

Abstract
Mutation in the clarin-1 gene (Clrn1) results in loss of hearing and vision in humans (Usher syndrome III), but the role of clarin-1 in the sensory hair cells is unknown. Clarin-1 is predicted to be a four transmembrane domain protein similar to members of the tetraspanin family. Mice carrying null mutation in the clarin-1 gene (Clrn1−/−) show loss of hair cell function and a possible defect in ribbon synapse. We investigated the role of clarin-1 using various in vitro and in vivo approaches. We show by immunohistochemistry and patch-clamp recordings of Ca2+ currents and membrane capacitance from inner hair cells that clarin-1 is not essential for formation or function of ribbon synapse. However, reduced cochlear microphonic potentials, FM1-43 [N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl) pyridinium dibromide] loading, and transduction currents pointed to diminished cochlear hair bundle function in Clrn1−/− mice. Electron microscopy of cochlear hair cells revealed loss of some tall stereocilia and gaps in the v-shaped bundle, although tip links and staircase arrangement of stereocilia were not primarily affected by Clrn1−/− mutation. Human clarin-1 protein expressed in transfected mouse cochlear hair cells localized to the bundle; however, the pathogenic variant p.N48K failed to localize to the bundle. The mouse model generated to study the in vivo consequence of p.N48K in clarin-1 (Clrn1N48K) supports our in vitro and Clrn1−/− mouse data and the conclusion that CLRN1 is an essential hair bundle protein. Furthermore, the ear phenotype in the Clrn1N48K mouse suggests that it is a valuable model for ear disease in CLRN1N48K, the most prevalent Usher syndrome III mutation in North America.
Introduction
Hearing and balance rely on hair cells, specialized epithelial cells in the inner ear that develop a set of stereocilia (hair bundle) at their apical surface. The hair bundle detects vibration-induced movements in the ear and transduces these stimuli into electrical signals. Investigating gene function in the bundle is critical to understanding hair cell biology and hearing loss linked to bundle pathology. Mouse mutants have played an important role; many of the deaf mutants have defects in the sensory hair bundle and provide in vivo models for investigating gene functions in the hair bundle (Leibovici et al., 2008).
Usher syndrome (USH), an autosomal recessive disorder, accounts for ∼50% of the cases of combined inherited blindness and deafness (Saihan et al., 2009). USH type III (USH3) is caused by mutation in the clarin-1 gene (CLRN1) and is characterized by progressive hearing loss and variable balance impairment (Joensuu et al., 2001; Ness et al., 2003). The primary structure of the clarin-1 protein (CLRN1) predicts four transmembrane domains similar to a large family of membrane proteins, including tetraspanins and claudins (Adato et al., 2002). Based on in vitro and biochemical assays, Tian et al. (2009) posited a possible role for CLRN1 in the regulation and homeostasis of actin filaments. We reported the first animal model for ear disease in USH3: a mouse carrying a null allele of Clrn1 (Clrn1−/−) (Geng et al., 2009). Clrn1−/− mice showed early-onset profound hearing loss and variable balance impairment. Elevated auditory-evoked brainstem response (ABR) and vestibular evoked potential thresholds in Clrn1−/− mice were associated with reduced amplitudes and delayed latencies of the compound action potential (CAP) of cochlear and vestibular ganglion neurons. The cochlear hair bundle structure was disrupted in Clrn1−/− mice at a young age [postnatal day 2 (P2) to P10] without concomitant loss of ganglion cells. We speculated that a hair bundle defect caused the elevated threshold, and the observed delay in peak latency is a secondary consequence of the hair bundle defect in Clrn1−/− mice. Alternatively, the mutant phenotype could be attributable to a defect in hair cell-to-afferent neuron communication (a ribbon synapse defect). These findings led us to two mutually nonexclusive hypotheses: (1) the primary role of CLRN1 is in maintenance of the structural integrity of the hair bundle, and/or (2) CLRN1 is essential for hair cell ribbon synapse formation or function. To test these hypotheses, we performed a series of experiments focusing on the hair bundle and ribbon synapses of the Clrn1−/− mice. Furthermore, we explored the consequence of a missense mutation CLRN1N48K (p.N48K) in the context of the hair cells in vitro and in vivo. Here we report, for the first time, the generation of the Clrn1N48K (knock-in) mouse model for ear phenotypes in CLRN1N48K patients. Our investigation has defined the role of clarin-1 in sensory hair cells and has uncovered a pathogenic mechanism for the most prevalent North American USH3 mutation (Adato et al., 2002; Fields et al., 2002).
Materials and Methods
The genes and proteins used in this study.
The following are the genes used: Clrn1−/−, homozygous knock-out allele; Clrn1+/−, heterozygous knock-out allele; Clrn1+/N48K, heterozygous N48K knock-in allele; Clrn1N48K/N48K, homozygous N48K knock-in allele; CLRN1, human clarin-1 gene; hCLRN1, human clarin-1 protein; Clrn1, mouse clarin-1 gene; mCLRN1, mouse clarin-1 protein; clarin-1, a general reference to the gene and protein in more than one species.
Mice.
The use of mice at Case Western Reserve University (CWRU) was approved by the Institutional Animal Care and Use Committee (protocol 2010-0074). Unless stated otherwise, all mice used in this study were derived from homozygous-by-heterozygous mating, and mice of either sex were used. Mice heterozygous for the clarin-1 mutation were indistinguishable from their wild-type siblings and, therefore, used as the control for all experiments.
ABRs.
ABR recording was conducted as described previously (Geng et al., 2009). Briefly, mice aged P18 and older were anesthetized with a dilute intraperitoneal injection of ketamine, xylazine, and acepromazine at doses of 40, 5, and 1 mg/kg, respectively. Their body temperature was maintained at 37–38°C by placing them on a homeothermic heating pad (Harvard Apparatus), and they were placed in a soundproof chamber during testing. ABR testing was performed using a SmartEP system from Intelligent Hearing Systems. Platinum subdermal needle electrodes were inserted at the vertex and ventrolaterally to the right ear and left ear. To test hearing function, mutant mice were presented with 16 kHz pure tone stimuli at a stimulus intensity starting at 100 decibel sound pressure level (dB SPL) and decrementing in 10 dB steps; this sequence was repeated in 5 dB steps until the lowest intensity that evoked a reproducible ABR waveform (peaks I–IV) was detected. The stimulus was presented for 100 ms duration and for at least 500 sweeps to both the left and right ear (one at a time) through high-frequency transducers (a closed system). For each mouse, ABR thresholds from the left and right were averaged and used for statistical analysis. Number of mice tested were as follows: Clrn1−/−, n = 12; Clrn1N48K/N48K, n = 25; and Clrn1+/N48K, n = 5.
Statistical method used to analyze ABR data.
A one-way ANOVA was used to determine whether the difference in hearing thresholds observed among the three groups of mice was significant. Briefly, the data were arranged in columns using Excel (Microsoft), and the statistics were conducted with Prism (GraphPad Software). A Bonferroni's multiple comparison test was then used to determine which groups were significantly different to account for the overall total difference.
Cochlear microphonics and CAP.
Cochlear microphonic (CM) and CAP recordings were conducted as a variant of ABR described previously. With this setup, we stimulated the ear with a 100 μs rectangular pure tone stimulus at a firing rate of 20 per second. Stimuli were presented through a rubber tube from a Tucker Davis Technology speaker directly to the studied ear. Then, 1024 sweeps were recorded and averaged in the condensation and rarefaction phases separately. The two resulting waves were then added to extract the CAP (Henry and Chole, 1979). The CM was visualized in the condensation and rarefaction tracings. The CM amplitude was measured as the difference between the maximum peak of a given polarity and the maximum peak of the opposite polarity (Santarelli et al., 2006). The maximum intensity of 110 dB SPL was then used and decreased by 10 dB steps until the threshold was reached. The threshold was assessed as the intensity reproduced a clearly identifiable CAP. The CM was measured from peak to trough at ∼1 ms after recording started for all the mice. The stimulus used to measure the CM is the condensation stimulus. Data were collected in Excel (Microsoft) and analyzed using a Mann–Whitney U test for nonparametric data in the MedCalc program. A one-tailed p value <0.05 was considered significant. This test was conducted at all three frequencies tested, namely 2, 4, and 8 kHz. For technical reasons linked to the setup of the Tucker Davis Technologies software/hardware, we could not record CM responses at frequencies >8 kHz. Clrn1−/− mice were tested at ages P18–P19 before the onset of profound hearing loss (P21–P25) (Clrn1+/−, n = 8; Clrn1−/−, n = 9). Therefore, we chose a younger time point (P18–P19), when some hearing function was retained.
The results are shown as a scatter plot, as well as a box and whisker plot. The scatter plot shows the spread of raw data. In the box and whisker plot, the central box represents the values from the 25th to 75th percentiles. The middle line is the median. A line extends from the minimum to the maximum value, excluding “outside” and “far-out” values, which are displayed outside the error bars. An outside value is defined as a value that is smaller than the lower quartile minus 1.5 times the interquartile range, or larger than the upper quartile plus 1.5 times the interquartile range. A far-out value is defined as a value that is smaller than the lower quartile minus 3 times the interquartile range, or larger than the upper quartile plus 3 times the interquartile range. The error bars within the whiskers are the 95% confidence interval of the mean.
Cochlear culture preparation.
Cochlear organ cultures were prepared from mutant and control mice at 0–3 d of age as described previously (Russell and Richardson, 1987) and maintained on Cell-Tak-coated glass coverslips in Maximow side assemblies at 37°C in a medium containing 90% MEM, 10% horse serum, 10 mm HEPES, pH 7.2, and 10 mg/ml ampicillin for 24 h before use.
FM1-43 labeling.
FM1-43 [N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl) pyridinium dibromide] dye loading experiments were performed as described previously (Gale et al., 2001; Alagramam et al., 2011). In brief, coverslips with adherent cochlear cultures were washed once with HEPES-buffered (10 mm, pH 7.2) HBSS (HBHBSS), dipped for 10 s in HBHBSS containing 3 mm FM1-43, and washed immediately three times in a large volume of HBHBSS (10 s for each wash). The coverslips were then placed on a glass slide and viewed with an upright microscope equipped with epifluorescence optics and FITC filters (488 nm excitation, 520 nm emission) using a 40× dry objective. Images were captured at fixed time points after a dye application using a 12-bit cooled CCD camera (Retiga Exi Aqua Blue; Q-Imaging). FM1-43 levels were quantified for Clrn1+/−, Clrn1−/−, cadherin 23 heterozygous (Cdh23+/−) or cadherin 23 homozygous (Cdh23−/−) hair cells. We focused on the basal turn for quantification because hair cells are more mature at the base compared with the apex in neonates. Images were imported into Adobe Photoshop CS3 (Adobe Systems), and gray levels were measured in 24 hair cells from each explant using a 400 pixel (20 × 20) region of interest. Data were collected from nine Clrn1+/− (n = 9), 10 Clrn1−/− (n = 10), three Cdh23+/− (n = 3), and three Cdh23−/− (n = 3) explants. Differential interference contrast (DIC) images were captured to map location of hair cells. Data was exported to Excel (Microsoft) and analyzed using the software PAST. Because the sample sizes within each group were small and unlikely to have a normal distribution, a Mann–Whitney U test was used to compare the two groups. Grayscale levels from Clrn1+/− and Clrn1−/− explants were plotted as a box and whisker plot as well as a scatter plot. The Cdh23−/− explants showed no uptake, and it was not plotted.
Tissue preparation for electrophysiology.
Cochleae and utricles were excised from wild-type and Clrn1−/− mouse pups at P1–P8 in accordance with protocols approved by the Animal Care and Use Committee of the University of Virginia (Protocol 3123). Briefly, pups were killed by rapid decapitation, and the temporal bone was removed and bathed in MEM supplemented with 10 mm HEPES and 0.05 mg/ml ampicillin, pH 7.4. Utricle sensory epithelia were carefully removed, treated briefly with subtilisin to facilitate removal of the otoconia, and mounted as described previously (Holt et al., 1997). Organs of Corti were gently isolated, and tectorial membranes were mechanically removed. The tissue was then divided in half turns (base and apex) and pinned flat beneath a pair of thin glass fibers adhered to a round glass coverslip. The hair cells were visualized from the apical surface using an upright Axioskop FS microscope (Carl Zeiss) equipped with a 63× water-immersion objective with differential interference contrast (DIC) optics. Images were acquired with a C2400 CCD camera and Argus image processor (Hamamatsu).
