

Abstract
Alternative splicing patterns are regulated by RNA binding proteins that assemble onto each pre-mRNA to form a complex RNP structure. The polypyrimidine tract binding protein, PTB, has served as an informative model for understanding how RNA binding proteins affect spliceosome assembly and how changes in the expression of these proteins can control complex programs of splicing in tissues. In this review, we describe the mechanisms of splicing regulation by PTB and its function, along with its paralog PTBP2, in neuronal development.
Keywords: Alternative splicing, spliceosome, RNA binding protein, neuronal development, RNP
Alternative splicing patterns are regulated by RNA binding proteins that package pre-mRNAs into complex RNP structures (Black, 2003; Matlin et al., 2005; Chen and Manley, 2009). The polypyrimidine tract binding protein, PTB, was among the first of these proteins to be discovered as a splicing regulator, perhaps because of its large number of targets and its efficient crosslinking to RNA with short-wave UV. It is now clear that PTB is one example of a large class of proteins that bind pre-mRNA to alter sites of splicing and polyadenylation. These proteins, including the SR proteins, many of the original hnRNP proteins (notably A1, H, L), the CELF, Nova, Rbfox, and Muscle blind families, and others, belong to a variety of structural families and have different RNA recognition properties and different patterns of expression in tissues (Graveley, 2000; Dreyfuss et al., 2002; Black, 2003; Chen and Manley, 2009; Kalsotra and Cooper, 2011). In addition to their nuclear role in RNA processing, these proteins, including PTB, also often affect cytoplasmic processes such as mRNA localization, translation or decay (Hellen et al., 1993; Cote et al., 1999; Kim et al., 2000; Tillmar and Welsh, 2002; Hamilton et al., 2003; de Hoog et al., 2004; Bushell et al., 2006; Fred et al., 2006; Sawicka et al., 2008; Babic et al., 2009; Besse et al., 2009; Kafasla et al., 2009; Cobbold et al., 2010; Matus-Nicodemos et al., 2011). Within the nucleus, these proteins affect large programs of alternative processing events (Ule et al., 2006; Boutz et al., 2007b; Kalsotra et al., 2008; Licatalosi et al., 2008; Xue et al., 2009; Yeo et al., 2009; Kalsotra and Cooper, 2011). Both the biological roles of the post-transcriptional regulatory networks controlled by these proteins, and the biochemical mechanisms by which they alter splicing choices are still largely not understood.
PTB has been extensively studied as an informative model for understanding how RNA binding proteins affect spliceosome assembly. In this review we focus on the mechanisms of splicing regulation by PTB and its function, along with its paralog PTBP2, in neuronal development.
Isoforms, paralogs, and conservation
PTB was first identified in HeLa nuclear extract as a factor that crosslinks to the pyrimidine tracts within 3′ splice sites (Garcia-Blanco et al., 1989; Patton et al., 1991). This gave the protein its name, but it later became clear that another protein, U2AF65, was responsible for recognizing the polypyrimidine tract at the 3′ splice site during spliceosome assembly (Zamore and Green, 1989; Gil et al., 1991; Singh et al., 1995). PTB in contrast had a negative effect on splice site recognition and served to regulate the alternative splicing patterns for a variety of pre-mRNAs (Ashiya and Grabowski, 1997; Chan and Black, 1997; Gooding et al., 1998; Mulligan et al., 1992; Pérez et al., 1997a; Carstens et al., 2000). PTB was also identified as one of the hnRNP group of proteins that crosslink to hnRNA in HeLa cell nuclei and thus called hnRNP I (Ghetti et al., 1992). Like the other hnRNPs, PTB is very abundant and can be present in ~10,000 copies per cell nucleus.
PTB has an N-terminal nuclear shuttling domain followed by four non-canonical RNA Recognition Motifs that act as RNA binding domains connected by flexible linker peptides (RRM, also called RNP consensus or RNPcs domains). This primary structure is conserved from flies to humans (Figure 1). The mouse, human, and rat orthologs are more than 95% identical, while the chicken, xenopus, and zebrafish proteins are only slightly less similar to the human protein. Overall, the Drosophila PTB (also called Hephaestos) is 40% identical to the human protein, but the RRM domains are more than 50% identical. Notably, the residues that contact RNA are conserved in human, mouse, rat, and chicken. The Xenopus, zebrafish, and Drosophila proteins contain a few conservative changes in these positions (Figure 1). One PTB isoform in Drosophila is male germline specific and is important for spermatid individualization. Thus, Drosophila PTB may have different or additional functions from mammalian PTB or may combine functions of mammalian PTB and PTBP2 (Robida and Singh, 2003; Robida et al., 2010). A gene in C. elegans named PTB does not exhibit the same level of sequence similarity as the PTBs found in other species, and whether it has a common function is not clear. The conservation of the PTB proteins indicates that their RNA recognition properties are likely very similar across diverse metazoan species.
Figure 1.

Protein sequence alignment of human, mouse, chicken, Xenopus, Zebrafish, and Drosophila PTB proteins and the human PTBP2 protein. Residues identical to human PTB are shown as dots. RRM domains are shaded light green. RNA interacting residues are shaded dark green. The black boxes indicate the RNP1 and RNP2 motifs. The arrowheads indicate the RNA interacting residues that are different in PTBP2. The N-terminal region of the Drosophila sequence is not shown and the sequence starts at residue 192.
The PTB locus on human chromosome 19 (gene name PTBP1) produces several spliced isoforms. Alternative splicing of exon 9 alters the length of the linker region between RRMs 2 and 3. PTB isoform 4 (PTB-4) includes exon 9 to produce a 557 amino acid protein. Skipping of exon 9 generates the shorter, 531 amino acid, PTB isoform 1 (PTB-1) (Gil et al., 1991; Ghetti et al., 1992; Wagner et al., 1999). These are the most abundant isoforms and usually appear as a nearly equimolar doublet on protein gels (Wagner et al., 1999). However, their relative abundance can vary with cell type and growth conditions and there is evidence that they can vary in their splicing repression activity (Wagner et al., 1999; Wollerton et al., 2001). There is also a minor, 550 amino acid, isoform 2 that is generated from an alternative 3′ splice site in exon 9. In addition, alternative splicing of PTB exon 11 produces an mRNA isoform (Gooding et al., 1998) that is subject to nonsense mediated mRNA decay, as discussed below (Wollerton et al., 2004).
In vertebrates, PTB is widely expressed across many although not all tissues. PTB is a member of a small gene family with several paralogs that exhibit more restricted tissue specific expression. PTBP2 (also called neuronal or brain PTB, nPTB, brPTB) is encoded on human chromosome 1 and expressed in neurons, testis and some other cell types. Smooth muscle PTB (smPTB) is a homolog found so far only in the mouse genome and expressed in smooth muscle (mouse chromosome 4) (Gooding et al., 2003). Human Rod1 (Regulator of Differentiation 1 recently named PTBP3) was first identified by its ability to complement mutations in the S. pombe gene Nrd1. Mammalian Rod1/PTBP3 is primarily expressed in hematopoietic cells (human chromosome 9) (Yamamoto et al., 1999; Spellman et al., 2007). All of these proteins, though encoded on separate genes and having different expression patterns, have similar domain structures and RNA binding properties. We will discuss PTBP2 below.
Structure
RRM domains have a common βαββαβ fold and are defined by two conserved RNP motifs. The RNP2 hexamer is on the β1 strand and the RNP1 octamer is on β3. The canonical RNP2 motif is I/L/V-F/Y- I/L/V-X-N-L and the canonical RNP1 motif is R/K-G-F/Y-G/A-F/Y-I/L/V-X-F/Y. The aromatic residues of these typical motifs engage in base stacking interactions during RNA recognition. The consensus of the four PTB RNP2 motifs is I/L-H/I/LI/V/L-R/E/S-K/N-L/I and the RNP1 consensus is K/Q/R-N/F/E/K-Q/N/M-A-F/L-I/L-E/Q. Notably, the PTB RNP motifs are non-canonical in replacing the aromatic residues with non-aromatic residues. Nevertheless, mutational and structural analyses of the PTB motifs indicate that the equivalent residues are still important for RNA binding and perform similar functions (Conte et al., 2000; Oberstrass et al., 2005).
NMR solution structures have been determined of each of the PTB RRM domains (Conte et al., 2000; Simpson et al., 2004; Oberstrass et al., 2005; Petoukhov et al., 2006; Auweter and Allain, 2008). RRMs 1 and 4 have the typical RNA binding domain (RBD) βαββαβ topology (Figures 2 and 3). The two alpha helices are packed against the 4-stranded antiparallel beta sheet that provides a surface for RNA binding. RRM domains 2 and 3 are non-canonical, in that they have an additional 5th strand of the β sheet derived from a C-terminal extension (Conte et al., 2000; Simpson et al., 2004; Oberstrass et al., 2005; Vitali et al., 2006). Interestingly, the linker between β4 and β5 of RRM3 is positioned above the RNA binding surface and plays a role in RNA recognition (Yuan et al., 2002; Oberstrass et al., 2005).
Figure 2.

A and B. Ribbon representations of PTB RRMs 1 and 2 bound to a CUCUCU hexamer. The alpha helices are colored cyan, beta strands magenta and loops beige. C and D. Base specific contacts made by RRMs 1 and 2. Amino acids and nucleotides are shown as stick models. The main chain cartoon traces are colored gray. Atomic contacts are indicated by dashed lines.
Figure 3.

A. Ribbon representation of PTB RRMs 3 and 4, each bound to a CUCUCU hexamer. B and C. Base specific contacts made by RRMs 3 and 4, respectively. Amino acids and nucleotides are shown as stick models. The main chain cartoon traces are colored gray. Atomic contacts are indicated by dashed lines.
NMR studies of individual PTB RRMs bound to a CUCUCU hexamer have given detailed information of RNA recognition by each domain (Oberstrass et al., 2005). RRM1 interacts with U2, C3 and U4, which are spread across the beta sheet surface of the protein (PDB 2AD9; Figure 2). U2 is stabilized by a stacking interaction with Arg64 and a hydrogen bond with the side chain of Gln129 (To allow alignment with Figure 1, this residue numbering is for human PTB-4. The PDB structure files are numbered according to PTB-1. Residues 1 through 298 are the same for PTB-1 and PTB-4. Residues 299 through 324 of PTB-4 are missing from PTB-1. Thus, for positions above residue 324 in PTB-4, the equivalent residue in PTB-1 or the PDB files is obtained by subtracting 26.). C3 is stacked between His62 and Asn132. Base specific interactions of this nucleotide include hydrogen bonds between the cytidine O2 and Ser131, and between the cytidine N3 and amino group and main chain of Asn132 and Phe130, respectively. U4 is positioned in a hydrophobic pocket formed by five protein side chains and is stabilized by H-bonds between N3 and O4 of U4 and the main chain of the C-terminus of the domain.
RRM2 interacts with C3, U4 and U6 in contacts similar to RRM1 (PDB 2ADB; Figure 2). C3 is stabilized by hydrogen bonds between C3 O2 and the side chain of Ser258, and between C3 N3 and the main chain of Lys259. Again, N3 and O4 of U4 H-bonds with the main chain of Asn264. U6 makes contacts with Lys266, Lys271 and Tyr267 residues located on the loop of the C-terminal extension before beta strand 5, although these interactions are variable between different structures within the NMR ensemble (Oberstrass et al., 2005).
RRM3 interacts with 5 nucleotides U2, C3, U4, C5 and U6 (PDB 2ADC; Figure 3). Again, sequence specific interactions are made to U2, C3 and U4. The 5′ phosphate of U2 interacts with Arg431. Again similar to RRMs 1 and 2, C3 engages in base specific H-bonds between C3 O2 and the side chain of Ser435 and between C3 N3 and the main chain of Lys436. RRM3 also makes additional RNA contacts. The Nε of His437 interacts with the 2′ OH of the C3 ribose. The base of C5 engages in stacking and Van der Waals interactions with Phe397 and Leu396. The side chain amino group of Lys394 interacts with phosphate oxygens of both C5 and U6. Additionally, U6 can be stabilized by an H-bond between the ribose 2′OH and the side chain of Arg444, but this contact is not present in all structures of the ensemble. This extended interaction with 5 nucleotides gives RRM3 more contacts with the CUCUCU hexamer than the other RRMs (Oberstrass et al., 2005).
RRM4 binds to 3 nucleotides, U4, C5 and U6 (PDB 2ADC; Figure 3) with interactions similar to those made by RRMs 1, 2 and 3. Specifically, the U4 N3 H-bonds with the amide of Asn474. Both U4 N1 and the U4 ribose interact with the side chain hydroxyl group of Ser485. His483 stacks on C5, while the main chain of Lys554 hydrogen bonds with the C5 base. U6 is stabilized by an H-bond between U6 O2 and the amino group of Lys511, and by a stacking interaction with Phe513.
The NMR structures indicate that (1) the overall fold of each RRM remains largely unaltered in the free and bound states, (2) the beta sheet surfaces and loops across these surfaces make specific base and backbone contacts to the RNA, and (3) base specificity is primarily to pyrimidine triplets containing CU or UC dinucleotides. Each of the domains uses similar interactions to define base specific binding, most notably domains 1, 2 and 3 each use a serine residue to hydrogen bond to the O2 moiety of a central C nucleotide. These Serines and most of the other amino acids making direct RNA contacts are conserved across multiple species, as well as in PTBP2, indicating that the binding specificity of these proteins is likely to be similar (Figure 1).
