

Abstract
Isoprenoid biosynthesis is essential for survival of all living organisms. More than 50,000 unique isoprenoids occur naturally, with each constructed from two simple five-carbon precursors: isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP). Two pathways for the biosynthesis of IPP and DMAPP are found in nature. Humans exclusively use the mevalonate (MVA) pathway, while most bacteria, including all Gram-negative and many Gram-positive species, use the unrelated methylerythritol phosphate (MEP) pathway. Here we report the development of a novel, whole-cell phenotypic screening platform to identify compounds that selectively inhibit the MEP pathway. Strains of Salmonella enterica serovar Typhimurium were engineered to have separately inducible MEP (native) and MVA (nonnative) pathways. These strains, RMC26 and CT31-7d, were then used to differentiate MVA pathway- and MEP pathway-specific perturbation. Compounds that inhibit MEP pathway-dependent bacterial growth but leave MVA-dependent growth unaffected represent MEP pathway-selective antibacterials. This screening platform offers three significant results. First, the compound is antibacterial and is therefore cell permeant, enabling access to the intracellular target. Second, the compound inhibits one or more MEP pathway enzymes. Third, the MVA pathway is unaffected, suggesting selectivity for targeting the bacterial versus host pathway. The cell lines also display increased sensitivity to two reported MEP pathway-specific inhibitors, further biasing the platform toward inhibitors selective for the MEP pathway. We demonstrate development of a robust, high-throughput screening platform that combines phenotypic and target-based screening that can identify MEP pathway-selective antibacterials simply by monitoring optical density as the readout for cell growth/inhibition.
INTRODUCTION
Antibiotic resistance, especially among Gram-negative bacteria, continues to be a serious public health concern. While considerable effort has been invested in developing new Gram-positive agents, significantly fewer programs or pipeline agents can be found for Gram-negative therapeutics. Carbapenems are among the top drugs for treating serious hospital-acquired (nosocomial) infections (NIs) caused by Gram-negative agents (39), but, unfortunately, the emergence of blaNDM-1, a plasmid expressing a metallo-ß-lactamase that confers resistance to virtually all β-lactam antibiotics, has increased the risk of resistance to members of this drug class (46). Multidrug-resistant Gram-negative bacteria now require the use of compounds that were long ago abandoned due to toxicity, such as colistin, which exhibits nephrotoxicity, due to a lack of anything better (27).
Aggravating the concern are increasing reports of antibiotic resistance in NIs, which add $4.5 billion to health care costs and are responsible for 90,000 deaths each year (27). More than 60% of NIs are caused by Gram-negative pathogens, with many of these resistant to most, if not all, available therapies. The Infectious Disease Society of America (IDSA) in its update of the “Bad Bugs, No Drugs” report discusses the therapeutic development needs for a group of pathogens that cause the majority of NIs (7). The IDSA authors lament the lack of new purely anti-Gram-negative agents in late stage clinical development and the correlative risk to patients. A separate report puts the concern plainly, saying “It is time to intensify attention to Gram-negative resistance” (39).
The methylerythritol phosphate (MEP) pathway for isoprenoid biosynthesis has been described as a promising new target for discovering novel Gram-negative antibacterial agents in this time, when there is an “innovation gap” for antibiotic discovery (42, 43). Isoprenoids are involved in numerous essential cellular processes in all organisms, such as cellular respiration, cell wall biosynthesis, photosynthesis, cell signaling, oxidative phosphorylation, and protein biosynthesis (6). All isoprenoids are produced from the essential precursors isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP). All Gram-negative bacteria, in addition to many Gram-positives and apicomplexan parasites, require the MEP pathway for IPP and DMAPP biosynthesis, while humans (and other eukaryotes) use the unrelated mevalonate (MVA) pathway. The MEP and MVA pathways have nonoverlapping chemical intermediates and biosynthetic enzymes (Fig. 1).
Fig 1.

MEP and MVA pathways for isoprenoid biosynthesis.
The MVA pathway has been well studied since the 1950s. The first two steps involve condensation reactions with acetyl-coenzyme A (acetyl-CoA) to produce acetoacetyl-CoA and hydroxymethyl glutaryl-CoA (HMG-CoA) (3, 30). HMG-CoA is reduced to create MVA, which is subsequently phosphorylated twice before it is decarboxylated to form IPP (1, 5). IPP isomerase (IDI) converts IPP to DMAPP (10). Unlike the MVA pathway, the MEP pathway was discovered only recently. DXP synthase was the first protein identified, in 1997 (40), and IspH (catalyzing the last two steps) was the last enzyme reported, in 2002 (2). The MEP pathway begins with the condensation of glyceraldehyde 3-phosphate (G3P) and pyruvate to produce 1-deoxy-d-xylulose 5-phosphate (DXP) by DXP synthase (DXS) followed by the MEP synthase (DXR)-catalyzed rearrangement and reduction of DXP to generate MEP (16, 20, 22, 25). MEP is converted to 2-C-methyl-d-erythritol 2,4-cyclodiphosphate (cMEDP) by the successive actions of 4-diphosphocytidyl-2-C-methyl-d-erythritol (CDP-ME) synthase (IspD), CDP-ME 2-phosphate (CDP-ME2P) synthase (IspE), and cMEDP synthase (IspF) (18, 26, 35). 1-Hydroxy 2-methyl-2-buten-4-yl diphosphate (HDMAPP) is formed by the IspG-catalyzed ring opening of cMEDP (34). IPP and DMAPP are ultimately formed in one step by IspH (34). Each of these enzymes has been demonstrated to be essential for cellular viability (41). A nonessential IDI likely maintains a balanced pool of IPP and DMAPP in organisms utilizing the MEP pathway (13).
The MEP pathway has been validated as an antibacterial target with fosmidomycin (Fos). In the 1980s, Fos was investigated clinically as an antibacterial, although its target was not known. In 1998, long after it was abandoned as an antibacterial candidate due to poor bioavailability, DXR was identified as the target (21). Fos has also demonstrated efficacy in treating the malaria parasite Plasmodium falciparum, which requires the MEP pathway for viability, with minimal side effects on humans (19, 24). However, since the discovery and elucidation of the MEP pathway, there has not been a single agent to have entered clinical development targeting it.
Each of the seven enzymes in the MEP pathway represents unexploited druggable targets that could yield new antibacterial classes. Here we report the development of a novel, whole-cell phenotypic screening platform for identifying compounds that selectively inhibit the MEP pathway. Here we report a genetically engineered Salmonella enterica serovar Typhimurium strain, CT31-7d, that has been constructed and formatted as part of a high-throughput screening (HTS) platform which was validated using two known MEP pathway-selective compounds: the previously described Fos and 5-ketoclomazone (5-KT), which inhibits DXS (15, 31).