Hair cell electrophysiology.
The sensory tissue was bathed in standard artificial perilymph solution containing the following (in mm): 144 NaCl, 0.7 NaH2PO4, 5.8 KCl, 1.3 CaCl2, 0.9 MgCl2, 5.6 d-glucose, and 10 HEPES, NaOH-adjusted to pH 7.4 (320 mOsm/kg). Vitamins (1:50; catalog #11120) and amino acids (1:100; catalog #11130) were added from concentrates (Invitrogen). Recording pipettes (3–5 MΩ) were pulled from R6 capillary glass (King Precision Glass) and filled with an intracellular solution containing the following (in mm): 135 KCl, 5 EGTA-KOH, 5 HEPES, 2.5 K2ATP, 2.5 MgCl2, and 0.1 CaCl2, pH 7.4. Currents were recorded under whole-cell voltage clamp (holding potential, −64 mV) at room temperature using an Axopatch 200B (Molecular Devices), filtered at 1–10 kHz with a low-pass Bessel filter, digitized at ≥20 kHz with a 12-bit acquisition board (Digidata 1322) and pClamp 8.2 (Molecular Devices), and stored on disk for offline analysis using OriginPro 7.1 (OriginLab). Data are presented as means ± SE unless otherwise noted.
Patch-clamp analysis of inner hair cell presynaptic function.
For this, we performed perforated patch-clamp recordings in apical coils of freshly dissected organs of Corti (Moser and Beutner, 2000) from Clrn1−/− mice and age-matched wild-type mice (C57BL/6N) at P14–P20. The pipette solution contained 130 mm Cs-gluconate, 10 mm TEA-Cl, 10 mm 4-AP (Merck), 1 mm MgCl2, 10 mm HEPES, pH adjusted with HCl to 7.17 (osmolarity of ∼290 mOsm), and 300 μg/ml amphotericin B (Calbiochem). The extracellular solution contained (in mm): 104 NaCl, 35 TEA-Cl, 2.8 KCl, 10 CaCl2, 1 MgCl2, 10 HEPES, 1 Cs-gluconate, 5 4-AP, 11.1 d-glucose, pH adjusted with NaOH to 7.2 (osmolarity of ∼300 mOsm). EPC-9 amplifier controlled by Pulse software (HEKA) was used for measurements. All voltages were corrected for liquid-junction potentials. Currents were sampled at 20 kHz and low-pass filtered at 2 kHz. Cells that displayed a holding current exceeding −50 pA were discarded from analysis. Ca2+ currents were further isolated using a P/n protocol. Series resistance (RS) was <30 MΩ. Patch-clamp data were analyzed with Igor (Wavemetrics).
Mechanical stimulation.
Vestibular type II hair cells were mechanically stimulated by coupling the kinocilium to a stimulus pipette filled with extracellular solution using negative pressure (Holt et al., 2002). A piezoelectric device with a 10–90% rise time of 0.6 ms was used to drive movement of the stimulus pipette and sensory hair bundle unit. Voltage steps were used to calibrate the motion of the stimulus probe ±2 μm relative to its rest position. Movement of the hair bundle and stimulus probe was monitored visually, and video images of the probe were recorded to confirm the absence of off-axis motion and to calibrate the probe motion (spatial resolution of ∼4 nm). Coupling quality of the stimulus pipette and conformity of hair bundle movement were monitored before, during, and after the recording using video microscopy.
Cochlear outer hair bundles were stimulated using an angled stiff glass probe with a fire-polished tip ∼3–5 μm in diameter as described previously (Stauffer and Holt, 2007). Briefly, the probe was mounted on a one-dimensional PICMA chip piezo actuator (Physik Instruments) driven by a 400 mA ENV400 amplifier (Piezosystem). Voltage steps were used to evoke bundle deflections with a stimulus filtered at 20 kHz by a low-pass eight-pole Bessel filter (Krohn-Hite) to eliminate residual probe resonance. Hair bundle deflections were continuously monitored via video microscopy during recording to ensure that the probe and stereocilia bundle moved in unison. Pictures taken of the probe at static voltage steps were used to calibrate the stimulus displacement relative to the probe rest position to 2 μm in both the positive and negative direction (spatial resolution of ∼4 nm). Video images (temporal resolution of ∼30 ms) of the probe were recorded to confirm on-axis motion. The 10–90% rise time of the probe was ∼20 μs.
Expression constructs.
Constructs designed to express human CLRN1 or CLRN1N48K mutant protein, each fused to an HA epitope sequence, were reported previously (Tian et al., 2009). The CMV–GFP–Myo15a construct was obtained from Dr. Thomas Friedman (National Institute on Deafness and Other Communication Disorders, Bethesda, MD). The CMV–YFP plasmid vector (mVenus–N1) and the CMV–mouse Prestin–YFP expression construct were obtained from Dr. Jian Zuo (St. Jude Children's Research Hospital, Memphis, TN) (Wu et al., 2007). Constructs designed to express human CLRN1–YFP or CLRN1N48K–YFP were generated as follows. Full-length human CLRN1 coding sequence (National Center for Biotechnology Information Reference Sequence NM_174878) was cloned from human retina RNA (Clontech) with primers 5-gtttctcatcatgccaagccaacagaag-3 and 5-gtgaccaaagcaagtctactcccttgta-3, and cloned into a mammalian expression vector containing a CMV promoter and a C-terminal YFP coding sequence. An infusion cloning protocol kit from Invitrogen was used to recombine cDNA into the expression vector (a schematic diagram of the constructs is shown in Results.)
Gene gun transfection.
Inner-ear sensory epithelium cultures were prepared from the organ of Corti of P1–P4 C57BL/6 as described previously (Belyantseva et al., 2003). Cultures were then transfected using a Helios gene gun (Bio-Rad). Gold particles (1.0 μm; Bio-Rad) were coated with plasmid DNA at a ratio of 2 μg plasmid DNA to 1 mg of gold particles and precipitated onto the inner wall of Tefzel tubing, which was cut into individual cartridges containing ∼1 μg of plasmid DNA. Samples were bombarded with the gold particles from one cartridge per culture, using 110 psi of helium. After an additional 18–24 h in culture, samples were fixed in 4% paraformaldehyde, stained with phalloidin-conjugated Alexa Fluor 546, and observed using a Leica confocal microscope. For each transfection, three to six transfected hair cells were observed.
Heterologous expression and detection of hCLRN1 or hCLRN1N48K protein in human embryonic kidney cells.
Human embryonic kidney 293 (HEK293) cell lines stably expressing either hCLRN1 or hCLRN1N48K were stained by the immunofluorescence labeling method as described previously (Tian et al., 2009). Hemagglutinin (HA) and FLAG epitope sequences were fused to the C-terminal tail of CLRN1 and CLRN1N48K to facilitate the detection of protein expression and localization. The stable cell lines were treated with proteasome inhibitor bortezomib (15 nm) for 16 h and then fixed with 4% paraformaldehyde in PBS. The fixed cells were costained with antibodies against HA tag (Covance) and an endoplasmic reticulum (ER) marker calreticulin (Sigma-Aldrich) or a plasma membrane marker Na/K ATPase (α5; Developmental Studies Hybridoma Bank at University of Iowa, Iowa City, IA) to visualize the localization of expressed proteins. Cells were then labeled with anti-mouse IgG conjugated with Alexa Fluor 488 and anti-rabbit IgG conjugated with Cy3. Samples were visualized by a Leica SP2 confocal microscope.
Generation of Clrn1N48K knock-in mice.
The transgenic mouse was generated in collaboration with inGenious Targeting Laboratory. Briefly, a bacterial artificial chromosome (BAC) clone harboring Clrn1 gene was isolated from RP23 mouse genomic BAC library. The genomic region containing promoter, exon 1, and intron 1 regions of Clrn1 were subcloned from the BAC clone. A QuikChange site-directed in vitro mutagenesis kit (Stratagene) was used to introduce a single nucleotide change in exon 1 that would result in the translation of lysine (K) in place of asparagine (N) at position 48 in the amino acid sequence of CLRN1 (referred to as “N48K” mutation). A cassette containing the neomycin resistance gene (Neo) flanked by loxP sites was cloned into the long homologous arm downstream of exon 1 of the targeting construct using the recombineering technology (Copeland et al., 2001). The targeting construct was subsequently electroporated into C57BL/6J and 129/Sv hybrid embryonic stem (ES) cells, and recombinant clones were selected using G418. Recombined ES cells were microinjected into BALB/c blastocysts and implanted into pseudo-pregnant mothers. Chimeric progeny were obtained, and highly chimeric animals were subsequently mated with C57BL/6J mice to identify germ-line transmission of the Clrn1 deletion. Founder animals were identified, and heterozygous progeny were delivered to CWRU. Clrn1+/N48K mice were crossed with B6.C-Tg(CMV–cre)1Cgn/J mice to delete the Neo cassette. Clrn1N48K/N48K mice were derived by mating Clrn1+/N48K mice. The new allele described in this study was maintained by crossing it to the C57BL/6J (B6) strain, which was used in all parts of this study.
Scanning electron microscopy.
Cochleae from Clrn1+/−, Clrn1−/−, Clrn1+/N48K, or Clrn1N48K/N48K mice at P3 were fixed by intra-labyrinthine perfusion with 2.5% glutaraldehyde in a 0.1 m sodium cacodylate buffer containing 2 mm CaCl2, pH 7.4, and then immersed in the fixative for 2 h. They were stored in 0.25% glutaraldehyde diluted with a buffer at 4°C until additional processing, subsequently washed in the buffer, dissected to reveal the organ of Corti, and postfixed with cacodylate-buffered 1% OsO4 for 1 h. After washing in the cacodylate buffer, samples were impregnated with osmium using the osmium-thiocarbohydride (OTOTO) method (for details, see Furness and Hackney, 1986). After OTOTO, they were dehydrated through a series of increasing ethanol concentrations up to 100% ethanol dried over a molecular sieve and critical point dried from liquid CO2 using a Polaron critical point dryer. Cochleae were affixed to specimen stubs using silver conducting paint (Agar Scientific) and examined in a Hitachi S4500 field emission scanning electron microscope operated at 5 kV. For each genotype, three cochlear specimens were examined by scanning the electron microscopy.
Immunofluorescence analysis of inner hair cell ribbon synapse.
Immunostaining was performed as described previously (Khimich et al., 2005; Meyer et al., 2009). Briefly, the freshly dissected organ of Corti (apical turn) was fixed with 2% paraformaldehyde for 15 min at room temperature [for staining with antibody to glutamate receptor subunits 2 and 3 (anti-GluR2/3) or anti-CtBP2/RIBEYE]. The following antibodies were used: mouse IgG1 anti-CtBP2 (also recognizing the ribbon protein RIBEYE; 1:150; BD Biosciences), rabbit anti-GluR2/3 (1:200; Millipore Bioscience Research Reagents), and secondary Alexa Fluor 488- and Alexa Fluor 594-labeled antibodies (1:200; Invitrogen). Samples were visualized by Leica SP2 confocal microscope.
Results
Loss of clarin-1 disrupts hair bundle function
The CM potential was significantly diminished in Clrn1−/− mice. Clrn1−/− mice show bundle morphology defects in cochlear hair cells and profound hearing loss by P21 (Geng et al., 2009). We hypothesized that hair bundle defect is the primary reason for loss of hair cell function and significantly elevated ABR thresholds in Clrn1−/− mice. However, ABR recordings reflect the electrical responses of both the cochlear ganglion neurons and the nuclei of the central auditory pathway to sound stimulation, making it difficult to evaluate loss of cochlear function in isolation in the mutant model. In contrast, CM responses can be used to evaluate cochlear function (Santarelli et al., 2006; Cheatham et al., 2011). CM response is an electrical potential arising from hair cell depolarization, and it reflects hair cell mechanoelectrical transduction (MET). CM was recorded from the round window. CM and ABR recording techniques depend on the presentation and transmission of sound stimuli through the ear, and mice acquire that capability at ∼2 weeks after birth; therefore, these recordings cannot be performed in neonates.