Collectively, the structures of the RNA-bound domains nicely explain the binding preference for extended pyrimidine tracts of mixed C and U nucleotides. Nevertheless, each PTB RRM has distinct structural features and how these differences affect binding to the wide variety of PTB binding sites is still an open question (Oberstrass et al., 2005; Auweter et al., 2007). It is not clear whether other oligopyrimidine sequences might show additional interactions with individual domains and have different domain preferences. It is also likely that each domain can make additional specific contacts with RNAs that are longer than the short oligonucleotides used for structure determination. What constitutes an optimal site for the full length protein and how the four domains are arranged when bound to extended regions of RNA is an area of current research (see below).
NMR studies of the full length protein demonstrated that the linker peptides between RRMs 1 and 2 and between RRMs 2 and 3 are flexible, allowing RRMs 1 and 2 to behave independently in solution. In contrast, RRMs 3 and 4 are in fixed contact with each other, requiring a particular orientation of the bound RNA substrate (see below). The α helical faces of these two domains and their linker form a hydrophobic interface that orients the two RNA binding beta sheet surfaces onto opposite faces (Figure 3) (Oberstrass et al., 2005; Vitali et al., 2006). With this orientation, the simultaneous engagement of two RNA elements by the two domains will create a RNA loop. To interact with the two domains, two CU tracts must be separated by at least 15 nucleotides (Lamichhane et al., 2010). The RNA is oriented on the two domains such that the 5′ CU element is bound by RRM 3 and the 3′ element by RRM 4. When the individual domain 3 or domain 4 fragments are examined, the RRM’s adopt folds similar to their structure in PTB3+4. However, their affinity for polypyrimidine tracts is much reduced from that of PTB3+4 (Pérez et al., 1997b; Maynard and Hall, 2010).
The modular nature of the protein with flexible peptides separating domains 1 and 2, and domains 2 and 3 make it likely that the arrangement of the RRMs will vary between different target RNAs and that additional contacts outside of the core pyrimidine motifs will contribute to the binding. A complete understanding of how PTB interacts with its targets will require structures of the full-length protein bound to long RNA substrates.
PTB assembly into pre-mRNP complexes
Early studies described PTB as a dimer (Pérez et al., 1997b; Oh et al., 1998). However, analytical ultracentrifugation experiments characterizing the molecular mass of PTB clearly indicate that it is a monomer in solution (Simpson et al., 2004; Amir-Ahmady et al., 2005). Small angle X-ray scattering studies further showed PTB to be an elongated molecule with an extended arrangement of the RRM’s (Simpson et al., 2004; Petoukhov et al., 2006). Nevertheless, PTB likely engages in homomeric or heteromeric protein–protein interactions when bound to RNA (Chou et al., 2000; Markovtsov et al., 2000; Polydorides et al., 2000; Huttelmaier et al., 2001; Amir-Ahmady et al., 2005; Henneberg et al., 2010). Covalent dimerization of PTB monomers was shown to occur when the protein is isolated under oxidizing conditions. This results from an inter-molecular disulphide bridge via Cys23 (Monie et al., 2005). In vivo cross linking and immunoprecipitation (CLIP) studies support the idea that disulphide linked dimers may form when RNA crosslinked products are isolated under non-reducing conditions (Xue et al., 2009). As discussed below, there is abundant evidence that multiple PTB monomers assemble onto extended splicing regulatory elements. The binding of the first PTB monomer on these RNAs can affect binding of subsequent proteins (Chou et al., 2000; Amir-Ahmady et al., 2005). However, it is not clear what kinds of direct PTB-PTB contacts might occur in these higher order assemblies.
There are a large number of PTB binding sites within the transcriptome (Xue et al., 2009). Known PTB binding sites can be classified into two groups. PTB can bind with high affinity to single stranded RNA regions containing multiple C and U residues that often alternate (Singh et al., 1995; Pérez et al., 1997b; Yuan et al., 2002; Simpson et al., 2004; Amir-Ahmady et al., 2005; Auweter et al., 2007). Such sequences are commonly found within splicing regulatory elements controlled by PTB. There are also more structured binding sites, where PTB makes specific interactions with bulged pyrimidine nucleotides within the paired stems and loops of a larger secondary structure. These are commonly found within the internal ribosome entry sites (IRES) bound by PTB, but are also found in other contexts (Mitchell et al., 2005; Bushell et al., 2006; Kafasla et al., 2009; Sharma et al., 2011).
Typical splicing regulatory elements that bind PTB are extended runs of pyrimidines. These are similar to high affinity binding sites isolated by in vitro selection (Singh et al., 1995; Pérez et al., 1997a). The pyrimidine tracts of native binding sites can vary from less than 6 to dozens of nucleotides in length. In general, the affinity for PTB depends on their length. Computational analysis of these binding sites indicates that individual G residues can be tolerated within the pyrimidine tracts, but A residues are deleterious for binding (A. Han and DLB, unpublished). The minimal high affinity binding site for PTB was characterized from the c-src pre-mRNA (Amir-Ahmady et al., 2005). High affinity binding required more than 30 nucleotides of RNA including two copies of a CUUCUCUCU element as well as additional adjacent pyrimidine nucleotides. Gel shift analyses with this sequence identified two PTB/RNA complexes. Complex 1 formed at lower protein concentration (Kd ~ 1 nM), while the larger complex 2 required more protein, indicating that the second binding event was of lower affinity (Kd ~ 140 nM). Complex 1 also formed on a short RNA (containing only 1 CUUCUCUCU element), albeit with lower affinity than a longer RNA. However, complex 2 required both CUUCUCUCU elements plus an adjacent pyrimidine region immediately upstream or downstream of the repeated element. Additional short polypyrimidine elements distant from the high affinity site stimulated complex 2 formation at lower PTB concentrations, presumably by increasing the affinity of the second PTB in the assembly through interaction of an additional RRM with the distant CU element.
Elements needed to mediate PTB-dependent splicing repression of an exon have also been examined. For a weakly spliced exon, a single high affinity binding site present either within the exon or the 3′ splice site can be sufficient to repress exon inclusion (Shen et al., 2004; Amir-Ahmady et al., 2005). However, for an efficiently spliced exon to be repressed, at least 2 PTB binding sites are required. Under these conditions, the upstream site must be long enough to form complex 2, but the second binding site can be of lower affinity. It is possible, in transcripts where only one high affinity site has been identified, that additional low affinity sites are present. In most systems multiple PTB binding sites are found in the region of the repressed exon. In exon 9 of the GABAA receptor γ2 pre-mRNA, four pyrimidine-rich repressor sites are clustered around the upstream branch site (Ashiya and Grabowski, 1997). FGF-R2 Exon IIIb is repressed by PTB binding to an intronic splicing silencer sequence upstream and to several sites downstream of the exon (Carstens et al., 2000; Wagner and Garcia-Blanco, 2001, 2002). Exon 3 of the α-tropomyosin transcript and c-src exon N1 are also repressed by PTB binding to sites both upstream and downstream of the exons (Chan and Black, 1997; Gooding et al., 1998; Chou et al., 2000). Thus for most exons, it appears that splicing repression requires multiple binding sites and the ability to form a multimeric PTB complex (Amir-Ahmady et al., 2005). The arrangement of PTB monomers and their individual RRM domains across these diverse sites is not yet clear.
Recently, PTB was found to interact with U1 snRNA within a PTB-repressed exon complex. In this complex, PTB could be crosslinked to stem loop 4 (SL4) of U1 (Sharma et al., 2011). SL4 is a stable GC rich stem with an internal pyrimidine bulge and a terminal pyrimidine-rich tetra loop. NMR and ITC studies show that PTB RRMs 1 and 2 each bind U1 SL4 with higher affinity than a short CU oligonucleotide. However, the affinity of the full length protein for an RNA containing CU elements is much higher than the SL4 interaction. The U1 SL4 interaction is interesting in its similarity to the interactions of PTB to structured IRES elements. This work led to a model where PTB is recruited to the pre-mRNA via high affinity binding to single stranded CU elements. This bound PTB can then interact with a nearby 5′ splice site-bound U1 to repress its function in splicing (see below and (Sharma et al., 2011)). The diversity of binding sites for PTB on pre-mRNAs, combined with its very different interaction with U1, indicate that the PTB-pre-mRNA structures responsible for exon repression will be more intricate than a single PTB/RNA contact blocking access by another factor (see below).
PTB interacting proteins
In addition to binding RNA, PTB makes specific interactions with other proteins. Interactions between components of pre-mRNPs will likely be important in determining their affect on splicing. PTB is very abundant and has often been identified copurifying with various proteins in mass spec and pull-down experiments. Although these interactions are interesting, the limited fraction of PTB engaged in them can make it difficult to assess their functional significance. Nevertheless, some proteins found to interact with PTB are already known splicing regulators.
The best characterized PTB interactor is Raver1, which binds PTB in yeast 2- hybrid, co-immunoprecipitation and immunofluorescent colocalization assays (Huttelmaier et al., 2001). The structural basis for this interaction has been elucidated. Raver1 is an RRM protein that also carries 4 copies of a motif ([S/G] [I/L] LGxxP), which acts as a PTB interaction peptide. These peptides bind to the alpha helical face of PTB RRM2, opposite to the RNA binding domain, allowing RRM2 to form a ternary complex that simultaneously binds Raver1 and RNA. A Y247Q mutation in the Raver1 interacting region of reduces splicing activity by a third (Joshi et al., 2011). In addition to Raver1 and its homolog Raver2, this PTB binding motif is found in other proteins that may also act as PTB cofactors. Raver1 acts as a corepressor with PTB for controlling α-tropomyosin exon 3 splicing (see below). However, the Raver proteins are apparently not universal cofactors for PTB regulation as not all PTB dependent exons are affected by Raver1 or 2 (Rideau et al., 2006; Henneberg et al., 2010).
Other known splicing regulators identified in yeast two hybrid screens and subsequently confirmed to bind PTB by co-IP or other assays include Sam68, Matrin3, and Nova1 (Polydorides et al., 2000; Chawla et al., 2009) (DLB and G. Chawla, unpublished observations). Matrin3 was recently shown to carry a PTB interacting peptide similar to Raver1 and to interact with PTB RRM2 in a similar manner (M Coelho and CWJ Smith, personal communication). These Matrin3 data are very interesting and indicate that this surface will be a common interface for multiple PTB cofactors.
Interactions of PTB with other proteins have been analyzed on a regulatory sequence downstream of the c-src N1 exon called the downstream control sequence (DCS). Gel shift studies indicate that PTB cooperates with the proteins hnRNP H/F and KSRP in assembling a multimeric complex on the DCS RNA (Chou et al., 1999; Markovtsov et al., 2000). The binding sites for these proteins are directly abutted on the DCS sequence and protein–protein interactions presumably exist within these RNP complexes. However, some of the apparent cooperativity in binding likely results from proteins affecting the structure of the RNA, and direct protein contacts between PTB and hnRNP H, hnRNP F, and KSRP have not been confirmed.
Elucidating the interactions of PTB with its cofactors and the role these interactions play in cooperative protein assembly onto RNA will be very important for understanding splicing regulation. Splicing regulatory sequences can be densely packed with binding elements, and PTB sites are usually adjacent to binding sites for multiple other proteins. The proteins that bind to these elements generally have multiple paralogs that vary in their tissue specific expression and which likely differ in their protein–protein interactions. Thus, the rules concerning the assembly of pre-mRNP complexes will determine the outcome of many splicing decisions (Ohno et al., 2008). We will discuss this further in relation to PTBP2 below.
In addition to its interaction with other RNA binding proteins, PTB also interacts with other kinds of regulators, some of which control its subcellular localization. PTB is a shuttling protein that moves between the nucleus and cytoplasm, a process controlled by nuclear export and import signals within the conserved 55 amino acid N-terminal domain (Pérez et al., 1997b; Li and Yen, 2002). This signal consists of two blocks of basic amino acids, including Lys13, Arg14 and downstream residues KKFK, that flank a phosphorylation site at Ser16. An additional sequence in RRM 2 enhances nuclear export (Kamath et al., 2001). The nucleocytoplasmic shuttling is controlled by protein kinase A (PKA) (Xie et al., 2003). Phosphorylation of Ser-16 by activated PKA stimulates accumulation of PTB in the cytoplasm. In contrast, mutation of Ser16 to Alanine eliminates shuttling by PTB and causes the protein to be strictly nuclear.
