exist within monomeric gp120. In this state, we propose that the V1/V2 stem, which would comprise half of the bridging sheet after CD4 binding, is in an “up” conformation, similar to the stem position observed in the b12-bound gp120 core crystal structure (PDB 2B4C), positioning V1/V2 next to V3 (81). This assignment is also supported by the HDX data for unliganded gp120, which revealed that the bridging sheet and CD4 binding site elements were surprisingly disordered (Fig. 7D). If the V1/V2 stem is “up,” the bridging sheet elements would be split and would be expected to exhibit a less-stable secondary structure and hence rapid deuterium exchange. When the CD4-bound crystal structures are docked into the SAXS envelope, the only major structural element that does not fall within the envelope is the V1/V2 stem. Atomic models of unliganded gp120 with V1/V2 loops oriented in the “up” position were more consistent with the SAXS pattern than models with the bridging sheet formed and V1/V2 “down,” further supporting the position of this loop in the crest density (Fig. 6A). This discrepancy also holds true for comparisons against the crystal structure of the SIV core gp120, which shows the V1/V2 stem forming a β-sheet running along the lower end of the inner domain, positioning V1/V2 and V3 on opposite ends of the molecule (6).
The disposition of V1/V2 and the conformational state of the bridging sheet elements contrast with the recently published structure of unliganded, extended gp120 core (28), which showed that the bridging sheet and CD4 binding site adopt a conformation nearly identical to that seen with CD4 and the stabilizing ligands bound. Based upon comparisons of various truncation constructs, epitope mapping, and thermodynamic measurements, the authors of the gp120 core study inferred that truncation of the V1/V2 and V3 loops causes the core to revert to the CD4-bound conformation (28). Our results are consistent with the hypothesis that the variable loops maintain gp120 in a native unliganded conformation that is distinct from that observed in the unliganded core structure.
The positioning of the variable loops atop the core confers a shape to the gp120 subunit that closely resembles the primary lobes of density observed in cryo-electron tomographic studies of Env on the surface of virions as well as soluble SOSIP gp140 trimers (Fig. 3) (19, 35, 73). This suggests that the gp120 subunit in isolation is largely similar to the structure of the subunit in the context of the trimer, although the variable loops in the trimer spike are likely drawn toward the axial center through intersubunit interactions. Interestingly, the crest density for each gp120 subunit more closely resembles the prominent bulges seen in the gp140 SOSIP soluble trimer reconstructions (19), more so than the rather flat-topped crown of the Env trimer on the virus (35, 73). It remains to be seen whether this reflects real structural differences or perhaps inherent limitations in resolution of the two approaches. Since the gp120 subunit occupies most of the upper portions of the trimeric spike, it appears that the base must be occupied by gp41 (Fig. 3). Overall, we were surprised that full-length gp120 isolated from its native oligomeric context still appeared to fairly faithfully maintain its overall morphology. This observation may have implications for the use of full-length gp120 subunits as immunogens.
While the crest feature observed in the SAXS models for unliganded gp120 appeared to indicate that V1/V2 and V3 form some degree of association, HDX-MS indicated these regions do not appear to exhibit stable secondary structure or order involving H-bonding of the backbone amide groups (Fig. 7B; see also Fig. S13 in the supplemental material). No significant protection was detected within V3 or V2 loop fragments, even upon CD4 binding. However, interactions may still exist within the variable loops that are perhaps mediated by side chain interactions, e.g., hydrophobic or ionic, or that may be too weak or intermittent to result in sufficient protection to be observed by HDX-MS. Previous examination of the isolated V3 loop by nuclear magnetic resonance (NMR) showed the C-terminal half of the loop forming a partial helical structure (70). The fragment 314–322, exactly at this position, showed no protection based on HDX, arguing against the presence of a stable helix in the context of the full-length gp120. One possible explanation for this discrepancy is that the two N-linked glycans, one within and one just adjacent to the V3 loop, which were absent from the NMR study, give rise to significantly more flexibility within this region. All-atom gp120 models incorporating the NMR coordinates of V3 with helical C-terminal segments were also overall less consistent with the SAXS data.