We recorded CM responses from Clrn1−/− mice at P18, before the onset of hair cell degeneration, which is reported to be at approximately P21 (Geng et al., 2009). The amplitude of the signal from the Clrn1−/− mice at P18 was lower compared with age-matched Clrn1+/− mice at 2 kHz (p = 0.01), 4 kHz (p = 0.04), and 8 kHz (p = 0.15) (Fig. 1). Although reduction in CM amplitude did not reach statistical significance at 8 kHz, it was clearly reduced in the mutant, with most values at or below the median value of the control. Although CM were not recorded at higher frequencies because of technical reasons (explained in Materials and Methods), we believe that CM amplitudes will be attenuated at all frequencies in the Clrn1−/− cochlea. This conclusion is supported by the fact that Clrn1−/− mutants showed hearing loss at 8, 16, and 32 kHz, and hair bundle defects were noted in apical, midbasal, and basal turns of Clrn1−/− cochlea from 2- to 3-week-old mice (Geng et al., 2009). Together, these data support our hypothesis that hair bundle defect is the cause of hair cell dysfunction in Clrn1−/− mice, suggesting a possible role for CLRN1 in maintaining bundle integrity. Loss of CLRN1 could lead to disintegration of the bundle over time, but this process may not be uniform along cochlear turn (especially early on), leading to the observed variation in the CM output noted above. However, it could also be argued that a reduced CM response could be caused by abnormalities of the tectorial membrane, MET channel, or other factors needed for proper hair cell depolarization. To test the bundle defect hypothesis directly, we examined FM1-43 dye loading in nonstimulated hair bundles and recorded transduction currents after mechanical stimulation of the bundles.
Figure 1.
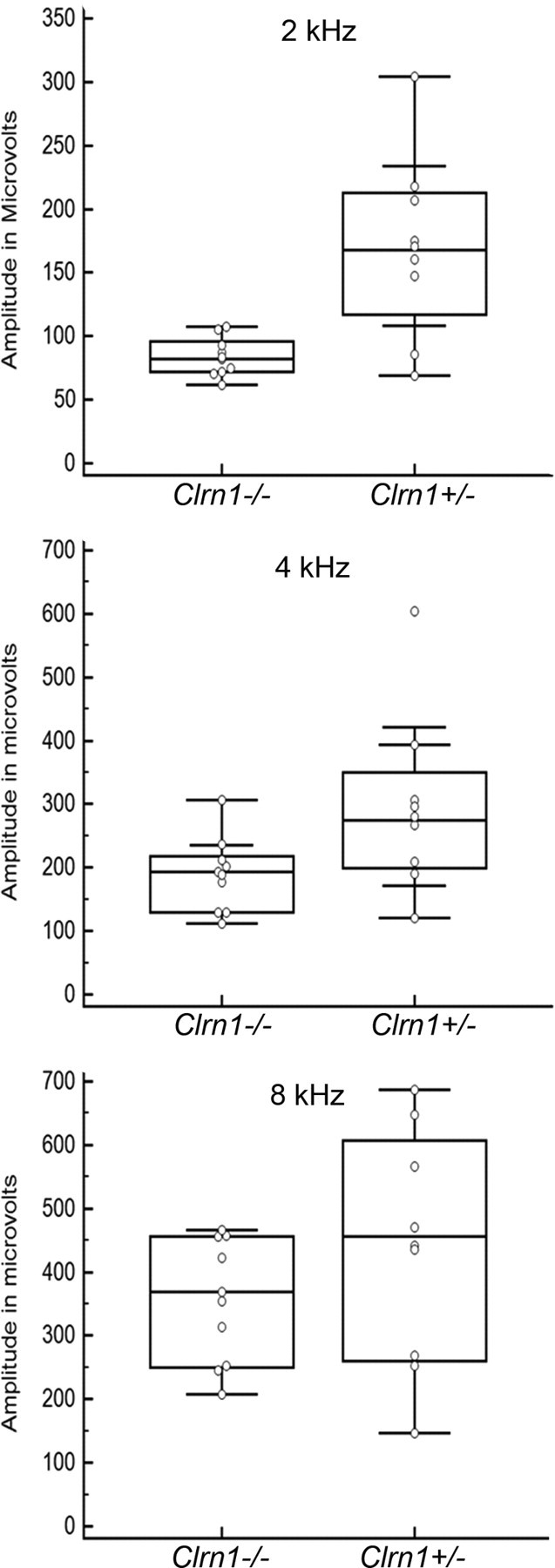
Cochlear microphonics. CM was recorded from Clrn1−/− or Clrn1+/− mice (P18–P19) at 2, 4, and 8 kHz. The graph shows the spread of the data at each frequency tested for Clrn1−/− (n = 9) and Clrn1+/− (n = 8) mice. The results show that, on average, the receptor (hair cell) potential was lower in the Clrn1−/− mice compared with the controls. Amplitude reduction in Clrn1−/− mice was statistically significant at 2 kHz (p = 0.01) and 4 kHz (p = 0.04) but not 8 kHz (p = 0.15).
Hair cells of Clrn1−/− mice show reduced FM1-43 loading
FM1-43 is an amphipathic styryl dye that is known to rapidly accumulate in sensory hair cells via the MET channels that are open at rest in nonstimulated hair bundles (Gale et al., 2001; Meyers et al., 2003). Cochlear cultures from Clrn1+/− and Clrn1−/− mice were examined for their ability to load with FM1-43 in response to a brief, 10 s exposure to the dye. Hair cells from Clrn1−/− mice showed dye loading in the apical and basal turns of the cochlea, although it was clearly reduced in both turns compared with the controls (images from midbasal section shown in Fig. 2A–D). Quantitative analysis of the basal turn of the Clrn1−/− mice indicated a 30–40% reduction in dye loading relative to that observed in the controls (Fig. 2E), and the difference between the two groups was statistically significant (p = 0.0205). The reduced uptake could be attributable to loss of tip links or MET channels, namely mutation in Clrn1 directly affects the formation of the tip-link complex or MET channels. Alternatively, reduced FM1-43 loading could be a consequence of bundle disruption, leading to a reduced number of tip links or functional MET channels open at rest in mutant hair cells. If mutation in Clrn1 directly affects the formation of the tip-link complex or MET channels, then hair cells carrying Clrn1−/− (a null) mutation would be expected to show no dye loading, but that is not the case. However, it is possible that FM1-43 enters hair cells through routes independent of the MET channels (during the 10 s exposure) and hence the observed loading. To address these issues and as an important reference control, cochlear cultures prepared from mice carrying a presumptive null allele of tip-link protein cadherin 23, Cdh23v2J/v2J (Di Palma et al., 2001), were examined for their ability to load FM1-43 under the same condition used to test the cochlear culture from Clrn1−/− mice. No dye loading was observed in Cdh23v2J/v2J culture (Fig. 2C,D), indicating that dye loading occurs exclusively through MET channels in our experimental condition. If CLRN1 was directly involved in the development of tip links or in the formation of the MET channel, then we would expect no dye loading in the Clrn1 knock-out background similar to that observed with Cdh23v2J/v2J cultures. However, these results (Fig. 2A,B,E) suggest that reduced FM1-43 loading in Clrn1−/− mice is more likely attributable to compromised MET channel function as a result of loss of hair bundle integrity.
Figure 2.
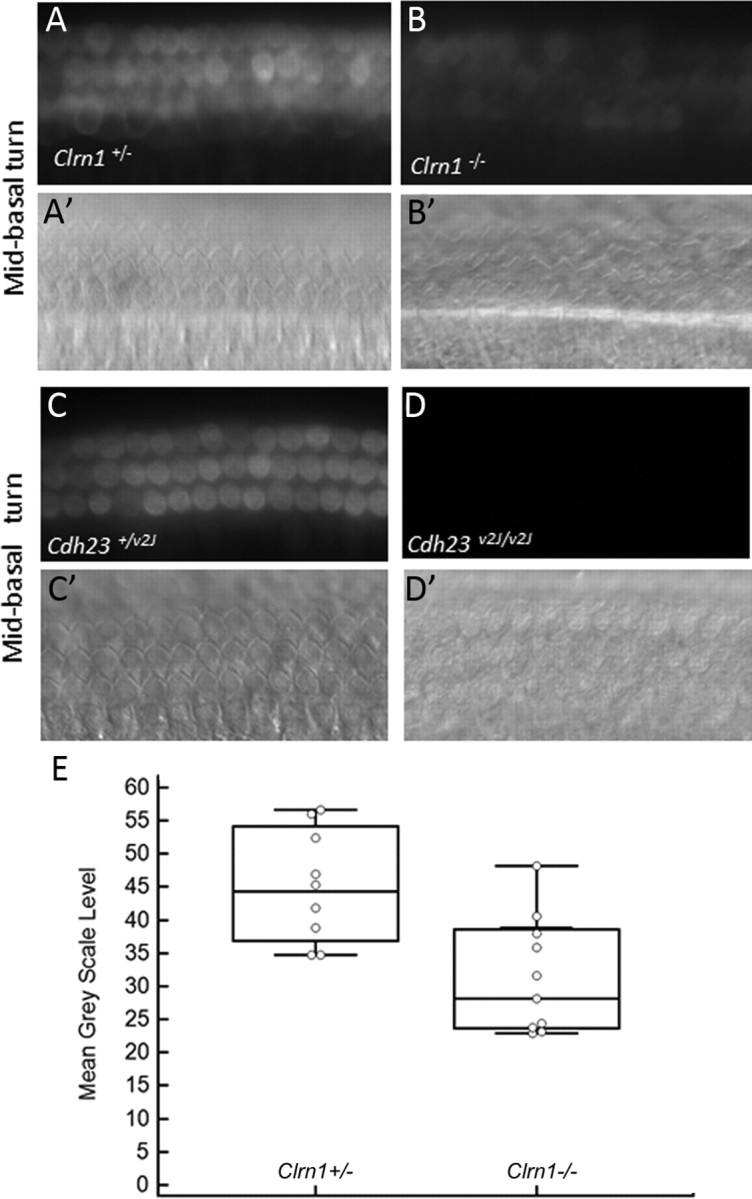
FM1-43 uptake in Clrn1−/− and Cdh23−/− cochlear hair cells in culture at P3. Uptake in Clrn1−/− (B) is diminished compared with the control (A). A′ and B′ are DIC images of A and B showing the location of the hair cells. Hair cells from Cdh23v2J/v2J mice hair cells fail to load with FM1-43 (C, D). C′ and D′ are DIC images of C and D showing the location of the hair cells. When there is complete loss of hair cell function, no dye uptake is observed (no background). E, Quantitative analysis of FM1-43 dye loading in the basal coils of the Clrn1+/− controls (n = 8) and Clrn1−/− mutants (n = 9). The number of hair cells included to determine the mean grayscale levels per tissue is 24. The reduction in dye loading was significant (p = 0.0205). The error bars represent SD.
Transduction currents of hair cells of Clrn1−/− mice had significantly reduced amplitudes and sensitivity
MET currents were recorded from 39 Clrn1+/+ hair cells and 34 Clrn1−/− hair cells. Clrn1−/− hair bundles with morphologies most similar to wild type were selected for analysis. Representative transduction currents are shown in Figure 3 for both cochlear and vestibular hair cells. In both cell types, the deletion of Clrn1 resulted in a reduction of the maximal transduction current that could be evoked by stiff probe deflection of the hair bundle. On average, the maximal transduction current was reduced by 32% in the cochlear outer hair cells (OHCs) and by 44% in utricular type II hair cells (Fig. 3A–E). To identify possible developmental contributions of Clarin-1, hair cell transduction was recorded during the first postnatal week, at time points when mouse transduction development has been well characterized (Lelli et al., 2009). When we subdivided the pooled cochlear data according to developmental age and cochlear region, we found similar reductions in transduction current amplitude over all ages and regions examined (Fig. 3F), suggesting that the effect of Clrn1 deletion is not tonotopically or developmentally regulated.
Figure 3.

MET currents in Clrn1+/+ and Clrn1−/− hair cells. A–D, Representative families of MET currents evoked by rapid hair bundle deflections that ranged in amplitude from −0.5 to 2 μm. Currents were recorded in voltage-clamp mode (Vhold = −64 mV) from the indicated organs and genotypes between P1 and P6. The scale bars in B and D also apply to A and C, respectively. E, Mean maximal transduction currents measured from 73 hair cells as the difference between the smallest currents evoked by negative bundle deflection and the largest currents evoked by positive deflections. The number of cells for each of the four groups is indicated above. F, Mean maximal transduction currents plotted for different cochlear regions and ages. The number of cells for each group is indicated above.