CT31-7d was derived from strain RMC26 (41), which was engineered to have both the MEP and MVA pathways, each independently inducible. Construction of RMC26, which was engineered to have both the MEP and MVA pathways, each independently inducible, has been described elsewhere (41). Briefly, RMC26 has a lethal disruption (dxs::MVAoperon) in the MEP pathway, which was accomplished by inserting a synthetic mevalonate operon (MVAoperon) into the chromosomal copy of the gene encoding DXS. The MVAoperon is under the control of an arabinose-inducible promoter (PBAD) and contains three genes encoding the proteins responsible for converting MVA to IPP: MVA kinase, phospho-MVA (PMVA) kinase, and MVA diphosphate decarboxylase. A kanamycin resistance (Kanr) cassette was included in the insertion to facilitate selection of cells harboring an insertion. Viability of RMC26 can be restored by supplementing the growth medium with 1-deoxy-d-xylulose (DX) or 2-C-methyl-d-erythritol (ME), which have been shown to be taken up by the cell and subsequently phosphorylated, thereby reconstituting the MEP pathway (4, 11, 33, 36, 38, 41). Alternatively, cell viability may be restored by utilizing the engineered MVA pathway by supplementation with MVA and arabinose (MVA/ara). In the absence of ara and MVA, RMC26 is unable to propagate, as the operon is not turned on. A compound that specifically inhibits the MEP pathway elicits a phenotype of growth with MVA/ara and no growth with DX.
RMC26 lacks DXS and as a result is unable to identify inhibitors of this step of the MEP pathway. While DXS enzymes exhibit high levels of sequence similarity to mammalian transketolases, the crystal structure has revealed that the structural domains have a novel arrangement in bacteria, suggesting a potential druggable target (45). As a result, CT31-7d was created to enable identification of DXS inhibitors, in addition to the six other MEP pathway enzymes, by the introduction of a plasmid-encoded DXS to RMC26. The gene encoding DXS was cloned from Bacillus anthracis and inserted into the isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible and ampicillin (Amp)-resistant plasmid pTrcHis2a, creating pCT25, which was subsequently introduced into RMC26, creating CT31-7d. CT31-7d is still unable to utilize the MVA pathway unless provided exogenous MVA and ara. Identification of MEP pathway-selective inhibitors can be accomplished by screening compound collections and evaluating their effects on MEP pathway growth compared to MVA pathway growth (Fig. 2). Compounds that inhibit MEP pathway growth but not MVA pathway growth represent MEP-selective antibacterials (Fig. 2, rows A and B). Compounds affecting growth of both pathways represent antibacterials that act on a target other than the MEP pathway (Fig. 2, rows C and D), while compounds not affecting the growth of either pathway are not antibacterial (Fig. 2, rows E to H). The screening platform enables identification of inhibitors of any of the seven steps of the MEP pathway. Importantly, hits in screens using our platform yielded three results: (i) the inhibitors are antibacterial and able to cross the S. Typhimurium cell wall; (ii) they target the MEP pathway selectively; and (iii) they do not affect the MVA pathway.
Fig 2.
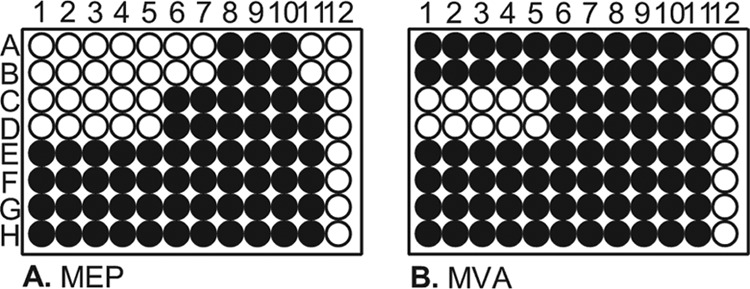
Expected screening results for the MEP screening platform. Rows A and B represent an MEP pathway-selective inhibitor and rows C and D an antibacterial that is not selective for the MEP pathway; rows E and F and rows G and H are not antibacterial. Wells A11 and B11 are Fos controls, C11 to H11 are no-inhibitor controls, and column 12 represents media blanks. ● = cell growth; ○ = no cell growth.
MATERIALS AND METHODS
Materials and general methods.
Medium (LB broth and agar, Mueller-Hinton broth), pTrcHis2a, an Easy-DNA kit, and Fos were purchased from Invitrogen (Carlsbad, CA). Mevalonolactone, l-arabinose (ara), IPTG, and antibiotics were purchased from Sigma-Aldrich (St. Louis, MO). 5-KT, DX, and ME are commercially available from Echelon Biosciences, Inc. (Salt Lake City, UT). MVA was prepared by hydrolysis of 1 vol of 1 M mevalonolactone with 1.02 vol of 1 M KOH followed by incubation of the mixture at 37°C for 30 min. MVA was used at a final concentration of 5 mM with 0.04% ara, except where noted. DX was used at 5 μM, except where noted. Kanamycin (Kan) was used at 40 μg/ml and ampicillin (Amp) at 100 μg/ml. AeraSeal sterile breathable sealing films were obtained from Excel Scientific (Victorville, CA). All assays were performed using clear 96-well microtiter plates (MTPs) from Nunc (Rochester, NY). MICs were determined using a slight modification of the broth microdilution method (32).
Construction of strain CT31-7d.
Genomic DNA was isolated from B. anthracis (Sterne 34F2 strain) using an Easy-DNA kit per the manufacturer's instructions and used for PCR amplification of dxs. Primers were designed to introduce NcoI and BglII sites at the 5′ and 3′ termini, respectively, for subcloning into the corresponding sites of pTrcHis2a, creating pCT25. S. Typhimurium strain RMC26 was transformed with pCT25 to create CT31-7d using standard protocols (37).
RMC26 growth assay optimization.
Well-isolated colonies of RMC26 were picked from LB/Kan/DX plates and used to inoculate 3 ml of LB/Kan/DX. The same colony was used to inoculate 3 ml of LB/Kan/MVA/ara. Cultures were grown for 18 h at 37°C with shaking at 250 rpm (1:10 dilution of cultures typically exhibited an absorbance at 600 nm [A600] of ∼0.250 and corresponded to ∼5 × 107 CFU/ml compared to the McFarland standard). Cell suspensions for screening were prepared by adding 1 μl overnight (o/n) culture per 1 ml LB/Kan (which makes a 2× cell suspension of 5 × 105 CFU/ml). DX concentrations (0, 1, 2, 5, 10, and 100 μM) for MEP pathway growth and MVA (0, 1, 5, 10 and 50 mM) plus ara (0, 0.001, 0.005, 0.01, 0.05 and 0.1%) concentrations were investigated for screening. Growth evaluation was performed in clear 96-well plates using a Biomek 2000 workstation for most pipetting steps. Plates are covered with sterile breathable seals and incubated at 37°C and 80% humidity with shaking at 350 rpm for 20 h using a Multitron II orbital shaker (Appropriate Technical Resources, Inc., Laurel, MD). Growth was determined by reading absorbance at 600 nm (A600) using a SpectraMax M2 plate reader (Molecular Devices, Sunnyvale, CA).