The recently reported crystal structure of broadly neutralizing antibody (bNAb) PG9 in complex with V1/V2 fused to a scaffold protein showed that this subdomain of gp120 adopts a 4-stranded β-sheet when complexed with the antibody (39). Although our HDX-MS study only had partial coverage of this region, fragment 158–173 corresponds to the position of the third β-strand and shows no protection in either unbound or CD4-bound states. Models incorporating the conformation of V1/V2 observed in the PG9-bound structure were less consistent with the SAXS patterns for unliganded gp120 than unrestrained loops. From these data, we conclude that while V1/V2 are localized atop the core in full-length, unliganded gp120, this motif is at most only transiently ordered in the absence of stabilizing ligands, such as PG9 antibody. It remains possible that the 4-stranded β-sheet motif may be present within trimeric Env and potentially stabilized by quaternary interactions.
CD4 binding dramatically alters the organization and dynamic properties of the gp120 subunit.
In the unliganded state, V1/V2 do not appear to occlude the CD4 binding site (Fig. 2 and 5B). This does not necessarily exclude the possibility that flexible glycans on the binding site periphery may act as entropic brushes to decrease accessibility (25). Upon CD4 binding, the SAXS results indicate that a repositioning of a significant mass, inferred to be V1/V2, takes place from the crest to a lateral position relative to the core. The apparent repositioning of V1/V2 was corroborated by the increased protection observed by HDX at the base of V1 (fragment 111–125 [Fig. 7A]), which forms part of the bridging sheet. Formation of the bridging sheet would swing the V1/V2 stem from something like the position observed in the b12 antibody-bound structure to that observed in all CD4-bound structures (Fig. 5A). All-atom models of sCD4-bound gp120 with this lateral occupancy of V1/V2 were in excellent agreement with the SAXS data (Fig. 6B and E).
In the SAXS-based model for the CD4-bound state, V1/V2 extend forward from the variable loop stem to occupy a position forming a collar around the base of the CD4/gp120 contact (Fig. 4; see also Movies S2 and S4 in the supplemental material). This region of the gp120 core does not bear significant glycosylation, unlike the immunologically silent face. We hypothesize that the variable loops in the CD4-bound state may thus mask underlying conserved epitopes that become ordered only after the receptor is engaged. Interestingly, antibody 21c binds in a very similar position, with a binding footprint that includes both CD4 and gp120 elements (10). 21c binding was shown to exhibit a striking dependence on whether V1/V2 was present. Our SAXS results, low resolution as they may be, appear to at least partially explain the influence of V1/V2 on 21c binding to full-length gp120.
In contrast to the approachable CD4 binding site in unliganded gp120, CD4-induced features, such as the 17b epitope, which largely overlaps with the coreceptor binding surface, appear to be obstructed by the crest density (V1/V2, V3) (Fig. 5B) (10, 53, 64, 77). Furthermore, our data also indicate that the bridging sheet subdomain that serves as a major portion of the 17b and coreceptor binding site is not well ordered in unliganded gp120. Upon CD4 binding, the bridging sheet forms and V1/V2 swing away from the crest position, generating the stable 17b-coreceptor binding face. The SAXS-derived shape of the gp120-sCD4-17b Fab complex is in good agreement with the available crystallographic data with the gp120 core (Fig. 5B), indicating that 17b binding does not appear to induce major structural changes beyond those elicited by CD4.
Implications for antigenicity and immunogenicity.
Due to its functional conservation among diverse isolates of HIV-1, the CD4 binding site is a highly sought target for eliciting bNAbs (58, 76). Based upon crystallographic data from diverse isolates, the structure at the CD4 binding site, when bound by receptor or CD4 binding site antibodies, also appears highly conserved. The current HDX data demonstrate that the discontinuous portions of gp120 that are drawn together to form the CD4 binding site are relatively disordered and dynamic in unliganded gp120, becoming ordered only after CD4 binding (Fig. 9). Surprisingly, this is also true for the CD4 binding loop itself, which is invariant between various gp120 core structures and thought to be the initial site of CD4 attachment. It appears that the CD4 binding loop is only conformationally rigid in the CD4-induced form, whereas in the unbound form observed in full-length gp120 it retains significantly more flexibility and possibly exhibits a range of conformational diversity between isolates (Davenport et al, unpublished data).