Hair cells of Clrn1−/− mice were significantly less sensitive than those of Clrn1+/+ cells. To investigate sensitivity, we examined the stimulus–response [I(X)] relationships of Clrn1+/+ and Clrn1−/− cells (Fig. 4). The I(X) data were extracted from those presented in Figure 3 and are plotted as the peak transduction current as a function of the hair bundle deflection. Figure 4, A and B, shows representative I(X) curves from vestibular cells as well as cochlear cells. The raw data were fit with a second-order Boltzmann equation. The I(X) curves revealed both reduced current amplitudes in the Clrn1−/− cells and reduced sensitivity, which is evident as the broader I(X) relationship, with a shallower slope. To highlight this finding, we normalized the I(X) relations in Figure 4, C and D. To quantify the change in sensitivity, we measured the displacement required to activate 10–90% of the response. The mean 10–90% operating ranges are presented for both utricle type II hair cells and cochlear OHCs from Clrn1+/+ and Clrn1−/− cells in Figure 4, E and F. Hair cells excised from Clrn1−/− mice had significantly broader I(X) relationships. In summary, vestibular and cochlear hair cells of Clrn1−/− mice had significantly reduced amplitudes and reduced sensitivity. Both effects are likely the consequence of disrupted hair bundle stereocilia.
Figure 4.

I(X) relationships from P1–P6 hair cells excised from Clrn1+/+ and Clrn1−/− mice. Peak current is plotted as a function of the stimulus amplitude for utricle (A) and cochlea (B) cells, respectively. The data points were fitted with second-order Boltzmann equations. The data shown in A and B were normalized to the maximal current and replotted to illustrate the broader relationships in hair cells from the utricle (C) and cochlea (D) of Clrn1−/− mice. To quantify the sensitivity of the cells, we measured the 10–90% operating range for each of the 73 cells. The mean operating ranges are plotted on the bar graphs for utricle (E) and cochlea (F) hair cells. The number of cells in each group is indicated on the graphs.
CLRN1 is a membrane protein that localizes to hair bundle stereocilia
Several attempts were made to detect endogenous mouse CLRN1 by immunolabeling, but we have not achieved specific labeling yet. We raised antibodies against two peptides from the extracellular loops, one peptide from cytoplasmic tail and full-length mCLRN1 protein. Although these antibodies recognize mCLRN1 expressed in cell lines, their specificity could not be confirmed in mouse inner ear tissue (data not shown). All antibodies mentioned in this paragraph were diligently tested in two different laboratories (those of K.N.A. and Y.I.), and we observed the same labeling pattern in cochlear tissue from Clrn1+/+ and Clrn1−/− mice, although Clrn1 mRNA is not detected in the cochlea of Clrn1−/− mice (Geng et al., 2009). This is different from Cosgrove and colleagues, who reported CLRN1 expression in the inner ear and retina using a rabbit CLRN1 antiserum they generated (Zallocchi et al., 2009, 2012). We suspect that the expression level of CLRN1 in the mouse ear is low, making it difficult to detect the endogenous protein. To circumvent this problem, we took the alternate approach described below, and all evidence presented here suggests that we were successful in determining the CLRN1 localization in hair cells.
CLRN1 localizes to the hair bundle
Data from the cochlear and hair cell physiology experiments led to the hypothesis that CLRN1 is a necessary hair bundle protein and, therefore, should be expressed in the hair bundle. Targeting of hCLRN1 to the stereocilia in wild-type hair cells was demonstrated by transfection of mouse organ of Corti explants using gene gun transfection protocol. hCLRN1–YFP accumulated in the stereocilia of hair cells that were transfected with a hCLRN1–YFP cDNA construct (Fig. 5A). The tissue was counterstained with phalloidin–Alexa Fluor 546. Phalloidin is a high-affinity filamentous actin (F-actin) probe. Fluorescently labeled phalloidin highlights F-actin-rich structures (stereocilia) within the hair bundle. Merged images showed localization of hCLRN1–YFP to the bundle (Fig. 5A,A′,merged image). In some of the transfected hair cells (n = 4), faint fluorescent labeling was observed in plasma membrane (Fig. 5A, arrow). All cells transfected with the hCLRN1–YFP construct showed hair bundle localization of the fusion protein (n = 27 hair cells).
Figure 5.
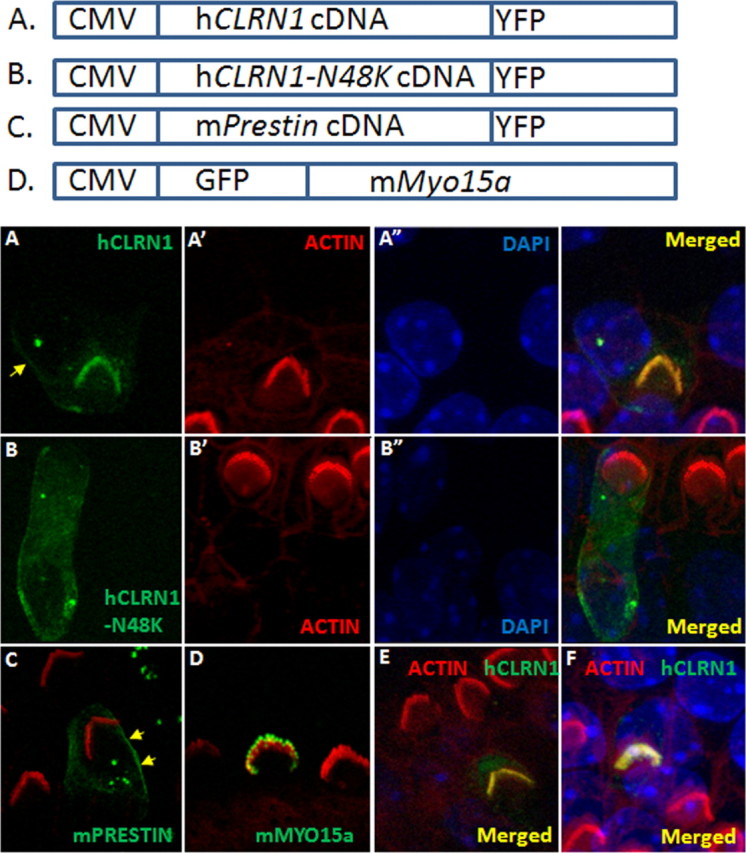
CLRN1 localizes to the hair bundle. P3 wild-type mouse cochlear epithelia explants transfected with a specific construct (using gene gun) and neighboring nontransfected hair cells after 1 d in culture and ∼20 h after transfection. Expression is driven by the CMV promoter in all cases, and detection of the fluorescent protein marks transfected cells; the tissue was counterstained with phalloidin-conjugated Alexa Fluor 546 (red) and DAPI (blue). A, CLRN1–YFP (green); A′ and A″ are phalloidin and DAPI counterstained images of A. CLRN1–YFP predominantly localized to the bundle with a small amount of the fusion protein localizing to the plasma membrane in the soma (arrow). B, CLRN1N48K–YFP (green); B′ and B″ are phalloidin and DAPI counterstained images of B. C, Merged image showing Prestin–YFP (green)-expressing hair cell and phalloidin-labeled bundles on transfected and nontransfected cells. Arrows point to membrane localization of Prestin–YFP. D, GFP–Myo15a (green)-expressing hair cell and phalloidin-labeled bundles on transfected and nontransfected cells. GFP–Myo15a appears at the tip of the stereocilia as expected. E, F, YFP/phalloidin merged images from two separate (from A) rounds of transfection with CLRN1–YFP construct to show reproducibility of results shown in A.
Next, we focused on one of the common USH3 mutations in North America, namely p.N48K. Here, asparagine at position 48 in CLRN1 is replaced by lysine. Bionformatic analysis shows that asparagine at position 48 is a conserved amino acid in clarin-1 across species and a conserved glycosylation site (Adato et al., 2002). It is known that lack of N-glycosylation could lead to abnormal posttranslational processing and localization of membrane proteins (Smith et al., 2011). Based on this information and the results presented previously (Figs. 1–5A), we hypothesized that hCLRN1N48K protein will not localize to the hair bundle. Here, we show that hCLRN1 carrying the pathogenic mutation N48K fails to reach the hair bundle and instead was retained in the soma of the hair cell (Fig. 5B). All cells transfected with the hCLRN1N48K–YFP construct showed localization of the fusion protein to the soma but not to the hair bundle (n = 24 hair cells). These data support our hypothesis that hCLRN1N48K fails to localize to the hair bundle and, for the first time in hair cells, reveals a possible mechanism underlying a common pathogenic mutation in CLRN1.
To negate the concern that the observed localization of hCLRN1 to the hair bundle is an artifact of the CMV-driven overexpression and/or YFP fusion, we performed the following control experiments. The mouse organ of Corti was transfected with mouse Prestin–YFP or GFP–Myo15a cDNA construct. Prestin–YFP localized to the hair cell plasma membrane, not to the hair bundle (Fig. 5C), and GFP–Myo15a accumulated at the tips of the stereocilia, as reported previously (Belyantseva et al., 2000, 2003), and not anywhere else in the hair cell (Fig. 5D). Results from these control experiments indicate that CMV-driven expression and YFP fusion do not interfere with native localization of the expressed protein in hair cells, nor is hair bundle localization an artifact of overexpression of a protein fused to YFP. The lack of bundle targeting of overexpressed hCLRN1N48K further supports the notion that the bundle localization of the hCLRN1 protein is not an artifact of overexpression.
Heterologous expression confirms membrane localization of hCLRN1 and mislocalization of the hCLRN1N48K protein
Clarin-1 is predicted to be a membrane protein (Adato et al., 2002). Gene gun transfection showed that hCLRN1 localizes to the hair bundle (Fig. 5), but the resolution of those images was not sufficient to discern membrane localization. To examine the subcellular localization of wild-type hCLRN1 and hCLRN1N48K mutant, we generated two HEK293 cell lines stably expressing either hCLRN1 or hCLRN1N48K fused with HA tag for detection. It has been reported that N48 is the only N-glycosylation site in CLRN1, and N48K mutation led to the mutant protein lacking glycosylation (Tian et al., 2009). Under normal conditions, the expression level of hCLRN1N48K is very low because of active degradation of this protein in HEK293 cells (Tian et al., 2009). We treated the two stable cell lines with bortezomib to enhance the protein level and to facilitate our detection of immunofluorescence. The cells were costained with antibodies against the HA tag and a plasma membrane marker Na/K ATPase (Fig. 6A). Wild-type hCLRN1 localized to the plasma membrane (Fig. 6A, top row) with a small amount observed in the ER. The cells were also costained with antibodies against the HA tag and calreticulin, a major Ca2+ binding (storage) chaperone in the ER (Fig. 6B). Unlike wild-type hCLRN1, the hCLRN1N48K mutant failed to localize to the plasma membrane (Fig. 6A, bottom row) and mainly colocalized with calreticulin (Fig. 6B, bottom row), suggesting that most of the mutated proteins were retained in the ER. Results described above show that N48K mutation in hCLRN1 alters normal plasma membrane localization of hCLRN1 and support an idea that hCLRN1 is localized to the plasma membrane compartment of the hair bundle.
Figure 6.
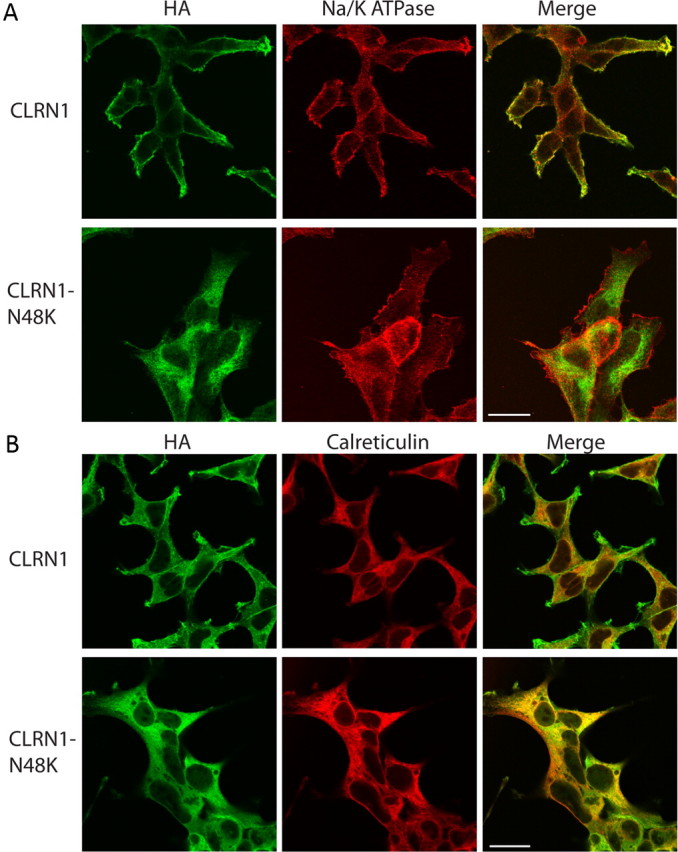
Localization of CLRN1 and its CLRN1N48K mutant heterologously expressed in HEK293 cells. The HEK cell lines stably expressing either wild-type human CLRN1 fused to HA tag (top rows of A and B) or CLRN1N48K fused to HA tag (bottom rows of A and B) were treated with bortezomib (15 nm) for 16 h before fixed by 4% paraformaldehyde. A, The cells were stained by antibodies against HA tag (green) and Na/K ATPase (red), which localizes to the plasma membrane. CLRN1 localizes to the plasma membrane, whereas CLRN1N48K fails to reach the plasma membrane. B, The cells were stained by antibodies against HA tag (green) and calreticulin (red). The majority of CLRN1N48K is observed in the ER and the cytoplasm. Scale bars, 20 μm.