At steady state, PTB shows the predominantly diThuse nuclear staining expected for a regulator of splicing and polyadenylation. There is also more concentrated localization in nuclear speckles, and particularly in the peri-nucleolar compartment (PNC) (Matera et al., 1995; Hall et al., 2004; Kopp and Huang, 2005). The PNC is a structure associated with the edge of the nucleolus, but having a distinct composition (Huang et al., 1997). The function of the PNC is unclear but it is enriched in transformed cells (Kopp and Huang, 2005; Slusarczyk et al., 2010). PTB can be induced to relocalize to the cytoplasm by several stimuli including viral infection, cellular adhesion, apoptosis, and stress conditions such as hypoxia and toxic agents (Hellen et al., 1993; de Hoog et al., 2004; Ma et al., 2007; Galban et al., 2008; Sawicka et al., 2008; Babic et al., 2009; Cobbold et al., 2010; Gorospe et al., 2011). PTB is also cytoplasmic in frog eggs and Drosophila early embryos (Cote et al., 1999; Lewis et al., 2008; Besse et al., 2009). It is not clear whether all of these relocalization events involve PKA activation (Ma et al., 2007). There are other phosphorylation sites on PTB in addition to Ser16, and other posttranslational modifications are also likely present. Cytoplasmic relocalization is presumably required for PTB’s known cytoplasmic functions such as the control of mRNA localization, translation, and mRNA stability (Hamilton et al., 2003; de Hoog et al., 2004; Fred et al., 2006; Lewis et al., 2008; Sawicka et al., 2008; Matus-Nicodemos et al., 2011).
PTB is also studied for its interaction with the PTB associated splicing factor (PSF), which copurifies with it during biochemical fractionation (Patton et al., 1991; Patton et al., 1993; Gozani et al., 1994; Meissner et al., 2000). PSF is thought to play a role in early spliceosome formation and has been implicated in multiple aspects of splicing as well as other cellular processes including transcription and histone deacetylation (Shav-Tal and Zipori, 2002). How its interaction with PTB might affect these functions is not yet clear. Another recent study found a possible interaction between PTB and the protein MRG15, which binds K36me3 modified histone H3, and identified changes in some PTB dependent exons in response to MRG15 knockdown (Luco et al., 2010). These splicing changes were small relative to those seen in response to PTB modulation or CU element mutation and may result from indirect effects of the knockdown. Nevertheless, the growing literature describing effects of chromatin and transcription elongation rate on splicing point to potential additional activities of PTB in gene expression (Pandya-Jones, 2011).
PTB as a splicing regulator
Among the first PTB regulated alternative splicing events to be identified were pairs of mutually exclusive exons in the α- and β-tropomyosin transcripts (Mulligan et al., 1992; Lin and Patton, 1995; Gooding et al., 1998). Other targets identified in early studies included the N1 exon of c-src pre-mRNA, exon 9 of GABA A receptor-γ2 subunit, exon IIIb of FGF-R2, exon SM of α-actinin, exon 4 of calcitonin/CGRP pre-mRNA and others (Ashiya and Grabowski, 1997; Chan and Black, 1997; Lou et al., 1999; Southby et al., 1999; Carstens et al., 2000; Wagner and Garcia-Blanco, 2001). Many of these transcripts showed muscle or neuron-specific patterns of regulation, and in most cases PTB seemed to act as a repressor to block one splicing choice. However, it is now known that PTB controls splicing in additional cell types and can also serve as a splicing activator (Boutz et al., 2007b; Xue et al., 2009; Llorian et al., 2010).
Variability in the location of PTB binding sites relative to its targeted splice sites has made it difficult to derive general rules for its mechanism of action. Nearly all studies so far have examined exons that are repressed by PTB. In these transcripts, functional PTB binding sites may be exonic or intronic. Intronic sites are often within the pyrimidine tract of the 3′ splice site, immediately upstream of the branchpoint, or downstream of the 5′ splice site of the repressed exon. However, more distant binding sites can also function (Gooding et al., 1998). Most often, multiple PTB binding sites are required for efficient repression, as described above. But in some cases, just one binding site within either the regulated exon (Fas exon 6, IgM exon M2) or a flanking intron (myosin phosphatase targeting subunit 1 exon 12) appears to be sufficient to repress splicing (Dirksen et al., 2003; Shen et al., 2004; Izquierdo et al., 2005). When a single site is sufficient, PTB may cooperate with additional factors that are not required in repressing exons with multiple PTB binding sites.
Activation of exon inclusion by PTB is much less studied. Studies of CT/CGRP exon 4 identified PTB as positively affecting exon inclusion (Lou et al., 1999). An intronic enhancer downstream of exon 4 was found to bind both PTB and the U1 snRNP. However, in this system it was difficult to separate effects on splicing from effects on alternative polyadenylation sites that change concurrently with the change in splicing (Lou et al., 1996; Lou et al., 1999). Like most splicing regulators, PTB also can affect polyadenylation site use (Castelo-Branco et al., 2004). Recent identification of the U1 snRNP as a negative regulator of polyadenylation is also interesting in this regard (Kaida et al., 2010).
Genomewide profiling of splicing after PTB depletion identified a group of exons whose splicing decreases with the loss of PTB (Boutz et al., 2007b; Llorian et al., 2010). CLIP and bioinformatic analyses indicated that these positively affected exons are likely direct targets of the PTB protein. However, there are conflicting results regarding the placement of binding sites needed for PTB enhancement of these exons (Xue et al., 2009; Llorian et al., 2010). Some exons whose splicing is enhanced by PTB contain binding sites close to the adjacent exons, as if PTB could stimulate exon use by slowing the competing exon skipping reaction (Xue et al., 2009). Other PTB enhanced exons have binding sites immediately downstream, a common position for intronic splicing enhancer elements (Ule et al., 2006; Yeo et al., 2009; Llorian et al., 2010). Larger scale analysis of the PTB-enhanced exon set is needed to assess whether there are clear rules for binding site placement that determine enhancement and repression, as is seen with some other splicing factors (Ule et al., 2006; Zhang et al., 2008; Yeo et al., 2009).
The variable placement of binding sites has led to a variety of models for the mechanism of splicing repression by PTB (Figure 4). In cases where high affinity PTB binding sites are found within the polypyrimidine tract of the 3′ splice site, it was suggested that PTB might simply compete with the splicing factor U2AF, whose binding of the polypyrimidine tract is required for normal spliceosome assembly (Lin and Patton, 1995; Singh et al., 1995; Wagner and Garcia-Blanco, 2001; Sauliere et al., 2006; Spellman and Smith, 2006). This mechanism likely contributes to some examples of PTB mediated exon skipping. However, a high affinity PTB binding site within the polypyrimidine tract is not always sufficient to repress an exon, unless exon recognition is somehow weakened, by making the exon shorter or mutation of the 5′ splice site (Chan and Black, 1997; Modafferi and Black, 1999; Amir-Ahmady et al., 2005). Moreover, exon repression does not require that a PTB binding site be within the 3′ splice site (Amir-Ahmady et al., 2005); Exons can be efficiently repressed by PTB binding sites upstream of the branchpoint and downstream of the 5′ splice site. The splice sites of some repressed exons have been shown to still assemble the initial spliceosomal components, U1 and U2AF, but then to fail in later steps of assembly (Izquierdo et al., 2005; Sharma et al., 2005; Sharma et al., 2008). Thus, PTB must be able to induce exon skipping by more complex mechanisms than simply blocking early spliceosomal factors from binding the pre-mRNA.
Figure 4.
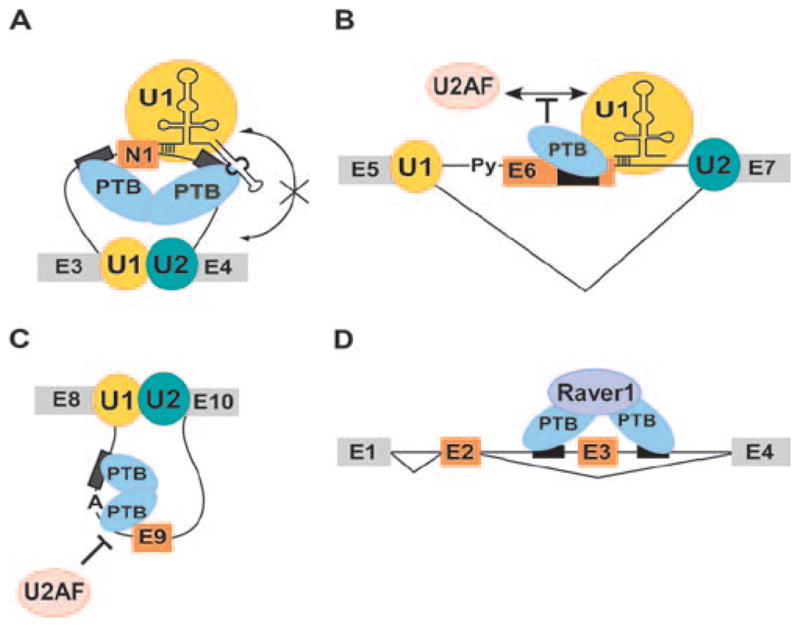
Models for mechanism of splicing repression by PTB. A. PTB binding to introns flanking the c-src N1 exon does not affect 5′ splice site recognition by U1 snRNP but prevents the bound U1 from making intron definition contacts with the components of the 3′ splice site complex at exon 4 downstream. B. In a mechanism similar to c-src N1 exon, binding of PTB to Fas exon 6 does not affect U1 binding. The bound U1 is not able to facilitate formation of the upstream 3′ splice site complex. C. During repression exon E9 of the GABA3 receptor γ2-subunit, PTB binds intronic CU-rich sequences flanking the branchpoint sequence in intron 8 and prevents binding of 3′ splice site factors. D. Repression of tropomyosin exon 3 by PTB requires a cofactor Raver-1, which may bridge the PTB molecules bound to sequences in the flanking introns.
A second model postulated that PTB binding to a high affinity site could nucleate its oligomerization along the RNA to envelop splice sites in an RNP structure that prevents their recognition (Wagner and Garcia-Blanco, 2001). A similar model has been proposed for splicing repression by the hnRNP A1 protein (Zhu et al., 2001; Okunola and Krainer, 2009). However, there is little evidence for PTB complexes that contain protein not bound to pyrimidine elements and which cover alternative exons. In some cases, it is clear that other components including the U1 snRNP continue to bind the exon when it is repressed by PTB (Izquierdo et al., 2005; Sharma et al., 2011).
A third model hypothesized, instead of oligomerization along adjacent RNA, that distant PTB molecules could interact and loop out a regulated exon, and that this might sequester it from spliceosome assembly (Chou et al., 2000). The domain structure of PTB will cause RNA looping even with a single bound monomer (Oberstrass et al., 2005; Lamichhane et al., 2010). PTB monomers may also engage in homomeric interactions when bound to the RNA. There are data indicating that PTB binding to one site can affect its binding at a more distant site (Chou et al., 2000; Liu et al., 2002; Wagner and Garcia-Blanco, 2002; Amir-Ahmady et al., 2005; Matlin et al., 2007). However, it is not clear whether a PTB–PTB interaction that generates a loop in the pre-mRNA is required for splicing repression. At this point, no model can explain PTB action on all of its target exons and it appears that its mechanism must be different in different systems. This is illustrated in the several examples below.
The c-src N1 exon is repressed in non-neuronal cells that contain high levels of PTB. In contrast, the N1 exon is included in the c-src mRNA in neurons, where PTB expression is low (Chan and Black, 1997; Chou et al., 2000; Markovtsov et al., 2000; Sharma et al., 2011). PTB-binding CU-rich elements both upstream and downstream of N1 are essential for this repression (Figure 4A). The repression of the N1 exon by PTB and its splicing in neurons has been reconstructed in extracts of neuronal and non-neuronal cells (Black, 1992; Chan and Black, 1995, 1997). This allowed the splicing complexes that assemble onto N1 exon containing pre-mRNAs to be isolated and characterized under the two regulatory conditions (Sharma et al., 2005; Sharma et al., 2008). These analyses revealed that binding of PTB to the pre-mRNA does not obstruct the binding of U1 snRNP to the N1 5′ splice site (Sharma et al., 2005). However, the U1 on the PTB-repressed exon does not interact with the exon complex downstream to progress further in spliceosome assembly (Sharma et al., 2008). It thus fails to be incorporated into an intron spanning spliceosome.
It was found that in repressing N1 splicing, PTB interacts with the stem-loop 4 (SL4) of U1 snRNA (Sharma et al., 2011) (Figure 4A). PTB RRMs 1 and 2 have significantly higher affinity for SL4 than for typical single stranded CU rich elements (see above). This work led to a model where PTB molecules bound to the pre-mRNA at the CU elements use one or more of their RRMs to make a specific U1 contact. It is not yet clear if this PTB/U1 interaction is required for splicing repression, but it is thought that the contact with SL4 could block interactions with other spliceosomal components, perhaps at the downstream 3′ splice site, that are required for further assembly. Alternatively, PTB might simply alter the conformation of the bound U1 to make its subsequent assembly steps less favored, and allowing a competing assembly pathway to be preferred.
The PTB-repressed N1 exon complexes contain multiple other proteins that are not found in complexes assembled under conditions where PTB is absent and splicing is allowed. Thus, PTB is likely assisted by cofactors in mediating N1 repression, but the roles of these factors are not yet understood. It is also unclear whether a higher order PTB complex must assemble to repress the exon and which PTB molecule within this assembly might contact U1. To mediate splicing repression, the upstream CU rich sequence must be long enough to bind more than one PTB molecule (Amir-Ahmady et al., 2005). Stoichiometry measurements indicate that up to three PTB molecules can bind to this upstream CU sequence (Clerte and Hall, 2009). The binding sites downstream of N1 are much shorter and bind PTB with lower affinity (Amir-Ahmady et al., 2005). They are nevertheless required for splicing repression and affect the binding of PTB to the upstream sites (Chou et al., 2000; Sharma et al., 2011). It is not clear if one or more of the upstream molecules directly contact the downstream elements through one of their RRMs, or whether separate PTBs bound at the upstream and downstream sites are bridged by other proteins. Also interesting is whether the upstream or downstream PTB proteins are contacting U1 snRNA.