Fig 9.

Local conformational dynamics of unliganded and CD4-bound gp120. (A) Model of unliganded SF162 gp120 (PDB 3JWD and 2B4C) lacking V1/V2 (62-residue) and V3 (25-residue) loops. The strands that form the bridging sheet and layer 1 of the inner domain along with the CD4 binding loop (red) were found to be relatively disordered. (B) Model of sCD4-bound gp120, shown in the same orientation. CD4 binding not only rigidifies the structure of the CD4 binding loop and the strands of the bridging sheet but also regions within the inner domain at the gp41-interacting region (blue). CD4 is shown in orange.
It has been proposed that the relative disorder and fragmented nature of the CD4 binding site may explain why, despite its conservation, the humoral response has difficulty in the generation of highly potent, broadly cross-reactive CD4 binding site-targeted bNAbs (8). Stabilized gp120 immunogens have shown promise at inducing a more effective humoral response (9), but this response was not specifically directed against the stabilized CD4 binding site. It is conceivable that stabilized immunogens may compromise neutralization potency by eliciting antibodies against only the fully formed, conformationally fixed CD4 binding site, rather than the more inchoate epitope as it exists prior to receptor engagement. The CD4 interaction is known to have high entropic cost and is thought to be driven by significant folding upon binding (43). Some of the most potent CD4 binding site-directed bNAbs also bind with a large change in entropy (80), although they do not necessarily induce the same conformational changes (12, 33). In light of this, both the nature of the CD4 binding site and the structural effects of antibody binding in trimeric Env and any prospective immunogens will have to be understood for the design of an effective immunogen.
Allosteric changes triggered by CD4 binding.
CD4 binding was observed to induce a significant degree of protection in regions that emanate from the binding site proper (Fig. 9), most notably in regions of the inner domain extending to the putative gp41-interactive portions of gp120 on the opposite side of the core. Mutagenesis studies have likewise recently indicated that alterations within the inner domain can influence CD4 binding, suggesting a link between the CD4 and gp41-interacting face of gp120 (14). The current HDX data directly resolve this coupling, revealing specific allosteric changes at the gp41-interacting region. Both layer 1 and the helix spanning residues 95–110, thought to be the elements that associate with gp41 in the trimeric spike (44), showed major changes upon CD4 binding. Interestingly, in the b12 antibody-bound core crystal structure, the segment from residue 95–110 does not adopt a complete, ordered helix, consistent with the higher degree of solvent exchange in unliganded gp120. Once CD4 binds, the segment becomes highly ordered, consistent with full helix formation. This region is particularly of interest, because the V1 loop and bridging sheet elements are directly C-terminal to this helix and it links the CD4 binding surface and the gp41-interactive face of gp120. This allosteric coupling of structural elements may thus alter the interaction between gp120 and gp41 in response to receptor binding, potentially serving as the first of several steps that trigger the fusion mechanism.
In summary, based upon our complementary solution-phase studies, we conclude that gp120 in its unliganded state has disordered CD4 and coreceptor binding elements and V1/V2 and V3 positioned atop the gp120 core, in a similar position to that observed in intact Env trimers. In response to CD4 binding, the CD4 binding site on gp120 becomes ordered and the bridging sheet subdomain forms, drawing the V1/V2 loops into a “down” orientation and positioning them alongside CD4. Lastly, structural ordering is transduced to the gp41-interactive face of gp120, reflecting changes in gp120 that may lead to priming of the fusogenic subunit, gp41.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge Hiro Tsuruta and Tsutomu Matsui at SSRL BL4-2 for valuable assistance with SAXS data collection and Patrick Weinkam, UC San Francisco, for assistance with all-atom modeling. We thank Zachary Caldwell, Noah Sather, and Leonidas Stamatatos for providing gp120 from 293E cells. CHO cell-expressed gp120 was a kind gift from Novartis. We also thank James Robinson and Peter Kwong for 17b IgG. The following reagent was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: sCD4-183 (2-domain) from Progenics.