Generation of Clrn1N48K knock-in mice
Results described thus far led to the hypothesis that CLRN1 is an essential hair bundle protein and that loss of hearing associated with the CLRN1N48K mutation in USH3 is attributable to defective intracellular trafficking of the CLRN1N48K protein to the hair bundle. To study the effect of N48K mutation in vivo, we generated a transgenic mouse carrying missense mutation in the first coding exon (exon 1) of the endogenous (mouse) Clrn1 gene. The amino acid sequence of mouse and human clarin-1 are highly conserved (>85%), including the conserved glycosylation site at position 48 (Adato et al., 2002), making the mouse an ideal in vivo model to generate the N48K knock-in mutation. The knock-in transgenic mouse was produced by a homologous recombination of engineered construct carrying the missense mutation; the wild-type exon 1 was replaced with exon 1 carrying N48K mutation (Fig. 7A). The knock-in allele was designated Clrn1N48K. Normal Mendelian segregation of the Clrn1N48K allele (wild-type Clrn1+/+, heterozygous Clrn1+/N48K, and homozygous Clrn1N48K/N48K) was observed. DNA sequence analysis of cDNA generated from Clrn1+/N48K mouse tissue confirmed mutation at the target site (Fig. 7B,C), and RT-PCR analysis confirmed that the expression of mouse Clrn1 mRNA and the sizes of splicing variants were indistinguishable from those observed in Clrn1+/+ mice (Fig. 7D). These experiments confirm that we have successfully generated a knock-in allele carrying the missense mutation in the mouse.
Figure 7.
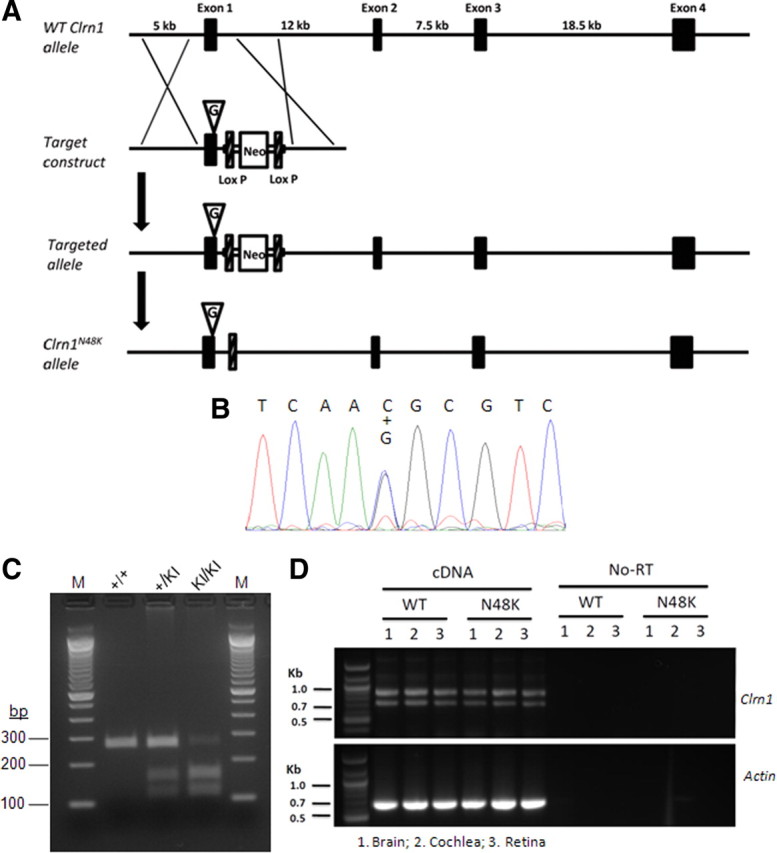
Generation of Clrn1N48K knock-in mouse. A, Targeting map for Clrn1N48K knock-in mice. Top, Mouse Clrn1 consists of four exons. Second row, The targeting vector contained 5.7 kb of 5′ and 2.2 kb of 3′ homologous sequence with C-to-G transversion at the codon 48. Third row, As a result of homologous recombination, codon 48 in exon 1 was changed to AAG, and the loxP flanked Neo gene was inserted after exon 1. Bottom row, Neo was removed from targeted locus by cre–loxP recombination. B, DNA sequence analysis confirms mutation at the target nucleotide in exon 1: both wild-type nucleotide C and mutant nucleotide G were observed in a cDNA sample from heterozygous knock-in mice (+/KI). C, The missense mutation introduced a BsaHI restriction site, which was used to distinguish the wild-type (+/+), heterozygous (+/KI), and homozygous (KI/KI) mutants. D, Mouse Clrn1 mRNA is expressed in brain, cochlea, and retina; the upper band (∼850 bp) and the lower band (∼750 bp) seen in the upper half is typical Clrn1 mRNA and alternative splicing expression pattern. Results show that Clrn1 mRNA expression is not affected by the missense mutation. WT, Wild type.
Clrn1N48K/N48K mice show early onset hearing loss that progresses to deafness around weaning age
To assess hearing function in Clrn1N48K/N48K mice, we recorded ABR from Clrn1N48K/N48K, Clrn1+/N48K, and Clrn1−/− mice at P18, P21, and P24. ABR recordings reflect the electrical responses of both the cochlear ganglion neurons and the nuclei of the central auditory pathway to sound stimulation. Clrn1+/N48K mice showed wild-type thresholds at all time points tested, confirming the recessive nature of the phenotype associated with clarin-1 mutation and, at the same time, arguing against dominant-negative effects by a single Clrn1N48K allele. Clrn1N48K/N48K mice displayed early-onset hearing loss that deteriorated to profound hearing loss after weaning. In contrast to Clrn1−/− mice, however, the Clrn1N48K/N48K mice displayed thresholds indicative of hearing loss but not profound hearing loss at P18 (p < 0.0001) or P21 (p < 0.0001). ABR thresholds of Clrn1N48K/N48K mice were quite variable at P18: although some mice showed threshold close to 40 dB SPL, others were in the 70–80 dB SPL range. The hearing function deteriorated to near deafness shortly after weaning age (p < 0.0001) (Fig. 8). Although ABR recordings can be performed in mice 2 weeks or older, it is not (yet) technically feasible to record transduction currents from cochlear hair cells at this age. We believe the ABR data to be a good reflection of the functional status of the cochlea in the Clrn1N48K/N48K mice. It is known that USH3 patients with CLRN1N48K mutation develop hearing loss with variable onset and severity (Sadeghi et al., 2005; Herrera et al., 2008). Here we report that N48K mutation in the mouse Clrn1 gene also causes hearing loss with variable levels of severity between mutants. However, all Clrn1N48K/N48K mice lose all hearing during the young-adult stage (P20–P30). The hearing loss phenotype observed in Clrn1N48K/N48K mutants, combined with data reported above (Figs. 1–6), suggest an important role for CLRN1 in the hair bundle.
Figure 8.

Assessment of hearing in Clrn1N48/K/N48K knock-in compared with the Clrn1−/− mice over time. The plot shows that mean ABR thresholds of Clrn1N48K/N48K mice (n = 25) are significantly elevated compared with the thresholds of Clrn1+/N48K mice (n = 6), which displayed wild-type thresholds at all time points tested. The plot also shows that ABR thresholds of Clrn1N48K/N48K mice are significantly lower compared with Clrn1−/− mice (n = 12) at P18 and P21; the difference is not significant at P24 because mice from both groups display profound hearing loss at P24. *p < 0.0001.
Mutation in Clrn1 disrupts hair bundle morphology, not tip links
Next, scanning electron microscopy was used to examine and compare the impact of null and missense mutation in Clrn1 on hair bundle development and structure. Cochlear hair bundles were studied by field emission scanning electron microscopy in the Clrn1−/− and Clrn1N48K/N48K mouse at P3–P4. Specifically, we looked at the overall organization of the hair bundle, bundle orientation, staircase arrangement, and tip links. The Clrn1−/− and Clrn1N48K/N48K both showed disruption of the hair bundle integrity compared with the control (Clrn1+/−) (Fig. 9). The bundle disturbance in these two alleles of Clrn1 was categorized into mild, moderate, and severe disturbance (marked by asterisks in Fig. 9). Of the 15 OHCs in each mutant panel, ∼40% from the Clrn1N48K/N48K mice showed mild bundle disturbance compared with ∼20% in the Clrn1−/− specimen; conversely, the organ of Corti from the Clrn1−/− mouse showed a greater number of moderately (50%) to severely (20%) disturbed hair bundles compared with that of the Clrn1N48/K/N48K mouse (30% moderate, 20% severe). Specifically, the disruption includes splits in the bundle and loss of some of the tall stereocilia. Some of the hair cells from the Clrn1−/− mice showed more severe disruption of the hair bundle arrangement compared with hair cells from Clrn1N48K/N48K mice (Fig. 10B,E). Hair cells from both mutants bear kinocilia, and the bundle orientation (vertex of the “v” facing the lateral edge) is generally not affected. Even in disrupted hair bundles, the graded heights of the stereocilia and tenting of the shorter stereocilia toward the taller stereocilia were apparent in both mutants (Fig. 10A,D,E). Tip links were identified in hair bundles examined from both mutants, with some exceptions. Overall, the profile of the hair bundle phenotype was the same in both mutants, although the degree of disruption in the bundle was more severe in Clrn1−/− mice compared with Clrn1N48K/N48K mice. Results from the Clrn1−/− mice show that Clrn1 is not required for the formation of tip links, development of graded heights of the stereocilia, or polarized orientation of the hair bundle. Rather, clear disruption in hair bundle structure in the Clrn1−/− mutants associated with loss of stereocilia and a milder phenotype of similar profile in Clrn1N48K/N48K mice suggests that mCLRN1 may be required to maintain the structure of the bundle after it is formed. However, it is likely that morphogenesis and maintenance of bundle stereocilia share common mechanisms to some degree, and, therefore, it is difficult to exclude CLRN1 from either process at this time. Although the mechanism by which mCLRN1 exerts its function in the bundle stereocilia remains to be elucidated, data presented here clearly demonstrate that CLRN1 is a protein essential for either maintenance or morphogenesis of hair bundles but is not essential for establishing planer cellular polarity.
Figure 9.
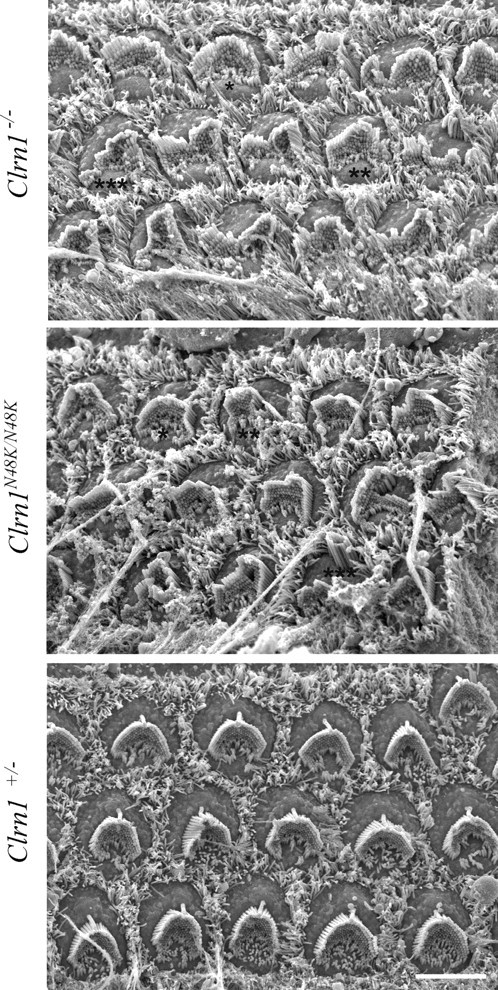
Scanning electron microscopy of the organ of Corti showing regions of ∼15 OHCs each from the Clrn1−/−, Clrn1N48K/N48K, and Clrn1+/− mice. Mild (*), moderate (**), and severe (***) bundle disruption can be seen in both the Clrn1−/− and Clrn1N48/K/N48K panels compared with the Clrn1+/− panel. However, the organ of Corti from the Clrn1−/− mouse showed a greater number of moderately to severely disturbed hair bundles compared with that of the Clrn1N48/K/N48K mouse. Scale bar, 5 μm.
