

Abstract
Voltage dependent anion channels (VDAC) are highly conserved proteins that are responsible for permeability of the mitochondrial outer membrane to hydrophilic metabolites like ATP, ADP and respiratory substrates. Although previously assumed to remain open, VDAC closure is emerging as an important mechanism for regulation of global mitochondrial metabolism in apoptotic cells and also in cells that are not dying. During hepatic ethanol oxidation to acetaldehyde, VDAC closure suppresses exchange of mitochondrial metabolites, resulting in inhibition of ureagenesis. In vivo, VDAC closure after ethanol occurs coordinately with mitochondrial uncoupling. Since acetaldehyde passes through membranes independently of channels and transporters, VDAC closure and uncoupling together foster selective and more rapid oxidative metabolism of toxic acetaldehyde to nontoxic acetate by mitochondrial aldehyde dehydrogenase. In single reconstituted VDAC, tubulin decreases VDAC conductance, and in HepG2 hepatoma cells, free tubulin negatively modulates mitochondrial membrane potential, an effect enhanced by protein kinase A. Tubulin-dependent closure of VDAC in cancer cells contributes to suppression of mitochondrial metabolism and may underlie the Warburg phenomenon of aerobic glycolysis.
Keywords: acetaldehyde; ethanol; glycogen synthase kinase-3β; hepatoma; liver, mitochondria; protein kinase A; tubulin; voltage dependent anion channel; Warburg phenomenon
INTRODUCTION
Voltage dependent anion channels
Voltage dependent anion channels (VDAC) are highly conserved proteins in the mitochondrial outer membrane (MOM) with three isoforms in mice and humans: VDAC1, 2 and 3, each of a molecular mass of about 30 kDa [12,23,74]. VDAC forms a barrel in the bilayer comprised of a transmembrane alpha helix and 13 (or more) transmembrane beta strands. This beta barrel encloses an aqueous channel of ~3 nm in internal diameter, which in the open state allows passage of non-electrolytes up to 5 kDa in size, although molecular weight is not the only determinant of permeance through VDAC. Similar models with additional beta strands have also been proposed, including recently published 3D structures of a 19-stranded β barrel, but these results have been challenged [7,25,29,39,82].
Except for a relatively few membrane-permeant lipophilic compounds, metabolites that enter and leave mitochondria must cross MOM through VDAC [23]. VDAC shows both ion selectivity and voltage dependence. In the open state, anions are favored over cations, but this selectivity is actually weak. Positive and negative membrane potentials (ΔΨ) close VDAC symmetrically with half maximal closure at ±50 mV. Whether Ψ is a physiological regulator of VDAC conductance remains a matter of conjecture, since in the closed state VDAC becomes a cation selective pore of 1.8 nm in diameter that still conducts small cations, such as K+, Na+ and Ca2+, as well as Cl−, whose movement through VDAC collapses electrical potentials [77]. One possibility is that a Donnan potential may form across the outer membrane that is large enough to gate VDAC [77]. Donnan potentials result from asymmetric distribution of impermeant charged species across a membrane, typically proteins. However, the issue is controversial because charged macromolecules reside on both sides of the outer membrane. Moreover, high ionic strength of the intracellular milieu would be also expected to decrease the magnitude of any Donnan potentials forming to below that causing VDAC to close. Nonetheless, a report of a pH across the outer membrane supports the existence of a Donnan potential of ~-40 mV, close to a gating potential for VDAC [65]. Metabolism-driven fluxes of charged metabolites are unlikely to contribute significantly to outer membrane potential formation, since such fluxes are limited by transporters in the inner membrane and are typically at least 2 orders of magnitude less than the flux of small ions through VDAC even in its closed state. Nonetheless, VDAC closure when it occurs would be expected to block movement of major anionic metabolites, including respiratory substrates, creatine phosphate, ATP, ADP and Pi during oxidative phosphorylation [67].
Each mammalian VDAC isoform confers permeability in liposomes with a similar molecular weight cutoff, but electrophysiological studies show differences. VDAC1 and VDAC2 have prototypic voltage gating, but VDAC2 also has a second discrete lower conductance and ion selective state. VDAC3, by contrast, does not show clear voltage dependency (reviewed in [27]). Superresolution microscopy reveals that VDAC1 and VDAC2 colocalize in distinct clusters in mitochondria of osteosarcoma cells, whereas VDAC3 is uniformly distributed [61].
In the past, the assumption has generally been that VDAC is open during normal metabolism to allow free diffusion of metabolites across MOM. In this view, MOM simply serves as a scaffold enclosing the mitochondrial inner membrane (MIM). Recent data, however, suggests that VDAC has the ability to close and inhibit exchange of metabolites between the cytosol and mitochondria [28,41,48,84]. For example, VDAC appears to close relatively early in the evolution of apoptosis with the consequence that mitochondria no longer release ATP to the cytosol or take up ADP, Pi and respiratory substrates [84]. Our evidence also indicates that VDAC closes after acute ethanol treatment of hepatocytes and in cancer cells displaying aerobic glycolysis (Warburg phenomenon) [41,48,52] (see below). VDAC conductance is regulated by a variety of factors, including hexokinase, bcl2 family members, protein kinase A (PKA), glycogen synthase 3β (GSK3β), protein C kinase ε (PKCε), NADH (which increases after ethanol exposure) and possibly others. [3,4,45,66,85,86].
VDAC AND ETHANOL METABOLISM
Alcoholic liver disease
Alcoholic liver disease (ALD) affects more than 2.5 million people in the U.S. alone. Heavy drinking causes ALD, but many heavy drinkers may never experience hepatic symptoms, although in patients with chronic hepatitis C infection, even moderate alcohol drinking causes hepatic cirrhosis and end stage liver disease. Single alcohol binges produce little liver injury except for reversible hepatic steatosis (fatty liver) [11,46,51,63]. Hepatic steatosis without alcohol abuse (non-alcoholic fatty liver disease, NAFLD) occurs in association with the metabolic syndrome, whose features include obesity, insulin resistance, type 2 diabetes and hypertension, which can progress to nonalcoholic steatohepatitis (NASH), cirrhosis and liver failure [19,22,91]. Remarkably, the histopathology of NADLD/NASH and ALD are indistinguishable with features of ballooning degeneration, apoptosis and necrosis of hepatocytes, necroinflammatory foci with neutrophilic infiltration, and hyaline inclusions (Mallory bodies). Steatohepatitis virtually identical to ALD also occurs from occupational exposure of non-obese, non-drinking industrial workers to vinyl chloride, a substance metabolized in the liver to chloroacetaldehyde (ClAcAld), an example of toxicant-associated steatohepatitis (TASH) [21].
Hepatic alcohol metabolism
Ethanol is metabolized predominantly by the liver and undergoes two-step oxidation first to acetaldehyde (AcAld) and then to acetate (Fig. 1) [15]. Alcohol dehydrogenase (ADH), cytochrome P450 2E1 (CYP450 2E1) and catalase in the cytosol, endoplasmic reticulum and peroxisomes, respectively, catalyze the first oxidation step, which converts ethanol to AcAld, a toxic and reactive chemical. Although ADH is quantitatively the most important first step enzyme, CYP450 2E1 metabolism is noteworthy in producing reactive oxygen species (ROS) [50]. Aldehyde dehydrogenase (ALDH) catalyzes the further oxidation of AcAld to acetate. Of 19 known mammalian ALDH genes, mitochondrial ALDH2 with high affinity for AcAld (Km < 1 μM) is most important for AcAld oxidation (and detoxification) to acetate [53]. Together, ADH and ALDH form two moles of NADH for each mole of ethanol oxidized to acetate. For ethanol oxidation to continue, NADH must be oxidized back to NAD+ by mitochondrial respiration.
Figure 1. Metabolism of ethanol.
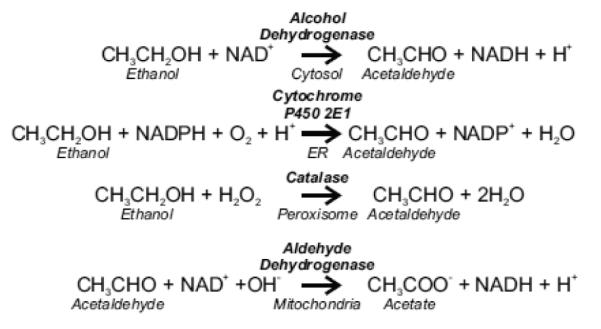
See text for details.