The effect of dimethyl sulfoxide (DMSO) was evaluated by performing serial dilutions using both MEP and MVA growth conditions. Aliquots (150 μl) of LB/Kan plus 4% DMSO were added to wells in column 1 in each of 2 MTPs. LB/Kan was added (75 μl) to all wells in columns 2 to 11, with 150 μl LB/Kan added to wells in column 12 (media blanks). The contents of wells in column 1 were mixed followed by transfer of 75 μl from column 1 to column 2; the contents of the wells in column 2 were mixed, and then 75 μl was transferred to column 3. This process is repeated until column 10 was reached, with the remaining 75 μl discarded. A MEP or MVA cell suspension (75 μl of a 2× concentration) was added to all wells in columns 1 to 11, and plates were incubated as described above. The concentrations of MVA evaluated were 1, 2, and 5 mM, with 0.01%, 0.04%, and 0.08% ara also evaluated for each MVA concentration examined. The DMSO concentration range evaluated for each combination of MVA plus ara was 0.0039% to 2%.
CT31-7d growth assay optimization.
Well-isolated colonies of CT31-7d were picked from LB/Kan/Amp plates and used to inoculate liquid cultures of both LB/Kan/Amp and LB/Kan/Amp/MVA/ara using the same inoculum. Cultures were grown and screened as described above, except MEP cell suspensions were prepared using LB/Kan/Amp. Various concentrations of IPTG created by serial dilution starting at 20 μM (final dilution of 0.0039 μM) and a preparation without IPTG were investigated for their effect on growth of CT31-7d. Various concentrations of IPTG (0, 2, 20, and 200 μM) were used to investigate the effect on the MIC of Fos against CT31-7d. Serial dilutions were performed using the indicated concentrations of IPTG starting at 0.2 μg/ml Fos. DMSO was evaluated as described for RMC26, using the corresponding CT31-7d growth media.
Activity of known MEP pathway inhibitors against RMC26 or CT31-7d.
MEP and MVA cultures of RMC26 were prepared as described above using final concentrations of 5 μM DX or 5 mM MVA plus 0.04% ara. MEP and MVA cultures of CT31-7d were prepared as described above. Compound stocks were prepared in DMSO, with a final DMSO concentration of ≤2%. Compound solutions (2×) were prepared by adding 25 μl compound stock to 600 μl medium (LB/Kan for RMC26 or LB/Kan/Amp for CT31-7d). To perform the screening, 150 μl of 2× compound solution was manually added to 2 wells in column 1 of a 96-well MTP (4 compounds/plate) for evaluation in duplicate. Medium (75 μl) was added to all wells in columns 2 to 10 of the MTP, and 150 μl was added to all wells in column 12 (media blank). Compounds were serially diluted by mixing column 1 followed by transfer of 75 μl to column 2. This was repeated through column 11. The remaining 75 μl of material from column 11 was discarded. The process was repeated for a second MTP. An MEP or MVA cell suspension (75 μl of 2×) was added to all wells in columns 1 to 11. 5-KT screening used a starting concentration of 2 μg/ml, while Fos screening used 0.2 μg/ml as the initial concentration (except in the investigation of MVA inhibition, which started at 1,000 μg/ml).
To further demonstrate the utility of our cell lines, DX (10 μM) and ME (5 and 25 μg/ml) were used to supplement growth media for CT31-7d for incubation with Fos (100 μg/ml starting concentration). Inhibition of growth was investigated as described above.
Activity of known antibacterials against RMC26 and CT31-7d.
Plates and cultures were prepared as described above for MEP pathway inhibitors. Fos was added to wells A11 and B11 as a positive control (3 μM final concentration). This concentration of Fos was selected as it inhibits CT31-7d and is ineffective at inhibiting the growth of any wild-type strains. Percent inhibition was determined by comparing bacterial growth to no-inhibitor controls. Values corresponding to the absence of inhibition were obtained by averaging the value for no-inhibitor wells (C11 to H11) and subtracting the value for the average for medium blank wells (A12 to H12). MIC values were defined as the lowest concentration causing >90% inhibition of bacterial growth for all wells, relative to no-inhibitor control results (measuring A600). The initial screening concentrations (column 1) are listed in Table 1.
Table 1.
Antibiotics screeneda
| Antibiotic | Initial concn (μg/ml) | MIC (μg/ml) |
||||
|---|---|---|---|---|---|---|
| RMC26 |
CT31-7d |
S. Typhimurium LT2 | ||||
| MEP | MVA | MEP | MVA | |||
| Ampicillin | 200 | 2.5 | 2.5 | NA | NA | 0.8–1.6 |
| Carbenicillin | 200 | 20 | 200 | NA | NA | 100 |
| Streptomycin | 200 | 50 | 100 | 100 | 100 | 12.5 |
| Neomycin | 200 | NA | NA | NA | NA | 25 |
| Azithromycin | 20 | 0.31–2.5 | 5–10 | 5 | 5 | 12.5 |
| Clindamycin | 200 | 0.16–0.63 | 2.5–5 | NA | NA | 2.5–5 |
| Erythromycin | 200 | 50–100 | 100–200 | 25–50 | 50 | 100–200 |
| Clarithromycin | 200 | 25–50 | 100 | 25–50 | 50 | 100–200 |
| Lincomycin | 200 | 12.5 | NA | 25–50 | 200 | NA |
| Tetracycline | 2 | 0.25–0.5 | 0.5–2 | 2 | 2 | 0.5 |
| Doxycycline | 20 | 0.08–0.6 | 0.63–1.25 | 1.25–2.5 | 1.25 | 0.5 |
| Trimethoprim | 20 | 2.5 | 20 | 1.25–2.5 | 1.25–2.5 | 0.5 |
| Chloramphenicol | 20 | 1.25 | 2.5 | 1.25–2.5 | 1.25–2.5 | 5 |
| Ciprofloxacin | 0.08 | 0.005–0.01 | 0.01–0.02 | 0.01 | 0.01 | 0.02 |
| Levofloxacin | 2 | 0.06–0.25 | 0.25–0.5 | 0.03 | 0.03 | 0.03 |
| Nalidixic acid | 20 | 1.25 | 2.5 | 2.5 | 2.5 | 1.25–5 |
| Fosfomycin | 100 | (-) | (-) | 1 | 1 | (-) |
| Fosmidomycin | Var. | 0.2 | NA | 0.01 | NA | 10 |
MICs were determined as the lowest concentration that inhibited bacterial growth > 90%, as determined by A600 values relative to no-inhibitor control results. A minimum of 3 independent experiments, each run in duplicate, were used to determine MICs. Plates were incubated at 37°C and 80% humidity with shaking at 250 rpm. Plates were read after 18 h. NA = no activity; (-) = not screened.