This work was supported by NIH grant R00-GM080352 (K.K.L.) and UAB CFAR grant P30-AI027767 through the CNIHR program for new HIV investigators (K.K.L.) and by P01-RR000166 (Washington National Primate Research Center) and R01-AI076170 (S.L.H.). N.K.G. was supported by NIH training grant T32-GM007750. Data collection at SSRL was supported by grant number P41-RR001209 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health.
Footnotes
Published ahead of print 6 June 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Ashish, Garg R, Anguita J, Krueger JK. 2006. Binding of full-length HIV-1 gp120 to CD4 induces structural reorientation around the gp120 core. Biophys. J. 91:L69–L71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bai Y, Milne JS, Mayne L, Englander SW. 1993. Primary structure effects on peptide group hydrogen exchange. Proteins 17:75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beddows S, Lister S, Cheingsong R, Bruck C, Weber J. 1999. Comparison of the antibody repertoire generated in healthy volunteers following immunization with a monomeric recombinant gp120 construct derived from a CCR5/CXCR4-using human immunodeficiency virus type 1 isolate with sera from naturally infected individuals. J. Virol. 73:1740–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bures R, et al. 2000. Immunization with recombinant canarypox vectors expressing membrane-anchored glycoprotein 120 followed by glycoprotein 160 boosting fails to generate antibodies that neutralize R5 primary isolates of human immunodeficiency virus type 1. AIDS Res. Hum. Retroviruses 16:2019–2035 [DOI] [PubMed] [Google Scholar]
- 5. Center RJ, Earl PL, Lebowitz J, Schuck P, Moss B. 2000. The human immunodeficiency virus type 1 gp120 V2 domain mediates gp41-independent intersubunit contacts. J. Virol. 74:4448–4455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen B, et al. 2005. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature 433:834–841 [DOI] [PubMed] [Google Scholar]
- 7. DeLano WL. 2002. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, CA [Google Scholar]
- 8. Dey B, et al. 2007. Characterization of human immunodeficiency virus type 1 monomeric and trimeric gp120 glycoproteins stabilized in the CD4-bound state: antigenicity, biophysics, and immunogenicity. J. Virol. 81:5579–5593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dey B, et al. 2009. Structure-based stabilization of HIV-1 gp120 enhances humoral immune responses to the induced co-receptor binding site. PLoS Pathog. 5:e1000445 doi:10.1371/journal.ppat.1000445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Diskin R, Marcovecchio PM, Bjorkman PJ. 2010. Structure of a clade C HIV-1 gp120 bound to CD4 and CD4-induced antibody reveals anti-CD4 polyreactivity. Nat. Struct. Mol. Biol. 17:608–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eswar N, et al. 2006. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics 5:Unit 5.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Falkowska E, et al. 2012. PGV04, an HIV-1 gp120 CD4 binding site antibody, is broad and potent in neutralization but does not induce conformational changes characteristic of CD4. J. Virol. 86:4394–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Finzi A, et al. 2010. Conformational characterization of aberrant disulfide-linked HIV-1 gp120 dimers secreted from overexpressing cells. J. Virol. Methods 168:155–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Finzi A, et al. 2010. Topological layers in the HIV-1 gp120 inner domain regulate gp41 interaction and CD4-triggered conformational transitions. Mol. Cell 37:656–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fischer H, de Oliveira Neto M, Napolitano HB, Craievich AF, Polikarpov I. 2010. Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. J. Appl. Crystallogr. 43:101–109 [Google Scholar]
- 16. Guvench O, et al. 2008. Additive empirical force field for hexopyranose monosaccharides. J. Comput. Chem. 29:2543–2564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guvench O, Hatcher ER, Venable RM, Pastor RW, Mackerell AD. 2009. CHARMM additive all-atom force field for glycosidic linkages between hexopyranoses. J. Chem. Theory Comput. 5:2353–2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hansen JE, Clausen H, Hu SL, Nielsen JO, Olofsson S. 1992. An O-linked carbohydrate neutralization epitope of HIV-1 gp 120 is expressed by HIV-1 env gene recombinant vaccinia virus. Arch. Virol. 126:11–20 [DOI] [PubMed] [Google Scholar]