Swift increase of alcohol metabolism
Ethanol ingestion produces an adaptive increase of hepatic ethanol metabolism called swift increase of alcohol metabolism (SIAM) [57,88], which occurs in as little as 2.5 h after a single bolus dose of ethanol (e.g., 5 g/kg i.g. in rats) [79,92]. Since ethanol oxidation is obligatorily linked to NAD+ supply, mitochondrial respiration causing NADH oxidation also nearly doubles in SIAM. Although increased mitochondrial oxygen consumption should in theory lead to increased ATP generation by oxidative phosphorylation, to the contrary ethanol treatment actually decreases hepatic ATP, stimulates glycolysis and depletes glycogen stores. Furthermore, mitochondrial β-oxidation of fatty acids becomes inhibited, promoting fat accumulation within hepatocytes (steatosis) [16,73]. Overall, SIAM is an adaptive response by the liver to oxidize ethanol more rapidly. The mitochondrial respiratory burst, in particular, promotes more rapid elimination of toxic AcAld by ALDH and increases NAD+ supply for ADH-dependent ethanol metabolism. Neither ethanol oxidation by CYP450 nor hepatic ADH activity is altered when SIAM first develops [92], but SIAM is partially blocked by 3-aminotriazole (AT), a CyP450 inhibitor, and more fully blocked by 4-methylpyrazole (MP), an ADH inhibitor [78]. However, AT also inhibits catalase, and other studies suggest that catalase may play a role in SIAM [14,16]. Adrenergic hormones release free fatty acids from adipose tissue, which serve as substrates for long-chain fatty acid peroxisomal β-oxidation. The ensuing peroxisomal H2O2 formation then can promote catalase-dependent alcohol metabolism.
Mitochondria and fatty liver disease
Hepatic ethanol metabolism, AcAld formation and oxidative stress are generally accepted as important initiators of ALD. Among the earliest manifestations of hepatocyte injury by alcohol are morphological and functional abnormalities of mitochondria [46]. Chronic ethanol treatment selectively depletes the mitochondrial antioxidant GSH [30]. AcAld is toxic to mitochondria and aggravates oxidative stress by binding to GSH and promoting GSH leakage [49]. NADH overproduction triggered by ethanol oxidation may also promote mitochondrial production of ROS. Impairment of oxidative phosphorylation and development of megamitochondria after ethanol are possibly related to lipid peroxidation [56]. Alcohol also causes oxidative mitochondrial DNA (mtDNA) damage [40], and mtDNA deletions that occur in 85% of alcoholic patients with steatosis may impair mitochondrial β-oxidation of fatty acids [32,33]. Exposure of cultured hepatocytes to ethanol causes formation of ROS, onset of the mitochondrial permeability transition (MPT) and apoptosis [2]. Overexpression of human CYPP450 2E1 in HepG2 hepatoma cells promotes ethanol-induced MPT onset and apoptosis, effects that are prevented by CYP450 2E1 inhibitors, antioxidants and the MPT inhibitor cyclosporin A (CsA) [20]. Ethanol-induced mitochondrial damage likely impairs ATP production and thus may enhance hepatic susceptibility to alcohol-induced centrolobular hypoxia [31]. Likewise in NASH, similar mitochondrial dysfunction seems to occur leading to impaired fat metabolism, overproduction of ROS, lipid peroxidation and cell death [71].
Hypothesis that Closure of Voltage Dependent Anions Channels Contributes to the Swift Increase of Alcohol Metabolism
How SIAM leads to enhanced mitochondrial respiration while simultaneously depleting ATP and inhibiting mitochondrial β-oxidation remains unknown. Here, we review data in support of the hypothesis that a relative closure of VDAC underlies many of the metabolic adaptations occurring in SIAM. By inhibiting entry of normal respiratory substrates like pyruvate and fatty acyl-CoA, VDAC closure allows the selective and more rapid oxidation of toxic AcAld, since AcAld crosses mitochondrial membranes freely without need for a transporter or channel. Although beneficial in promoting detoxification of aldehydes, VDAC closure has the potential of becoming maladaptive by promoting ATP depletion and steatosis.
Decreased Mitochondrial Outer Membrane Permeability in Ethanol-Treated Hepatocytes
VDAC is the only channel known that allows hydrophilic metabolites to cross MOM in normal viable cells. By contrast, the mitochondrial protein import apparatus is specific for polypeptides, and the Bax/Bak-dependent cytochrome c release channel opens irreversibly only after activation of apoptotic pathways [69,76]. To assess changes of MOM permeability after ethanol, rat hepatocytes were first treated with ethanol and then exposed to a low concentration of digitonin, which selectively permeabilizes (skins) the plasma membrane to allow entry of adenine nucleotides and exogenous respiratory substrates like succinate [41]. In such skinned hepatocytes, ethanol pretreatment decreased succinate-supported respiration and suppressed accessibility of ADP to adenylate kinase in the mitochondrial intermembrane space (IMS) (Fig. 2). MOM permeabilization with high digitonin overcame these effects and restored respiration and adenylate kinase activity to that of skinned hepatocytes not pretreated with ethanol. Moreover in skinned hepatocytes not treated with ethanol, MOM permeabilization with high digitonin did not produce an additional increase of succinate oxidation or adenylate kinase activity. These results are consistent with the conclusion that MOM permeability to hydrophilic metabolites decreases after ethanol treatment and becomes rate-limiting for mitochondrial metabolism.
Figure 2. Ethanol limits suppresses respiration and accessibility to adenylate kinase in the mitochondrial intermembrane space.
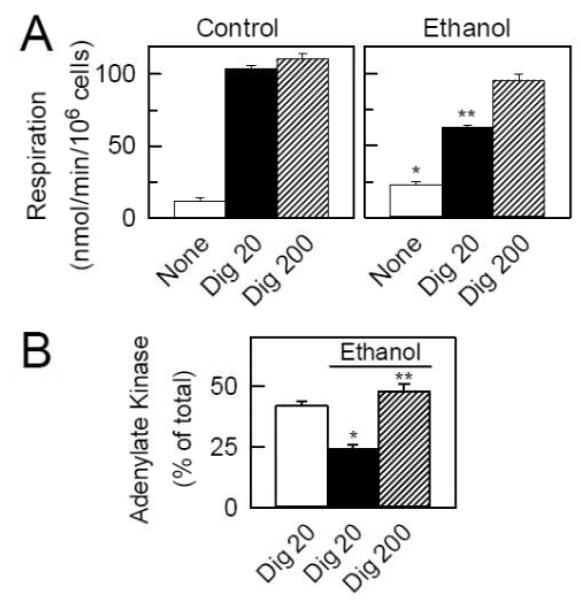
In A, succinate-supported respiration of untreated and ethanol-treated rat hepatocytes was measured before and after sequential addition of 8 and 80 μM digitonin to permeabilize the plasma membrane and MOM, respectively. Plasmalemmal permeabilization allowed entry of succinate and stimulation of respiration, which was less in ethanol-treated hepatocytes. After MOM permeabilization with high digitonin, respiration of ethanol-treated and untreated hepatocytes was not different. In B, untreated and ethanol-treated rat hepatocytes were exposed to 8 μM digitonin, centrifuged and resuspended to remove cytosolic enzymes. AK was measured with and without 80 μM digitonin to permeabilize MOM, as indicated. After ethanol, AK was decreased. Suppression of AK activity was restored by high digitonin, which showed that AK inhibition was due to decreased access of substrates for AK into the IMS. Adapted from [41].
