

Abstract
Chagas disease, caused by the protozoan pathogen Trypanosoma cruzi, remains a challenging infection due to the unavailability of safe and efficacious drugs. Inhibitors of the trypanosome sterol 14α-demethylase enzyme (CYP51), including azole antifungal drugs, are promising candidates for development as anti-Chagas disease drugs. Posaconazole is under clinical investigation for Chagas disease, although the high cost of this drug may limit its widespread use. We have previously reported that the human protein farnesyltransferase (PFT) inhibitor tipifarnib has potent anti-T. cruzi activity by inhibiting the CYP51 enzyme. Furthermore, we have developed analogs that minimize the PFT-inhibitory activity and enhance the CYP51 inhibition. In this paper, we describe the efficacy of the lead tipifarnib analog compared to that of posaconazole in a murine model of T. cruzi infection. The plasma exposure profiles for each compound following a single oral dose in mice and estimated exposure parameters after repeated twice-daily dosing for 20 days are also presented. The lead tipifarnib analog had potent suppressive activity on parasitemia in mice but was unsuccessful at curing mice, whereas posaconazole as well as benznidazole cured 3 of 5 and 4 of 6 mice, respectively. The efficacy results are consistent with posaconazole having substantially higher predicted exposure than that of the tipifarnib analog after repeat twice-daily administration. Further changes to the tipifarnib analogs to reduce plasma clearance are therefore likely to be important. A crystal structure of a trypanosomal CYP51 bound to a tipifarnib analog is reported here and provides new insights to guide structure-based drug design for further optimized compounds.
INTRODUCTION
Although Chagas disease is primarily associated with Latin American countries, the recognition that substantial numbers of immigrants to Europe and North America are infected with Trypanosoma cruzi has brought additional attention to this neglected tropical disease (7, 18). In fact, the first clinical trial in decades of a new anti-Chagas disease drug candidate, posaconazole, is under way in Spain (4). The existing drugs for Chagas disease, benznidazole and nifurtimox, developed in the 1960s and 1970s, are limited by toxicity and inadequate efficacy (1, 27). Posaconazole, developed by Schering-Plough, was approved in 2006 for treating fungal infections. It acts by inhibiting the sterol 14α-demethylase enzyme (CYP51) that is part of the ergosterol biosynthesis pathway. Among the azole antifungal drugs, posaconazole appears to be the most efficacious in animal models of Chagas disease, largely due to its potent trypanocidal activity and excellent pharmacological characteristics (26). If successful in clinical trials with Chagas disease patients, posaconazole would be a much-needed addition to the limited armamentarium of anti-Chagas disease drugs. Unfortunately, the extremely high cost of posaconazole may limit its use in resource-limited countries where the disease is most prevalent (4). If a less-expensive alternative to posaconazole could be discovered, such a compound could potentially reach larger groups of patients in need of treatment for Chagas disease.
Previously, we reported that the preclinical anticancer drug tipifarnib (Fig. 1) has highly potent in vitro activity against T. cruzi (11–13). Although tipifarnib is an inhibitor of human protein farnesyltransferase (PFT), we discovered that the target of activity in T. cruzi was CYP51, as demonstrated by inhibition of endogenous sterol biosynthesis (11) and binding to recombinant T. cruzi CYP51 (13). Importantly, tipifarnib does not bind the human CYP51 (13). Since inhibitory activity against human PFT would lead to undesirable side effects such as bone marrow suppression, we designed and synthesized analogs of tipifarnib that avoided PFT-inhibitory activity while retaining CYP51 inhibition. The best compound, compound 1 (Fig. 1), had subnanomolar inhibitory activity on intracellular T. cruzi amastigotes and was devoid of inhibitory activity on human PFT. It was also shown to have highly potent activity in the murine model of T. cruzi infection (13). However, due to a 2-methyl-phenyl group attached to the main quinolone ring, compound 1 was compromised by the existence of stable rotamers (atropisomerism [14]) that we believed would complicate further drug development. In this study, new analogs that cannot form atropisomers were designed. Compounds 2 and 3 (Fig. 1) were synthesized and shown to be similar in potency to compound 1 against T. cruzi amastigotes (50% effective concentrations [EC50] of 0.5 and 0.3 nM, respectively) (12). Compound 2 proved to have better pharmacokinetic properties than did compound 3; thus, we performed a murine efficacy study comparing compound 2 to posaconazole, as described here. In addition, crystallographic studies were conducted with tipifarnib analogs and trypanosomal CYP51, resulting in a 2.0-Å-resolution structure of compound 3 bound in the enzyme active site. The structure provides insights into the binding mode of the compound that differ from the previous predictions made by molecular modeling. As a chemically simple compound, compound 2 appears to have features that are desirable in a new Chagas disease drug.
Fig 1.
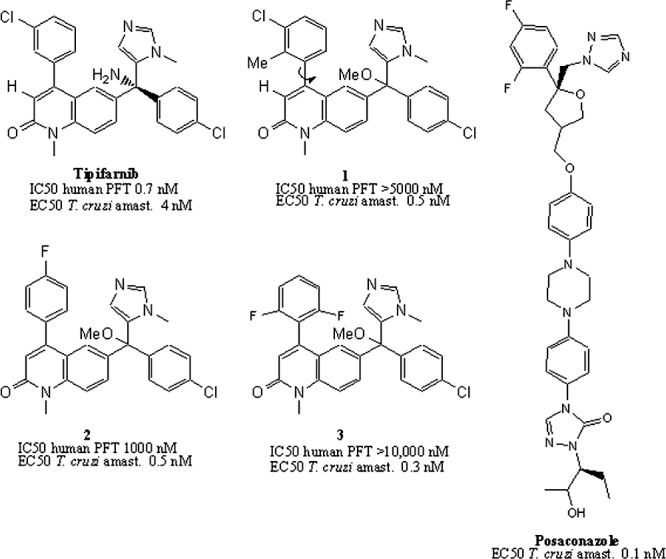
Structures of tipifarnib and analogs with potent anti-T. cruzi activity. Compound 1 exhibits atropisomerism around the bond marked with the arrow. Compounds 2 and 3 solve the rotamer problem while retaining highly potent T. cruzi activity and low human PFT inhibition. The crystal structure of Trypanosoma brucei CYP51 was solved with compound 3. The structure of posaconazole is provided for comparison. amast., amastigotes.