Figure 10.
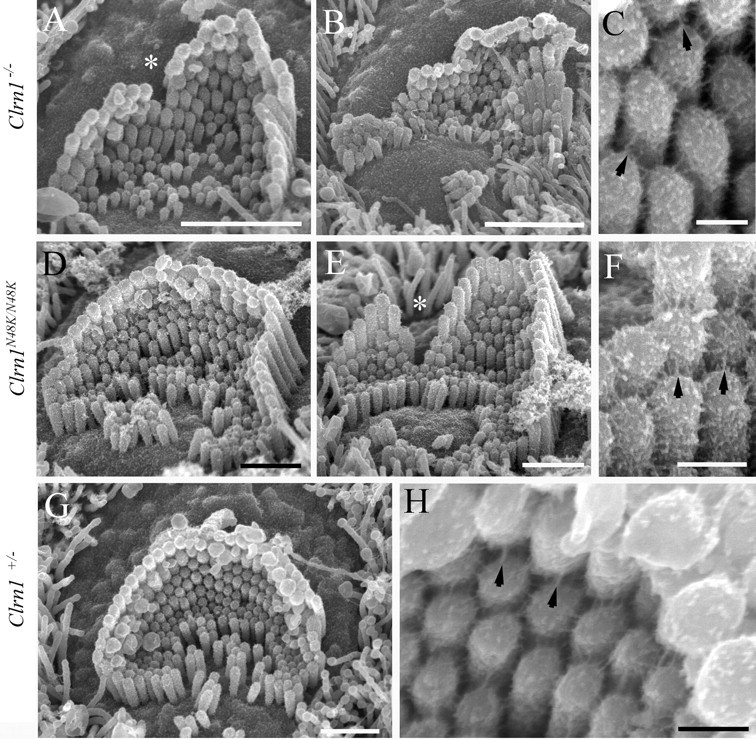
Scanning electron microscopy of P3 clarin knock-out (top row), knock-in (middle row), and heterozygote (bottom row) organ of Corti. The Clrn1−/− and Clrn1N48K /N48K both show substantial but variable disruption of the bundle, the former to a greater degree than the latter as exemplified by B and E. The disruption includes splits in the bundle (*) and loss of some of the tallest stereocilia in particular. At a higher magnification (C, F), tip-link-like links (arrows) and abundant lateral links typical of this age are evident in both. The heterozygote shows normal bundle features (G) and links (H; tip-link-like links highlighted with arrows). Scale bars: A, 2 μm; B, 1.5 μm; D, E, G, 1 μm; C, F, H, 200 nm.
Clrn1 is not essential for hair cell ribbon synapse development or function
Previous reports suggested a possible role for clarin-1 in the sensory synapses based on limited amino acid sequence homology to cerebellar synapse protein stargazin (Adato et al., 2002). Also, Geng et al. (2009) reported a delay in peak latencies in ABRs and vestibular evoked potential recordings. In the case of the ABR, peaks I–IV represent the response time (of the electrical responses) of both the cochlear ganglion neurons (or hair cell to afferent neurons) (peak I) and various nuclei of the central auditory pathway (peaks II–IV) to sound stimulation. Reevaluation of Clrn1−/− mice since the 2009 study confirmed delay in the peak latencies, but closer examination showed that a delay in the initial response or peak I could be associated with the mutant phenotype; whereas delays in the timing of peaks II–IV were secondary to the delay in peak I since interpeak latency (IPL) value was the same in Clrn1+/+ and Clrn1−/− mice (Table 1). A delay in initial response suggested a hair cell-to-afferent communication delay or ribbon synapse defect. We used two approaches to determine whether Clrn1 is essential for hair cell presynaptic function and/or development.
Table 1.
ABR from Clrn1−/− and Clrn1+/+ mice at P18
| Averages | Peak Ia | Peak II | Peak III | IPL I–III |
|---|---|---|---|---|
| Clrn1−/− | 1.81 | 2.51 | 3.44 | 1.63 |
| Clrn1+/+ | 1.46 | 2.30 | 3.06 | 1.62 |
aPeak latency measured in milliseconds.
Inner hair cell ribbon synapse develops normally in Clrn1−/− mice
We explored whether null mutation in Clrn1−/− affects development of the inner hair cell (IHC) ribbon synapse in Clrn1−/− mice using presynaptic and postsynaptic marker antibodies. The afferent ribbon synapses of P15 Clrn1+/− and Clrn1−/− mice were examined by immunofluorescence microscopy, using both an antibody directed against the nuclear protein CtBP2, which also detects RIBEYE, a major structural component of the ribbon (Schmitz et al., 2000), and an antibody against GluR2/3, associated with the dendrites of afferent neurons. Intact ribbon synapses were defined by the presence of juxtaposed RIBEYE and GluR2/3 fluorescent spots (Khimich et al., 2005). Whole mounts of organs of Corti from P15 Clrn1+/− and Clrn1−/− mice double immunolabeled for CtBP2 and GluR2/3 were analyzed by confocal microscopy. Quantitative analysis showed that the IHCs of Clrn1−/− had ∼90% the number of ribbon synapses observed in the Clrn1+/− specimen at P15 (Fig. 11A,B; Table 2). The observed reduction, ∼10% compared with control, is not considered significant and is probably secondary to the deteriorating condition of the hair cells in the Clrn1−/− background. Our results show that Clrn1 is not required for the development or maintenance of ribbon synapse, and the observed phenotype, namely delayed initial response in evoked potentials, is secondary to the compromised hair bundle structure/function.
Figure 11.

Light microscopy analysis of ribbon synapse in Clrn1−/− mice. Whole mounts of organs of Corti from P15 mice are double immunolabeled for CtBP2 and GluR2/3 and analyzed by confocal microscopy, followed by deconvolution and surface reconstruction. The difference in the number of double-labeled spots in Clrn1+/− (A) and Clrn1−/− (B) mice was not significant (for quantitative analysis, see Table 2).
Table 2.
Quantitative analysis of IHC ribbon synapses
| Clrn1+/− | Clrn1−/− | Reduction in Clrn1−/− | |
|---|---|---|---|
| Number of IHCs examined | 50 | 50 | |
| RIBEYE and GluR2/3 | 738 (14.76/cell) | 662 (13.24/cell) | 10% |
| RIBEYE spots | 795 (15.90/cell) | 720 (14.40/cell) | 9.4% |
| GluR2/3 spots | 782 (15.64/cell) | 712 (14.24/cell) | 8.9% |
Clrn1 is not essential in hair cell presynaptic function
Next, we explored the physiological impact of null mutation in Clrn1 on exocytic membrane capacitance changes (ΔCm) in response to Ca2+ influx (ICa) evoked by step depolarizations in perforated-patch recordings from IHCs at P14–P20. As illustrated for representative (Fig. 12A) and averaged (Fig. 12B,C) data, Clrn1−/− IHCs showed normal ICa and ΔCm. Neither the amplitude nor the voltage dependence of CaV1.3-mediated Ca2+ influx was altered. We did not find any obvious changes in exocytosis. This argues against an essential function for clarin-1 in hair cell presynaptic function.
Figure 12.
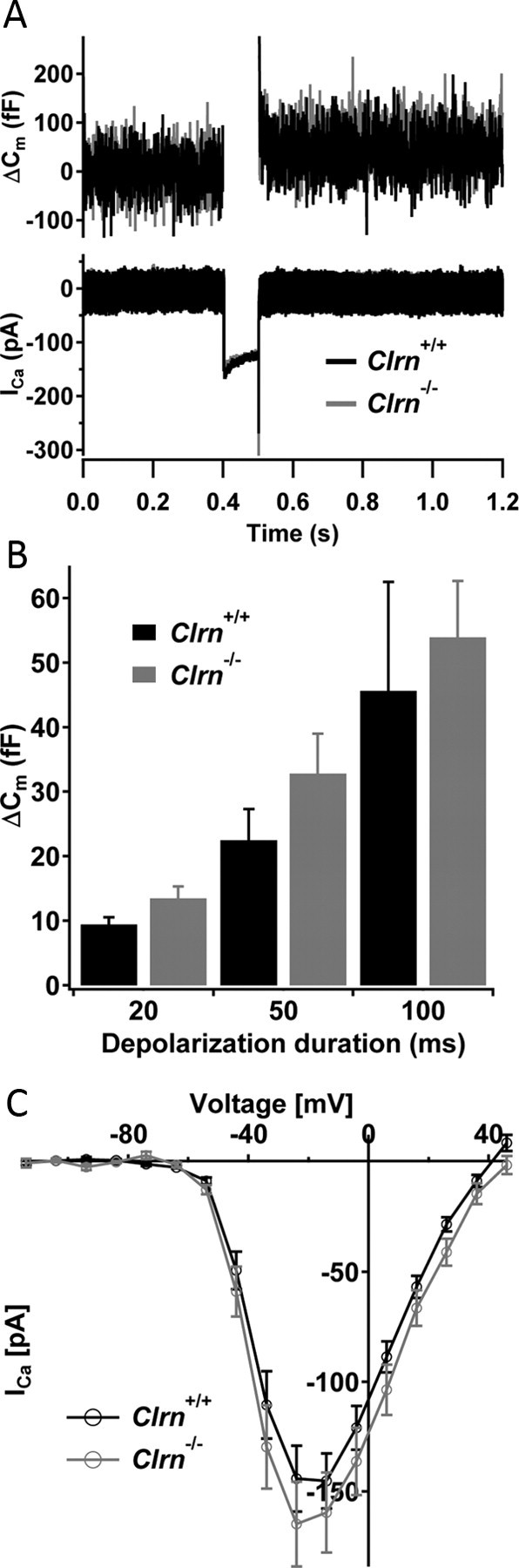
Patch-clamp analysis of presynaptic IHC function. A, Representative exocytic membrane capacitance changes (ΔCm, top) and Ca2+ currents (ICa, bottom) in response to 50 ms depolarization to −14 mV in Clrn1−/− (gray) and Clrn1+/+ (black) IHCs. B, Current–voltage relationship for Cav1.3 Ba2+ currents in control (black, n = 12 IHCs) and mutant (n = 10 IHCs) IHCs. Currents were evoked by 10 ms step depolarizations from a resting potential of −84 mV to variable potentials, and their average of the last 2 ms of current (excluding the tail current) was plotted against the test potential. C, Average ΔCm responses of control IHCs (7, 8, and 10 IHCs for 20, 50, and 100 ms depolarizations) and mutant IHCs (5, 6, and 8 IHCs for 20, 50, and 100 ms depolarizations) to 50 ms depolarizations to −14 mV. All responses of B and C are given as grand averages (calculated from the means of the individual cells) ± SEM.
Discussion
Here, we show that (1) CLRN1 is a hair bundle protein needed for development or maintenance of the normal shape of the bundle, and (2) the pathogenic mechanism underlying CLRN1N48K mutation in USH3 is a likely failure to express the mutated protein in the bundle. These findings have been confirmed using a novel mouse model carrying a Clrn1N48K knock-in mutation to investigate the consequence of the missense mutation N48K in mCLRN1 in vivo.