MOM permeability was also independently assessed by confocal microscopy from entrapment of 3 kDa rhodamine-dextran (RhDex) in mitochondria of mechanically permeabilized hepatocytes [41,48]. Hepatocytes were first loaded with MitoTracker Green, which covalently labels mitochondria with green fluorescence, and then incubated with 3 kDa RhDex in an artificial intracellular buffer (ICB). The plasma membranes were then torn open by drawing a micropipette tip across the hepatocytes. Because 3 kDa is below the size exclusion limit of VDAC [24,26,75], added RhDex entered the IMS provided, of course, that VDAC was open. After RhDex entry, 4,4′-diisothiocyano-2,2′-stilbenedisulfonic acid (DIDS), an inhibitor of anion channels including VDAC [8,24,75], was added to close VDAC and entrap RhDex within the IMS. The cells were then washed with ICB containing DIDS but no RhDex. After the final wash, RhDex fluorescence disappeared entirely from the extracellular space and cytosol but was retained by MTG-labeled mitochondria and to a lesser extent by endosomes and nuclei (Fig. 3). In this procedure, fluorescent RhDex freely enters the IMS of mitochondria with open VDAC to be entrapped after DIDS treatment, but as VDAC closes less RhDex can enter the IMS and become entrapped. Using this procedure, ethanol decreased RhDex entrapment in mitochondria by 35% compared to untreated hepatocytes (Fig. 3) [41]. Overall, these results demonstrate that acute ethanol exposure decreases mitochondrial outer membrane permeability most likely by inhibition of VDAC.
Figure 3. Decreased entry of 3 kDa rhodamine-dextran into the mitochondrial intermembrane space after ethanol treatment.

Hepatocytes were pretreated with vehicle (Control) or 50 mM ethanol subjected to a RhoDex entrapment protocol. Note that ethanol pretreatment decreases mitochondrial retention of red-fluorescing RhoDex. Adapted from [41].
Suppression by ethanol of ureagenic respiration in cultured rat hepatocytes
In hepatocytes, ureagenesis comprises a sequence of biochemical reactions occurring in separate intracellular compartments – the mitochondrial matrix and cytosol [90]. Ureagenesis is highly energy-requiring, and four moles of high energy phosphate are required to synthesize one mole of urea from ammonia and bicarbonate precursors. In particular, ureagenesis requires flux of ornithine, citrulline, adenine nucleotides, respiratory substrates and other metabolites across MOM into and out of mitochondria. Thus, if VDAC were to begin to close after ethanol, ureagenesis would be one of the first mitochondrial processes to be affected. Indeed, ethanol suppresses ureagenesis by isolated perfused rat livers, an effect associated with decreased hepatic ATP levels [1]. In cultured rat hepatocytes, respiration nearly doubled in parallel with increased urea formation after addition of ureagenic substrates (Fig. 4). Ethanol dose-dependently inhibited the stimulation of respiration by ureagenic substrates (ureagenic respiration), but ethanol did not inhibit uncoupled respiration or basal respiration [41,42]. Half maximal suppression of respiration stimulated by ureagenic substrates occurred at ~50 mM ethanol, and as little as 10 mM ethanol had a clear effect (Fig. 4). These concentrations are comparable to those for ethanol entering the liver via the portal vein in drinkers. Ethanol also inhibited urea formation in proportion to the inhibition of ureagenic respiration. Suppression of ureagenic respiration by ethanol was partially reversed by inhibitors of ADH, CYP450 2E1 and catalase. By contrast, ALDH inhibition enhanced suppression of ureagenic respiration. These observations were consistent with the conclusion that AcAld was responsible, at least in part, for ethanol-induced suppression of ureagenesis.
Figure 4. Suppression of ureagenic respiration by ethanol and acetaldehyde.

Respiration by cultured rat hepatocytes was measured before and after sequential addition of ureagenic substrates (NH4Cl, ornithine, lactate) and KCN, as indicated, in the presence and absence of ethanol (left panel) or acetaldehyde (right panel). Note inhibition of respiration by ethanol and acetaldehyde. Adapted from [42].
Suppression of ureagenic respiration by exogenous acetaldehyde
When exogenous AcAld was added to cultured hepatocytes, ureagenic respiration was inhibited with half maximal inhibition at ~100 M after correction for evaporative loss of the volatile AcAld, but AcAld did not inhibit uncoupled respiration (Fig. 4) [42]. Other aldehydes, such as MDA and ClAcAld, also inhibited ureagenic respiration with half maximal inhibition of 5 and 250 μM, respectively (uncorrected for evaporation). MDA was a particularly strong inhibitor of ureagenic respiration, but did not inhibit uncoupled respiration . Taken together, these findings support the conclusion that ethanol oxidation to AcAld underlies ethanol-dependent suppression of ureagenic respiration and that other aldehydes can produce similar effects. Since ethanol oxidation causes oxidative stress via CYP450 2E1 metabolism, aldehydes like MDA formed after lipid peroxidation may also contribute to suppression of ureagenesis.
To verify directly that AcAld decreases MOM permeability, uptake of RhDex into the IMS was assessed in skinned hepatocytes. AcAld (500 μM, uncorrected for evaporation) prevented RhDex entrapment to an even greater extent than ethanol pretreatment [42]. Since VDAC conductance solely determines the permeability of the MOM to RhDex, this experiment provides direct evidence that AcAld causes VDAC closure, which likely explains inhibition of ureagenesis by ethanol and AcAld.
Ethanol-induced mitochondrial depolarization in vivo
VDAC closure induced by ethanol metabolism promotes selective oxidation of AcAld and helps explain ATP depletion and steatosis after ethanol, since VDAC closure blocks mitochondrial ATP release and uptake of fatty acids required for β-oxidation. The VDAC hypothesis also accounts for why rats fed short and medium chain fatty acids in their diet do not develop steatosis after ethanol treatment [60], since short and medium chain fatty acids cross mitochondrial membranes freely without using the carnitine shuttle or other transporter system [6]. However, VDAC closure cannot explain the stimulation of mitochondrial respiration in SIAM. Indeed, inhibition of uptake of cytosolic ADP would be expected to suppress respiration that is ordinarily stimulated by ADP. These considerations imply an additional mitochondrial adaptation to ethanol, namely mitochondrial uncoupling to cause futile dissipation of the protonmotive force and respiratory stimulation [48].
Such respiratory stimulation does not occur in vitro in hepatocytes treated with ethanol, consistent with observations that the respiratory burst of SIAM is an in vivo response to ethanol that does not occur in isolated systems. To better characterize hepatic mitochondrial responses to acute ethanol treatment in vivo, intravital confocal/multiphoton microscopy was performed on the livers of anesthetized mice. After a single intragastric dose of ethanol (up to 6 g/kg), mitochondria depolarized within hepatocytes in an all-or-nothing fashion in as little one hour, as shown by lack of uptake of Ψ-indicating fluorophores like rhodamine 123 and tetramethylrhodamine methylester (TMRM) [93]. Ethanol-induced mitochondrial depolarization was dose- and time-dependent, maximally affecting more than 90% of hepatocytes and peaking after 6 to 12 h of the ethanol dosing [93]. After 24 h, mitochondria of most hepatocytes recovered normal polarization, and after a week the livers appeared entirely normal. Necrotic and apoptotic cell death was minimal throughout. The time course of ethanol-induced mitochondrial depolarization was mirrored that for SIAM reported previously.
In mitochondria of hepatocytes and most eukaryotic cells, NADH autofluorescence arises primarily from mitochondria, whereas cytosolic NADH fluorescence is quenched [62]. NAD+ is nonfluorescent. After ethanol treatment in vivo, intravital multiphoton microscopy revealed a decrease of NADH autofluorescence specifically in hepatocytes with depolarized mitochondria and not in hepatocytes with polarized mitochondria. With respiratory inhibition, such as that caused by anoxia, NADH fluorescence increases and NAD+ decreases, whereas the opposite occurs after mitochondrial uncoupling. Thus, mitochondrial depolarization occurring with oxidation of NADH after in vivo ethanol treatment signified an uncoupling event rather than anoxia or respiratory inhibition. Mitochondrial depolarization was linked to ethanol metabolism to AcAld, since mitochondrial depolarization after ethanol was decreased in rodents deficient in alcohol dehydrogenase or CYP2E1. Moreover activation of ALDH decreased depolarization, whereas ALDH inhibition enhanced depolarization [94].
Overall, these findings are consistent with the conclusion that ethanol has two rapid effects on mitochondria. The first is a relative closure of VDAC, and the second is opening a protonophoric uncoupling circuit that dissipates mitochondrial Ψ and stimulates respiration. Together these changes promote rapid and selective mitochondrial oxidation of acetaldehyde. Nonetheless, future studies are needed to determine the mechanisms underlying AcAld-mediated VDAC closure and protonophoric uncoupling. AcAld may not be the only aldehyde causing these effects, and aldehydes like MDA produced after lipid peroxidation and ClAcAld formed during vinyl-chloride metabolism may produce similar VDAC closure and mitochondrial uncoupling. The aldehyde-dependent disruptions of normal mitochondrial functioning may be an early common effect leading to the virtually identical pathologies of ALD, NASH and TASH.