MATERIALS AND METHODS
Chiral separation of compound 2.
The syntheses of compounds 2 and 3 were previously described (referred to as compounds 12 and 16 in the previous paper) (12). The racemic mixture of compound 2 was separated into its two enantiomers by chiral high-pressure liquid chromatography (HPLC) using a Chiralpak AD-H column (250 by 4.6 mm; Chiral Technologies, Inc., West Chester, PA). The eluent was isopropyl alcohol and hexane (1:20) run at a flow rate of 1 ml/min. The first eluted peak, at a retention time (tR) of 164.3 min, is termed compound 2.1, and a second peak, eluting at a tR of 185.4 min, is termed compound 2.2.
Trypanosoma cruzi amastigote growth inhibition assay.
Compounds were screened against the mammalian stages of the Tulahuen strain of T. cruzi grown in coculture with 3T3 fibroblast cells using the β-galactosidase reporter system as previously described (2).
PK studies in mice.
Pharmacokinetic (PK) studies in mice were performed in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, and study protocols were approved by the Monash Institute of Pharmaceutical Sciences Animal Ethics Committee. The experiments were conducted using nonfasted 5-week-old female Swiss outbred mice weighing 20 to 24 g. Mice had access to food and water ad libitum throughout the pre- and postdose sampling period. A single dose of compound 2 (40 mg/kg of body weight) or posaconazole (20 mg/kg) was administered orally by gavage in an aqueous suspension vehicle containing 20% (wt/vol) Trappsol cyclodextrins (CTD, Inc., Alachua, FL) (0.2 ml per mouse). Blood samples were collected up to 24 h postdose with up to two samples collected from each animal at two separate time points, the first via submandibular bleed (approximately 120 μl; conscious sampling) and the second via terminal cardiac puncture (0.6 ml; under isofluorane anesthesia). Blood was collected directly into polypropylene Eppendorf tubes containing heparin, Complete (a protease inhibitor cocktail; Roche), potassium fluoride, and EDTA. Once collected, samples were centrifuged, and plasma was removed and stored at −20°C until analysis by liquid chromatography-mass spectrometry. The lower limits of quantitation for compound 2 and posaconazole were 0.001 μM and 0.071 μM, respectively. A single-compartment model assuming first-order absorption and first-order elimination was fitted to the measured single-dose data by nonlinear regression (WinNonlin software version 5.2.1; Pharsight Corporation, Mountain View, CA). The broad range of plasma concentrations observed for compound 2 necessitated that a weighting scheme of 1/Y2 be used, whereas no weighting of data was required to fit the data for posaconazole. To estimate the exposure after repeat oral administration in the efficacy model as described below, the fitted single-compartment pharmacokinetic model was used to simulate 20-day repeat-dose (twice-daily [b.i.d.], every-12-h [q12h]) profiles, assuming no changes in the pharmacokinetic properties (i.e., no induction or inhibition of clearance and no change in compound distribution) with repeat dosing. Exposure parameters (maximum concentration of drug in serum [Cmax] and area under the concentration-time curve from 0 h to infinity [AUC0–∞]) and average steady-state concentration (Cav,ss calculated as AUC0–∞/12 h) for both compounds were calculated using noncompartmental analysis of the simulated plasma concentration-time profiles after the first administration on day 1 and after the second administration on day 20.
Microsome stability and cytochrome P450 enzyme inhibition assays.
Liver microsome metabolism assays were performed with CD-1 female mouse microsomes or pooled human liver microsomes (BD Biosciences, San Jose, CA). The reaction mixtures (400 μl) contained 0.1 M potassium phosphate buffer (pH 7.4), 3 mM MgCl2, 1 mM EDTA, 1 mM NADP, 5 mM glucose-6-phosphate, 1 U/ml glucose-6-phosphate dehydrogenase (Sigma), and 0.5 mg/ml liver microsomes. Each reaction mixture was incubated at 37°C for 10 min, and then 2 μl of the test compound (at 300 μM in dimethyl sulfoxide [DMSO]) was added to give a final concentration of 1.5 μM. At each time point, samples of the reaction mixtures were stopped with 3× the volume of acetonitrile. An extraction standard was added to determine extraction yields. The samples were then centrifuged to remove microsomal solids, and the supernatants were dried under a vacuum. These samples were redissolved in acetonitrile and injected into an Agilent C18 column using a standard reverse-phase gradient followed by mass spectrometry performed on a Waters Quattro Micro spectrometer in tandem mass spectrometry (MS/MS) mode. The loss of the parent compound over time was used to calculate the half-life (t1/2) using Prism software (GraphPad Software, Inc., San Diego, CA). The experiments were performed twice with similar results.
Inhibition of cytochrome P450 enzymes was conducted in human liver microsomes (BD Gentest; Discovery Labware Inc., Woburn, MA) using an isoform-specific approach. Microsomes were suspended in phosphate buffer at a protein concentration of 0.4 mg/ml and incubated independently at 37°C in the presence of specific substrates [CYP1A2, 40 μM phenacetin; CYP2C9, 140 μM tolbutamide; CYP2C19, 30 μM (S)-mephenytoin; CYP2D6, 3 μM dextromethorphan; CYP3A4/5, 2.5 μM midazolam or 50 μM testosterone] and various concentrations of compound or positive-control inhibitor. Reactions were initiated by the addition of an NADPH-regenerating system as described above and were quenched after 4 to 40 min using ice-cold acetonitrile. Samples were centrifuged, and concentrations of isoform-specific metabolites of each substrate were assessed by liquid chromatography-mass spectrometry (LC-MS). The 50% inhibitory concentration (IC50) was taken as the concentration at which there was a 50% reduction in metabolite formation relative to the maximal formation in the absence of inhibitor.