Comparison of MICs to wild-type S. Typhimurium.
Activities of known antibacterials against RMC26 and CT31-7d were compared to those against S. Typhimurium LT2 (ATCC 19585; parent strain of RMC26). Cultures grown on LB agar plates were used to inoculate 3 ml of medium (LB or cation-adjusted Mueller-Hinton broth [CAMHB]). Liquid cultures were grown overnight (18 to 20 h) at 37°C and then added to fresh medium to produce a 2× screening culture (1 × 106 CFU/ml compared to the McFarland standard). Serial dilutions were performed as described above. Antibacterials were evaluated in duplicate, and experiments were performed independently at least three times.
RESULTS
Construction of an MEP pathway mutant cell line for antibacterial screening.
As previously reported, RMC26 is unable to propagate unless the growth medium is supplemented to circumvent the lack of DXS either by restoring the MEP pathway (DX or ME) or by utilizing the MVA pathway (MVA and ara). CT31-7d was constructed by transforming RMC26 with pCT25, a pTrcHis-derived plasmid bearing dxs from B. anthracis. The plasmid pCT25 was constructed so that transcription of the dxs gene was under the control of IPTG-inducible trc promoter, facilitating growth through the MEP pathway. Alternatively, the MVA pathway originally engineered into RMC26 may be turned on by adding both MVA and ara. The presence of the pTrcHis plasmid also confers ampicillin resistance in addition to kanamycin resistance of RMC26.
Optimization and validation of MEP pathway mutant cell lines for antibacterial screening.
While RMC26 was previously described, optimization of growth conditions was not reported and its use in antibacterial screening has not been described. Therefore, optimal concentrations of DX or ME for MEP pathway growth and MVA and ara for MVA pathway growth were determined. Substrate concentrations, of 5 μM DX and 5 mM MVA plus 0.04% ara, were selected as they were the lowest concentrations that afforded growth at an A600 of ∼0.9 and <10% inhibition by 2% DMSO under both sets of growth conditions (data not shown).
MEP pathway growth for CT31-7d was evaluated by adjusting the concentration of IPTG (Fig. 3). We found that even in the absence of IPTG, CT31-7d was viable. This was likely due to the leakiness of the trc promoter from the pTrcHis plasmid (47), whereby endogenous levels of induction are suitable for cell growth. Adjusting the level of IPTG induction had a significant effect on the MIC of Fos against CT31-7d (Fig. 4). As the level of IPTG was increased, thereby increasing the level of growth through the MEP pathway, the MIC of Fos increased. DXS precedes DXR and has been implicated as the rate-limiting step of the MEP pathway (23), so this result would be expected, especially given the report by DeVito et al. of a study where they engineered antibacterial screening strains with increased susceptibility to target-selective compounds by modulating expression of target proteins (9). As there were no changes to the engineered MVA operon for CT31-7d, the optimal concentrations of MVA and ara (5 mM and 0.04%, respectively) determined for RMC26 were used. In the absence of MVA and ara, both RMC26 and CT31-7d were unable to propagate using the MVA pathway. DMSO was well tolerated by both cell lines, with no effect at up to a 2% final concentration for both MEP and MVA pathway growth (data not shown).
Fig 3.
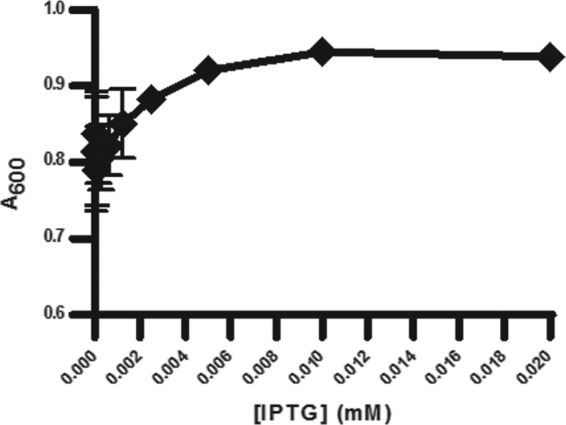
Effect of adjusting [IPTG] on CT31-7d growth. Cultures were grown in LB/Kan/Amp with various concentrations of IPTG (serial dilutions starting at 0.02 mM and ending at 0.00039 mM) and no IPTG. Plates were incubated at 37°C and 80% humidity with shaking at 250 rpm for 18 h, at which point A600 values were determined. Error bars represent the averages of four independent data points.
Fig 4.
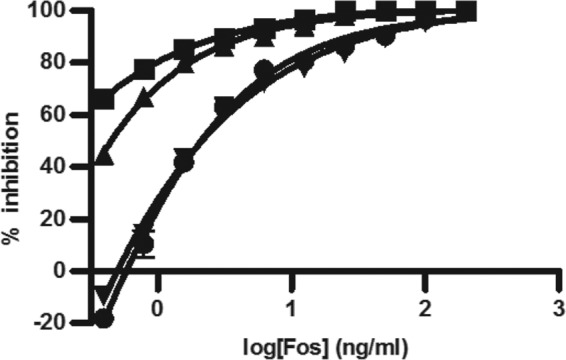
Adjusting [IPTG] for CT31-7d growth affects Fos MIC. Cultures were grown in LB/Kan/Amp with various concentrations of Fos (serial dilutions starting at 200 ng/ml and ending at 0.39 ng/ml). Plates were incubated at 37°C and 80% humidity with shaking at 250 rpm for 18 h, at which point A600 values were determined. Error bars represent the averages of four independent data points. ■ = 0 mM IPTG, ▲ = 0.002 mM IPTG, ● = 0.02 mM IPTG, ▼ = 0. 2 mM IPTG.
For screening chemical libraries, the platform is capable of evaluating 2,560 compounds in duplicate (32 plates) per day.
Activity of known MEP pathway inhibitors against RMC26 or CT31-7d.