VOLTAGE DEPENDENT ANION CHANNELS AND WARBURG METABOLISM IN CANCER CELLS
Warburg phenomenon
As recognized by Otto Warburg 75 years ago, malignant cancer cells display high rates of glycolysis even when fully oxygenated [36,89]. Ordinarily, cells with mitochondria metabolize glucose to pyruvate, which is further oxidized to CO2 and H2O by the tricarboxylic acid cycle and mitochondrial respiratory chain. In this aerobic metabolism, glycolysis accounts for only about 5% of ATP production with the remainder produced by oxidative phosphorylation. In cancer cells, however, high glycolytic rates and net formation of lactate persist despite adequate oxygenation, a fact fully confirmed in cancer patients by positron emission tomography of the glucose analog 18fluorodeoxyglucose [35]. Consequently, glycolysis accounts for 50 to 70% of ATP formation in tumor cells [55,83].
The advantage of aerobic glycolysis to tumor cells remains a matter of conjecture. Enhanced glycolytic capacity may be beneficial for tumors whose rapid growth often exceeds their blood and hence oxygen supply. Furthermore, although glycolysis is less efficient, glycolysis may still produce ATP faster than oxidative phosphorylation to support more rapid growth, an advantage to rapidly proliferating cells, especially in an hypoxic environment [38]. Additionally, glycolytic intermediates are needed as precursors for anabolic biosynthesis by proliferating cells. The Warburg phenomenon is more than an epiphenomenon, since pharmacologic interventions to promote oxidative phosphorylation and/or inhibit glycolysis (e.g., dichloroacetic acid, 3-bromopyruvate) cause tumor cell death both in vitro and in vivo [55,58].
Aerobic glycolysis in cancer cells is associated with increased expression of hexokinase (HK), especially the HK-2 isoform, and plasmalemmal glucose transporters (e.g., GLUT1) [18,37]. Although Warburg suggested that aerobic glycolysis in tumor cells might be the consequence of a defect in what we now know is mitochondrial oxidative phosphorylation, studies show that isolated tumor mitochondria are fully functional with regards to respiration and ATP synthesis [59]. The mystery persists as to why fully functional mitochondria inside tumor cells are nonetheless relatively inactive in terms of respiration and ATP generation.
A preference for glycolysis also occurs for yeast growing aerobically on glucose. When glucose and oxygen are plentiful, yeast cells prefer glucose fermentation to aerobic respiration even though aerobic respiration represents a much more efficient use of glucose. Only after glucose is exhausted does a so-called diauxic shift occur, at which time new enzymes are expressed for aerobic mitochondrial metabolism of fermentation products like ethanol [34]. However, growth rates after the diauxic shift are slower. The preference in yeast for aerobic glycolysis is analogous to the Warburg phenomenon and indicates that aerobic glycolysis supports more rapid cell proliferation, a selective growth advantage for both yeast and cancer cells [34,83].
Voltage dependent anion channels, tubulin and protein kinases
Nanomolar dimeric tubulin closes VDAC reconstituted into planar phospholipid bilayers and appears to decrease MOM permeability to adenine nucleotides in isolated brain mitochondria and permeabilized synaptosomes and cardiac myocytes [68,81]. These effects of tubulin are enhanced when VDAC is phosphorylated by protein kinase A (PKA) [72]. Purified VDAC1 is a substrate for PKA in vitro, and PKA phosphorylation of VDAC inhibits association of VDAC with other proteins, such as Bax and tBid [5,10], where tBid but not Bax causes VDAC closure [66]. By contrast, VDAC2 phosphorylation by GSK3β appears to promote channel opening [28]. In isolated rat liver mitochondria, PKA phosphorylation inhibits onset of the MPT, an effect possibly associated with phosphorylation of VDAC [64].
VDAC hypothesis for suppression of mitochondria metabolism in the Warburg phenomenon
Much research on cancer cell metabolism and the Warburg phenomenon focuses on enhancement of glycolysis, and mechanisms underlying suppression of mitochondrial metabolism in cancer cells have received less attention. Previously, we proposed that relative but not absolute VDAC closure occurs in mitochondria of cancer cells [48]. Such VDAC closure limits exchange of mitochondrial metabolites and globally suppresses mitochondrial metabolism in favor of glycolysis. Here, we review recent data that high levels of free tubulin in cancer cells are responsible for this VDAC closure and that protein kinase signaling pathways, such as those involving PKA and GSK3β, further modulate tubulin-dependent VDAC closure.
Maintenance of mitochondrial membrane potential through both respiration and ATP hydrolysis in HepG2 cells
Myxothiazol (10 μM), a Complex III respiratory inhibitor, caused little change of mitochondrial polarization to HepG2 human hepatoma cells, as assessed by uptake of the potential–indicating cationic fluorophore, TMRM. Only after addition of oligomycin, an inhibitor of the F1F0-ATP synthase, was TMRM released, signifying mitochondrial depolarization [52]. These results illustrate that ATP from other sources, such as glycolysis, can maintain mitochondrial Ψ in the presence of respiratory inhibition by reversal of the F1F0-ATP synthase. By contrast, when oligomycin was added before myxothiazol, mitochondria hyperpolarized and only depolarized when myxothiazol was added subsequently. Hyperpolarization after oligomycin in the absence of respiratory inhibitor signified that respiration was driving ATP formation, because oligomycin reverses the depolarizing effect of ATP synthesis on Ψ. Overall, these results show that both respiration and ATP hydrolysis can support mitochondrial polarization in HepG2 cells.
Rotenone, colchicine and nocodazole decrease mitochondrial membrane potential
Rotenone, a Complex I inhibitor, should in theory exert changes very much like myxothiazol. However, when HepG2 cells were exposed to rotenone, they depolarized quickly [52]. Rotenone also caused cellular rounding unlike myxothiazol which did not. In previous studies, rotenone depolymerized microtubules [17,54]. Accordingly, two structurally unrelated microtubule destabilizers, colchicine and nocodazole, were examined for their effect on mitochondrial Ψ. Both caused mitochondrial depolarization and cell rounding to a similar extent as rotenone (Fig. 5). Thus, each structurally distinct microtubule destabilizer caused mitochondrial depolarization. To assess whether mitochondrial depolarization depended specifically on increased free tubulin inside cells, HepG2 cells were exposed to the microtubule stabilizer, paclitaxel [43,44]. In contrast to the depolarizing effect of colchicine, rotenone and nocodazole, paclitaxel hyperpolarized mitochondria. Moreover, paclitaxel prevented mitochondrial depolarization when the HepG2 cells were subsequently treated with rotenone, colchicine or nocodazole (Fig. 5). In parallel, depolymerized (free) to polymerized tubulin ratios were assessed using a Western blot procedure. As expected, the microtubule-depolymerizing agents, colchicine, nocodazole and rotenone, increased free to polymerized tubulin ratios by more than 8-fold, whereas paclitaxel, a microtubule stabilizer, decreased the free to polymerized tubulin ratio and prevented microtubule depolymerization by colchicine, nocodazole and rotenone (Fig. 6) [52]. Myxothiazol, unlike rotenone, produced no change of the free to polymerized tubulin ratio. Overall, free to polymerized tubulin ratios showed a strong inverse correlation with mitochondrial polarization.
Figure 5. Modulation of mitochondrial membrane potential by free tubulin and protein kinase.