Efficacy studies in mice.
Swiss mice (4- to 6-week-old females) were obtained from the Animal Facility at the Universidade Federal de Ouro Preto (UFOP), Minas Gerais, Brazil, and maintained in a temperature-controlled room with access to water and food ad libitum. All procedures and experimental protocols were conducted in accordance with the guidelines issued by the Brazilian College of Animal Experimentation (COBEA) and approved by the Ethics Committee in Animal Research at UFOP (approval no. 2009/17). Animals were inoculated with 5 × 103 trypomastigotes of the Y strain of T. cruzi by intraperitoneal (i.p.) injection. Five experimental groups were established: (i) 7 mice treated with compound 2 at 40 mg/kg b.i.d. (q12h) for 20 days, (ii) 7 mice treated with posaconazole at 20 mg/kg b.i.d. (q12h) for 20 days, (iii) 7 mice treated with benznidazole at 100 mg/kg for 20 days, (iv) 7 mice maintained as infected and untreated control, and (v) another 7 mice maintained as the noninfected and untreated control group. Posaconazole and compound 2 were suspended in 20% (wt/vol) Trappsol cyclodextrins (CTD, Inc., Alachua, FL). Benznidazole was suspended in distilled water using 4% methylcellulose (Sigma), and each animal received 0.2 ml of drug suspension by gavage. In all therapeutic schemes, oral treatment was started at 4 days postinfection, immediately after the appearance of parasitemia detected by fresh blood examination.
Parasitemia was evaluated by microscopic examination of fresh blood samples up to 30 days posttreatment. In addition, real-time PCR was performed 30 days posttreatment in all surviving mice. Animals that did not show microscopic reactivation of parasitemia were subjected to immunosuppression, which consisted of three cycles of 50 mg of cyclophosphamide/kg of body weight, for four consecutive days, with intervals of 3 days between each cycle (3). Microscopic parasitemia of these animals was evaluated during the cyclophosphamide immunosuppression, as well as for the following 10 days after immunosuppression.
Real-time PCR.
Mice were bled (200 μl) from the orbital venous sinus 30 days after the end of drug treatment. The extraction of total genomic DNA was performed using a commercial kit (Wizard genomic DNA purification kit; Promega). The PCR mixture contained 50 ng of genomic DNA; 0.35 μM T. cruzi 195-bp repeat DNA-specific primers TCZ-F (5′-GCTCTTGCCCACAMGGGTGC-3′, where M = A or C) and TCZ-R (5′-CCAAGCAGCGGATAGTTCAGG-3′) (Invitrogen) (8), modified from reference 21, which amplify a 182-bp product; 5 μl of SYBR green PCR Mastermix (Applied Biosystems); and water to a final total volume of 10 μl. Separately, reaction mixtures containing 50 ng of genomic DNA; 0.50 μM tumor necrosis factor alpha (TNF-α) primers TNF-5241 (5′-TCCCTCTCATCAGTTCTATGGCCCA-3′) and TNF-5411 (5′-CAGCAAGCATCTATGCACTTAGACCCC-3′) (Invitrogen) (8), which amplify a 170-bp product; 5 μl of SYBR green PCR Mastermix; and water (QSP, 10 μl) (Applied Biosystems) were used as loading controls. The reaction mixtures were loaded into a Fast 96-well reaction plate (MicroAmp; Applied Biosystems), capped, centrifuged for 2 min at 200 × g, and placed in the StepOnePlus real-time PCR system (Applied Biosystems). The cycling program consisted of an initial denaturation at 95°C for 10 min, followed by 40 cycles of 94°C for 15 s and 64.3°C for 1 min with fluorescence acquisition at 60°C. Amplification was immediately followed by a melt program with an initial denaturation of 15 s at 95°C, cooling to 60°C for 1 min, and then a stepwise temperature increase of 0.3°C/s from 60 to 95°C. Each 96-well reaction plate contained standard curve and two negative controls. Negative controls consisted of a reaction mixture with T. cruzi-specific or mouse-specific primers without DNA and also with blood or tissue DNA from noninfected mice. Each DNA sample was quantified in duplicate. The mean values for T. cruzi-specific DNA samples were normalized by the ratio with the average values for mouse-specific DNA samples. The efficiencies of amplification were determined automatically by the StepOneSoftware v2.0 by the following calculation: efficiency (E) = 10 (−1/slope) (23).
X-ray crystallography.
The N-terminus-truncated expression construct of Trypanosoma brucei CYP51 (16), described in reference 17, was used for cocrystallization with compound 3 (molar ratio of inhibitor to enzyme, 2:1). Crystals were obtained by hanging-drop vapor diffusion at 24°C, with a 285 μM P450 solution in 20 mM potassium phosphate buffer, pH 7.4, containing 100 mM NaCl, 0.1 mM EDTA, 10% glycerol, and 0.02 mM n-tetradecyl-β-d-maltoside, against a well solution containing 50 mM potassium phosphate buffer, pH 7.4, and 15% (wt/vol) polyethylene glycol (PEG) 5000. Crystals were soaked briefly in a 40% cryo-buffer and flash-cooled in liquid nitrogen. Data were collected at the Life Sciences Collaborative Access Team (LS-CAT), Advanced Photon Source, Argonne National Laboratory, beamline 21ID-F, and processed with the HKL2000 software package (22). The solvent content was estimated using a Matthews probability calculator. The structure was determined with Phaser-MR of the CCP4 suite (6) using ligand-free T. brucei CYP51 (Protein Data Bank [PDB] code 3g1q) as a search model, which yielded a single solution with four monomers in the asymmetric unit, an initial Rfactor of 0.32, and a log-likelihood gain of 11,180. Model building and refinement were performed with COOT (10) and REFMAC5 (CCP4 suite), respectively. Table S1 in the supplemental material summarizes the diffraction and refinement statistics; an electron density map for the inhibitor binding site is presented as Fig. S1. Structure superposition was done in LSQkab of the CCP4 suite using a secondary-structure-matching algorithm.