The use of the MEP pathway inhibitor Fos or 5-KT requires activation of the MVA pathway to restore viability of CT31-7d. For both RMC26 and CT31-7d, DX supplementation of the growth medium was unable to restore viability when Fos was included in the growth medium, and yet both cell lines were able to grow when provided ME (Fig. 5). As expected, inhibition of CT31-7d by 5-KT was restored by supplementation with ME.
Fig 5.
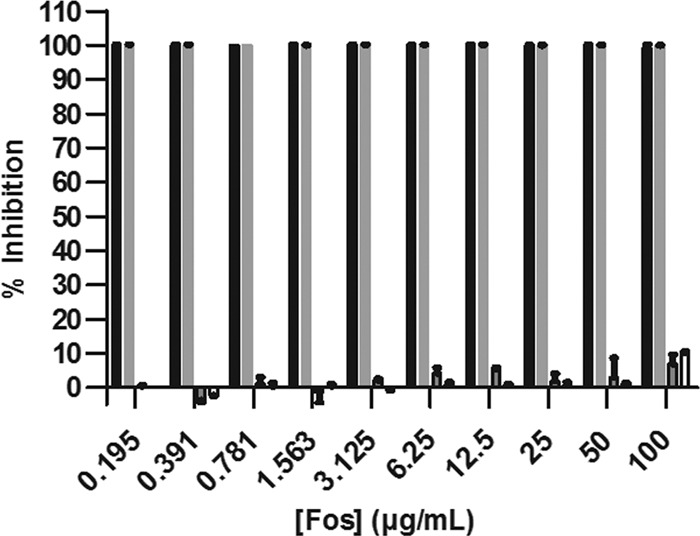
Growth of CT31-7d with or without 100 μg/ml Fos in LB/Kan/Amp and with or without DX and ME supplementation. Black bars = no supplements plus Fos; gray bars = 10 μM DX plus Fos; boxed gray bars = 25 μg/ml ME plus Fos; boxed white bars = 5 mM MVA plus 0.04% ara plus Fos. Cultures were grown in LB/Kan/Amp with the indicated supplements at 37°C and 80% humidity with shaking at 250 rpm for 18 h, at which point A600 values were determined. Values represent the means ± standard errors of duplicate data points from two independent experiments.
Activity of common antibiotics and comparison to wild-type S. Typhimurium results.
To validate the performance of CT31-7d, common antibiotics among several classes were examined. MICs for all compounds were the same for all organisms when LB or CAMHB was used as the growth medium. In most cases, the antibiotics demonstrated MICs against RMC26 and CT31-7d similar to those seen with S. Typhimurium LT2 (Table 1). Perhaps more importantly, MICs for MEP versus MVA growth for the two cell lines were equivalent. Additionally, the MEP pathway inhibitors Fos and 5-KT were evaluated. We observed that both RMC26 and CT31-7d were more sensitive to Fos than LT2, as reflected in the MIC values. MICs for RMC26 and CT31-7d MEP growth were 0.2 and 0.01 μg/ml, respectively, while the MIC for LT2 was 10 μg/ml. The MIC for 5-KT against CT31-7d was 0.06 μg/ml, and 5-KT was inactive against LT2 (up to 1,000 μg/ml). Activity of 5-KT against RMC26 was not investigated, as it lacks the intracellular target, DXS. CT31-7d, even with constitutive MEP pathway activation due to the leaky trc promoter, was more sensitive to Fos and 5-KT than wild-type LT2 and even RMC26. The quality of the screen of antibacterials and MEP pathway inhibitors was excellent, as reflected by average z′ values of 0.917 ± 0.094 (n = 128) for RMC26 and 0.927 ± 0.051 for CT31-7d (n = 114).
DISCUSSION
Whole-cell phenotypic screens offer the ability to identify inhibitors of a pathway of interest that are able to permeate cells and affect the target. We have developed such a system for identifying inhibitors that are selective against any step in the MEP pathway for isoprenoid biosynthesis using S. Typhimurium strain CT31-7d. The MEP pathway is an attractive target for development of new antimicrobial agents. It is an essential pathway in most bacteria and apicomplexan parasites such as P. falciparum, the causative agent of most human malaria. Of significance to the platform developed is that it utilizes a genetically engineered cell line that is capable of using both the MEP pathway and the nonnative MVA pathway. The only difference between the growth conditions is simply the pathway that is activated. The platform is a simple, robust system relying on optical density for bacterial growth as the readout, thereby facilitating broad utility. We developed the platform using 96-well plates, but this system could easily be adapted to higher-density MTPs.
Two related cell lines are described that can be used for high-throughput screening of chemical collections. Both were validated using the two available known MEP pathway inhibitors; 5-KT and Fos. Both of the MEP pathway mutant cell lines demonstrated increased sensitivity to Fos and 5-KT. Clomazone is an herbicide that has been used for several years, and yet its metabolite 5-KT is responsible for activity (15, 31). Clomazone is converted to 5-KT by the action of plant cytochrome P450 enzymes, which are likely responsible for herbicidal activity (12). While 5-KT has been known to inhibit plant growth, it has very little activity against bacterial cells. A recent report has shown that it has modest activity against Haemophilus influenzae, with a 12.5 μg/ml MIC (28). Those authors report an MIC of 800 μg/ml against Escherichia coli, which correlates with the lack of activity that we observed for S. Typhimurium reported above. We have also not seen any activity of 5-KT against E. coli (unpublished data). A known efflux pump inhibitor, phenylalanine-arginine β-naphthylamide, did not affect the activity against LT2, suggesting that 5-KT resistance is imparted by a mechanism other than efflux (data not shown).
Fos was evaluated as an antibacterial in the 1970s and 1980s, long before the first report of an MEP pathway enzyme (DXP synthase was reported in 1997 [40]). Kinetic analysis of Fos activity against DXR has revealed that it acts as a competitive inhibitor against DXP (20). A recent publication reports that E. coli is also inhibited at IspD when incubated with Fos, but the authors suggest that this may be indirectly due to Fos (48). They reported that Fos was effective against IspD only at very high concentrations, as reflected by a 50% inhibitory concentration (IC50) of 20.4 mM, and that IspD inhibition may be due to accumulation of a DXR reaction intermediate. In our platform, the appearance of slight (<10%) inhibition of MVA growth at 100 μg/ml Fos as shown in Fig. 5 was likely due to the DMSO used as a solvent. This interpretation is supported by evaluation of Fos concentrations of up to 1,000 μg/ml, which demonstrated <10% inhibition of MVA growth as well (data not shown). Growth conditions for the cell lines were adjusted so that the MEP pathway expression level was attenuated. This rendered them more susceptible to MEP pathway inhibitors, as demonstrated in Fig. 4, where adjusting levels of IPTG induction of DXS affected MICs of Fos.