- 19. Harris A, et al. 2011. Trimeric HIV-1 glycoprotein gp140 immunogens and native HIV-1 envelope glycoproteins display the same closed and open quaternary molecular architectures. Proc. Natl. Acad. Sci. U. S. A. 108:11440–11445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hoxie JA. 2010. Toward an antibody-based HIV-1 vaccine. Annu. Rev. Med. 61:135–152 [DOI] [PubMed] [Google Scholar]
- 21. Hu SL, Stamatatos L. 2007. Prospects of HIV Env modification as an approach to HIV vaccine design. Curr. HIV Res. 5:507–513 [DOI] [PubMed] [Google Scholar]
- 22. Huang CC, et al. 2005. Structure of a V3-containing HIV-1 gp120 core. Science 310:1025–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang CC, et al. 2004. Structural basis of tyrosine sulfation and VH-gene usage in antibodies that recognize the HIV type 1 coreceptor-binding site on gp120. Proc. Natl. Acad. Sci. U. S. A. 101:2706–2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Humbert M, et al. 2008. Inducing cross-clade neutralizing antibodies against HIV-1 by immunofocusing. PLoS One 3:e3937 doi:10.1371/journal.pone.0003937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koch M, et al. 2003. Structure-based, targeted deglycosylation of HIV-1 gp120 and effects on neutralization sensitivity and antibody recognition. Virology 313:387–400 [DOI] [PubMed] [Google Scholar]
- 26. Kong L, et al. 2010. Local conformational stability of HIV-1 gp120 in unliganded and CD4-bound states as defined by amide hydrogen/deuterium exchange. J. Virol. 84:10311–10321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kozin MB, Svergun DI. 2001. Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34:33–41 [Google Scholar]
- 28. Kwon YD, et al. 2012. Unliganded HIV-1 gp120 core structures assume the CD4-bound conformation with regulation by quaternary interactions and variable loops. Proc. Natl. Acad. Sci. U. S. A. 109:5663–5668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kwong PD, et al. 2002. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 420:678–682 [DOI] [PubMed] [Google Scholar]
- 30. Kwong PD, et al. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leonard CK, et al. 1990. Assignment of intrachain disulfide bonds and characterization of potential glycosylation sites of the type 1 recombinant human immunodeficiency virus envelope glycoprotein (gp120) expressed in Chinese hamster ovary cells. J. Biol. Chem. 265:10373–10382 [PubMed] [Google Scholar]
- 32. Lewis MS, Junghans RP. 2000. Ultracentrifugal analysis of molecular mass of glycoproteins of unknown or ill-defined carbohydrate composition. Methods Enzymol. 321:136–149 [DOI] [PubMed] [Google Scholar]
- 33. Li Y, et al. 2011. Mechanism of neutralization by the broadly neutralizing HIV-1 monoclonal antibody VRC01. J. Virol. 85:8954–8967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lisal J, et al. 2005. Functional visualization of viral molecular motor by hydrogen-deuterium exchange reveals transient states. Nat. Struct. Mol. Biol. 12:460–466 [DOI] [PubMed] [Google Scholar]
- 35. Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. 2008. Molecular architecture of native HIV-1 gp120 trimers. Nature 455:109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu L, Cimbro R, Lusso P, Berger EA. 2011. Intraprotomer masking of third variable loop (V3) epitopes by the first and second variable loops (V1V2) within the native HIV-1 envelope glycoprotein trimer. Proc. Natl. Acad. Sci. U. S. A. 108:20148–20153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lusso P, et al. 2005. Cryptic nature of a conserved, CD4-inducible V3 loop neutralization epitope in the native envelope glycoprotein oligomer of CCR5-restricted, but not CXCR4-using, primary human immunodeficiency virus type 1 strains. J. Virol. 79:6957–6968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mascola JR, et al. 1996. Immunization with envelope subunit vaccine products elicits neutralizing antibodies against laboratory-adapted but not primary isolates of human immunodeficiency virus type 1. The National Institute of Allergy and Infectious Diseases AIDS Vaccine Evaluation Group. J. Infect. Dis. 173:340–348 [DOI] [PubMed] [Google Scholar]