Protein structure accession number.
The coordinates and structure factors of CYP51 in complex with compound 3 (PDB ligand code JKF) have been deposited at the RCSB Protein Data Bank under PDB identification (ID) code 3TIK.
RESULTS
Pharmacokinetic characterization of compound 2 and posaconazole in mice.
Following a single oral dose of 40 mg/kg, compound 2 had a moderate rate of absorption (with time to maximum concentration of drug in serum [Tmax] observed 2 h postdose), after which concentrations declined gradually over the 24-h postdose period (Fig. 2A). Following a single oral dose of 20 mg/kg posaconazole, maximum plasma concentrations were observed 1 h postdose, but concentrations remained relatively flat for up to 7.5 h postdose (Fig. 2B). Based on the available data, the elimination half-lives for compound 2 and posaconazole were approximately 2 h and 18 h, respectively. The exposure to posaconazole at 20 mg/kg was higher than that of compound 2 after a single oral dose of 40 mg/kg based on both Cmax and AUC0–∞ (Table 1). Consistent with the shorter estimated half-life for compound 2, the simulated repeat (b.i.d.) dose profile suggests that there would be no significant accumulation after 20 days In contrast, posaconazole would be expected to exhibit substantial accumulation with repeat b.i.d. dosing and steady-state exposure parameters would be approximately 3-fold higher than those observed after a single dose.
Fig 2.
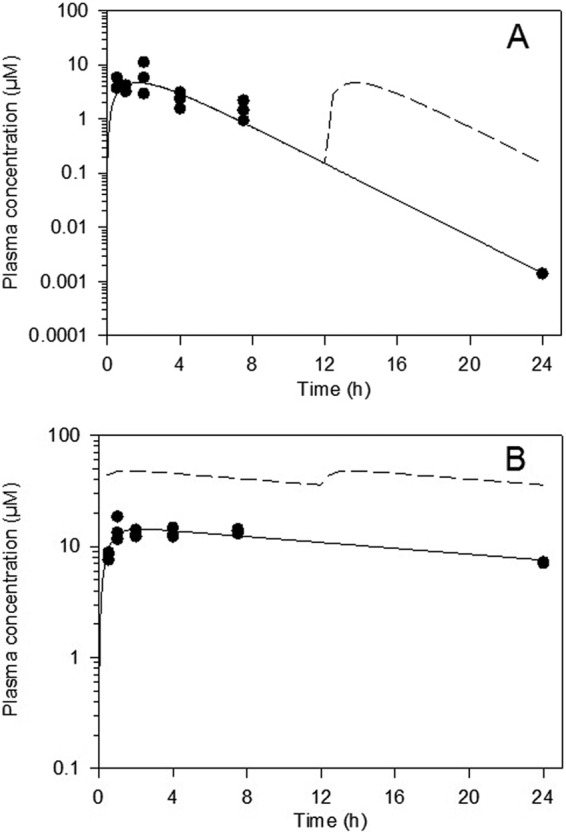
Plasma concentrations of compound 2 (40 mg/kg) (A) and posaconazole (20 mg/kg) (B) following single oral administration to mice. Filled symbols represent measured concentrations (n = 3 samples per time point), while the solid line represents profiles fitted with a single-compartment model. The dashed line represents estimated plasma concentrations on day 20 after b.i.d. (q12h) administration.
Table 1.
Pharmacokinetic parameters following oral administration of compound 2 and posaconazole to mice
| Parameterc | Compound 2 |
Posaconazole |
||
|---|---|---|---|---|
| Day 1a | Estimated day 20b | Day 1a | Estimated day 20b | |
| Dose (mg/kg) | 40 | 40 | 20 | 20 |
| Cmax (μM) | 4.7 | 4.8 | 14.3 | 48.2 |
| dn-Cmax (μM/mg/kg) | 0.1 | 0.1 | 0.7 | 2.4 |
| AUC0–∞ (μM · h) | 22.9 | 23.3 | 509 | 1700 |
| dn-AUC0–∞ (μM · h/mg/kg) | 0.57 | 0.58 | 25.5 | 85.0 |
| Cav,ss (μM) | 1.9 | 1.9 | 42.4 | 141 |
Day 1 data were based on the fit of a single-compartment pharmacokinetic model to experimental single-oral-dose data.
Day 20 b.i.d. data (after the second dose on day 20) were estimated using simulated profiles based on the fitted data after a single dose.
dn, dose normalized.
Comparison of compound 2 (racemic) with posaconazole and benznidazole in the murine model of T. cruzi infection.
Mice were treated orally with test compounds or vehicle for 20 days beginning 4 days postinfection. Vehicle-treated mice demonstrated high levels of parasitemia, peaking around day 7 postinfection. All 7 vehicle-treated mice died between days 12 and 24 postinfection. Microscopic parasitemia was dramatically suppressed in mice treated with compound 2 (Fig. 3). However, low-level reactivation was observed microscopically in 4 mice after completion of the compound 2 treatments. Five of 5 surviving mice that were treated with compound 2 had positive PCR tests 30 days after treatment, indicating that they were not parasitologically cured. In comparison, 2 of 5 mice treated with posaconazole were PCR positive and 2 of 6 mice treated with benznidazole were PCR positive, indicating that none of these treatments yielded 100% cures with this model (Table 2). Two mice that received treatment with compound 2 died (days 18 and 34 postinfection). Each was free of parasitemia at the time of death, so these deaths were due either to complications from repeated oral gavage treatments or to side effects from the compound. Similarly, two mice receiving posaconazole died (days 5 and 7 postinfection) and one mouse receiving benznidazole died (day 5 postinfection). The average weight gained by mice treated with compound 2 was below that of untreated controls on the last day of treatment; however, the weights rebounded to normal by 30 days posttreatment (Fig. 4).