Both RMC26 and CT31-7d were further evaluated using several known antibiotics, selected from commonly used classes, and compared for activity against S. Typhimurium strain LT2, the parent strain of RMC26 and CT31-7d. Lower or no susceptibility of RMC26 and CT31-7d to aminoglycosides was expected given the engineered resistance to the aminoglycoside kanamycin as part of the MVA operon. Resistance of CT31-7d to ampicillin and carbenicillin is conferred by pCT25, carrying the pTrcHis-encoded dxs gene. It is interesting that our engineered cell lines showed susceptibilities to the macrolides tested that were slightly different from those seen with LT2 under the growth conditions used. This observation requires additional investigation. With this exception, all of the antibiotics tested demonstrated MICs comparable to those seen with wild-type S. Typhimurium. Increased susceptibility to Fos and 5-KT is not surprising, given that growth through the MEP pathway has been attenuated and selected so that it is turned on at a very low level, as determined by the relative levels of cell growth. This is similar to the reported example by DeVito et al. discussed above (9), whose strains had target proteins downregulated, thereby rendering them more sensitive to inhibitors with that enzyme as their target. The cell lines described in this work have the MEP pathway downregulated by limiting the level at which DXP is available. DXS has been reported to represent a rate-limiting step of the MEP pathway in several organisms (8, 14, 17, 29). As a result, altering the levels of DXP available for DXR would be expected to have significant effects on cell viability. In the case of RMC26, the DXS enzyme is lacking due to its disruption with the MVA operon. As a result, the only source of DXP is that of supplementation with DX, which has been shown to be transported into bacterial cells and subsequently phosphorylated by the protein encoded by xylB to produce DXP independently of the MEP pathway (44). As a result, the pool of available DXP for the enzyme target of Fos, DXR, is limited to what is supplied in the growth medium. This situation is related to that described by Devito et al., except that the available substrate was limited instead of enzyme levels. For CT31-7d, DXP levels are limited by the level to which DXS is expressed, which is directly related to the amount of IPTG provided. Higher concentrations of IPTG corresponded to higher levels of DXS expression and thereby higher levels of DXP available for DXR. The heightened sensitivity of CT31-7d to Fos relative to RMC26 was surprising, given that its MEP pathway growth is dependent solely on endogenous induction of the pTrc plasmid. It is possible that lower levels of DX supplementation of RMC26 would render this cell line as sensitive as CT31-7d to Fos. ME supplementation results in conversion to MEP, which bypasses the inhibition of Fos. It is possible that attenuating levels of ME would have a similar effect on inhibitors downstream of DXR. However, there are no available inhibitors with antibacterial activity that are active against downstream enzymes.
Supplementation experiments with DX and ME have the ability to narrow the target of a selected MEP pathway inhibitor. DX conversion to DXP is discussed above, while ME has been shown to be transported into bacterial cells and phosphorylated by the sorbitol phosphotransferase system (41). MEP pathway inhibitors that have DXR as their target can be identified by feeding experiments using DX and ME (Fig. 6). DX supplementation was unable to restore viability when Fos was presented, as the MEP pathway is restored prior to the target enzyme DXR. ME, however, is capable of restoring cell growth, as it is converted to MEP independently of the MEP pathway (via the sorbitol transferase system). The ability to restore viability with ME supplementation of the growth medium and the lack of growth seen with DX supplementation in the presence of Fos are phenomena that are expected for any DXR inhibitor.
Fig 6.

Schematic of feeding experiments with a DXR inhibitor.
Identification of MEP pathway-selective inhibitors from compound collections using the cell lines can be accomplished by the following process. Large compound collections should be screened first with only the MEP pathway active to identify any antibacterial compound in a primary screen. This would eliminate ∼99% of the collection for secondary screening against both pathways, thereby conserving time and resources. Our internal discovery efforts have found an antibacterial hit rate of ∼1% in screening ∼275,000 compounds (data to be reported elsewhere). Antibacterial hits from the primary screen can then be cherry-picked and subjected to a secondary screen for identifying MEP pathway-selective agents using RMC26 and/or CT31-7d and evaluating both MEP and MVA growth conditions. In this secondary screen, hits are evaluated in one plate with only the MEP pathway activated and separately in a second plate with the MVA pathway turned on. For MEP pathway selectivity, compounds can be evaluated for dose response (DR) by serial dilution of hits. Those that inhibit MEP but not MVA pathway growth represent MEP-selective antibacterials. Compounds affecting growth of both pathways represent antibacterials that act on a target other than the MEP pathway. Subsequent screening with RMC26 enables filtering out any compounds that interfere with the trc promoter rather than inhibition of the MEP pathway.
Given the presence of a functional DXS enzyme, CT31-7d was determined to be the optimal cell line to use for antibacterial screening. The use of DX supplementation with RMC26 was able to identify inhibitors of the MEP pathway for all steps except for DXS, which may also be a viable antibacterial target as discussed above. In addition, CT31-7d is more sensitive to the MEP pathway inhibitors Fos and 5-KT than RMC26 and wild-type S. Typhimurium, indicating that it may be able to select less-potent inhibitors that could be optimized using medicinal chemistry to generate leads that could otherwise be missed if the less-sensitive RMC26 were used for screening.
The screening platform enables identification of inhibitors of any of the seven steps of the MEP pathway. The specific enzyme target can subsequently be identified by substrate feeding experiments (using DX and/or ME) and/or biochemical assays for the individual enzyme. Importantly, hits in screens using our platform yield three results: (i) they are antibacterial and able to permeate the S. Typhimurium cell; (ii) they target the MEP pathway selectively; and (iii) they do not affect the MVA pathway. The increased sensitivity of our screening platform to MEP pathway-specific compounds is expected to facilitate identification of weak inhibitors that are able to permeate the robust Gram-negative cell wall and would likely be missed using wild-type organisms. In fact, we have identified one such scaffold that we are currently pursuing (data to be reported elsewhere). The fact that the same cell line is used for both growth conditions with the only difference being the supplements utilized for growth is expected to minimize the frequency of identifying common promiscuous inhibitors, thereby saving development time.
This report describes development of a sensitive, robust whole-cell screening platform that is expected to provide a powerful tool to identify antibacterial leads against an exciting new antibacterial target pathway. In addition, the platform will be valuable for guiding hit-to-lead optimization efforts of MEP pathway inhibitors.
ACKNOWLEDGMENTS
We thank Glenn Prestwich for helpful discussion and Mark Nelson for critical review of the manuscript.