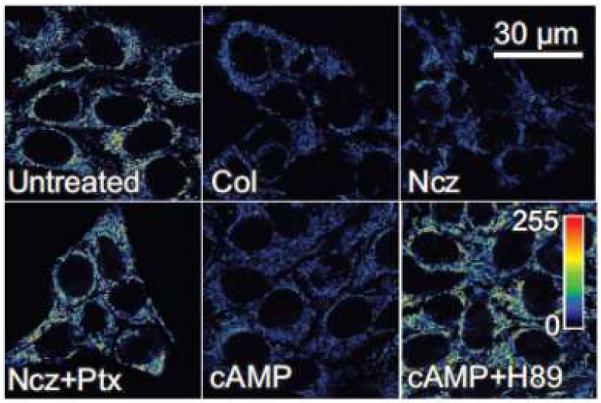
A. Pseudocolor of the fluorescence of TMRM was used to monitor changes of mitochondrial Ψ in HepG2 human hepatoma cells. Ψ decreased after microtubule depolymerization with colchicine (Col) and nocodazole (Ncz). Paclitaxel (Ptx), microtubule stabilizer, hyperpolarized mitochondria even in the presence of nocodazole. Dibutyrl cAMP (cAMP), a PKA agonist, also depolarized mitochondria, whereas H89, a PKA inhibitor, hyperpolarized mitochondria even in the presence of dibutyrl cAMP. Adapted from [52].
Figure 6. Inverse relationship between free to polymerized tubulin and mitochondrial membrane potential.
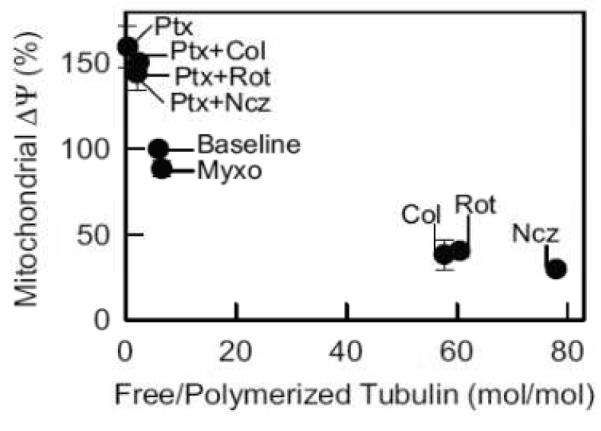
Mitochondrial Ψ and free to polymerized tubulin were measured after exposure of HepG2 cells to colchicine (Col), myxothiazol (Myxo), nocodazole (Ncz), paclitaxel (Ptx) and rotenone (Rot) in various combinations. Adapted from [52].
In primary rat hepatocytes, free to polymerized tubulin ratios were much smaller than in HepG2 hepatocarcinoma cells. Consistent with these low ratios, microtubule stabilization with paclitaxel did not cause hyperpolarization. However, depolymerization of microtubules with nocodazole or colchicine caused mitochondrial depolarization [52].
Lack of change of plasma membrane potential after microtubule depolymerization
Plasmalemmal potential also drives TMRM uptake into cells and ultimately into mitochondria. Thus, plasmalemmal hyperpolarization or depolarization might increase or decrease cellular uptake of TMRM. However as by monitored with DiBAC4(3) fluorescence, plasmalemmal potential did not change after exposure of HepG2 cells to colchicine, nocodazole, or rotenone and does not account for loss of TMRM fluorescence in the presence of microtubule-depolymerizing agents [52].
Regulation of tubulin-dependent changes of mitochondrial membrane potential by protein kinase A and glycogen synthase-3β
PKA phosphorylates VDAC [9], and a recent single channel study shows that PKA phosphorylation sensitizes VDAC to inhibition by tubulin [72]. Similarly in intact HepG2 cells, the PKA agonist, dibutyryl cAMP, caused mitochondrial depolarization, consistent with PKA augmentation of tubulin-dependent VDAC closure (Fig. 5). Okadaic acid, a protein phosphatase inhibitor that indirectly promotes phosphorylation, also depolarized mitochondria. By contrast, the PKA inhibitor, H89, hyperpolarized mitochondria and prevented depolarization induced by dibutyryl cAMP (Fig. 5). Moreover, H89 prevented and reversed the depolarizing effect of colchicine [52,72].
A recent study indicates that GSK3β-mediated phosphorylation promotes VDAC opening [28]. Thus, GSK3β inhibition should promote VDAC closure and mitochondrial depolarization. This expectation was confirmed with three structurally distinct GSK3β inhibitors, each of which caused release of TMRM fluorescence from HepG2 cells signifying mitochondrial depolarization [52].
Taken together, these experiments support the conclusion that free tubulin dynamically regulates mitochondrial function in cancer cells through regulation of VDAC conductance. When tubulin increases, increased VDAC closure occurs and mitochondrial function becomes suppressed, as reflected by decreased mitochondrial Ψ. By contrast, as free tubulin decreases, VDAC inhibition is lifted and mitochondrial metabolism becomes enhanced. Thus in the steady state, both an increase and a decrease of free tubulin will alter mitochondrial function. These effects are further modulated by PKA, GSK3β and possibly other signaling pathways implicated in Warburg metabolism. HIF-1α, for example, upregulates glucose transporters, increases expression of several glycolytic enzymes and inhibits pyruvate dehydrogenase via upregulation of pyruvate dehydrogenase kinase (PDK) to contribute to the aerobic glycolysis of the Warburg effect [13,58,70], but how HIF-1α may modulate tubulin-dependent VDAC closure remains to be determined.
Possible physiological role of tubulin regulation of voltage dependent anion channels
As shown in yeast, aerobic glycolysis supports a higher rate of cell growth than aerobic oxidative phosphorylation, and rapidly dividing cells maintain a free tubulin reserve for spindle formation at the mitosis. Thus, high free tubulin is one signature of cell proliferation, but why should tubulin have evolved as an inhibitor of VDAC and inducer of Warburg metabolism? One possibility is the need by cells for increased ATP supply during the energy-demanding events of chromosome separation and cytokinesis at mitosis. As the spindle apparatus forms in prophase, free tubulin decreases. As a consequence, VDAC inhibition by tubulin is reversed, and aerobic ATP-generating mitochondrial metabolism becomes upregulated. In this way, tumor and other rapidly proliferating cells may suspend Warburg metabolism (aerobic glycolysis) in favor of oxidative phosphorylation to meet the uniquely high bioenergetic demands of cell division. Then as microtubules of the spindle apparatus depolymerize in telophase, free tubulin increases and Warburg metabolism is restored.
CONCLUSIONS AND FUTURE PROSPECTS
Overall, the findings reviewed here support the conclusion that VDAC can dynamically regulate mitochondrial function as a kind of adjustable governor, or governator, which limits maximal rates of mitochondrial metabolism. In livers of animals exposed to ethanol, VDAC closure in MOM and opening of a protonophoric uncoupling pathway in MIM caused by aldehyde generation leads to selective and more rapid oxidation of AcAld (Fig. 7). In cancer cells, high free tubulin causes a relative closure of VDAC to suppress mitochondrial function in Warburg metabolism, an effect augmented by PKA but antagonized by GSK3β (Fig. 7). Notably, mitochondrial metabolism is poised to increase or decrease as free tubulin decreases or increases. Although not reviewed in detail here, VDAC closure seems to occur in other settings, including metabolic suppression in early apoptosis and suppression of mitochondrial ATP hydrolysis in ischemia [28,87]. Other examples where VDAC closure may also be important include the inhibition of respiration in the cytopathic hypoxia” of sepsis and hemorrhage/resuscitation and the inhibition of mitochondrial ATP release in unstimulated pancreatic beta cells [47,48]. Another potentially beneficial consequence of VDAC closure is blockade of the release of toxic superoxide from mitochondria in conditions of oxidative stress [80].
Figure 7. Schemes of voltage dependent anion channel closure.

In A, ethanol metabolism by ADH and P450 2E1 generates AcAld, which promotes VDAC closure in MOM. P450 2E1 also generates ROS to cause lipid peroxidation and formation of MDA, which also closes VDAC. Simultaneously in vivo, a proton-conducting uncoupling pathway opens in MIM. These two events together promote increased mitochondrial respiration and selective, more rapid oxidation and detoxification of AcAld and MDA by ALDH. In B, rotenone, colchicine and nocodazole increase free tubulin by depolmerizing microtubules. Tubulin binds to and inhibits VDAC, which decreases uptake of respiratory substrates and produces a relative mitochondrial depolarization. Paclitaxel stabilizes microtubules, which promotes VDAC opening and mitochondrial hyperpolarization. VDAC phosphorylation by PKA augments tubulin-dependent VDAC closure, whereas GSK3β promotes opening. High free tubulin in tumor cells causes a relative closure of VDAC, which suppresses mitochondrial metabolism and contributes to the Warburg phenomenon of aerobic glycolysis.