Fig 3.

Parasitemia of mice infected with Trypanosoma cruzi Y strain and treated with compound 2, posaconazole, or benznidazole for 20 consecutive days. Arrows indicate the first and the last day of treatment and the first day of cyclophosphamide immunosuppression.
Table 2.
Efficacy experiment with mice infected with Trypanosoma cruzi Y strain and treated daily with compound 2, posaconazole, or benznidazole for 20 consecutive daysa
| Parameter | Treatment group |
||
|---|---|---|---|
| Compound 2 (40 mg/kg) | Posaconazole (20 mg/kg) | Benznidazole (100 mg/kg) | |
| No. of drug doses for clearance of parasitemia (mean ± SD) | 1.71 ± 1.11 | 1.5 ± 0.58 | 1.17 ± 0.41 |
| No. of mice with natural parasitemia reactivation after treatment/total no. | 4/5 | 0/5 | 1/6 |
| No. of mice with parasitemia reactivation after immunosuppression/total no. | 3/5 | 0/5 | 1/6 |
| No. of mice positive by real-time PCRb (30 days after treatment)/total no. | 5/5 | 2/5 | 2/6 |
| Total no. of positive mice/total no. | 5/5 | 2/5 | 2/6 |
Treatments were started at the 4th day after infection.
Real-time PCR was performed in blood of all animals 30 days after treatment.
Fig 4.
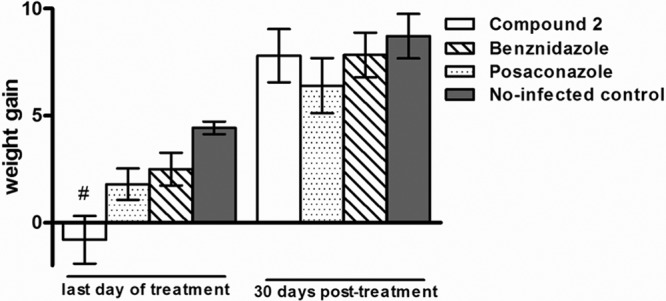
Weight gain in grams of animals infected with Trypanosoma cruzi strain and treated with compound 2, posaconazole, or benznidazole in comparison with the noninfected-control groups. The weight gain was calculated by the difference between the weights of animals obtained on days 1 and 20 of the treatment and on days 1 and 30 posttreatment. The weights of animals included in control groups were obtained at the same times. The statistical differences among treated and noninfected groups were determined by the nonparametric Tukey multiple comparison test. #, significant difference (P < 0.001) in relation to the noninfected group.
Separation and testing of enantiomers of compound 2.
The preclinical drug candidate tipifarnib is an R enantiomer, having >100-fold-greater PFT-inhibitory activity than the corresponding S enantiomer (29). The tipifarnib analogs that we previously described were synthesized and tested as racemic mixtures (11–13). To further evaluate the activities of compound 2, the two enantiomers were separated using chiral chromatography. The two enantiomers were tested for the following biological activities: (i) T. cruzi intracellular amastigote growth inhibition, (ii) rat PFT inhibition, (iii) T. cruzi CYP51 binding, (iv) CYP450 inhibition, and (v) stability upon incubation with liver microsomes. As can be seen in Table 3, the different enantiomers have significantly different activities. We deduce that the enantiomer (compound 2.1) with more potent activity on T. cruzi amastigotes and the lower Kd (dissociation constant) on T. cruzi CYP51 is the R enantiomer since the crystal structure of the closely related compound 3 (discussed below) demonstrates that the R enantiomer is bound in the CYP51 active site.
Table 3.
Comparisons of tipifarnib, compound 2 racemic mixture, enantiomers of compound 2, and posaconazole
| Assay | Tipifarnib | Compound 2 racemic mixture | Enantiomer 2.1 | Enantiomer 2.2 | Posaconazole |
|---|---|---|---|---|---|
| T. cruzi amastigote; EC50 (μM) | 0.006 | 0.0005 | 0.00031 | 0.0263 | 0.00016 |
| Rat PFT; IC50 (μM) | 0.0013 | 0.991 | |||
| T. cruzi CYP51 binding; Kd (μM)a | 0.11 | 0.18 | 3.8 | 0.06 | |
| Liver microsome stability; t1/2 (min) | |||||
| Human | 100 | 270 | >360 | ||
| Mouse | >180 | 28 | >180 | ||
| CYP450 inhibition (μM) | |||||
| CYP1A2 | 13.3 | >30 | >39 | ||
| CYP2C9 | 0.29 | 0.08 | 9.5 | ||
| CYP2C19 | 4.2 | 3.2 | 20.9 | ||
| CYP2D6 | 6.5 | 25 | >30 | ||
| CYP3A4/5 midazolam | 2.3 | 0.65 | 0.09 | ||
| CYP3A4/5 testosterone | 2.1 | 0.9 | 0.04 |
The apparent Kds were calculated by plotting T. cruzi CYP51 spectral responses (ΔA420–390) against free ligand concentration (15).
Since the racemic mixture of compound 2 was relatively inactive against rat PFT (IC50 of ∼1 μM), the separate enantiomers were not tested for PFT inhibition. The activity of the two enantiomers on human hepatic P450 enzymes demonstrated some differences, with lower inhibition fortuitously associated with compound 2.1. In particular, compound 2.1 inhibited CYP3A4/5 with an IC50 in the 2.1 to 2.3 μM range, which is more than twice as high as the IC50 for compound 2.2 and >20 times higher than the IC50 for posaconazole against this enzyme. Finally, assays of in vitro metabolic stability demonstrated that compound 2.1 was very stable in human liver microsomes with a t1/2 of 270 min, which is even longer than that of the parent drug, tipifarnib. Of particular interest is that compound 2.1 is metabolized considerably faster in mouse liver microsomes (t1/2 of 28 min) than in human liver microsomes.