- 39. McLellan JS, et al. 2011. Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature 480:336–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Medzihradszky KF, Burlingame AL. 2005. Characterization of protein N-glycosylation. Methods Enzymol. 405:116–138 [DOI] [PubMed] [Google Scholar]
- 41. Morgan CR, Engen JR. 2009. Investigating solution-phase protein structure and dynamics by hydrogen exchange mass spectrometry. Curr. Protoc. Protein Sci. 17:Unit 17.16.11–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mylonas E, Svergun DI. 2007. Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. J. Appl. Crystallogr. 40:s245–s249 [Google Scholar]
- 43. Myszka DG, et al. 2000. Energetics of the HIV gp120-CD4 binding reaction. Proc. Natl. Acad. Sci. U. S. A. 97:9026–9031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pancera M, et al. 2010. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc. Natl. Acad. Sci. U. S. A. 107:1166–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pantophlet R, Burton DR. 2006. GP120: target for neutralizing HIV-1 antibodies. Annu. Rev. Immunol. 24:739–769 [DOI] [PubMed] [Google Scholar]
- 46. Pelikan M, Hura GL, Hammel M. 2009. Structure and flexibility within proteins as identified through small angle X-ray scattering. Gen. Physiol. Biophys. 28:174–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Petoukhov MV, Konarev PV, Kikhney AG, Svergun DI. 2007. ATSAS 2.1: towards automated and web-supported small-angle scattering data analysis. J. Appl. Crystallogr. 40:s223–s228 [Google Scholar]
- 48. Petoukhov MV, Svergun DI. 2007. Analysis of X-ray and neutron scattering from biomolecular solutions. Curr. Opin. Struct. Biol. 17:562–571 [DOI] [PubMed] [Google Scholar]
- 49. Pettersen EF, et al. 2004. UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 [DOI] [PubMed] [Google Scholar]
- 50. Putnam CD, Hammel M, Hura GL, Tainer JA. 2007. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 40:191–285 [DOI] [PubMed] [Google Scholar]
- 51. Raska M, et al. 2010. Glycosylation patterns of HIV-1 gp120 depend on the type of expressing cells and affect antibody recognition. J. Biol. Chem. 285:20860–20869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rerks-Ngarm S, et al. 2009. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 361:2209–2220 [DOI] [PubMed] [Google Scholar]
- 53. Rizzuto CD, et al. 1998. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science 280:1949–1953 [DOI] [PubMed] [Google Scholar]
- 54. Rusert P, et al. 2011. Interaction of the gp120 V1V2 loop with a neighboring gp120 unit shields the HIV envelope trimer against cross-neutralizing antibodies. J. Exp. Med. 208:1419–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sather DN, et al. 2009. Factors associated with the development of cross-reactive neutralizing antibodies during human immunodeficiency virus type 1 infection. J. Virol. 83:757–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Scandella CJ, et al. 1993. Nonaffinity purification of recombinant gp120 for use in AIDS vaccine development. AIDS Res. Hum. Retroviruses 9:1233–1244 [DOI] [PubMed] [Google Scholar]
- 57. Scanlan CN, Offer J, Zitzmann N, Dwek RA. 2007. Exploiting the defensive sugars of HIV-1 for drug and vaccine design. Nature 446:1038–1045 [DOI] [PubMed] [Google Scholar]
- 58. Scheid JF, et al. 2011. Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science 333:1633–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schneidman-Duhovny D, Hammel M, Sali A. 2010. FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 38:W540–W544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Smirnovas V, et al. 2009. Distinct structures of scrapie prion protein (PrPSc)-seeded versus spontaneous recombinant prion protein fibrils revealed by hydrogen/deuterium exchange. J. Biol. Chem. 284:24233–24241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Smith DL, Deng Y, Zhang Z. 1997. Probing the non-covalent structure of proteins by amide hydrogen exchange and mass spectrometry. J. Mass Spectrom. 32:135–146 [DOI] [PubMed] [Google Scholar]