The three different VDAC isoforms are widely distributed in various tissues, which raises the possibility that the isoforms are differentially regulated. For example, a specific isoform(s) but not others may be responsible for suppression of the mitochondrial function in Warburg metabolism or for decreased outer membrane permeability in hepatocytes due to aldehydes. An intriguing possibility is that various protein kinases act differentially on VDAC isoforms and that other posttranslational modifications (e.g., acetylation) regulate VDAC. The avenues for future research on VDAC are rich and diverse.
Highlights.
Voltage dependent anion channels (VDAC) required for mitochondrial outer membrane permeability
VDAC emerging as important regulator of mitochondrial metabolism
Selective oxidation of toxic acetaldehyde after ethanol fostered byVDAC closure and uncoupling
Tubulin-dependent VDAC closure enhanced by protein kinase A and antagonized by GSK3β.
Contribution of VDAC closure to suppression of mitochondrial metabolism in Warburg effect
ACKNOWLEDGMENTS
This work was supported, in part, by NIH Grants DK073336 (JJL), DK37034 (JJL), AA017756 (ZZ and JJL) and HL101240-01 (ELH). Imaging facilities for this research were supported, in part, by Cancer Center Support Grant P30 CA138313 to the Hollings Cancer Center, Medical University of South Carolina.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Adachi K, Matsuhashi T, Nishizawa Y, Usukura J, Popinigis J, Wakabayashi T. Studies on urea synthesis in the liver of rats treated chronically with ethanol using perfused livers, isolated hepatocytes, and mitochondria. Biochem. Pharmacol. 1995;50:1391–1399. doi: 10.1016/0006-2952(95)02023-3. [DOI] [PubMed] [Google Scholar]
- 2.Adachi M, Ishii H. Role of mitochondria in alcoholic liver injury. Free Radic. Biol. Med. 2002;32:487–491. doi: 10.1016/s0891-5849(02)00740-2. [DOI] [PubMed] [Google Scholar]
- 3.Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V. In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem. J. 2004;377:347–355. doi: 10.1042/BJ20031465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baines CP, Song CX, Zheng YT, Wang GW, Zhang J, Wang OL, Guo Y, Bolli R, Cardwell EM, Ping P. Protein kinase Cepsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ. Res. 2003;92:873–880. doi: 10.1161/01.RES.0000069215.36389.8D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banerjee J, Ghosh S. Bax increases the pore size of rat brain mitochondrial voltage-dependent anion channel in the presence of tBid. Biochem. Biophys. Res. Commun. 2004;323:310–314. doi: 10.1016/j.bbrc.2004.08.094. [DOI] [PubMed] [Google Scholar]
- 6.Bartlett K, Eaton S. Mitochondrial beta-oxidation. Eur. J. Biochem. 2004;271:462–469. doi: 10.1046/j.1432-1033.2003.03947.x. [DOI] [PubMed] [Google Scholar]
- 7.Bayrhuber M, Meins T, Habeck M, Becker S, Giller K, Villinger S, Vonrhein C, Griesinger C, Zweckstetter M, Zeth K. Structure of the human voltage-dependent anion channel. Proc. Natl. Acad. Sci. U. S. A. 2008;105:15370–15375. doi: 10.1073/pnas.0808115105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beavis AD, vatol-Hag H. The mitochondrial inner membrane anion channel is inhibited by DIDS. J. Bioenerg. Biomembr. 1996;28:207–214. doi: 10.1007/BF02110652. [DOI] [PubMed] [Google Scholar]
- 9.Bera AK, Ghosh S. Dual mode of gating of voltage-dependent anion channel as revealed by phosphorylation. J. Struct. Biol. 2001;135:67–72. doi: 10.1006/jsbi.2001.4399. [DOI] [PubMed] [Google Scholar]
- 10.A K. Bera, Ghosh S, Das S. Mitochondrial VDAC can be phosphorylated by cyclic AMP-dependent protein kinase. Biochem. Biophys. Res. Commun. 1995;209:213–217. doi: 10.1006/bbrc.1995.1491. [DOI] [PubMed] [Google Scholar]
- 11.Bergheim I, McClain CJ, Arteel GE. Treatment of alcoholic liver disease. Dig. Dis. 2005;23:275–284. doi: 10.1159/000090175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blachly-Dyson E, Forte M. VDAC channels. IUBMB. Life. 2001;52:113–118. doi: 10.1080/15216540152845902. [DOI] [PubMed] [Google Scholar]
- 13.Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 14.Bradford BU, Enomoto N, Ikejima K, Rose ML, Bojes HK, Forman DT, Thurman RG. Peroxisomes are involved in the swift increase in alcohol metabolism. J. Pharmacol. Exp. Ther. 1999;288:254–259. [PubMed] [Google Scholar]
- 15.Bradford BU, Rusyn I. Swift increase in alcohol metabolism (SIAM): understanding the phenomenon of hypermetabolism in liver. Alcohol. 2005;35:13–17. doi: 10.1016/j.alcohol.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 16.Bradford BU, Rusyn I. Swift increase in alcohol metabolism (SIAM): understanding the phenomenon of hypermetabolism in liver. Alcohol. 2005;35:13–17. doi: 10.1016/j.alcohol.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 17.Brinkley BR, Barham SS, Barranco SC, Fuller GM. Rotenone inhibition of spindle microtubule assembly in mammalian cells. Exp. Cell Res. 1974;85:41–46. doi: 10.1016/0014-4827(74)90210-9. [DOI] [PubMed] [Google Scholar]
- 18.Bustamante E, Morris HP, Pedersen PL. Energy metabolism of tumor cells. Requirement for a form of hexokinase with a propensity for mitochondrial binding. J. Biol. Chem. 1981;256:8699–8704. [PubMed] [Google Scholar]
- 19.Caldwell S, Argo C. The natural history of non-alcoholic fatty liver disease. Dig. Dis. 2010;28:162–168. doi: 10.1159/000282081. [DOI] [PubMed] [Google Scholar]
- 20.Caro AA, Cederbaum AI. Oxidative stress, toxicology, and pharmacology of CYP2E1. Annu. Rev. Pharmacol. Toxicol. 2004;44:27–42. doi: 10.1146/annurev.pharmtox.44.101802.121704. [DOI] [PubMed] [Google Scholar]
- 21.Cave M, Falkner KC, Ray M, Joshi-Barve S, Brock G, Khan R, Bon HM, McClain CJ. Toxicant-associated steatohepatitis in vinyl chloride workers. Hepatology. 2010;51:474–481. doi: 10.1002/hep.23321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiang DJ, Pritchard MT, Nagy LE. Obesity, diabetes mellitus, and liver fibrosis. Am. J. Physiol Gastrointest. Liver Physiol. 2011;300:G697–G702. doi: 10.1152/ajpgi.00426.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colombini M. VDAC: the channel at the interface between mitochondria and the cytosol. Mol. Cell Biochem. 2004;256-257:107–115. doi: 10.1023/b:mcbi.0000009862.17396.8d. [DOI] [PubMed] [Google Scholar]
- 24.Colombini M. VDAC: the channel at the interface between mitochondria and the cytosol. Mol. Cell Biochem. 2004;256-257:107–115. doi: 10.1023/b:mcbi.0000009862.17396.8d. [DOI] [PubMed] [Google Scholar]
- 25.Colombini M. The published 3D structure of the VDAC channel: native or not? Trends Biochem. Sci. 2009;34:382–389. doi: 10.1016/j.tibs.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 26.Colombini M, Yeung CL, Tung J, Konig T. The mitochondrial outer membrane channel, VDAC, is regulated by a synthetic polyanion. Biochim. Biophys. Acta. 1987;905:279–286. doi: 10.1016/0005-2736(87)90456-1. [DOI] [PubMed] [Google Scholar]
- 27.Craigen WJ, Graham BH. Genetic strategies for dissecting mammalian and Drosophila voltage-dependent anion channel functions. J. Bioenerg. Biomembr. 2008;40:207–212. doi: 10.1007/s10863-008-9146-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das S, Wong R, Rajapakse N, Murphy E, Steenbergen C. Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation. Circ. Res. 2008;103:983–991. doi: 10.1161/CIRCRESAHA.108.178970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De P, V, Prezioso G, Thinnes F, Link TA, Palmieri F. Peptide-specific antibodies and proteases as probes of the transmembrane topology of the bovine heart mitochondrial porin. Biochemistry. 1991;30:10191–10200. doi: 10.1021/bi00106a017. [DOI] [PubMed] [Google Scholar]