Studies of Fas exon 6 regulation have uncovered both differences and interesting similarities to c-src exon N1 (Figure 4B). Fas splicing is mediated by antagonistic regulation by PTB and TIA-1 protein (Izquierdo et al., 2005). TIA-1 binds to a U-rich sequence immediately downstream of exon 6 and stabilizes U1 snRNP binding to the 5′ splice site (Forch et al., 2002). This promotes exon definition and assembly of the U2AF/U2 complex at the upstream 3′ splice site, and leads to splicing of exon 6. Unlike the N1 exon, PTB binds to a single silencer element within the exon to block assembly of the 3′ splice site complex and exon 6 splicing (Izquierdo et al., 2005). Interestingly, PTB also appears to stabilize U1 binding to exon 6. This has led to a model that PTB, through its interaction with U1, can block the cross-exon interactions needed to allow upstream spliceosome assembly (Izquierdo et al., 2005) (Figure 4B). Thus for Fas exon 6, although it is acting through an exonic site and blocking cross exon interactions, PTB is apparently targeting the U1 snRNP and its interactions with the 3′ splice site complex, very similar to what is proposed for c-src exon N1.
The ability of the different RRMs of PTB to play separate roles in splicing repression has been examined in studies of 24-nt exon E9 of the GABAA receptor γ2 subunit (Liu et al., 2002) (Figure 4C). PTB binds to a long pyrimidine tract upstream of the E9 branchpoint sequence. It was found that deletion of RRM4 did not affect RNA binding by the protein to the upstream element, but nevertheless led to a loss of splicing repression both in vitro and in vivo. It was further found that nucleotides downstream of the main PTB binding site, within the polypyrimidine tract of the 3′ splice site were protected from modification by PTB binding. Deletion of the last beta strand in RRM4 eliminated this protection. These results led to the hypothesis that the first three RRMs of PTB are responsible for initial binding to the upstream silencer element (Figure 4C). This initial binding allows an RRM4 interaction with the Py tract to block recognition of the branch-point. Given the structure of RRMs 3 and 4, the contacts upstream and downstream of the branchpoint presumably form a loop. The 5′ to 3′ orientation of this loop, with RRM3 interacting with the 5′ binding site, is consistent with that observed in FRET analyses (Oberstrass et al., 2005; Lamichhane et al., 2010). However, other studies find that this intronic region can bind multiple PTB molecules and a more complex assembly may act on this exon (Clerte and Hall, 2006). A reverse arrangement of multiple PTB binding sites is found in the intron between the mutually exclusive SM and NM exons of α-actinin. In this case, the binding of PTB at the polypyrimidine tract is promoted by binding sites downstream (Matlin et al., 2007).
Studies of the α-tropomyosin (Tpm1) pre-mRNA have identified the first cofactor that functions with PTB, and have shown that PTB is clearly acting by mechanisms more complex than simply blocking access to splice sites (Gromak et al., 2003) (Figure 4D). Exons 2 and 3 of Tpm1 are mutually exclusive, with exon 3 repressed by PTB (Gooding et al., 1998). PTB interacts with long pyrimidine-rich elements in the introns flanking exon 3, and it is estimated that up to 5 to 6 PTB molecules might bind these sequences in nuclear extracts (Cherny et al., 2010). It was found that the downstream elements could be substituted with a MS2 stem loop to induce exon 3 repression, when a PTB-MS2 stem loop-binding-protein fusion is expressed in trans (Wagner and Garcia-Blanco, 2002; Rideau et al., 2006). Examination of additional fusion proteins showed that a tethered PTB fragment containing only RRM2 plus the following linker is sufficient to repress splicing (Rideau et al., 2006; Robinson and Smith, 2006).
In additional studies of Tpm1 exon 3, it was found that PTB requires the cofactor Raver1 to mediate its effect (Gromak et al., 2003) (Figure 4D). Raver1 was identified in a two hybrid screen for PTB interactors and its contacts with PTB have now been studied in detail (see above) (Huttelmaier et al., 2001). Raver1 binds through a specific peptide interaction site on the alpha helical face of PTB RRM2 (Rideau et al., 2006). This contact is needed for RRM2-MS2 fusions to be active, and tethering MS2-Raver1 fusions also repress splicing. Thus, the activity of the tethered PTB seems to come from its ability to recruit Raver1. Additional interactions of Raver1 required for splicing repression are not yet known. Given that Raver1 has 4 PTB interacting motifs and that the PTB binding elements upstream and downstream of Tpm1 exon 3 are separated by 460 nucleotides, an appealing model is that Raver1 can form a bridge between two or more PTB molecules at these two sites (Rideau et al., 2006) (Figure 4D). It is not yet clear whether the splicing machinery is assembling on the Tpm exon 3 splice sites during PTB repression but there are similarities between this system and the c-src N1 exon. Raver1 is found within the PTB repressed N1 exon complex, but does not appear to be required for repression (Sharma et al., 2005; Robinson and Smith, 2006). Similarly, PTB repression of other exons does not seem to require Raver1. However, the PTB interacting peptide found in Raver1 is found in other proteins that have recently been shown to also interact with PTB and to affect splicing (M. Coelho and CJW Smith, personal communication). Thus, different PTB regulated exons may use similar mechanisms but different cofactors to silence splicing. These results are very interesting in relation to how PTB activity might be modulated on different exon subsets in different cell types.
PTBP2
PTBP2 was first identified as a PTB immunoreactive protein with slightly different gel mobility seen in extracts of rat brain or retinoblastoma cells (Ashiya and Grabowski, 1997; Chan and Black, 1997). This protein was subsequently cloned by purification from extract, isolated in a two-hybrid screen for interactions with the splicing regulator Nova, and identified in other cDNA libraries (Kikuchi et al., 2000; Markovtsov et al., 2000; Polydorides et al., 2000). It was originally called neuronal PTB (nPTB) or brain PTB (brPTB) because of its restricted expression and its effects on neuronally regulated exons. However, although its expression is not as broad as PTB, PTBP2 is not limited to neurons or the brain, being found in testis, myoblasts, lymphocytes, and likely other cell types (Lillevali et al., 2001; Boutz et al., 2007b; Xu and Hecht, 2007, 2008; Cheung et al., 2009; Nowak et al., 2011). We will use the official gene name PTBP2 to describe this protein and the traditional name of PTB for PTBP1. It should be noted that PTB1 and PTB4 describe alternatively spliced isoforms of PTBP1, whereas PTBP2 is an independent paralogous gene.
Physical and functional differences between PTB and PTBP2
PTBP2 is about 74% identical in peptide sequence to PTB, with the same domain structure and noncanonical RNP motifs (Figure 1). The two proteins copurify from nuclear extract over multiple chromatographic steps but can be separated on Blue Sepharose. There is near identity in the residues that make direct contact with RNA in PTB, with one lysine to arginine and one phenylalanine to tyrosine change (Markovtsov et al., 2000). The length of the linker region between RRM’s 2 and 3 of PTBP2 is 23aa shorter than the longer PTB4 isoform of PTBP1 and similar to that of PTB1. PTBP2 can interact with Raver1 similarly to PTB (Joshi et al., 2011). The RNA binding properties of the two proteins are quite similar (Markovtsov et al., 2000). PTB and PTBP2 have approximately equal affinity for the pyrimidine rich sequence upstream from the c-src N1 exon. In binding the downstream control sequence (DCS) of the N1 exon, PTBP2 showed somewhat higher affinity than PTB. PTBP2 also formed a multiprotein complex with hnRNP H and KSRP on the DCS more readily than PTB (Markovtsov et al., 2000).
PTB and PTBP2 also clearly differ in their effects on splicing. Several cassette exons that are repressed by PTB, show weaker repression by PTBP2, including the c-src N1 exon, GABAA receptor γ2 exon 9, and exon 8A of the L-type calcium channel (Grabowski, 1998; Markovtsov et al., 2000; Tang et al., 2011). In contrast, other exons respond to the two proteins roughly equally, including PSD95 exon 18 (Zheng et al., 2012), and α-tropomyosin (Tpm1) exon 2 (Spellman et al., 2007). How these two proteins can direct different splicing outcomes is a subject of current studies. One likely possibility is that differences in their protein–protein interactions will allow PTB and PTBP2 to respond to different co-factors. One candidate for such a cofactor is Nova-1, which activates splicing of exon 3A in the GlyRα2 pre-mRNA. When PTBP2 is co-expressed with Nova, it antagonizes Nova’s enhancement of exon 3A splicing through its own potential binding site nearby (Polydorides et al., 2000). However, it is not yet clear whether PTB and PTBP2 can affect the activity of Nova differently. Other proteins that might interact differently with the two PTB’s are the Rbfox proteins. The DCS sequence downstream of the c-src N1 exon contains an Rbfox binding site essential for its enhancer activity in neurons and abutting a PTB binding site. However, possible interactions between the Rbfox proteins and the PTB proteins on this sequence have not yet been examined and could be antagonistic. Genomewide analyses of PTB and PTBP2 targets makes clear that some exons are more strongly affected by one protein or the other, whereas other exons seem to respond equally to both (Boutz et al., 2007b).
Regulation of PTB and PTBP2 expression
PTB is abundant in most cell lines and is broadly expressed in tissues, but absent in mature neurons, muscle, and other cell types. PTB plays a role in cellular metabolism and proliferation. It has been studied for its effects on cancer biology, including aerobic glucose metabolism, apoptosis, cell migration and adhesion, and loss of growth factor responsiveness (He et al., 2007; Wang et al., 2008; Cheung et al., 2009; Clower et al., 2010; David et al., 2010). PTB has been shown to play specific roles in ovarian and glial cell tumors (McCutcheon et al., 2004; Cheung et al., 2006; He et al., 2007). In some of these contexts, PTB may be affecting aspects of mRNA metabolism other than splicing, such as mRNA localization or translation. However, in some cases its splicing regulatory function is clearly needed for tumor cell growth (Jin et al., 2003; Clower et al., 2010; David et al., 2010). It is notable that in normal development, PTB is downregulated when proliferating neuronal progenitors and myoblasts are induced to exit the cell cycle and begin to differentiate into postmitotic neurons and myotubes (Charlet et al., 2002; Ladd et al., 2005; Boutz et al., 2007a; Makeyev et al., 2007).
As neuronal progenitors differentiate, PTB expression is repressed. This is, at least in part, due to the action of the microRNA miR124, whose expression is stimulated during differentiation (Makeyev et al., 2007). PTBP2 is weakly expressed in neuronal progenitors but increases with differentiation. The decrease of PTB coincides with the loss of proliferative marker Nestin, and the increase of PTBP2 coincides with the gain of TuJ antigen, an early marker of post mitotic neurons (Makeyev et al., 2007; Boutz et al., 2007b). PTBP2 levels remain high in mouse cortex during the first 2 postnatal weeks when active neurite growth and synaptogenesis occur, but then decrease after 3 weeks. Neurons in adult brain retain moderate levels of PTBP2. In the adult brain, PTB expression is not seen in neurons, but is found in GFAP positive glia, cells of the vasculature, ependymal cells of the choroid plexi, and in a layer of cells along the inner edge of the dentate gyrus that may correspond to adult neuronal precursor cells (QL and DLB, unpublished). The changes in expression of the two PTBs, define two transitions in splicing regulation, one early in neuronal differentiation when PTB is replaced with PTBP2, and one later in neuronal maturation when PTBP2 expression is reduced (Zheng et al., 2012).
Interestingly, PTB and PTBP2 are both expressed in C2C12 myoblasts in culture (Boutz et al., 2007a). They are also both seen in sporadic cells distributed across skeletal muscle and heart that may correspond to satellite cells, although this has not been confirmed with additional markers (Z Tang and DBL, unpublished). When C2C12 cells are induced to differentiate into myotubes, both PTB and PTBP2 are repressed. Like in neurons, this is in part controlled by the induction of muscle specific microRNAs miR1 and miR133 (Boutz et al., 2007a). Also as seen in neurons, muscle and heart differentiation are accompanied by the reprogramming of an ensemble of alternative splicing events characteristic of post-mitotic myotubes, including muscle specific exons in NCAM, CAPN3 and MEF2A (Bland et al., 2010). This program is affected by both loss of splicing regulators seen in myoblasts, such as PTB and PTBP2, and the expression of new splicing regulators specific to mature muscle (Charlet et al., 2002; Ladd et al., 2005; Lin et al., 2006; Kalsotra et al., 2008).
The expression of both PTB and PTBP2 is controlled through multiple transcriptional and posttranscriptional processes (Figure 5). PTB transcription is activated by Myc in cell culture (David et al., 2010). This is important for its upregulation in cancer cells, although the contribution of myc to PTB control in neurons is not yet clear. PTB contains an alternative exon (exon 11) that is regulated by PTB itself. The autoregulated skipping of this exon leads to a translational frameshift, premature translational termination, and nonsense mediated decay (NMD) of the PTB mRNA (Wollerton et al., 2004; Spellman et al., 2005). This kind of autoregulatory loop through splicing and mRNA decay contributes to the homeostatic control of expression for many RNA binding proteins (Sureau et al., 2001; Lejeune and Maquat, 2005; Lareau et al., 2007; Ni et al., 2007; McGlincy and Smith, 2008; Saltzman et al., 2008). It is also likely that PTB exon 11 is affected by other proteins to allow control of PTB expression in particular physiological settings.