X-ray crystal structure of CYP51 bound to compound 3.
In the crystallographic studies, we worked with the Trypanosoma brucei CYP51 (83.4% amino acid sequence identity to the T. cruzi CYP51 ortholog). These two trypanosomal enzymes have high overall structural similarity (15) and only one amino acid difference within the binding cavity (Fig. 5). Formation of the enzyme/inhibitor complex does not cause significant structural rearrangements in the CYP51 molecule. The root mean square deviation between all Cα-atoms in the ligand-free structure (PDB ID, 3G1Q [17]) and compound 3-bound structures is only 0.63 Å, which is comparable to the deviations between the four compound 3-bound T. brucei CYP51 molecules in the asymmetric unit (0.548 ± 0.07 Å) and is just slightly exceeded by the deviation between the compound 3-bound T. brucei CYP51 and posaconazole-bound T. cruzi (PDB ID, 3K1O; 0.857 Å). Although compound 3 was used for cocrystallization as a racemic mixture, only the R enantiomer is present in the structure. In all four molecules in the asymmetric unit, the inhibitor adopts the same orientation within the CYP51 active site, no amino acid side chain repositioning affecting the heme-protein interactions (15, 17) being observed. The imidazole ring of compound 3 is coordinated with the heme iron. The length of the N3-Fe coordination bond (2.08 ± 0.04 Å) is slightly shorter than the average length observed in the structures of P450-azole complexes (∼2.15 to 2.3 Å) and similar to that found in T. brucei CYP51 complexed with N-1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethyl-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide (17), which may be indicative of stronger interaction. The nonligated portion of the inhibitor molecule is surrounded by 20 protein residues; 17 of them are located within the van der Waals bond distances (<4.5 Å). Six residues are provided by the B′-helix/B′-C loop (Y103, F105, M106, F110, A115, and Y116); four are contributed by helix I (A287, F290, A291, and T295), two are from the αK/β1-4 loop (L356 and M358), two are from the β4 hairpin (M460 and V461), and one residue (L127) is from helix C. Unique for this inhibitory scaffold is the involvement in the interaction of the αF/FF″ loop region (E205 and L208). This is due to the positioning of the phenyl ring (attached to the main quinolone group), which occupies an additional CYP51 active-site cavity bordered by helices F and I and the β4-hairpin. This ring has served as the subject for the hydrogen atom substitutions in this study (Fig. 1). Altogether, the structural data imply that the high potency of this inhibitory scaffold is largely due to the precise hand-in-glove fit between the ligand molecule and the enzyme active-site geometry, and therefore, minor chemical modifications that would optimize the scaffold pharmacokinetic/pharmacodynamic properties may significantly enhance its curative potential.
Fig 5.
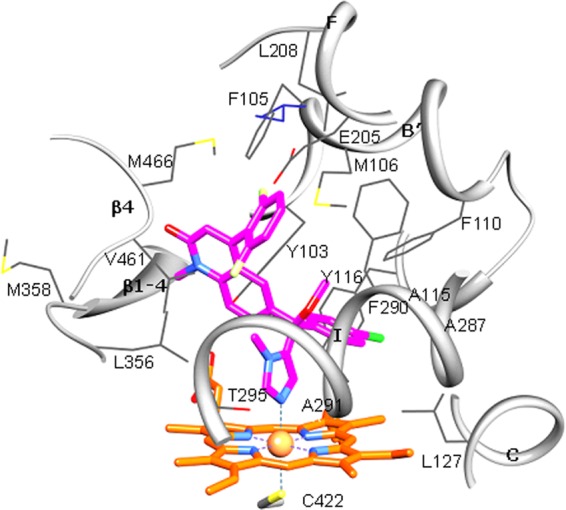
Active site of Trypanosoma brucei CYP51 with bound compound 3. The residues located within van der Waals contacts (<4.5 Å) with the inhibitor are shown in wire representation and labeled, carbon atoms are colored in gray, and the corresponding secondary structural elements of the protein are presented as ribbons. The single active-site amino acid residue that differs in T. cruzi CYP51 (I105) is colored in purple. Coordinated with the heme (orange), compound 3 is colored in magenta. Nitrogens are blue, oxygens are red, chlorine is green, and fluorines are light yellow. A stereoview of the active site in the superimposed T. brucei and T. cruzi (3K1O) CYP51 structures can be seen as Fig. S2 in the supplemental material.
DISCUSSION
The anti-Trypanosoma cruzi activity of sterol 14α-demethylase inhibitors has been recognized since the early 1980s (9). The early imidazole and triazole antifungal drugs such as ketoconazole, itraconazole, and fluconazole appeared to lack sufficient potency and/or pharmacological properties to cure mice with chronic T. cruzi infection (26). However, newer azole antifungal drugs, including D0870 (discontinued in clinical development) and posaconazole, have been shown to have curative activity in mouse models of Chagas disease (20, 28). The outcome of the ongoing clinical trial with posaconazole will help validate (or invalidate) CYP51 as a drug target for Chagas disease (4). If CYP51 is validated, additional CYP51 inhibitors with lower costs of goods may be necessary to bring treatment to the large numbers of Chagas disease patients who live in resource-limited settings. The investigations with compound 2 have been pursued with this framework in mind.
The efficacy study in mice demonstrated that the racemic mixture of compound 2 had strong suppressive activity on parasitemia; however, the ability to cure mice was lower than that of posaconazole. Of 5 surviving mice, all 5 treated with compound 2 remained parasitologically positive by sensitive testing methods, whereas only 2 of 5 mice treated with posaconazole were positive after treatment. The finding that 2 of 6 benznidazole-treated mice remained PCR positive demonstrates the high stringency of the mouse model with Y strain parasites used in these experiments. In previous work using a different strain of T. cruzi (Tulahuen), we observed that posaconazole led to negative parasitemia by PCR (25). In addition to using different strains, we also tested for cure by performing PCR after inducing immunosuppression, which further enhances the ability to uncover low-level residual infections.