- 30.Fernandez-Checa JC, Kaplowitz N. Hepatic mitochondrial glutathione: transport and role in disease and toxicity. Toxicol. Appl. Pharmacol. 2005;204:263–273. doi: 10.1016/j.taap.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 31.French SW, Benson NC, Sun PS. Centrilobular liver necrosis induced by hypoxia in chronic ethanol-fed rats. Hepatology. 1984;4:912–917. doi: 10.1002/hep.1840040521. [DOI] [PubMed] [Google Scholar]
- 32.Fromenty B, Grimbert S, Mansouri A, Beaugrand M, Erlinger S, Rotig A, Pessayre D. Hepatic mitochondrial DNA deletion in alcoholics: association with microvesicular steatosis. Gastroenterology. 1995;108:193–200. doi: 10.1016/0016-5085(95)90024-1. [DOI] [PubMed] [Google Scholar]
- 33.Fromenty B, Pessayre D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol. Ther. 1995;67:101–154. doi: 10.1016/0163-7258(95)00012-6. [DOI] [PubMed] [Google Scholar]
- 34.Galdieri L, Mehrotra S, Yu S, Vancura A. Transcriptional regulation in yeast during diauxic shift and stationary phase. OMICS. 2010;14:629–638. doi: 10.1089/omi.2010.0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gambhir SS. Molecular imaging of cancer with positron emission tomography. Nat. Rev. Cancer. 2002;2:683–693. doi: 10.1038/nrc882. [DOI] [PubMed] [Google Scholar]
- 36.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 37.Geschwind JF, Georgiades CS, Ko YH, Pedersen PL. Recently elucidated energy catabolism pathways provide opportunities for novel treatments in hepatocellular carcinoma. Expert. Rev. Anticancer Ther. 2004;4:449–457. doi: 10.1586/14737140.4.3.449. [DOI] [PubMed] [Google Scholar]
- 38.Harvey AJ, Kind KL, Thompson JG. REDOX regulation of early embryo development. Reproduction. 2002;123:479–486. doi: 10.1530/rep.0.1230479. [DOI] [PubMed] [Google Scholar]
- 39.Hiller S, Garces RG, Malia TJ, Orekhov VY, Colombini M, Wagner G. Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science. 2008;321:1206–1210. doi: 10.1126/science.1161302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoek JB, Cahill A, Pastorino JG. Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology. 2002;122:2049–2063. doi: 10.1053/gast.2002.33613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holmuhamedov E, Lemasters JJ. Ethanol exposure decreases mitochondrial outer membrane permeability in cultured rat hepatocytes. Arch. Biochem. Biophys. 2009;481:226–233. doi: 10.1016/j.abb.2008.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holmuhamedov EL, Czerny C, Beeson CC, Lemasters JJ. Ethanol Suppresses Ureagenesis in Rat Hepatocytes: Role of Acetaldehyde. 2011. [DOI] [PMC free article] [PubMed]
- 43.Horwitz SB. Mechanism of action of taxol. Trends Pharmacol. Sci. 1992;13:134–136. doi: 10.1016/0165-6147(92)90048-b. [DOI] [PubMed] [Google Scholar]
- 44.Horwitz SB, Lothstein L, Manfredi JJ, Mellado W, Parness J, Roy SN, Schiff PB, Sorbara L, Zeheb R. Taxol: mechanisms of action and resistance. Ann. N. Y. Acad. Sci. 1986;466:733–744. doi: 10.1111/j.1749-6632.1986.tb38455.x. [DOI] [PubMed] [Google Scholar]
- 45.Lee AC, Zizi M, Colombini M. Beta-NADH decreases the permeability of the mitochondrial outer membrane to ADP by a factor of 6. J. Biol. Chem. 1994;269:30974–30980. [PubMed] [Google Scholar]
- 46.Lefkowitch JH. Morphology of alcoholic liver disease. Clin. Liver Dis. 2005;9:37–53. doi: 10.1016/j.cld.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 47.Lemasters JJ. Modulation of mitochondrial membrane permeability in pathogenesis, autophagy and control of metabolism. J. Gastroenterol. Hepatol. 2007;22(Suppl 1):S31–7. doi: 10.1111/j.1440-1746.2006.04643.x. S31-S37. [DOI] [PubMed] [Google Scholar]
- 48.Lemasters JJ, Holmuhamedov E. Voltage-dependent anion channel (VDAC) as mitochondrial governator--thinking outside the box. Biochim. Biophys. Acta. 2006;1762:181–190. doi: 10.1016/j.bbadis.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 49.Lieber CS. Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol. 2004;34:9–19. doi: 10.1016/j.alcohol.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 50.Lu Y, Cederbaum AI. CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008;44:723–738. doi: 10.1016/j.freeradbiomed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lucey MR, Mathurin P, Morgan TR. Alcoholic hepatitis. N. Engl. J. Med. 2009;360:2758–2769. doi: 10.1056/NEJMra0805786. [DOI] [PubMed] [Google Scholar]
- 52.Maldonado EN, Patnaik J, Mullins MR, Lemasters JJ. Free tubulin modulates mitochondrial membrane potential in cancer cells. Cancer Res. 2010;70:10192–10201. doi: 10.1158/0008-5472.CAN-10-2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marchitti SA, Brocker C, Stagos D, Vasiliou V. Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert. Opin. Drug Metab Toxicol. 2008;4:697–720. doi: 10.1517/17425250802102627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marshall LE, Himes RH. Rotenone inhibition of tubulin self-assembly. Biochim. Biophys. Acta. 1978;543:590–594. doi: 10.1016/0304-4165(78)90315-x. [DOI] [PubMed] [Google Scholar]
- 55.Mathupala SP, Ko YH, Pedersen PL. Hexokinase-2 bound to mitochondria: cancer’s stygian link to the “Warburg Effect” and a pivotal target for effective therapy. Semin. Cancer Biol. 2009;19:17–24. doi: 10.1016/j.semcancer.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matsuhashi T, Karbowski M, Liu X, Usukura J, Wozniak M, Wakabayashi T. Complete suppresion of ethanol-induced formation of megamitochondria by 4-hydroxy-2,2,6,6-tetramethyl-piperidine-1-oxyl (4-OH-TEMPO) Free Radic. Biol. Med. 1998;24:139–147. doi: 10.1016/s0891-5849(97)00210-4. [DOI] [PubMed] [Google Scholar]
- 57.Mendelson JH, Mello NK. Experimental analysis of drinking behavior of chronic alcoholics. Ann. N. Y. Acad. Sci. 1966;133:828–845. doi: 10.1111/j.1749-6632.1966.tb50930.x. [DOI] [PubMed] [Google Scholar]
- 58.Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010;2:31ra34. doi: 10.1126/scitranslmed.3000677. [DOI] [PubMed] [Google Scholar]
- 59.Nakashima RA, Paggi MG, Pedersen PL. Contributions of glycolysis and oxidative phosphorylation to adenosine 5′-triphosphate production in AS-30D hepatoma cells. Cancer Res. 1984;44:5702–5706. [PubMed] [Google Scholar]
- 60.Nanji AA, Yang EK, Fogt F, Sadrzadeh SM, Dannenberg AJ. Medium chain triglycerides and vitamin E reduce the severity of established experimental alcoholic liver disease. J. Pharmacol. Exp. Ther. 1996;277:1694–1700. [PubMed] [Google Scholar]
- 61.Neumann D, Buckers J, Kastrup L, Hell SW, Jakobs S. Two-color STED microscopy reveals different degrees of colocalization between hexokinase-I and the three human VDAC isoforms. PMC Biophys. 2010;3:4. doi: 10.1186/1757-5036-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nieminen AL, Byrne AM, Herman B, Lemasters JJ. Mitochondrial permeability transition in hepatocytes induced by t-BuOOH: NAD(P)H and reactive oxygen species. Am. J. Physiol. 1997;272:C1286–C1294. doi: 10.1152/ajpcell.1997.272.4.C1286. [DOI] [PubMed] [Google Scholar]