Figure 5.

Complex post-transcriptional regulation of PTBP1 and PTBP2. Each gene contains an autoregulated exon whose repression leads to nonsense mediated decay of the transcript. These exons can also be crossregulated by the opposite protein. This is most evident in the repression of PTBP2 exon 10 by PTBP1. Both transcripts are targeted by miRNAs in neurons and muscle cells.
The control of PTBP2 expression is similarly complex (Figure 5). The PTBP2 gene contains a nearby binding site for the Neural Restricted Silencing Factor (NRSF/REST). Part of its induction is thus likely to be transcriptional, in response to the changes in activity of NRSF during neuronal development. The splicing regulator NSR100 controls the splicing and activity of NRSF/REST during differentiation and NSR100 can also affect the expression of PTBP2 (Calarco et al., 2009; Raj et al., 2011). However, neuronal progenitors do express some PTBP2 protein, and many non-neuronal cell lines express PTBP2 mRNA, without significant PTBP2 protein.
PTBP2 contains an alternatively spliced cassette exon (Exon10) that is equivalent to the autoregulated exon 11 of PTB (Rahman et al., 2002; Rahman et al., 2004) (Figure 5). Interestingly, this exon in PTBP2 is within one of the most conserved sequences in mammalian genomes (Bejerano et al., 2004). The approximately 2kb intronic region encompassing PTBP2 exon 10 is nearly 100% identical between human and mouse. The reason for this stringent selection is not clear, but like PTB exon 11, PTBP2 induces skipping of its own exon 10, resulting in a reading frame shift, premature translation termination, and NMD (Boutz et al., 2007b; Spellman et al., 2007). Interestingly, PTBP2 exon 10 is strongly cross-regulated by PTB, in addition to being autoregulated (Figure 5). RNAi mediated depletion of PTB from most cell lines, neuronal or non-neuronal, leads to increased splicing of PTBP2 exon 10 and induction of PTBP2 protein expression (Boutz et al., 2007b; Spellman et al., 2007). Reexpression of PTB in mature neuronal cultures represses PTBP2 expression through changes in exon 10 splicing (Zheng et al., 2012). These results indicate that PTB plays a key role in maintaining the repression of PTBP2 across a wide range of cells. However it should be noted that specific knockout of PTB in mouse brain does not lead to increased PTBP2 expression (Sika Zheng and DLB, unpublished). Thus, in vivo there must be additional mechanisms enforcing its proper expression. During neuronal differentiation, the increased expression of miR124, leads to repression of PTB and induction of PTBP2 (Figure 5) (Makeyev et al., 2007). PTBP2 also represses PTB exon 11 and reduces PTB protein levels. Although this effect is weaker than the action of PTB on PTBP2, it may help reinforce the switch from one protein to the other in neurons.
PTBP2 expression is also affected by reduced translational efficiency resulting from an unusual codon bias in its open reading frame (Robinson et al., 2008). Widespread replacement of the rare codons with synonymous common codons to produce ‘optimized’ PTBP2 is needed to allow efficient protein expression from the transfected cDNA. In developing neurons, PTBP2 is efficiently expressed and there must be mechanisms in place that allow use of the rare codons. Similarly, depletion of PTB from almost any cell line leads to PTBP2 protein expression, indicating that the loss of PTB allows PTBP2 translation. Mechanisms that allow PTBP2 translation are unknown.
Programs of splicing regulation by PTB and PTBP2
PTB and PTBP2 each target large sets of exons to coordinate programs of splicing events during development. In initial studies to characterize their target exon sets, RNA from cells depleted of PTB, PTBP2 or both was used to probe splicing sensitive exon junction microarrays (Boutz et al., 2007b; Makeyev et al., 2007; Spellman et al., 2007). This identified exons that increase in splicing after PTB depletion alone or only after the double knockdown. This work also identified exons whose splicing decreased with the loss of PTB and were apparently positively regulated by the protein (Boutz et al., 2007b). Exons that show a strong response to PTB knockdown alone are apparently more responsive to PTB than to PTBP2, which replaces PTB in the single knockdown. Many of these exons were found to switch their splicing during neuronal differentiation when PTB is replaced with PTBP2 (Figure 6). Thus, the switch in these two factors activates a network of new spliced isoforms in developing neurons. There are also exons that do not respond to either PTB or PTBP2 single knockdown, but are induced by double PTB/PTBP2 knockdown. These exons are repressed by both proteins. Several exons that are induced in muscle differentiation are equally responsive to PTB and PTBP2 (Spellman et al., 2007; Llorian et al., 2010). More recently it was found that a reduction in PTBP2 expression occurs late in neuronal maturation and this controls a second transition in splicing regulation (Figure 6) (Zheng et al., 2012). When PTBP2 expression is reduced, exons that had been repressed by both PTB and PTBP2 are now allowed to splice. In PTBP2 knockout mouse embryos these exons are increased in splicing to cause the precocious expression of many adult mRNA isoforms (QL and DLB, unpublished data).
Figure 6.

The sequential down regulation of PTB and PTBP2 controls two transitions in splicing programs during neuronal differentiation. Cycling neuronal progenitor cells express PTB but only limited PTBP2. This maintains repression of PTB dependent exons. When PTB is repressed at the onset of differentiation, in part through the action of miR124, PTBP2 is induced. This alters the splicing of transcripts that are more sensitive to PTB than PTBP2 early in neuronal differentiation. PTBP2 expression remains high in differentiating cells. As cells mature and undergo synaptogenesis, PTBP2 expression is reduced. This leads to another splicing regulatory transition, where exons that are effected by both PTB and PTBP2 undergo changes in their splicing.
The lists of potential PTB targets have been expanded by additional genomewide studies using chromatin immunoprecipitation, microarray analysis of immunoprecipitated RNA, and most notably crosslinking-immunoprecipitation (CLIPseq) analysis of genomewide PTB binding sites (Gama-Carvalho et al., 2006; Moore and Silver, 2008; Xue et al., 2009). CLIPseq analysis in HeLa cells indicated that about one fourth of annotated alternative splicing events have associated PTB binding sites. PTB crosslinking to pyrimidine-rich RNA is very efficient, and it is not clear that all of the cross linked sites identified by CLIP are functionally important. Nevertheless, these data demonstrate the broad reach of the PTB regulatory network. As seen with many other splicing regulators, the CLIPseq data also identify many PTB binding sites in 3′ UTRs, presumably indicating PTB regulation of mRNA function subsequent to splicing (Babic et al., 2009; Xue et al., 2009).
The alignment of CLIPseq reads to the genome and their clustering into prominent peaks of crosslinking allows the creation of a map of protein binding sites for the cell being examined (Witten and Ule, 2011). Comparing the location of these binding sites relative to alternative exons can identify key regulatory elements for that exon. For several RNA binding proteins, the location of CLIP clusters adjacent to alternative exons show an interesting correlation of the position of protein binding with the direction of the regulatory effect. This is perhaps most clear with the Nova and Rbfox proteins, where binding sites are generally found upstream from exons that are repressed by the proteins and binding sites downstream of exons correlates with their activation (Licatalosi et al., 2008; Zhang et al., 2008; Yeo et al., 2009). These observations provide predictive rules for direction of splicing regulation, and the unknown mechanistic basis for the change from positive to negative regulation with binding site placement is quite interesting.
Similar analyses of the PTB CLIP map have not been as clear. As has long been known, exons repressed by PTB often have PTB sites both upstream and downstream. However, exons positively regulated by PTB were found to have CLIP clusters near the adjacent exons rather than the regulated exon itself (Xue et al., 2009). In contrast, another study found exons stimulated by PTB to have downstream binding sites without the upstream site seen with many PTB repressed exons. The PTB binding sites for these exons look similar to exons enhanced by Nova or Rbfox. However, the number of PTB enhanced exons studied so far is relatively small. It does not appear that rules correlating binding site position with the regulatory sign will be as clear for PTB as for other proteins.
Although there is significant overlap between the sets of PTB and PTBP2 targets, the proteins clearly serve different roles. It will be very interesting to compare genome-wide PTB and PTBP2 binding by CLIPseq, particularly in cells such as myoblasts or neuronal progenitors where both proteins are present. Such analyses are underway. Genetic studies of the proteins are also ongoing. PTB null mutations cause embryonic lethality in mouse, whereas conditional knockout specifically in the brain has minimal phenotype (Shibayama et al., 2009; Suckale et al., 2011; Sika Zheng and DLB, unpublished). Both a full PTBP2 null mutation and its conditional knockout in brain show a phenotype of neonatal lethality from an apparent breathing deficit (QL and DLB, unpublished; Donny Licatalosi and Robert Darnell, unpublished). The null mutation may be more severe than the brain specific knockout due to additional heart defects. Neurons carrying PTBP2 null mutations begin to differentiate normally from progenitor cells and initiate neurite outgrowth. However, they then undergo a catastrophic failure to mature and die late in development (Qin Li and DLB, unpublished). Thus, PTBP2 is clearly required for proper neuronal differentiation.
The phenotypes of the PTB and PTBP2 mutant mice point to the different programs of splicing controlled by them. Individual exon targets within these networks will need to be examined to understand the cellular and developmental roles of these proteins. PTBP2 knockout embryos exhibit precocious expression of a number of adult mRNA isoforms (Qin Li and DLB, unpublished). These often involve exons that are normally repressed by PTBP2 until its expression is reduced late in neuronal maturation. Adult mRNA isoforms that are expressed too early in these mice include many proteins important for mature synaptic function such as CamK IIβ, Dynamin2, Drebrin and NCAM. The controlled splicing events within these mRNAs are conserved across vertebrate species, but why these splicing changes are important for neuronal maturation is not always clear.
One PTB/PTBP2 target with a clear role in neuronal maturation is Postsynaptic Density protein 95 (PSD95) (Zheng et al., 2012). PSD95 is an important scaffold protein of the postsynaptic density where among other activities it controls membrane insertion of AMPA receptors. PSD95 contains an alternative exon (exon 18) whose splicing determines the overall expression of the protein. Exon 18 is repressed by both PTB and PTBP2 and its splicing is thus not induced until PTBP2 expression is reduced late in neuronal maturation (Figure 6). The skipping of exon 18 leads to a reading frameshift, premature translation termination, and NMD of the PSD95 mRNA. Thus, the PTB proteins through their effects on exon 18 are controlling the expression of the PSD95 protein during development.
The switch in expression from PTB to PTBP2 early in neuronal development affects an earlier splicing transition involving exons that are more responsive to PTB than to PTBP2 (Figure 6) (Boutz et al., 2007b). These exons include the c-src N1 exon and exon 8A of the L-type Calcium Channel, CaV1.2 (Boutz et al., 2007b; Tang et al., 2011). Both of these exons are highly conserved and are switched early in neuronal differentiation. Exon 8A is also interesting as the site of human mutations causing Timothy syndrome (TS) (Splawski et al., 2004; Splawski et al., 2005). The mutually exclusive exon 8 (also the site of TS mutations) is expressed early in development but is replaced by exon 8A when PTB protein is repressed (Tang et al., 2011). Thus understanding this splicing transition, and the roles of the PTB proteins in directing it, will shed light on the phenotype of this human disease.
We have learned a great deal about the mechanisms of splicing regulation from studies of PTB and PTBP2. Genetic studies are beginning to describe the essential biological roles of these proteins. Genomic analyses of their targets have elucidated large programs of splicing controlled by these proteins. These genomic data are lending insight into both mechanisms of regulation and the biological significance of splicing regulatory networks. The challenge for the coming years will be to connect mechanism to biology. We want to understand how PTB can have different effects than PTBP2, how the two proteins can alter splicing by apparently different mechanisms on different exons, how they interact with their cofactors, and how these activities lead to different biological outcomes during development.
Acknowledgments
We thank our many colleagues in the PTB field for illuminating discussions and our anonymous reviewers for helpful corrections to the manuscript. We apologize for unreferenced important results.
Footnotes
Declaration of interest
Our own work has been supported by NIH Grants RO1 GM49662, R24 GM070857 (to D.L.B.), fellowship and training grant support from an NRSA and 5T32 NS07449 (to N.K.) and from NARSAD (to Q.L.), a grant from the California Institute for Regenerative Medicine (RFA 09-02), and funding from the Howard Hughes Medical Institute. D.L.B. is an investigator of the HHMI. We declare no financial interests in any of the work described.