The estimated plasma concentrations of compound 2 are expected to be substantially lower than those for posaconazole after b.i.d. dosing for 20 days, attributable both to posaconazole's higher single-dose exposure and to its longer half-life, leading to substantial accumulation upon repeat administration. Estimated Cav,ss values on day 20 are approximately 1.9 μM for compound 2 and 141 μM for posaconazole. The simulated profiles (Fig. 2) suggest that posaconazole concentrations remained above approximately 30 μM for the full 20-day dosing period, whereas concentrations of compound 2 were below 10 μM, and frequently below 1 μM, throughout the dosing period, which may partly account for the differences in efficacy. Other factors, such as plasma protein binding and the volume of distribution (not available in mice), may also play a role in the different efficacy profiles.
The active enantiomer (compound 2.1) is considerably more metabolically stable in human microsomes (t1/2 of 270 min) than in mouse microsomes (t1/2 of 28 min) (Table 3). The lower activity of compound 2 than of posaconazole in mice could therefore be due to the higher clearance of compound 2, translating to lower exposure of the parasites to the compound (Fig. 2). It may be necessary to make additional changes to the tipifarnib scaffold that further improve metabolic stability and reduce plasma clearance rates. As we know that the pharmacokinetic profile of tipifarnib in humans is quite good (5, 30), these results suggest that the rodent model may underestimate the potential activity of tipifarnib analogs in humans.
There were similar numbers of deaths in each group of mice treated with compound 2, posaconazole, and benznidazole (2, 2, and 1, respectively). The deaths are best attributed to complications from repeated oral gavages over the 20-day treatment course rather than to side effects from the drugs. The average weight of mice at the end of treatment with compound 2 was lower than that for untreated mice, but the weight rebounded to normal levels by 30 days posttreatment (Fig. 4). Since the mice fully recover their weight to levels of the control mice by 30 days posttreatment, this indicates that the side effects are reversible. It seems unlikely that the toxicity is due to PFT inhibition, given the low level of activity of compound 2 on the rodent PFT enzyme (IC50, ∼1 μM), particularly compared to that of tipifarnib (IC50, ∼1 nM), which is tolerated by rodents. It is also unlikely that compound 2 inhibits the rodent CYP51 since tipifarnib and several analogs were previously shown to not inhibit human CYP51 (13). Superposition of the experimental structures of human CYP51 (24) on the T. brucei CYP51 (an excellent proxy for the T. cruzi enzyme) complex with compound 3 reveals several clashes that make it impossible for compound 3 to bind to the human CYP51. The lack of weight gain during treatment could be due to inhibition of one of the other CYP enzymes (19) or to a completely unrelated mechanism that will require additional investigation to elucidate.
Compound 2 was further characterized by separating the racemic mixture into its two enantiomers. Enantiomer 2.1 was observed to be a tighter T. cruzi CYP51 ligand and more active against T. cruzi cells than enantiomer 2.2. As discussed above, we concluded that enantiomer 2.1 corresponds to the R enantiomer. The CYP inhibition studies showed that compound 2.1 had much less inhibitory activity on CYP3A4/5 than did posaconazole. Given the important role of CYP3A4 in drug metabolism, the observation suggests that compound 2.1 has an advantage over posaconazole with respect to the potential for drug-drug interactions. It is fortuitous that compound 2.1 had lower inhibitory activity against most of the CYP isoforms than did the 2.2 enantiomer. For an imidazole-containing molecule, compound 2.1 has a good profile against hepatic CYP enzymes, consistent with what is known about tipifarnib (29).
The crystal structure of the Trypanosoma brucei CYP51 bound to compound 3 reveals a significant difference between the binding mode of the tipifarnib-related compounds and that predicted from a structural model of the enzyme that was based on the mycobacterial CYP51 structure, which is only 30% identical in sequence (11). This underscores the value of experimentally determining crystal structures rather than depending on molecular models built on homology. The binding mode of compound 3, discussed in Results, provides insights for our group to design and synthesize new analogs to further explore this structural series as potential antitrypanosomal agents.
In conclusion, the analog of tipifarnib (compound 2) is highly potent against T. cruzi cultures and demonstrated strong suppressive anti-T. cruzi activity in the mouse model. It had a lower cure rate than that of the drug posaconazole, which could possibly be the result of higher plasma clearance, and hence lower exposure, of compound 2 than of posaconazole. The data suggest that further changes to the tipifarnib scaffold, guided by the recent crystal structure of CYP51, are needed to optimize the pharmacokinetic properties while retaining anti-T. cruzi activity. The very good pharmacokinetic attributes of tipifarnib in humans give us confidence that optimized tipifarnib analogs can be achieved.
Supplementary Material
ACKNOWLEDGMENTS
Funding to F. S. Buckner, C. L. M. J. Verlinde, and M. H. Gelb came from NIH (grants AI070218 and AI054384) and the Drugs for Neglected Diseases Initiative. Funding to M. T. Bahia and S. A. Charman came from the Drugs for Neglected Diseases Initiative. Funding to G. I. Lepesheva came from NIH (GM067871).