The work described was supported in part by grants R43AI077220 and R43AI078554 from the National Institute of Allergy and Infectious Diseases.
The content is solely our responsibility and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
Footnotes
Published ahead of print 9 July 2012
REFERENCES
- 1. Agranoff BW, Eggerer H, Henning U, Lynen F. 1960. Biosynthesis of terpenes. VII. Isopentenyl pyrophosphate isomerase. J. Biol. Chem. 235:326–332 [PubMed] [Google Scholar]
- 2. Altincicek B, et al. 2002. LytB protein catalyzes the terminal step of the 2-C-methyl-D-erythritol-4-phosphate pathway of isoprenoid biosynthesis. FEBS Lett. 532:437–440 doi:10.1016/S0014-5793(02)03726-2 [DOI] [PubMed] [Google Scholar]
- 3. Amdur BH, Rilling H, Bloch K. 1957. The enzymatic conversion of mevalonic acid to squalene. J. Am. Chem. Soc. 79:2646–2647 [Google Scholar]
- 4. Arigoni D, et al. 1997. Terpenoid biosynthesis from 1-deoxy-D-xylulose in higher plants by intramolecular skeletal rearrangement. Proc. Natl. Acad. Sci. U. S. A. 94:10600–10605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bloch K, Chaykin S, Phillips AH, de Waard A. 1959. Mevalonic acid pyrophosphate and isopentenyl pyrophosphate. J. Biol. Chem. 234:2595–2604 [PubMed] [Google Scholar]
- 6. Bochar DA, Freisen JA, Stauffacher CV, Rodwell VW. 1999. Biosynthesis of mevalonic acid from acetyl-CoA, p 15–44 In Cane D. (ed), Comprehensive natural products chemistry. Pergamon Press, Oxford, United Kingdom [Google Scholar]
- 7. Boucher HW, et al. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48:1–12 [DOI] [PubMed] [Google Scholar]
- 8. Brown AC, Eberl M, Crick DC, Jomaa H, Parish T. 2010. The nonmevalonate pathway of isoprenoid biosynthesis in Mycobacterium tuberculosis is essential and transcriptionally regulated by Dxs. J. Bacteriol. 192:2424–2433 doi:10.1128/JB.01402-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DeVito JA, et al. 2002. An array of target-specific screening strains for antibacterial discovery. Nat. Biotechnol. 20:478–483 doi:10.1038/nbt0502-478 [DOI] [PubMed] [Google Scholar]
- 10. Dhe-Paganon S, Magrath J, Abeles RH. 1994. Mechanism of mevalonate pyrophosphate decarboxylase: evidence for a carbocationic transition state. Biochemistry 33:13355–13362 doi:10.1021/bi00249a023 [DOI] [PubMed] [Google Scholar]
- 11. Duvold T, Cali P, Bravo JM, Rohmer M. 1997. Incorporation of 2-C-methyl-D-erythritol, a putative isoprenoid intermediate in the mevalonate-independent pathway, into ubiquinone and menaquinone of Escherichia coli. Tetrahedron Lett. 38:4769–4772 doi:10.1016/S0040-4039(97)01045-9 [Google Scholar]
- 12. ElNaggar SP, Creekmore RW, Schocken MJ, Rosen RT, Robinson RA. 1992. Metabolism of clomazone herbicide in soybean. J. Agric. Food Chem. 40:880–883 doi:10.1021/jf00017a036 [Google Scholar]
- 13. Eoh H, Brennan PJ, Crick DC. 2009. The Mycobacterium tuberculosis MEP (2C-methyl-D-erythritol 4-phosphate) pathway as a new drug target. Tuberculosis (Edinb.) 89:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Estévez JM, Cantero A, Reindl A, Reichler S, Leon P. 2001. 1-Deoxy-D-xylulose-5-phosphate synthase, a limiting enzyme for plastidic isoprenoid biosynthesis in plants. J. Biol. Chem. 276:22901–22909 [DOI] [PubMed] [Google Scholar]
- 15. Ferhatoglu Y, Barrett M. 2006. Studies of clomazone mode of action. Pesticide Biochem. Physiol. 85:7–14 [Google Scholar]
- 16. Hahn FM, et al. 2001. 1-Deoxy-D-xylulose 5-phosphate synthase, the gene product of open reading frame (ORF) 2816 and ORF 2895 in Rhodobacter capsulatus. J. Bacteriol. 183:1–11 doi:10.1128/JB.183.1.1-11.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harker M, Bramley PM. 1999. Expression of prokaryotic 1-deoxy-D-xylulose-5-phosphatases in Escherichia coli increases carotenoid and ubiquinone biosynthesis. FEBS Lett. 448:115–119 doi:10.1016/S0014-5793(99)00360-9 [DOI] [PubMed] [Google Scholar]
- 18. Herz S, et al. 2000. Biosynthesis of terpenoids: YgbB protein converts 4-diphosphocytidyl-2-C-methyl-D-erythritol 2-phosphate to 2-C-methyl-D-erythritol 2,4-cyclodiphosphate. Proc. Natl. Acad. Sci. U. S. A. 97:2486–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jomaa H, et al. 1999. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science 285:1573–1576 doi:10.1126/science.285.5433.1573 [DOI] [PubMed] [Google Scholar]
- 20. Koppisch AT, Fox DT, Blagg BS, Poulter CD. 2002. E. coli MEP synthase: steady-state kinetic analysis and substrate binding. Biochemistry 41:236–243 doi:10.1021/bi0118207 [DOI] [PubMed] [Google Scholar]
- 21. Kuzuyama T, Shimizu T, Takahashi S, Seto H. 1998. Fosmidomycin, a specific inhibitor of 1-deoxy-D-xylulose 5-phosphate reductoisomerase in the nonmevalonate pathway for terpenoid biosynthesis. Tetrahedron Lett. 39:7913–7916 doi:10.1016/S0040-4039(98)01755-9 [Google Scholar]
- 22. Kuzuyama T, Takahashi S, Watanabe H, Seto H. 1998. Direct formation of 2-C-methyl-D-erythritol 4-phosphate from 1-deoxy-D-xylulose 5-phosphate from 1-deoxy-D-xylulose 5-phosphate reductoisomerase, a new enzyme in the non-mevalonate pathway to isopentenyl diphosphate. Tetrahedron Lett. 39:4509–4512 doi:10.1016/S0040-4039(98)00802-8 [Google Scholar]