References
- Amir-Ahmady B, Boutz PL, Markovtsov V, Phillips ML, Black DL. Exon repression by polypyrimidine tract binding protein. RNA. 2005;11:699–716. doi: 10.1261/rna.2250405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashiya M, Grabowski PJ. A neuron-specific splicing switch mediated by an array of pre-mRNA repressor sites: evidence of a regulatory role for the polypyrimidine tract binding protein and a brain-specific PTB counterpart. RNA. 1997;3:996–1015. [PMC free article] [PubMed] [Google Scholar]
- Auweter SD, Allain FH. Structure-function relationships of the polypyrimidine tract binding protein. Cell Mol Life Sci. 2008;65:516–527. doi: 10.1007/s00018-007-7378-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auweter SD, Oberstrass FC, Allain FH. Solving the structure of PTB in complex with pyrimidine tracts: an NMR study of protein-RNA complexes of weak affinities. J Mol Biol. 2007;367:174–186. doi: 10.1016/j.jmb.2006.12.053. [DOI] [PubMed] [Google Scholar]
- Babic I, Sharma S, Black DL. A role for polypyrimidine tract binding protein in the establishment of focal adhesions. Mol Cell Biol. 2009;29:5564–5577. doi: 10.1128/MCB.00590-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, Haussler D. Ultraconserved elements in the human genome. Science. 2004;304:1321–1325. doi: 10.1126/science.1098119. [DOI] [PubMed] [Google Scholar]
- Besse F, López de Quinto S, Marchand V, Trucco A, Ephrussi A. Drosophila PTB promotes formation of high-order RNP particles and represses oskar translation. Genes Dev. 2009;23:195–207. doi: 10.1101/gad.505709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black DL. Activation of c-src neuron-specific splicing by an unusual RNA element in vivo and in vitro. Cell. 1992;69:795–807. doi: 10.1016/0092-8674(92)90291-j. [DOI] [PubMed] [Google Scholar]
- Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- Bland CS, Wang ET, Vu A, David MP, Castle JC, Johnson JM, Burge CB, Cooper TA. Global regulation of alternative splicing during myogenic differentiation. Nucleic Acids Res. 2010;38:7651–7664. doi: 10.1093/nar/gkq614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutz PL, Chawla G, Stoilov P, Black DL. MicroRNAs regulate the expression of the alternative splicing factor nPTB during muscle development. Genes Dev. 2007a;21:71–84. doi: 10.1101/gad.1500707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutz PL, Stoilov P, Li Q, Lin CH, Chawla G, Ostrow K, Shiue L, Ares M, Jr, Black DL. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 2007b;21:1636–1652. doi: 10.1101/gad.1558107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushell M, Stoneley M, Kong YW, Hamilton TL, Spriggs KA, Dobbyn HC, Qin X, Sarnow P, Willis AE. Polypyrimidine tract binding protein regulates IRES-mediated gene expression during apoptosis. Mol Cell. 2006;23:401–412. doi: 10.1016/j.molcel.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Calarco JA, Superina S, O’Hanlon D, Gabut M, Raj B, Pan Q, Skalska U, Clarke L, Gelinas D, van der Kooy D, Zhen M, Ciruna B, Blencowe BJ. Regulation of vertebrate nervous system alternative splicing and development by an SR-related protein. Cell. 2009;138:898–910. doi: 10.1016/j.cell.2009.06.012. [DOI] [PubMed] [Google Scholar]
- Carstens RP, Wagner EJ, Garcia-Blanco MA. An intronic splicing silencer causes skipping of the IIIb exon of fibroblast growth factor receptor 2 through involvement of polypyrimidine tract binding protein. Mol Cell Biol. 2000;20:7388–7400. doi: 10.1128/mcb.20.19.7388-7400.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelo-Branco P, Furger A, Wollerton M, Smith C, Moreira A, Proudfoot N. Polypyrimidine tract binding protein modulates efficiency of polyadenylation. Mol Cell Biol. 2004;24:4174–4183. doi: 10.1128/MCB.24.10.4174-4183.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RC, Black DL. Conserved intron elements repress splicing of a neuron-specific c-src exon in vitro. Mol Cell Biol. 1995;15:6377–6385. doi: 10.1128/mcb.15.11.6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RC, Black DL. The polypyrimidine tract binding protein binds upstream of neural cell-specific c-src exon N1 to repress the splicing of the intron downstream. Mol Cell Biol. 1997;17:4667–4676. doi: 10.1128/mcb.17.8.4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlet-B N, Logan P, Singh G, Cooper TA. Dynamic antagonism between ETR-3 and PTB regulates cell type-specific alternative splicing. Mol Cell. 2002;9:649–658. doi: 10.1016/s1097-2765(02)00479-3. [DOI] [PubMed] [Google Scholar]
- Chawla G, Lin CH, Han A, Shiue L, Ares M, Jr, Black DL. Sam68 regulates a set of alternatively spliced exons during neurogenesis. Mol Cell Biol. 2009;29:201–213. doi: 10.1128/MCB.01349-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009;10:741–754. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherny D, Gooding C, Eperon GE, Coelho MB, Bagshaw CR, Smith CW, Eperon IC. Stoichiometry of a regulatory splicing complex revealed by single-molecule analyses. EMBO J. 2010;29:2161–2172. doi: 10.1038/emboj.2010.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung HC, Corley LJ, Fuller GN, McCutcheon IE, Cote GJ. Polypyrimidine tract binding protein and Notch1 are independently re-expressed in glioma. Mod Pathol. 2006;19:1034–1041. doi: 10.1038/modpathol.3800635. [DOI] [PubMed] [Google Scholar]
- Cheung HC, Hai T, Zhu W, Baggerly KA, Tsavachidis S, Krahe R, Cote GJ. Splicing factors PTBP1 and PTBP2 promote proliferation and migration of glioma cell lines. Brain. 2009;132:2277–2288. doi: 10.1093/brain/awp153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou MY, Rooke N, Turck CW, Black DL. hnRNP H is a component of a splicing enhancer complex that activates a c-src alternative exon in neuronal cells. Mol Cell Biol. 1999;19:69–77. doi: 10.1128/mcb.19.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou MY, Underwood JG, Nikolic J, Luu MH, Black DL. Multisite RNA binding and release of polypyrimidine tract binding protein during the regulation of c-src neural-specific splicing. Mol Cell. 2000;5:949–957. doi: 10.1016/s1097-2765(00)80260-9. [DOI] [PubMed] [Google Scholar]
- Clerte C, Hall KB. Characterization of multimeric complexes formed by the human PTB1 protein on RNA. RNA. 2006;12:457–475. doi: 10.1261/rna.2178406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerte C, Hall KB. The domains of polypyrimidine tract binding protein have distinct RNA structural preferences. Biochemistry. 2009;48:2063–2074. doi: 10.1021/bi8016872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, Krainer AR. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci USA. 2010;107:1894–1899. doi: 10.1073/pnas.0914845107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobbold LC, Wilson LA, Sawicka K, King HA, Kondrashov AV, Spriggs KA, Bushell M, Willis AE. Upregulated c-myc expression in multiple myeloma by internal ribosome entry results from increased interactions with and expression of PTB-1 and YB-1. Oncogene. 2010;29:2884–2891. doi: 10.1038/onc.2010.31. [DOI] [PubMed] [Google Scholar]
- Conte MR, Grüne T, Ghuman J, Kelly G, Ladas A, Matthews S, Curry S. Structure of tandem RNA recognition motifs from polypyrimidine tract binding protein reveals novel features of the RRM fold. EMBO J. 2000;19:3132–3141. doi: 10.1093/emboj/19.12.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote CA, Gautreau D, Denegre JM, Kress TL, Terry NA, Mowry KL. A Xenopus protein related to hnRNP I has a role in cytoplasmic RNA localization. Mol Cell. 1999;4:431–437. doi: 10.1016/s1097-2765(00)80345-7. [DOI] [PubMed] [Google Scholar]
- David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364–368. doi: 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoog CL, Foster LJ, Mann M. RNA and RNA binding proteins participate in early stages of cell spreading through spreading initiation centers. Cell. 2004;117:649–662. doi: 10.1016/s0092-8674(04)00456-8. [DOI] [PubMed] [Google Scholar]
- Dirksen WP, Mohamed SA, Fisher SA. Splicing of a myosin phosphatase targeting subunit 1 alternative exon is regulated by intronic cis-elements and a novel bipartite exonic enhancer/silencer element. J Biol Chem. 2003;278:9722–9732. doi: 10.1074/jbc.M207969200. [DOI] [PubMed] [Google Scholar]
- Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol. 2002;3:195–205. doi: 10.1038/nrm760. [DOI] [PubMed] [Google Scholar]
- Förch P, Puig O, Martínez C, Séraphin B, Valcárcel J. The splicing regulator TIA-1 interacts with U1-C to promote U1 snRNP recruitment to 5′ splice sites. EMBO J. 2002;21:6882–6892. doi: 10.1093/emboj/cdf668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fred RG, Tillmar L, Welsh N. The role of PTB in insulin mRNA stability control. Curr Diabetes Rev. 2006;2:363–366. doi: 10.2174/157339906777950570. [DOI] [PubMed] [Google Scholar]
- Galbán S, Kuwano Y, Pullmann R, Jr, Martindale JL, Kim HH, Lal A, Abdelmohsen K, Yang X, Dang Y, Liu JO, Lewis SM, Holcik M, Gorospe M. RNA-binding proteins HuR and PTB promote the translation of hypoxia-inducible factor 1alpha. Mol Cell Biol. 2008;28:93–107. doi: 10.1128/MCB.00973-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gama-Carvalho M, Barbosa-Morais NL, Brodsky AS, Silver PA, Carmo-Fonseca M. Genome-wide identification of functionally distinct subsets of cellular mRNAs associated with two nucleocytoplasmic-shuttling mammalian splicing factors. Genome Biol. 2006;7:R113. doi: 10.1186/gb-2006-7-11-r113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Blanco MA, Jamison SF, Sharp PA. Identification and purification of a 62,000-dalton protein that binds specifically to the polypyrimidine tract of introns. Genes Dev. 1989;3:1874–1886. doi: 10.1101/gad.3.12a.1874. [DOI] [PubMed] [Google Scholar]
- Ghetti A, Piñol-Roma S, Michael WM, Morandi C, Dreyfuss G. hnRNP I, the polypyrimidine tract-binding protein: distinct nuclear localization and association with hnRNAs. Nucleic Acids Res. 1992;20:3671–3678. doi: 10.1093/nar/20.14.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil A, Sharp PA, Jamison SF, Garcia-Blanco MA. Characterization of cDNAs encoding the polypyrimidine tract-binding protein. Genes Dev. 1991;5:1224–1236. doi: 10.1101/gad.5.7.1224. [DOI] [PubMed] [Google Scholar]
- Gooding C, Kemp P, Smith CW. A novel polypyrimidine tract-binding protein paralog expressed in smooth muscle cells. J Biol Chem. 2003;278:15201–15207. doi: 10.1074/jbc.M210131200. [DOI] [PubMed] [Google Scholar]
- Gooding C, Roberts GC, Smith CW. Role of an inhibitory pyrimidine element and polypyrimidine tract binding protein in repression of a regulated alpha-tropomyosin exon. RNA. 1998;4:85–100. [PMC free article] [PubMed] [Google Scholar]
- Gorospe M, Tominaga K, Wu X, Fähling M, Ivan M. Post-Transcriptional Control of the Hypoxic Response by RNA-Binding Proteins and MicroRNAs. Front Mol Neurosci. 2011;4:7. doi: 10.3389/fnmol.2011.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozani O, Patton JG, Reed R. A novel set of spliceosome-associated proteins and the essential splicing factor PSF bind stably to pre-mRNA prior to catalytic step II of the splicing reaction. EMBO J. 1994;13:3356–3367. doi: 10.1002/j.1460-2075.1994.tb06638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski PJ. Splicing regulation in neurons: tinkering with cell-specific control. Cell. 1998;92:709–712. doi: 10.1016/s0092-8674(00)81399-9. [DOI] [PubMed] [Google Scholar]
- Graveley BR. Sorting out the complexity of SR protein functions. RNA. 2000;6:1197–1211. doi: 10.1017/s1355838200000960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromak N, Rideau A, Southby J, Scadden AD, Gooding C, Hüttelmaier S, Singer RH, Smith CW. The PTB interacting protein raver1 regulates alpha-tropomyosin alternative splicing. EMBO J. 2003;22:6356–6364. doi: 10.1093/emboj/cdg609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MP, Huang S, Black DL. Differentiation-induced colocalization of the KH-type splicing regulatory protein with polypyrimidine tract binding protein and the c-src pre-mRNA. Mol Biol Cell. 2004;15:774–786. doi: 10.1091/mbc.E03-09-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton BJ, Genin A, Cron RQ, Rigby WF. Delineation of a novel pathway that regulates CD154 (CD40 ligand) expression. Mol Cell Biol. 2003;23:510–525. doi: 10.1128/MCB.23.2.510-525.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Pool M, Darcy KM, Lim SB, Auersperg N, Coon JS, Beck WT. Knockdown of polypyrimidine tract-binding protein suppresses ovarian tumor cell growth and invasiveness in vitro. Oncogene. 2007;26:4961–4968. doi: 10.1038/sj.onc.1210307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellen CU, Witherell GW, Schmid M, Shin SH, Pestova TV, Gil A, Wimmer E. A cytoplasmic 57-kDa protein that is required for translation of picornavirus RNA by internal ribosomal entry is identical to the nuclear pyrimidine tract-binding protein. Proc Natl Acad Sci USA. 1993;90:7642–7646. doi: 10.1073/pnas.90.16.7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneberg B, Swiniarski S, Becke Sabine, Illenberger S. A conserved peptide motif in Raver2 mediates its interaction with the polypyrimidine tract-binding protein. Exp Cell Res. 2010;316:966–979. doi: 10.1016/j.yexcr.2009.11.023. [DOI] [PubMed] [Google Scholar]