For the work described in this paper, DNDi received financial support from the following donors: Department for International Development (DFID) (United Kingdom), GiZ on behalf of the Government of the Federal Republic of Germany (Germany), Ministry of Foreign and European Affairs (MAEE) (France), Spanish Agency for International Development Cooperation (AECID) (Spain), Médecins Sans Frontières (Doctors without Borders) (international), and a Swiss foundation. The donors had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Published ahead of print 9 July 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Bern C. 2011. Antitrypanosomal therapy for chronic Chagas' disease. N. Engl. J. Med. 364:2527–2534 [DOI] [PubMed] [Google Scholar]
- 2. Buckner FS, Verlinde CLMJ, La Flamme AC, Van Voorhis WC. 1996. Efficient technique for screening drugs for activity against Trypanosoma cruzi using parasites expressing β-galactosidase. Antimicrob. Agents Chemother. 40:2592–2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Caldas S, et al. 2008. Trypanosoma cruzi: acute and long-term infection in the vertebrate host can modify the response to benznidazole. Exp. Parasitol. 118:315–323 [DOI] [PubMed] [Google Scholar]
- 4. Clayton J. 2010. Chagas disease: pushing through the pipeline. Nature 465:S12–S15 [DOI] [PubMed] [Google Scholar]
- 5. Cohen SJ, et al. 2003. Phase II and pharmacodynamic study of the farnesyltransferase inhibitor R115777 as initial therapy in patients with metastatic pancreatic adenocarcinoma. J. Clin. Oncol. 21:1301–1306 [DOI] [PubMed] [Google Scholar]
- 6. Collaborative Computational Project N4 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760–763 [DOI] [PubMed] [Google Scholar]
- 7. Coura JR, Vinas PA. 2010. Chagas disease: a new worldwide challenge. Nature 465:S6–S7 [DOI] [PubMed] [Google Scholar]
- 8. Cummings KL, Tarleton RL. 2003. Rapid quantitation of Trypanosoma cruzi in host tissue by real-time PCR. Mol. Biochem. Parasitol. 129:53–59 [DOI] [PubMed] [Google Scholar]
- 9. Docampo R. 1981. Biochemical and ultrastructural alterations produced by miconazole and econazole in Trypanosoma cruzi. Mol. Biochem. Parasitol. 3:169–180 [DOI] [PubMed] [Google Scholar]
- 10. Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 11. Hucke O, Gelb MH, Verlinde CL, Buckner FS. 2005. The protein farnesyltransferase inhibitor tipifarnib as a new lead for the development of drugs against Chagas disease. J. Med. Chem. 48:5415–5418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kraus JM, et al. 2010. Second generation analogues of the cancer drug clinical candidate tipifarnib for anti-Chagas disease drug discovery. J. Med. Chem. 53:3887–3898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kraus JM, et al. 2009. Rational modification of a candidate cancer drug for use against Chagas disease. J. Med. Chem. 52:1639–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. LaPlante SR, Edwards PJ, Fader LD, Jakalian A, Hucke O. 2011. Revealing atropisomer axial chirality in drug discovery. ChemMedChem 6:505–513 [DOI] [PubMed] [Google Scholar]
- 15. Lepesheva GI, et al. 2010. Structural insights into inhibition of sterol 14alpha-demethylase in the human pathogen Trypanosoma cruzi. J. Biol. Chem. 285:25582–25590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lepesheva GI, Nes WD, Zhou W, Hill GC, Waterman MR. 2004. CYP51 from Trypanosoma brucei is obtusifoliol-specific. Biochemistry 43:10789–10799 [DOI] [PubMed] [Google Scholar]
- 17. Lepesheva GI, et al. 2010. Crystal structures of Trypanosoma brucei sterol 14alpha-demethylase and implications for selective treatment of human infections. J. Biol. Chem. 285:1773–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leslie M. 2011. Infectious diseases. A tropical disease hits the road. Science 333:934. [DOI] [PubMed] [Google Scholar]
- 19. Lewis DF. 2004. 57 varieties: the human cytochromes P450. Pharmacogenomics 5:305–318 [DOI] [PubMed] [Google Scholar]
- 20. Molina J, et al. 2000. Activities of the triazole derivative SCH 56592 (posaconazole) against drug-resistant strains of the protozoan parasite Trypanosoma (Schizotrypanum) cruzi in immunocompetent and immunosuppressed murine hosts. Antimicrob. Agents Chemother. 44:150–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moser DR, Kirchhoff LV, Donelson JE. 1989. Detection of Trypanosoma cruzi by DNA amplification using the polymerase chain reaction. J. Clin. Microbiol. 27:1477–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Otwinowski Z, Minor W. 1995. The HKL manual. Yale University Press, New Haven, CT [Google Scholar]
- 23. Stordeur P, et al. 2002. Cytokine mRNA quantification by real-time PCR. J. Immunol. Methods 259:55–64 [DOI] [PubMed] [Google Scholar]
- 24. Strushkevich N, Usanov SA, Park HW. 2010. Structural basis of human CYP51 inhibition by antifungal azoles. J. Mol. Biol. 397:1067–1078 [DOI] [PubMed] [Google Scholar]
- 25. Suryadevara PK, et al. 2009. Structurally simple inhibitors of lanosterol 14alpha-demethylase are efficacious in a rodent model of acute Chagas disease. J. Med. Chem. 52:3703–3715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Urbina JA. 2009. Ergosterol biosynthesis and drug development for Chagas disease. Mem. Inst. Oswaldo Cruz 104(Suppl 1):311–318 [DOI] [PubMed] [Google Scholar]
- 27. Urbina JA. 2010. Specific chemotherapy of Chagas disease: relevance, current limitations and new approaches. Acta Trop. 115:55–68 [DOI] [PubMed] [Google Scholar]
- 28. Urbina JA, et al. 1996. Cure of short- and long-term experimental Chagas' disease using D0870. Science 273:969–971 [DOI] [PubMed] [Google Scholar]
- 29. Venet M, End D, Angibaud P. 2003. Farnesyl protein transferase inhibitor ZARNESTRA R115. Curr. Top. Med. Chem. 3:1095–1102 [DOI] [PubMed] [Google Scholar]
- 30. Zujewski J, et al. 2000. Phase I and pharmacokinetic study of farnesyl protein transferase inhibitor R115777 in advanced cancer. J. Clin. Oncol. 18:927–941 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.