- 62. Smolsky IL, et al. 2007. Biological small-angle X-ray scattering facility at the Stanford Synchrotron Radiation Laboratory. J. Appl. Crystallogr. 40:s453–s458 [Google Scholar]
- 63. Sullivan N, et al. 1998. Determinants of human immunodeficiency virus type 1 envelope glycoprotein activation by soluble CD4 and monoclonal antibodies. J. Virol. 72:6332–6338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sullivan N, et al. 1998. CD4-induced conformational changes in the human immunodeficiency virus type 1 gp120 glycoprotein: consequences for virus entry and neutralization. J. Virol. 72:4694–4703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Svergun DI. 1992. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25:495–503 [Google Scholar]
- 66. Svergun DI. 1991. Mathematical methods in small-angle scattering data analysis. J. Appl. Crystallogr. 24:485–492 [Google Scholar]
- 67. Svergun DI. 1999. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76:2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Svergun DI, Petoukhov MV, Koch MH. 2001. Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 80:2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Volkov VV, Svergun DI. 2003. Uniqueness of ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36:860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Vranken WF, Budesinsky M, Fant F, Boulez K, Borremans FA. 1995. The complete consensus V3 loop peptide of the envelope protein gp120 of HIV-1 shows pronounced helical character in solution. FEBS Lett. 374:117–121 [DOI] [PubMed] [Google Scholar]
- 71. Weis DD, Engen JR, Kass IJ. 2006. Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J. Am. Soc. Mass Spectrom. 17:1700–1703 [DOI] [PubMed] [Google Scholar]
- 72. Weis DD, Wales TE, Engen JR, Hotchko M, Ten Eyck LF. 2006. Identification and characterization of EX1 kinetics in H/D exchange mass spectrometry by peak width analysis. J. Am. Soc. Mass Spectrom. 17:1498–1509 [DOI] [PubMed] [Google Scholar]
- 73. White TA, et al. 2010. Molecular architectures of trimeric SIV and HIV-1 envelope glycoproteins on intact viruses: strain-dependent variation in quaternary structure. PLoS Pathog. 6:e1001249 doi:10.1371/journal.ppat.1001249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wriggers W, Birmanns S. 2001. Using situs for flexible and rigid-body fitting of multiresolution single-molecule data. J. Struct. Biol. 133:193–202 [DOI] [PubMed] [Google Scholar]
- 75. Wu SR, et al. 2010. Single-particle cryoelectron microscopy analysis reveals the HIV-1 spike as a tripod structure. Proc. Natl. Acad. Sci. U. S. A. 107:18844–18849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wu X, et al. 2010. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329:856–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wyatt R, et al. 1995. Involvement of the V1/V2 variable loop structure in the exposure of human immunodeficiency virus type 1 gp120 epitopes induced by receptor binding. J. Virol. 69:5723–5733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zanetti G, Briggs JA, Grunewald K, Sattentau QJ, Fuller SD. 2006. Cryo-electron tomographic structure of an immunodeficiency virus envelope complex in situ. PLoS Pathog. 2:e83 doi:10.1371/journal.ppat.0020083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhang Z, Post CB, Smith DL. 1996. Amide hydrogen exchange determined by mass spectrometry: application to rabbit muscle aldolase. Biochemistry 35:779–791 [DOI] [PubMed] [Google Scholar]
- 80. Zhou T, et al. 2010. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science 329:811–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhou T, et al. 2007. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature 445:732–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhu P, et al. 2006. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature 441:847–852 [DOI] [PubMed] [Google Scholar]
- 83. Zwick MB, et al. 2003. A novel human antibody against human immunodeficiency virus type 1 gp120 is V1, V2, and V3 loop dependent and helps delimit the epitope of the broadly neutralizing antibody immunoglobulin G1 b12. J. Virol. 77:6965–6978 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.