- Huang S, Deerinck TJ, Ellisman MH, Spector DL. The dynamic organization of the perinucleolar compartment in the cell nucleus. J Cell Biol. 1997;137:965–974. doi: 10.1083/jcb.137.5.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüttelmaier S, Illenberger S, Grosheva I, Rüdiger M, Singer RH, Jockusch BM. Raver1, a dual compartment protein, is a ligand for PTB/hnRNPI and microfilament attachment proteins. J Cell Biol. 2001;155:775–786. doi: 10.1083/jcb.200105044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo JM, Majós N, Bonnal S, Martínez C, Castelo R, Guigó R, Bilbao D, Valcárcel J. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol Cell. 2005;19:475–484. doi: 10.1016/j.molcel.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Jin W, Bruno IG, Xie TX, Sanger LJ, Cote GJ. Polypyrimidine tract-binding protein down-regulates fibroblast growth factor receptor 1 alpha-exon inclusion. Cancer Res. 2003;63:6154–6157. [PubMed] [Google Scholar]
- Joshi A, Coelho MB, Kotik-Kogan O, Simpson PJ, Matthews SJ, Smith CW, Curry S. Crystallographic analysis of polypyrimidine tract-binding protein-Raver1 interactions involved in regulation of alternative splicing. Structure. 2011;19:1816–1825. doi: 10.1016/j.str.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafasla P, Morgner N, Pöyry TA, Curry S, Robinson CV, Jackson RJ. Polypyrimidine tract binding protein stabilizes the encephalomyocarditis virus IRES structure via binding multiple sites in a unique orientation. Mol Cell. 2009;34:556–568. doi: 10.1016/j.molcel.2009.04.015. [DOI] [PubMed] [Google Scholar]
- Kaida D, Berg MG, Younis I, Kasim M, Singh LN, Wan L, Dreyfuss G. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature. 2010;468:664–668. doi: 10.1038/nature09479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A, Cooper TA. Functional consequences of developmentally regulated alternative splicing. Nat Rev Genet. 2011;12:715–729. doi: 10.1038/nrg3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB, Cooper TA. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc Natl Acad Sci USA. 2008;105:20333–20338. doi: 10.1073/pnas.0809045105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RV, Leary DJ, Huang S. Nucleocytoplasmic shuttling of polypyrimidine tract-binding protein is uncoupled from RNA export. Mol Biol Cell. 2001;12:3808–3820. doi: 10.1091/mbc.12.12.3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi T, Ichikawa M, Arai J, Tateiwa H, Fu L, Higuchi K, Yoshimura N. Molecular cloning and characterization of a new neuron-specific homologue of rat polypyrimidine tract binding protein. J Biochem. 2000;128:811–821. doi: 10.1093/oxfordjournals.jbchem.a022819. [DOI] [PubMed] [Google Scholar]
- Kim YK, Hahm B, Jang SK. Polypyrimidine tract-binding protein inhibits translation of bip mRNA. J Mol Biol. 2000;304:119–133. doi: 10.1006/jmbi.2000.4179. [DOI] [PubMed] [Google Scholar]
- Kopp K, Huang S. Perinucleolar compartment and transformation. J Cell Biochem. 2005;95:217–225. doi: 10.1002/jcb.20403. [DOI] [PubMed] [Google Scholar]
- Ladd AN, Stenberg MG, Swanson MS, Cooper TA. Dynamic balance between activation and repression regulates pre-mRNA alternative splicing during heart development. Dev Dyn. 2005;233:783–793. doi: 10.1002/dvdy.20382. [DOI] [PubMed] [Google Scholar]
- Lamichhane R, Daubner GM, Thomas-Crusells J, Auweter SD, Manatschal C, Austin KS, Valniuk O, Allain FH, Rueda D. RNA looping by PTB: Evidence using FRET and NMR spectroscopy for a role in splicing repression. Proc Natl Acad Sci USA. 2010;107:4105–4110. doi: 10.1073/pnas.0907072107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau LF, Brooks AN, Soergel DA, Meng Q, Brenner SE. The coupling of alternative splicing and nonsense-mediated mRNA decay. Adv Exp Med Biol. 2007;623:190–211. doi: 10.1007/978-0-387-77374-2_12. [DOI] [PubMed] [Google Scholar]
- Lejeune F, Maquat LE. Mechanistic links between nonsense-mediated mRNA decay and pre-mRNA splicing in mammalian cells. Curr Opin Cell Biol. 2005;17:309–315. doi: 10.1016/j.ceb.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Lewis RA, Gagnon JA, Mowry KL. PTB/hnRNP I is required for RNP remodeling during RNA localization in Xenopus oocytes. Mol Cell Biol. 2008;28:678–686. doi: 10.1128/MCB.00999-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Yen TS. Characterization of the nuclear export signal of polypyrimidine tract-binding protein. J Biol Chem. 2002;277:10306–10314. doi: 10.1074/jbc.M109686200. [DOI] [PubMed] [Google Scholar]
- Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X, Darnell JC, Darnell RB. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–469. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilleväli K, Kulla A, Ord T. Comparative expression analysis of the genes encoding polypyrimidine tract binding protein (PTB) and its neural homologue (brPTB) in prenatal and postnatal mouse brain. Mech Dev. 2001;101:217–220. doi: 10.1016/s0925-4773(00)00566-9. [DOI] [PubMed] [Google Scholar]
- Lin CH, Patton JG. Regulation of alternative 3′ splice site selection by constitutive splicing factors. RNA. 1995;1:234–245. [PMC free article] [PubMed] [Google Scholar]
- Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swanson MS, Thornton CA. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet. 2006;15:2087–2097. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]
- Liu H, Zhang W, Reed RB, Liu W, Grabowski PJ. Mutations in RRM4 uncouple the splicing repression and RNA-binding activities of polypyrimidine tract binding protein. RNA. 2002;8:137–149. doi: 10.1017/s1355838202015029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorian M, Schwartz S, Clark TA, Hollander D, Tan LY, Spellman R, Gordon A, Schweitzer AC, de la Grange P, Ast G, Smith CW. Position-dependent alternative splicing activity revealed by global profiling of alternative splicing events regulated by PTB. Nat Struct Mol Biol. 2010;17:1114–1123. doi: 10.1038/nsmb.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou H, Gagel RF, Berget SM. An intron enhancer recognized by splicing factors activates polyadenylation. Genes Dev. 1996;10:208–219. doi: 10.1101/gad.10.2.208. [DOI] [PubMed] [Google Scholar]
- Lou H, Helfman DM, Gagel RF, Berget SM. Polypyrimidine tract-binding protein positively regulates inclusion of an alternative 3′-terminal exon. Mol Cell Biol. 1999;19:78–85. doi: 10.1128/mcb.19.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science. 2010;327:996–1000. doi: 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Liu G, Sun Y, Xie J. Relocalization of the polypyrimidine tract-binding protein during PKA-induced neurite growth. Biochim Biophys Acta. 2007;1773:912–923. doi: 10.1016/j.bbamcr.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Makeyev EV, Zhang J, Carrasco MA, Maniatis T. The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol Cell. 2007;27:435–448. doi: 10.1016/j.molcel.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovtsov V, Nikolic JM, Goldman JA, Turck CW, Chou MY, Black DL. Cooperative assembly of an hnRNP complex induced by a tissue-specific homolog of polypyrimidine tract binding protein. Mol Cell Biol. 2000;20:7463–7479. doi: 10.1128/mcb.20.20.7463-7479.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera AG, Frey MR, Margelot K, Wolin SL. A perinucleolar compartment contains several RNA polymerase III transcripts as well as the polypyrimidine tract-binding protein, hnRNP I. J Cell Biol. 1995;129:1181–1193. doi: 10.1083/jcb.129.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlin AJ, Clark F, Smith CW. Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol. 2005;6:386–398. doi: 10.1038/nrm1645. [DOI] [PubMed] [Google Scholar]
- Matlin AJ, Southby J, Gooding C, Smith CW. Repression of alpha-actinin SM exon splicing by assisted binding of PTB to the polypyrimidine tract. RNA. 2007;13:1214–1223. doi: 10.1261/rna.219607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus-Nicodemos R, Vavassori S, Castro-Faix M, Valentin-Acevedo A, Singh K, Marcelli V, Covey LR. Polypyrimidine tract-binding protein is critical for the turnover and subcellular distribution of CD40 ligand mRNA in CD4+ T cells. J Immunol. 2011;186:2164–2171. doi: 10.4049/jimmunol.1003236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard CM, Hall KB. Interactions between PTB RRMs induce slow motions and increase RNA binding affinity. J Mol Biol. 2010;397:260–277. doi: 10.1016/j.jmb.2009.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon IE, Hentschel SJ, Fuller GN, Jin W, Cote GJ. Expression of the splicing regulator polypyrimidine tract-binding protein in normal and neoplastic brain. Neuro-oncology. 2004;6:9–14. doi: 10.1215/S1152851703000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlincy NJ, Smith CW. Alternative splicing resulting in nonsense-mediated mRNA decay: what is the meaning of nonsense? Trends Biochem Sci. 2008;33:385–393. doi: 10.1016/j.tibs.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Meissner M, Dechat T, Gerner C, Grimm R, Foisner R, Sauermann G. Differential nuclear localization and nuclear matrix association of the splicing factors PSF and PTB. J Cell Biochem. 2000;76:559–566. [PubMed] [Google Scholar]
- Mitchell SA, Spriggs KA, Bushell M, Evans JR, Stoneley M, Le Quesne JP, Spriggs RV, Willis AE. Identification of a motif that mediates polypyrimidine tract-binding protein-dependent internal ribosome entry. Genes Dev. 2005;19:1556–1571. doi: 10.1101/gad.339105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modafferi EF, Black DL. Combinatorial control of a neuron-specific exon. RNA. 1999;5:687–706. doi: 10.1017/s1355838299990155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monie TP, Hernandez H, Robinson CV, Simpson P, Matthews S, Curry S. The polypyrimidine tract binding protein is a monomer. RNA. 2005;11:1803–1808. doi: 10.1261/rna.2214405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Silver PA. Global analysis of mRNA splicing. RNA. 2008;14:197–203. doi: 10.1261/rna.868008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan GJ, Guo W, Wormsley S, Helfman DM. Polypyrimidine tract binding protein interacts with sequences involved in alternative splicing of beta-tropomyosin pre-mRNA. J Biol Chem. 1992;267:25480–25487. [PubMed] [Google Scholar]
- Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O’Brien G, Shiue L, Clark TA, Blume JE, Ares M., Jr Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev. 2007;21:708–718. doi: 10.1101/gad.1525507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak U, Matthews AJ, Zheng S, Chaudhuri J. The splicing regulator PTBP2 interacts with the cytidine deaminase AID and promotes binding of AID to switch-region DNA. Nat Immunol. 2011;12:160–166. doi: 10.1038/ni.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberstrass FC, Auweter SD, Erat M, Hargous Y, Henning A, Wenter P, Reymond L, Amir-Ahmady B, Pitsch S, Black DL, Allain FH. Structure of PTB bound to RNA: specific binding and implications for splicing regulation. Science. 2005;309:2054–2057. doi: 10.1126/science.1114066. [DOI] [PubMed] [Google Scholar]
- Oh YL, Hahm B, Kim YK, Lee HK, Lee JW, Song O, Tsukiyama-Kohara K, Kohara M, Nomoto A, Jang SK. Determination of functional domains in polypyrimidine-tract-binding protein. Biochem J. 1998;331 (Pt 1):169–175. doi: 10.1042/bj3310169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno G, Hagiwara M, Kuroyanagi H. STAR family RNA-binding protein ASD-2 regulates developmental switching of mutually exclusive alternative splicing in vivo. Genes Dev. 2008;22:360–374. doi: 10.1101/gad.1620608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okunola HL, Krainer AR. Cooperative-binding and splicing-repressive properties of hnRNP A1. Mol Cell Biol. 2009;29:5620–5631. doi: 10.1128/MCB.01678-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya-Jones A. Pre-mRNA splicing during transcription in the mammalian system. Wiley Interdiscip Rev RNA. 2011;2:700–717. doi: 10.1002/wrna.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton JG, Mayer SA, Tempst P, Nadal-Ginard B. Characterization and molecular cloning of polypyrimidine tract-binding protein: a component of a complex necessary for pre-mRNA splicing. Genes Dev. 1991;5:1237–1251. doi: 10.1101/gad.5.7.1237. [DOI] [PubMed] [Google Scholar]
- Patton JG, Porro EB, Galceran J, Tempst P, Nadal-Ginard B. Cloning and characterization of PSF, a novel pre-mRNA splicing factor. Genes Dev. 1993;7:393–406. doi: 10.1101/gad.7.3.393. [DOI] [PubMed] [Google Scholar]