- 23. Kuzuyama T, Takagi M, Takahashi S, Seto H. 2000. Cloning and characterization of 1-deoxy-D-xylulose 5-phosphate synthase from Streptomyces sp. strain CL190, which uses both the mevalonate and nonmevalonate pathways for isopentenyl diphosphate biosynthesis. J. Bacteriol. 182:891–897 doi:10.1128/JB.182.4.891-897.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lell B, et al. 2003. Fosmidomycin, a novel chemotherapeutic agent for malaria. Antimicrob. Agents Chemother. 47:735–738 doi:10.1128/AAC.47.2.735-738.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lois LM, et al. 1998. Cloning and characterization of a gene from Escherichia coli encoding a transketolase-like enzyme that catalyzes the synthesis of D-1-deoxyxylulose 5-phosphate, a common precursor for isoprenoid, thiamin, and pyridoxol biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 95:2105–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lüttgen H, et al. 2000. Biosynthesis of terpenoids: YchB protein of Escherichia coli phosphorylates the 2-hydroxy group of 4-diphosphocytidyl-2-C-methyl-D-erythritol. Proc. Natl. Acad. Sci. U. S. A. 97:1062–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maragakis LL. 2010. Recognition and prevention of multidrug-resistant Gram-negative bacteria in the intensive care unit. Crit. Care Med. 38:S345–S351 doi:10.1097/CCM.0b013e3181e6cbc5 [DOI] [PubMed] [Google Scholar]
- 28. Matsue Y, et al. 2010. The herbicide ketoclomazone inhibits 1-deoxy-D-xylulose 5-phosphate synthase in the 2-C-methyl-D-erythritol 4-phosphate pathway and shows antibacterial activity against Haemophilus influenzae. J. Antibiot. (Tokyo) 63:583–588 [DOI] [PubMed] [Google Scholar]
- 29. Matthews PD, Wurtzel ET. 2000. Metabolic engineering of carotenoid accumulation in Escherichia coli by modulation of the isoprenoid precursor pool with expression of deoxyxylulose phosphate synthase. Appl. Microbiol. Biotechnol. 53:396–400 [DOI] [PubMed] [Google Scholar]
- 30. Miziorko HM, Lane MD. 1977. 3-Hydroxy-3-methylgutaryl-CoA synthase. Participation of acetyl-S-enzyme and enzyme-S-hydroxymethylgutaryl-SCoA intermediates in the reaction. J. Biol. Chem. 252:1414–1420 [PubMed] [Google Scholar]
- 31. Mueller C, Schwender J, Zeidler J, Lichtenthaler HK. 2000. Properties and inhibition of the first two enzymes of the non-mevalonate pathway of isoprenoid biosynthesis. Biochem. Soc. Trans. 28:792–793 [PubMed] [Google Scholar]
- 32. NCCLS/CLSI 2006. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard—6th ed. NCCLS document M7-A6. NCCLS/CLSI, Wayne, PA [Google Scholar]
- 33. Putra SR, Lois LM, Campos N, Boronat A, Rohmer M. 1998. Incorporation of [2,3-13C2]- and [2,4-13C2]-D-1-deoxyxylulose into ubiquinone of Escherichia coli via the mevalonate-independent pathway for isoprenoid biosynthesis. Tetrahedron Lett. 39:23–26 doi:10.1016/S0040-4039(97)10458-0 [Google Scholar]
- 34. Rohdich F, et al. 2003. The deoxyxylulose phosphate pathway of isoprenoid biosynthesis: studies on the mechanisms of the reactions catalyzed by IspG and IspH protein. Proc. Natl. Acad. Sci. U. S. A. 100:1586–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rohdich F, et al. 1999. Cytidine 5′-triphosphate-dependent biosynthesis of isoprenoids: YgbP protein of Escherichia coli catalyzes the formation of 4-diphosphocytidyl-2-C-methylerythritol. Proc. Natl. Acad. Sci. U. S. A. 96:11758–11763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sagner S, Latzel C, Eisenreich W, Bacher A, Zenk MH. 1998. Differential incorporation of 1-deoxy-D-xylulose into monoterpenes and carotenoids in higher plants. Chem. Commun. (Camb.) 2:221–222 [Google Scholar]
- 37. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 38. Schwender J, et al. 1997. Incorporation of 1-deoxy-D-xylulose into isoprene and phytol by higher plants and algae. FEBS Lett. 414:129–134 doi:10.1016/S0014-5793(97)01002-8 [DOI] [PubMed] [Google Scholar]
- 39. Siegel RE. 2008. Emerging gram-negative antibiotic resistance: daunting challenges, declining sensitivities, and dire consequences. Respir. Care 53:471–479 [PubMed] [Google Scholar]
- 40. Sprenger GA, et al. 1997. Identification of a thiamin-dependent synthase in Escherichia coli required for the formation of the 1-deoxy-D-xylulose 5-phosphate precursor to isoprenoids, thiamin, and pyridoxol. Proc. Natl. Acad. Sci. U. S. A. 94:12857–12862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Testa CA, Cornish RM, Poulter CD. 2004. The sorbitol phosphotransferase system is responsible for transport of 2-C-methyl-D-erythritol into Salmonella enterica serovar Typhimurium. J. Bacteriol. 186:473–480 doi:10.1128/JB.186.2.473-480.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Testa CA, Brown MJ. 2003. The methylerythritol phosphate pathway and its significance as a novel drug target. Curr. Pharm. Biotechnol. 4:248–259 [DOI] [PubMed] [Google Scholar]
- 43. Walsh C. 2003. Where will new antibiotics come from? Nat. Rev. Microbiol. 1:65–70 [DOI] [PubMed] [Google Scholar]
- 44. Wungsintaweekul J, et al. 2001. Phosphorylation of 1-deoxy-D-xylulose by D-xylulokinase of Escherichia coli. Eur. J. Biochem. 268:310–316 [DOI] [PubMed] [Google Scholar]
- 45. Xiang S, Usunow G, Lange G, Busch M, Tong L. 2007. Crystal structure of 1-deoxy-D-xylulose 5-phosphate synthase, a crucial enzyme for isoprenoids biosynthesis. J. Biol. Chem. 282:2676–2682 [DOI] [PubMed] [Google Scholar]
- 46. Yong D, et al. 2009. Characterization of a new metallo-beta-lactamase gene, bla(NDM-1), and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob. Agents Chemother. 53:5046–5054 doi:10.1128/AAC.00774-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yoon SH, et al. 2005. Production of vanillin by metabolically engineered Escherichia coli. Biotechnol. Lett. 27:1829–1832 [DOI] [PubMed] [Google Scholar]
- 48. Zhang B, et al. 2011. A second target of the antimalarial and antibacterial agent fosmidomycin revealed by cellular metabolic profiling. Biochemistry 50:3570–3577 doi:10.1021/bi200113y [DOI] [PMC free article] [PubMed] [Google Scholar]