Our previous report showed that mice homozygous for the targeted deletion (or knock-out) of first coding exon in Clrn1 displayed inner ear dysfunction and loss of Clrn1 mRNA expression in the inner ear (Geng et al., 2009); hence, the knock-out allele is considered a null allele of Clrn1 and is represented as Clrn1−/−. To investigate the specific role of clarin-1 in hair cells, we started with an in-depth analysis of hair cell function in a Clrn1−/− mice. Measures of hair cell function (CM, FM1-43 uptake, and MET currents) from Clrn1−/− mice show diminished transducer function but not total loss. Clrn1−/− hair cells have tip links and rows of stereocilia showing gradation in height, but there is disruption of the v-shaped hair bundle along with loss of some of the tallest stereocilia; the missense mutation Clrn1N48K had a similar but less severe phenotype. These data suggest that the reduced output or sensitivity of hair cell function in Clrn1 −/− mice is likely a secondary consequence of disrupted stereocilia within the bundle and that Clrn1 is not required for the formation of the channel or the transducer apparatus. In contrast to Clrn1 mutant hair cells, presumptive null mutations in protocadherin 15 and cadherin 23 genes result in loss of tip links, severe disruption of hair bundle morphology, and abnormal MET currents (Ahmed et al., 2006; Pawlowski et al., 2006; Kikkawa et al., 2008; Alagramam et al., 2011). Together, the phenotypic analysis of Clrn1 mutant hair cells shows loss of hair bundle integrity and shape, strongly suggesting that CLRN1 is an essential hair bundle protein.
Genes carrying a missense mutation in coding sequence often provide powerful tools to understand the pathogenic mechanism, as well as the normal localization and function of the corresponding wild-type protein they encode, as illustrated by many examples in the literature (Smit et al., 1995; Ward et al., 1995; Guo et al., 2007; Laurila and Vihinen, 2009; Pangrsic et al., 2010). Similarly, we reasoned that valuable insights could be obtained by comparing the localization of hCLRN1–YFP to hCLRN1N48K–YFP in hair cells. In the transfected mouse cochlear hair cells, hCLRN1–YFP reproducibly localized to the hair bundle. In contrast, hCLRN1N48K–YFP failed to localize to the hair bundle. This was not attributable to failure of the expression system because we were able to replicate targeting of Prestin and Myo15A to their native location in hair cells, as shown previously (Belyantseva et al., 2000, 2003, 2005). We also confirmed that the N48K mutation in hCLRN1 prevented targeting to the membrane and retention of the mutant protein in the ER in a stably transfected human epithelial cell line. Thus, replacement of asparagine at position 48 with lysine in hCLRN1 affects hair bundle localization, as well as cell membrane localization, which we believe is the probable pathogenic mechanism associated with human CLRN1N48K mutation in hair cells.
Clrn1N48K/N48K mice show measurable hearing function (40–80 dB SPL thresholds) at P18 compared with age-matched Clrn1−/− mice, but this function is all but lost by P25. Hair cells from Clrn1−/− neonates do function, albeit at reduced capacity compared with controls, but Clrn1−/− mice show profound hearing loss by P18. How do we explain the observed phenotypes in these two mutants in the context of the data discussed above? One possibility under the hypothesis that CLRN1 is an essential bundle protein is as follows. Disintegration of the bundle starts early in the absence of CLRN1 (knock-out scenario), but weak expression of CLRN1N48K in the stereocilia provides partial but unsustainable rescue. What is the possible mechanism for the latter? It is known that lack of N-glycosylation could result in degradation of the misfolded protein by the ER-associated degradation (ERAD) pathway. However, it is also known that accumulation of misfolded proteins in the ER could result in ER stress attributable to perturbations in ER folding capacity (Schroder and Kaufman, 2005; Claessen et al., 2012). ER stress signals are known to activate unconventional (Golgi-independent) trafficking of membrane proteins. For example, an unconventional trafficking pathway has been implicated in the weak surface expression of a mutant (misfolded) cystic fibrosis transmembrane conductance regulator (ΔF508–CFTR) protein on airway epithelial cells and restoration of chloride function to target cells (Luo et al., 2009; Heon et al., 2011); however, the mouse model, as well as humans with the ΔF508–CFTR mutation, eventually succumbs to the disease. We know that CLRN1N48K accumulates in the ER (Fig. 6). Therefore, it is possible that a small proportion of the newly synthesized CLRN1N48K in developing hair cells “escapes” ERAD and reaches the plasma membrane via the unconventional trafficking pathway. It is then made available for translocation to the stereocilia. However, similar to the CFTR example, the expression of CLRN1N48K in the stereocilia may be weak, resulting in partial but unsustainable “rescue” of hair cell function, and it is well known from the studies of various Usher models that hair cells with compromised bundle function degenerate and die.
USH type I mouse models are deaf and show severe balance deficits by weaning age (Gibson et al., 1995; Alagramam et al., 2001, 2005; Jones et al., 2005). In contrast, Clrn1N48K/N48K mice show early (P18)-onset variable hearing loss that progresses to profound hearing loss by P30 with no obvious sign of balance deficit by that age. It is possible that Clrn1N48K/N48K mice may develop vestibular dysfunction more gradually than the Clrn1−/− mice. A lag between the cochlear and vestibular phenotype was noted in the Clrn1−/− mice (Geng et al., 2009), although the overall inner ear phenotype of Clrn1−/− mice was much more disruptive compared with the Clrn1N48K/N48K mice, consistent with the severity of mutation in these two lines. The reason for the differential expression of the cochlear and vestibular phenotypes in Clrn1 mutants is unknown, and it certainly warrants additional investigation. Interestingly, however, some of the CLRN1N48K patients exhibit hearing loss with either normal vestibular responses or mild vestibular dysfunction (Plantinga et al., 2005; Sadeghi et al., 2005), suggesting that ear phenotype in Clrn1N48K/N48K mice is similar to that observed in CLRN1N48K patients.
A role for CLRN1 in the actin-rich bundle stereocilia is further supported by observations in the literature. First, other Usher proteins (five related to type I and three related to type II), although functionally heterogeneous (unconventional myosin, cadherins, scaffold proteins, etc), are also essential for the normal development and/or function of the hair bundle (Leibovici et al., 2008; Williams, 2008). Second, Tian et al. (2009) showed that hCLRN1, but not hCLRN1N48K, reorganized actin filament structures and induced cellular protrusions in transfected kidney cell lines. Third, genetic interaction between CLRN1 and MYO7A was proposed based on patient data (Adato et al., 1999): patients homozygous for null allele of CLRN1 and carrying a single null of allele of MYO7A exhibited USH type I hearing loss (Adato et al., 1999). In other reports, direct interaction of myosin 7a and actin filaments has been demonstrated (Udovichenko et al., 2002; Inoue and Ikebe, 2003). Furthermore, a regulatory role for MYO7A in actin dynamics of stereocilia has been reported (Prosser et al., 2008). These observations provide a compelling link between CLRN1, actin, and other USH proteins and support our conclusion that CLRN1 is an essential hair bundle protein.
Based on limited amino acid sequence homology to stargazin, a cerebellar synapse four-transmembrane domain protein, a possible role of clarin-1 in sensory synapse was proposed (Adato et al., 2002). Delayed response times in evoked potential recording from the Clrn1−/− mice reported by Geng et al. (2009) also added some credence to that speculation. However, closer examination of the Clrn1−/− mice here suggests that the observed delays in peak response times are probably secondary to hair bundle defect in the mutant mice. There is no evidence of synaptic dysfunction according to the measures used here, arguing against a role for clarin-1 in hair cell-afferent communication. Because the null mutant did not show an effect, we found no reason to investigate mice carrying less severe mutation Clrn1N48K.
In summary, we have defined a role for clarin-1 in hair cells and revealed the pathogenic mechanism associated with human CLRN1N48K mutation in hair cells. We believe the Clrn1N48K knock-in mouse is a valuable animal model for ear disease in USH3 patients with CLRN1N48K mutation.
Footnotes
This work was supported by Usher III Initiative (Y.I. and K.N.A.), Elden foundation (Y.I.), National Institutes of Health Grants R01-DC010816 (K.N.A.) and R01-DC05439 (J.R.H.), and Deafness Research UK funds (D.F.), and a grant from the German Research Foundation (T.M.) via the Collaborative Research Center 889, and the state of Lower Saxony (through a “Neurosenses” PhD fellowship to T.O.-T.). We are grateful to Richard Lee and Corrine Thai for their help in the process of generating N48K knock-in mice and Chie Iioka for her help in generating the CLRN1–YFP construct. The monoclonal antibody α5 developed by D. M. Fambrough was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa, Department of Biological Sciences (Iowa City, IA).
The authors declare no competing financial interests.
References
- Adato A, Kalinski H, Weil D, Chaib H, Korostishevsky M, Bonne-Tamir B. Possible interaction between USH1B and USH3 gene products as implied by apparent digenic deafness inheritance. Am J Hum Genet. 1999;65:261–265. doi: 10.1086/302438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adato A, Vreugde S, Joensuu T, Avidan N, Hamalainen R, Belenkiy O, Olender T, Bonne-Tamir B, Ben-Asher E, Espinos C, Millán JM, Lehesjoki AE, Flannery JG, Avraham KB, Pietrokovski S, Sankila EM, Beckmann JS, Lancet D. USH3A transcripts encode clarin-1, a four-transmembranedomain protein with a possible role in sensory synapses. Eur J Hum Genet. 2002;10:339–350. doi: 10.1038/sj.ejhg.5200831. [DOI] [PubMed] [Google Scholar]
- Ahmed ZM, Goodyear R, Riazuddin S, Lagziel A, Legan PK, Behra M, Burgess SM, Lilley KS, Wilcox ER, Riazuddin S, Griffith AJ, Frolenkov GI, Belyantseva IA, Richardson GP, Friedman TB. The tip-link antigen, a protein associated with the transduction complex of sensory hair cells, is protocadherin-15. J Neurosci. 2006;26:7022–7034. doi: 10.1523/JNEUROSCI.1163-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alagramam KN, Murcia CL, Kwon HY, Pawlowski KS, Wright CG, Woychik RP. The mouse Ames waltzer hearing-loss mutant is caused by mutation of Pcdh15, a novel protocadherin gene. Nat Genet. 2001;27:99–102. doi: 10.1038/83837. [DOI] [PubMed] [Google Scholar]
- Alagramam KN, Stahl JS, Jones SM, Pawlowski KS, Wright CG. Characterization of vestibular dysfunction in the mouse model for Usher syndrome 1F. J Assoc Res Otolaryngol. 2005;6:106–118. doi: 10.1007/s10162-004-5032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alagramam KN, Goodyear RJ, Geng R, Furness DN, van Aken AF, Marcotti W, Kros CJ, Richardson GP. Mutations in protocadherin 15 and cadherin 23 affect tip links and mechanotransduction in mammalian sensory hair cells. PLoS One. 2011;6:e19183. doi: 10.1371/journal.pone.0019183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyantseva IA, Adler HJ, Curi R, Frolenkov GI, Kachar B. Expression and localization of prestin and the sugar transporter GLUT-5 during development of electromotility in cochlear outer hair cells. J Neurosci. 2000;20:RC116. doi: 10.1523/JNEUROSCI.20-24-j0002.2000. (1–5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyantseva IA, Boger ET, Friedman TB. Myosin XVa localizes to the tips of inner ear sensory cell stereocilia and is essential for staircase formation of the hair bundle. Proc Natl Acad Sci U S A. 2003;100:13958–13963. doi: 10.1073/pnas.2334417100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyantseva IA, Boger ET, Naz S, Frolenkov GI, Sellers JR, Ahmed ZM, Griffith AJ, Friedman TB. Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat Cell Biol. 2005;7:148–156. doi: 10.1038/ncb1219. [DOI] [PubMed] [Google Scholar]