- Pérez I, Lin CH, McAfee JG, Patton JG. Mutation of PTB binding sites causes misregulation of alternative 3′ splice site selection in vivo. RNA. 1997a;3:764–778. [PMC free article] [PubMed] [Google Scholar]
- Pérez I, McAfee JG, Patton JG. Multiple RRMs contribute to RNA binding specificity and affinity for polypyrimidine tract binding protein. Biochemistry. 1997b;36:11881–11890. doi: 10.1021/bi9711745. [DOI] [PubMed] [Google Scholar]
- Petoukhov MV, Monie TP, Allain FH, Matthews S, Curry S, Svergun DI. Conformation of polypyrimidine tract binding protein in solution. Structure. 2006;14:1021–1027. doi: 10.1016/j.str.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Polydorides AD, Okano HJ, Yang YY, Stefani G, Darnell RB. A brain-enriched polypyrimidine tract-binding protein antagonizes the ability of Nova to regulate neuron-specific alternative splicing. Proc Natl Acad Sci USA. 2000;97:6350–6355. doi: 10.1073/pnas.110128397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman L, Bliskovski V, Kaye FJ, Zajac-Kaye M. Evolutionary conservation of a 2-kb intronic sequence flanking a tissue-specific alternative exon in the PTBP2 gene. Genomics. 2004;83:76–84. doi: 10.1016/s0888-7543(03)00207-6. [DOI] [PubMed] [Google Scholar]
- Rahman L, Bliskovski V, Reinhold W, Zajac-Kaye M. Alternative splicing of brain-specific PTB defines a tissue-specific isoform pattern that predicts distinct functional roles. Genomics. 2002;80:245–249. doi: 10.1006/geno.2002.6826. [DOI] [PubMed] [Google Scholar]
- Raj B, O’Hanlon D, Vessey JP, Pan Q, Ray D, Buckley NJ, Miller FD, Blencowe BJ. Cross-regulation between an alternative splicing activator and a transcription repressor controls neurogenesis. Mol Cell. 2011;43:843–850. doi: 10.1016/j.molcel.2011.08.014. [DOI] [PubMed] [Google Scholar]
- Rideau AP, Gooding C, Simpson PJ, Monie TP, Lorenz M, Hüttelmaier S, Singer RH, Matthews S, Curry S, Smith CW. A peptide motif in Raver1 mediates splicing repression by interaction with the PTB RRM2 domain. Nat Struct Mol Biol. 2006;13:839–848. doi: 10.1038/nsmb1137. [DOI] [PubMed] [Google Scholar]
- Robida M, Sridharan V, Morgan S, Rao T, Singh R. Drosophila polypyrimidine tract-binding protein is necessary for spermatid individualization. Proc Natl Acad Sci USA. 2010;107:12570–12575. doi: 10.1073/pnas.1007935107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robida MD, Singh R. Drosophila polypyrimidine-tract binding protein (PTB) functions specifically in the male germline. EMBO J. 2003;22:2924–2933. doi: 10.1093/emboj/cdg301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson F, Jackson RJ, Smith CW. Expression of human nPTB is limited by extreme suboptimal codon content. PLoS ONE. 2008;3:e1801. doi: 10.1371/journal.pone.0001801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson F, Smith CW. A splicing repressor domain in polypyrimidine tract-binding protein. J Biol Chem. 2006;281:800–806. doi: 10.1074/jbc.M510578200. [DOI] [PubMed] [Google Scholar]
- Saltzman AL, Pan Q, Blencowe BJ. Regulation of alternative splicing by the core spliceosomal machinery. Genes Dev. 2011;25:373–384. doi: 10.1101/gad.2004811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saulière J, Sureau A, Expert-Bezançon A, Marie J. The polypyrimidine tract binding protein (PTB) represses splicing of exon 6B from the beta-tropomyosin pre-mRNA by directly interfering with the binding of the U2AF65 subunit. Mol Cell Biol. 2006;26:8755–8769. doi: 10.1128/MCB.00893-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicka K, Bushell M, Spriggs KA, Willis AE. Polypyrimidine-tract-binding protein: a multifunctional RNA-binding protein. Biochem Soc Trans. 2008;36:641–647. doi: 10.1042/BST0360641. [DOI] [PubMed] [Google Scholar]
- Sharma S, Falick AM, Black DL. Polypyrimidine tract binding protein blocks the 5′ splice site-dependent assembly of U2AF and the prespliceosomal E complex. Mol Cell. 2005;19:485–496. doi: 10.1016/j.molcel.2005.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Kohlstaedt LA, Damianov A, Rio DC, Black DL. Polypyrimidine tract binding protein controls the transition from exon definition to an intron defined spliceosome. Nat Struct Mol Biol. 2008;15:183–191. doi: 10.1038/nsmb.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Maris C, Allain FH, Black DL. U1 snRNA directly interacts with polypyrimidine tract-binding protein during splicing repression. Mol Cell. 2011;41:579–588. doi: 10.1016/j.molcel.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shav-Tal Y, Zipori D. PSF and p54(nrb)/NonO–multi-functional nuclear proteins. FEBS Lett. 2002;531:109–114. doi: 10.1016/s0014-5793(02)03447-6. [DOI] [PubMed] [Google Scholar]
- Shen H, Kan JL, Ghigna C, Biamonti G, Green MR. A single polypyrimidine tract binding protein (PTB) binding site mediates splicing inhibition at mouse IgM exons M1 and M2. RNA. 2004;10:787–794. doi: 10.1261/rna.5229704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibayama M, Ohno S, Osaka T, Sakamoto R, Tokunaga A, Nakatake Y, Sato M, Yoshida N. Polypyrimidine tract-binding protein is essential for early mouse development and embryonic stem cell proliferation. FEBS J. 2009;276:6658–6668. doi: 10.1111/j.1742-4658.2009.07380.x. [DOI] [PubMed] [Google Scholar]
- Simpson PJ, Monie TP, Szendröi A, Davydova N, Tyzack JK, Conte MR, Read CM, Cary PD, Svergun DI, Konarev PV, Curry S, Matthews S. Structure and RNA interactions of the N-terminal RRM domains of PTB. Structure. 2004;12:1631–1643. doi: 10.1016/j.str.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Singh R, Valcárcel J, Green MR. Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science. 1995;268:1173–1176. doi: 10.1126/science.7761834. [DOI] [PubMed] [Google Scholar]
- Slusarczyk A, Kamath R, Wang C, Anchel D, Pollock C, Lewandowska MA, Fitzpatrick T, Bazett-Jones DP, Huang S. Structure and function of the perinucleolar compartment in cancer cells. Cold Spring Harb Symp Quant Biol. 2010;75:599–605. doi: 10.1101/sqb.2010.75.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southby J, Gooding C, Smith CW. Polypyrimidine tract binding protein functions as a repressor to regulate alternative splicing of alpha-actinin mutally exclusive exons. Mol Cell Biol. 1999;19:2699–2711. doi: 10.1128/mcb.19.4.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman R, Llorian M, Smith CW. Crossregulation and functional redundancy between the splicing regulator PTB and its paralogs nPTB and ROD1. Mol Cell. 2007;27:420–434. doi: 10.1016/j.molcel.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman R, Rideau A, Matlin A, Gooding C, Robinson F, McGlincy N, Grellscheid SN, Southby J, Wollerton M, Smith CW. Regulation of alternative splicing by PTB and associated factors. Biochem Soc Trans. 2005;33:457–460. doi: 10.1042/BST0330457. [DOI] [PubMed] [Google Scholar]
- Spellman R, Smith CW. Novel modes of splicing repression by PTB. Trends Biochem Sci. 2006;31:73–76. doi: 10.1016/j.tibs.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, Sanguinetti MC, Keating MT. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci USA. 2005;102:8089–96. doi: 10.1073/pnas.0502506102. discussion 8086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Suckale J, Wendling O, Masjkur J, Jäger M, Münster C, Anastassiadis K, Stewart AF, Solimena M. PTBP1 is required for embryonic development before gastrulation. PLoS ONE. 2011;6:e16992. doi: 10.1371/journal.pone.0016992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sureau A, Gattoni R, Dooghe Y, Stévenin J, Soret J. SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J. 2001;20:1785–1796. doi: 10.1093/emboj/20.7.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang ZZ, Sharma S, Zheng S, Chawla G, Nikolic J, Black DL. Regulation of the mutually exclusive exons 8a and 8 in the CaV1.2 calcium channel transcript by polypyrimidine tract-binding protein. J Biol Chem. 2011;286:10007–10016. doi: 10.1074/jbc.M110.208116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillmar L, Welsh N. Hypoxia may increase rat insulin mRNA levels by promoting binding of the polypyrimidine tract-binding protein (PTB) to the pyrimidine-rich insulin mRNA 3′-untranslated region. Mol Med. 2002;8:263–272. [PMC free article] [PubMed] [Google Scholar]
- Ule J, Stefani G, Mele A, Ruggiu M, Wang X, Taneri B, Gaasterland T, Blencowe BJ, Darnell RB. An RNA map predicting Nova-dependent splicing regulation. Nature. 2006;444:580–586. doi: 10.1038/nature05304. [DOI] [PubMed] [Google Scholar]
- Vitali F, Henning A, Oberstrass FC, Hargous Y, Auweter SD, Erat M, Allain FH. Structure of the two most C-terminal RNA recognition motifs of PTB using segmental isotope labeling. EMBO J. 2006;25:150–162. doi: 10.1038/sj.emboj.7600911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EJ, Carstens RP, Garcia-Blanco MA. A novel isoform ratio switch of the polypyrimidine tract binding protein. Electrophoresis. 1999;20:1082–1086. doi: 10.1002/(SICI)1522-2683(19990101)20:4/5<1082::AID-ELPS1082>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Wagner EJ, Garcia-Blanco MA. Polypyrimidine tract binding protein antagonizes exon definition. Mol Cell Biol. 2001;21:3281–3288. doi: 10.1128/MCB.21.10.3281-3288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EJ, Garcia-Blanco MA. RNAi-mediated PTB depletion leads to enhanced exon definition. Mol Cell. 2002;10:943–949. doi: 10.1016/s1097-2765(02)00645-7. [DOI] [PubMed] [Google Scholar]
- Wang C, Norton JT, Ghosh S, Kim J, Fushimi K, Wu JY, Stack MS, Huang S. Polypyrimidine tract-binding protein (PTB) differentially affects malignancy in a cell line-dependent manner. J Biol Chem. 2008;283:20277–20287. doi: 10.1074/jbc.M803682200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten JT, Ule J. Understanding splicing regulation through RNA splicing maps. Trends Genet. 2011;27:89–97. doi: 10.1016/j.tig.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollerton MC, Gooding C, Robinson F, Brown EC, Jackson RJ, Smith CW. Differential alternative splicing activity of isoforms of polypyrimidine tract binding protein (PTB) RNA. 2001;7:819–832. doi: 10.1017/s1355838201010214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollerton MC, Gooding C, Wagner EJ, Garcia-Blanco MA, Smith CW. Autoregulation of polypyrimidine tract binding protein by alternative splicing leading to nonsense-mediated decay. Mol Cell. 2004;13:91–100. doi: 10.1016/s1097-2765(03)00502-1. [DOI] [PubMed] [Google Scholar]
- Xie J, Lee JA, Kress TL, Mowry KL, Black DL. Protein kinase A phosphorylation modulates transport of the polypyrimidine tract-binding protein. Proc Natl Acad Sci USA. 2003;100:8776–8781. doi: 10.1073/pnas.1432696100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Hecht NB. Polypyrimidine tract binding protein 2 stabilizes phosphoglycerate kinase 2 mRNA in murine male germ cells by binding to its 3′UTR. Biol Reprod. 2007;76:1025–1033. doi: 10.1095/biolreprod.107.060079. [DOI] [PubMed] [Google Scholar]
- Xu M, Hecht NB. MSY2 and polypyrimidine tract binding protein 2 stabilize mRNAs in the mammalian testis. Int J Androl. 2008;31:457–461. doi: 10.1111/j.1365-2605.2008.00885.x. [DOI] [PubMed] [Google Scholar]
- Xue Y, Zhou Y, Wu T, Zhu T, Ji X, Kwon YS, Zhang C, Yeo G, Black DL, Sun H, Fu XD, Zhang Y. Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol Cell. 2009;36:996–1006. doi: 10.1016/j.molcel.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Tsukahara K, Kanaoka Y, Jinno S, Okayama H. Isolation of a mammalian homologue of a fission yeast differentiation regulator. Mol Cell Biol. 1999;19:3829–3841. doi: 10.1128/mcb.19.5.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo GW, Coufal NG, Liang TY, Peng GE, Fu XD, Gage FH. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat Struct Mol Biol. 2009;16:130–137. doi: 10.1038/nsmb.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Davydova N, Conte MR, Curry S, Matthews S. Chemical shift mapping of RNA interactions with the polypyrimidine tract binding protein. Nucleic Acids Res. 2002;30:456–462. doi: 10.1093/nar/30.2.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamore PD, Green MR. Identification, purification, and biochemical characterization of U2 small nuclear ribonucleoprotein auxiliary factor. Proc Natl Acad Sci USA. 1989;86:9243–9247. doi: 10.1073/pnas.86.23.9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Zhang Z, Castle J, Sun S, Johnson J, Krainer AR, Zhang MQ. Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev. 2008;22:2550–2563. doi: 10.1101/gad.1703108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Gray EE, Chawla G, Porse BT, O’Dell TJ, Black DL. PSD-95 is post-transcriptionally repressed during early neural development by PTBP1 and PTBP2. Nat Neurosci. 2012;15:381–8. S1. doi: 10.1038/nn.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Mayeda A, Krainer AR. Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Mol Cell. 2001;8:1351–1361. doi: 10.1016/s1097-2765(01)00409-9. [DOI] [PubMed] [Google Scholar]