- 63.O’Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Hepatology. 2010;51:307–328. doi: 10.1002/hep.23258. [DOI] [PubMed] [Google Scholar]
- 64.Pediaditakis P, Kim JS, He L, Zhang X, Graves LM, Lemasters JJ. Inhibition of the mitochondrial permeability transition by protein kinase A in rat liver mitochondria and hepatocytes. Biochem. J. 2010;431:411–421. doi: 10.1042/BJ20091741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Porcelli AM, Ghelli A, Zanna C, Pinton P, Rizzuto R, Rugolo M. pH difference across the outer mitochondrial membrane measured with a green fluorescent protein mutant. Biochem. Biophys. Res. Commun. 2005;326:799–804. doi: 10.1016/j.bbrc.2004.11.105. [DOI] [PubMed] [Google Scholar]
- 66.Rostovtseva TK, Antonsson B, Suzuki M, Youle RJ, Colombini M, Bezrukov SM. Bid, but not Bax, regulates VDAC channels. J. Biol. Chem. 2004;279:13575–13583. doi: 10.1074/jbc.M310593200. [DOI] [PubMed] [Google Scholar]
- 67.Rostovtseva TK, Komarov A, Bezrukov SM, Colombini M. VDAC channels differentiate between natural metabolites and synthetic molecules. J. Membr. Biol. 2002;187:147–156. doi: 10.1007/s00232-001-0159-1. [DOI] [PubMed] [Google Scholar]
- 68.Rostovtseva TK, Sheldon KL, Hassanzadeh E, Monge C, Saks V, Bezrukov SM, Sackett DL. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc. Natl. Acad. Sci. U. S. A. 2008;105:18746–18751. doi: 10.1073/pnas.0806303105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schmidt O, Pfanner N, Meisinger C. Mitochondrial protein import: from proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 2010;11:655–667. doi: 10.1038/nrm2959. [DOI] [PubMed] [Google Scholar]
- 70.Semenza GL. HIF-1 mediates the Warburg effect in clear cell renal carcinoma. J. Bioenerg. Biomembr. 2007;39:231–234. doi: 10.1007/s10863-007-9081-2. [DOI] [PubMed] [Google Scholar]
- 71.Serviddio G, Bellanti F, Vendemiale G, Altomare E. Mitochondrial dysfunction in nonalcoholic steatohepatitis. Expert. Rev. Gastroenterol. Hepatol. 2011;5:233–244. doi: 10.1586/egh.11.11. [DOI] [PubMed] [Google Scholar]
- 72.Sheldon KL, Maldonado EN, Lemasters JJ, Rostovtseva T, Bezrukov SM. Phosphorylation of Voltage-Dependent Anion Channel by Serine/Threonine Kinases Governs Its Interaction With Tubulin. 2011. [DOI] [PMC free article] [PubMed]
- 73.Shelmet JJ, Reichard GA, Skutches CL, Hoeldtke RD, Owen OE, Boden G. Ethanol causes acute inhibition of carbohydrate, fat, and protein oxidation and insulin resistance. J. Clin. Invest. 1988;81:1137–1145. doi: 10.1172/JCI113428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shoshan-Barmatz V, Gincel D. The voltage-dependent anion channel: characterization, modulation, and role in mitochondrial function in cell life and death. Cell Biochem. Biophys. 2003;39:279–292. doi: 10.1385/CBB:39:3:279. [DOI] [PubMed] [Google Scholar]
- 75.Shoshan-Barmatz V, Gincel D. The voltage-dependent anion channel: characterization, modulation, and role in mitochondrial function in cell life and death. Cell Biochem. Biophys. 2003;39:279–292. doi: 10.1385/CBB:39:3:279. [DOI] [PubMed] [Google Scholar]
- 76.Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 77.Tan W, Colombini M. VDAC closure increases calcium ion flux. Biochim. Biophys. Acta. 2007;1768:2510–2515. doi: 10.1016/j.bbamem.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thurman RG, McKenna WR, McCaffrey TB. Pathways responsible for the adaptive increase in ethanol utilization following chronic treatment with ethanol: inhibitor studies with the hemoglobin-free perfused rat liver. Molec. Pharmacol. 1976;12:156–166. [PubMed] [Google Scholar]
- 79.Thurman RG, Yuki T, Bleyman MA, Wendell G. The adaptive increase in ethanol metabolism due to pretreatment with ethanol: a rapid phenomenon. Drug Alcohol Depend. 1979;4:119–129. doi: 10.1016/0376-8716(79)90052-8. [DOI] [PubMed] [Google Scholar]
- 80.Tikunov A, Johnson CB, Pediaditakis P, Markevich N, Macdonald JM, Lemasters JJ, Holmuhamedov E. Closure of VDAC causes oxidative stress and accelerates the Ca(2+)-induced mitochondrial permeability transition in rat liver mitochondria. Arch. Biochem. Biophys. 2010;495:174–181. doi: 10.1016/j.abb.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Timohhina N, Guzun R, Tepp K, Monge C, Varikmaa M, Vija H, Sikk P, Kaambre T, Sackett D, Saks V. Direct measurement of energy fluxes from mitochondria into cytoplasm in permeabilized cardiac cells in situ: some evidence for Mitochondrial Interactosome. J. Bioenerg. Biomembr. 2009;41:259–275. doi: 10.1007/s10863-009-9224-8. [DOI] [PubMed] [Google Scholar]
- 82.Ujwal R, Cascio D, Colletier JP, Faham S, Zhang J, Toro L, Ping P, Abramson J. The crystal structure of mouse VDAC1 at 2.3 A resolution reveals mechanistic insights into metabolite gating. Proc. Natl. Acad. Sci. U. S. A. 2008;105:17742–17747. doi: 10.1073/pnas.0809634105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vander Heiden MG, Chandel NS, Li XX, Schumacker PT, Colombini M, Thompson CB. Outer mitochondrial membrane permeability can regulate coupled respiration and cell survival. Proc. Natl. Acad. Sci. U. S. A. 2000;97:4666–4671. doi: 10.1073/pnas.090082297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vander Heiden MG, Chandel NS, Li XX, Schumacker PT, Colombini M, Thompson CB. Outer mitochondrial membrane permeability can regulate coupled respiration and cell survival. Proc. Natl. Acad. Sci. U. S. A. 2000;97:4666–4671. doi: 10.1073/pnas.090082297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vander Heiden MG, Li XX, Gottleib E, Hill RB, Thompson CB, Colombini M. Bcl-xL promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. J. Biol. Chem. 2001;276:19414–19419. doi: 10.1074/jbc.M101590200. [DOI] [PubMed] [Google Scholar]
- 87.Vander Heiden MG, Li XX, Gottleib E, Hill RB, Thompson CB, Colombini M. Bcl-xL promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. J. Biol. Chem. 2001;276:19414–19419. doi: 10.1074/jbc.M101590200. [DOI] [PubMed] [Google Scholar]
- 88.Videla L, Israel Y. Factors that modify the metabolism of ethanol in rat liver and adaptive changes produced by its chronic administration. Biochem. J. 1970;118:275–281. doi: 10.1042/bj1180275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Warburg O. Ueber Den Stoffwechsel Der Tumoren. Constable; London: 1930. [Google Scholar]
- 90.Watford M. The urea cycle: a two-compartment system. Essays. Biochem. 1991;26:49–58. 49-58. [PubMed] [Google Scholar]
- 91.Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M, Landt CL, Harrison SA. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140:124–131. doi: 10.1053/j.gastro.2010.09.038. [DOI] [PubMed] [Google Scholar]
- 92.Yuki T, Thurman RG. Swift increase in alcohol metabolism: time course and involvement of glycolysis. Biochem. J. 1980;186:119–126. doi: 10.1042/bj1860119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhong Z, Ramshesh VK, Rehman H, Lemasters JJ. Acute ethanol causes liver mitochondrial depolarization in vivo independent of the mitochondrial permeability transition (MPT) and potassium channel opening. Hepatology. 2007;46(Suppl. 1):253A–254A. [Google Scholar]
- 94.Zhong Z, Ramshesh VK, Rehman H, Lemasters JJ. Acute ethanol-induced liver mitochondrial depolarization in vivo is dependent on alcohol dehydrogenase (ADH) and cytochrome-P450 2E1. Toxicol. Sci. 2009;108:237. [Google Scholar]