- Cheatham MA, Naik K, Dallos P. Using the cochlear microphonic as a tool to evaluate cochlear function in mouse models of hearing. J Assoc Res Otolaryngol. 2011;12:113–125. doi: 10.1007/s10162-010-0240-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claessen JH, Kundrat L, Ploegh HL. Protein quality control in the ER: balancing the ubiquitin checkbook. Trends Cell Biol. 2012;22:22–32. doi: 10.1016/j.tcb.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland NG, Jenkins NA, Court DL. Recombineering: a powerful new tool for mouse functional genomics. Nat Rev Genet. 2001;2:769–779. doi: 10.1038/35093556. [DOI] [PubMed] [Google Scholar]
- Di Palma F, Holme RH, Bryda EC, Belyantseva IA, Pellegrino R, Kachar B, Steel KP, Noben-Trauth K. Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat Genet. 2001;27:103–107. doi: 10.1038/83660. [DOI] [PubMed] [Google Scholar]
- Fields RR, Zhou G, Huang D, Davis JR, Möller C, Jacobson SG, Kimberling WJ, Sumegi J. Usher syndrome type III: revised genomic structure of the USH3 gene and identification of novel mutations. Am J Hum Genet. 2002;71:607–617. doi: 10.1086/342098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furness DN, Hackney CM. High-resolution scanning-electron microscopy of stereocilia using the osmium-thiocarbohydrazide coating technique. Hear Res. 1986;21:243–249. doi: 10.1016/0378-5955(86)90222-4. [DOI] [PubMed] [Google Scholar]
- Gale JE, Marcotti W, Kennedy HJ, Kros CJ, Richardson GP. FM1-43 dye behaves as a permeant blocker of the hair-cell mechanotransducer channel. J Neurosci. 2001;21:7013–7025. doi: 10.1523/JNEUROSCI.21-18-07013.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng R, Geller SF, Hayashi T, Ray CA, Reh TA, Bermingham-McDonogh O, Jones SM, Wright CG, Melki S, Imanishi Y, Palczewski K, Alagramam KN, Flannery JG. Usher syndrome IIIA gene clarin-1 is essential for hair cell function and associated neural activation. Hum Mol Genet. 2009;18:2748–2760. doi: 10.1093/hmg/ddp210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson F, Walsh J, Mburu P, Varela A, Brown KA, Antonio M, Beisel KW, Steel KP, Brown SD. A type VII myosin encoded by the mouse deafness gene shaker-1. Nature. 1995;374:62–64. doi: 10.1038/374062a0. [DOI] [PubMed] [Google Scholar]
- Guo RB, Rigolet P, Ren H, Zhang B, Zhang XD, Dou SX, Wang PY, Amor-Gueret M, Xi XG. Structural and functional analyses of disease-causing missense mutations in Bloom syndrome protein. Nucleic Acids Res. 2007;35:6297–6310. doi: 10.1093/nar/gkm536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry KR, Chole RA. Cochlear electrical activity in the C57BL/6 laboratory mouse: volume-conducted vertex and round window responses. Acta Otolaryngol. 1979;87:61–68. doi: 10.3109/00016487909126387. [DOI] [PubMed] [Google Scholar]
- Heon YG, Noh SH, Tang BL, Kim KH, Le MG. Rescue of delta F508-CFTR trafficking via a GRASP-Dependent Unconventional Secretion Pathway. Cell. 2011;146:746–760. doi: 10.1016/j.cell.2011.07.021. [DOI] [PubMed] [Google Scholar]
- Herrera W, Aleman TS, Cideciyan AV, Roman AJ, Banin E, Ben-Yosef T, Gardner LM, Sumaroka A, Windsor EA, Schwartz SB, Stone EM, Liu XZ, Kimberling WJ, Jacobson SG. Retinal disease in Usher syndrome III caused by mutations in the clarin-1 gene. Invest Ophthalmol Vis Sci. 2008;49:2651–2660. doi: 10.1167/iovs.07-1505. [DOI] [PubMed] [Google Scholar]
- Holt JR, Corey DP, Eatock RA. Mechanoelectrical transduction and adaptation in hair cells of the mouse utricle, a low-frequency vestibular organ. J Neurosci. 1997;17:8739–8748. doi: 10.1523/JNEUROSCI.17-22-08739.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt JR, Gillespie SK, Provance DW, Shah K, Shokat KM, Corey DP, Mercer JA, Gillespie PG. A chemical-genetic strategy implicates myosin-1c in adaptation by hair cells. Cell. 2002;108:371–381. doi: 10.1016/s0092-8674(02)00629-3. [DOI] [PubMed] [Google Scholar]
- Inoue A, Ikebe M. Characterization of the motor activity of mammalian myosin VIIA. J Biol Chem. 2003;278:5478–5487. doi: 10.1074/jbc.M210489200. [DOI] [PubMed] [Google Scholar]
- Joensuu T, Hämäläinen R, Yuan B, Johnson C, Tegelberg S, Gasparini P, Zelante L, Pirvola U, Pakarinen L, Lehesjoki AE, de la Chapelle A, Sankila EM. Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am J Hum Genet. 2001;69:673–684. doi: 10.1086/323610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SM, Johnson KR, Yu H, Erway LC, Alagramam KN, Pollak N, Jones TA. A quantitative survey of gravity receptor function in mutant mouse strains. J Assoc Res Otolaryngol. 2005;6:297–310. doi: 10.1007/s10162-005-0009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khimich D, Nouvian R, Pujol R, Tom Dieck S, Egner A, Gundelfinger ED, Moser T. Hair cell synaptic ribbons are essential for synchronous auditory signalling. Nature. 2005;434:889–894. doi: 10.1038/nature03418. [DOI] [PubMed] [Google Scholar]
- Kikkawa YS, Pawlowski KS, Wright CG, Alagramam KN. Development of outer hair cells in Ames waltzer mice: mutation in protocadherin 15 affects development of cuticular plate and associated structures. Anat Rec (Hoboken) 2008;291:224–232. doi: 10.1002/ar.20632. [DOI] [PubMed] [Google Scholar]
- Laurila K, Vihinen M. Prediction of disease-related mutations affecting protein localization. BMC Genomics. 2009;10:122. doi: 10.1186/1471-2164-10-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibovici M, Safieddine S, Petit C. Mouse models for human hereditary deafness. Curr Top Dev Biol. 2008;84:385–429. doi: 10.1016/S0070-2153(08)00608-X. [DOI] [PubMed] [Google Scholar]
- Lelli A, Asai Y, Forge A, Holt JR, Géléoc GS. Tonotopic gradient in the developmental acquisition of sensory transduction in outer hair cells of the mouse cochlea. J Neurophysiol. 2009;101:2961–2973. doi: 10.1152/jn.00136.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, McDonald K, Hanrahan JW. Trafficking of immature ΔF508-CFTR to the plasma membrane and its detection by biotinylation. Biochem J. 2009;419:211–219. doi: 10.1042/BJ20081869. 2 p following 219. [DOI] [PubMed] [Google Scholar]
- Meyer AC, Frank T, Khimich D, Hoch G, Riedel D, Chapochnikov NM, Yarin YM, Harke B, Hell SW, Egner A, Moser T. Tuning of synapse number, structure and function in the cochlea. Nat Neurosci. 2009;12:444–453. doi: 10.1038/nn.2293. [DOI] [PubMed] [Google Scholar]
- Meyers JR, MacDonald RB, Duggan A, Lenzi D, Standaert DG, Corwin JT, Corey DP. Lighting up the senses: FM1-43 loading of sensory cells through nonselective ion channels. J Neurosci. 2003;23:4054–4065. doi: 10.1523/JNEUROSCI.23-10-04054.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser T, Beutner D. Kinetics of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse of the mouse. Proc Natl Acad Sci U S A. 2000;97:883–888. doi: 10.1073/pnas.97.2.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness SL, Ben-Yosef T, Bar-Lev A, Madeo AC, Brewer CC, Avraham KB, Kornreich R, Desnick RJ, Willner JP, Friedman TB, Griffith AJ. Genetic homogeneity and phenotypic variability among Ashkenazi Jews with Usher syndrome type III. J Med Genet. 2003;40:767–772. doi: 10.1136/jmg.40.10.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangrsic T, Lasarow L, Reuter K, Takago H, Schwander M, Riedel D, Frank T, Tarantino LM, Bailey JS, Strenzke N, Brose N, Müller U, Reisinger E, Moser T. Hearing requires otoferlin-dependent efficient replenishment of synaptic vesicles in hair cells. Nat Neurosci. 2010;13:869–876. doi: 10.1038/nn.2578. [DOI] [PubMed] [Google Scholar]
- Pawlowski KS, Kikkawa YS, Wright CG, Alagramam KN. Progression of inner ear pathology in Ames waltzer mice and the role of protocadherin 15 in hair cell development. J Assoc Res Otolaryngol. 2006;7:83–94. doi: 10.1007/s10162-005-0024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plantinga RF, Kleemola L, Huygen PL, Joensuu T, Sankila EM, Pennings RJ, Cremers CW. Serial audiometry and speech recognition findings in Finnish Usher syndrome type III patients. Audiol Neurootol. 2005;10:79–89. doi: 10.1159/000083363. [DOI] [PubMed] [Google Scholar]
- Prosser HM, Rzadzinska AK, Steel KP, Bradley A. Mosaic complementation demonstrates a regulatory role for myosin VIIa in actin dynamics of stereocilia. Mol Cell Biol. 2008;28:1702–1712. doi: 10.1128/MCB.01282-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell IJ, Richardson GP. The morphology and physiology of hair cells in organotypic cultures of the mouse cochlea. Hear Res. 1987;31:9–24. doi: 10.1016/0378-5955(87)90210-3. [DOI] [PubMed] [Google Scholar]
- Sadeghi M, Cohn ES, Kimberling WJ, Tranebjaerg L, Möller C. Audiological and vestibular features in affected subjects with USH3: a genotype/phenotype correlation. Int J Audiol. 2005;44:307–316. doi: 10.1080/14992020500060610. [DOI] [PubMed] [Google Scholar]
- Saihan Z, Webster AR, Luxon L, Bitner-Glindzicz M. Update on Usher syndrome. Curr Opin Neurol. 2009;22:19–27. doi: 10.1097/wco.0b013e3283218807. [DOI] [PubMed] [Google Scholar]
- Santarelli R, Scimemi P, Dal Monte E, Arslan E. Cochlear microphonic potential recorded by transtympanic electrocochleography in normally-hearing and hearing-impaired ears. Acta Otorhinolaryngol Ital. 2006;26:78–95. [PMC free article] [PubMed] [Google Scholar]
- Schmitz F, Königstorfer A, Südhof TC. RIBEYE, a component of synaptic ribbons: a protein's journey through evolution provides insight into synaptic ribbon function. Neuron. 2000;28:857–872. doi: 10.1016/s0896-6273(00)00159-8. [DOI] [PubMed] [Google Scholar]
- Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Smit LS, Strong TV, Wilkinson DJ, Macek M, Jr, Mansoura MK, Wood DL, Cole JL, Cutting GR, Cohn JA, Dawson DC, et al. Missense mutation (G480C) in the CFTR gene associated with protein mislocalization but normal chloride channel activity. Hum Mol Genet. 1995;4:269–273. doi: 10.1093/hmg/4.2.269. [DOI] [PubMed] [Google Scholar]
- Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334:1086–1090. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer EA, Holt JR. Sensory transduction and adaptation in inner and outer hair cells of the mouse auditory system. J Neurophysiol. 2007;98:3360–3369. doi: 10.1152/jn.00914.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian G, Zhou Y, Hajkova D, Miyagi M, Dinculescu A, Hauswirth WW, Palczewski K, Geng R, Alagramam KN, Isosomppi J, Sankila EM, Flannery JG, Imanishi Y. Clarin-1, encoded by the Usher Syndrome III causative gene, forms a membranous microdomain: possible role of clarin-1 in organizing the actin cytoskeleton. J Biol Chem. 2009;284:18980–18993. doi: 10.1074/jbc.M109.003160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udovichenko IP, Gibbs D, Williams DS. Actin-based motor properties of native myosin VIIa. J Cell Sci. 2002;115:445–450. doi: 10.1242/jcs.115.2.445. [DOI] [PubMed] [Google Scholar]
- Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- Williams DS. Usher syndrome: animal models, retinal function of Usher proteins, and prospects for gene therapy. Vision Res. 2008;48:433–441. doi: 10.1016/j.visres.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Currall B, Yamashita T, Parker LL, Hallworth R, Zuo J. Prestin-prestin and prestin-GLUT5 interactions in HEK293T cells. Dev Neurobiol. 2007;67:483–497. doi: 10.1002/dneu.20357. [DOI] [PubMed] [Google Scholar]
- Zallocchi M, Meehan DT, Delimont D, Askew C, Garige S, Gratton MA, Rothermund-Franklin CA, Cosgrove D. Localization and expression of clarin-1, the Clrn1 gene product, in auditory hair cells and photoreceptors. Hear Res. 2009;255:109–120. doi: 10.1016/j.heares.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallocchi M, Meehan DT, Delimont D, Rutledge J, Gratton MA, Flannery J, Cosgrove D. Role for a novel usher protein complex in hair cell synaptic maturation. PLoS One. 2012;7:e30573. doi: 10.1371/journal.pone.0030573. [DOI] [PMC free article] [PubMed] [Google Scholar]