

Abstract
Screening for inherited thrombophilia (IT) is controversial; persons at high risk for venous thromboembolism (VTE) who benefit from screening need to be identified. We tested 533 first- and second-degree relatives of 206 pediatric VTE patients for IT (antithrombin, protein C, protein S, factor V G1691A, factor II G20210A) and determined the incidence of symptomatic VTE relative to their IT status. The risk for VTE was significantly increased among family members with, versus without, IT (hazard ratio = 7.6; 95% confidence interval [CI], 4.0-14.5; P < .001) and highest among carriers of antithrombin, protein C, or protein S deficiency (hazard ratio = 25.7; 95% CI, 12.2-54.2; P < .001). Annual incidences of VTE were 2.82% (95% CI, 1.63%-4.80%) among family members found to be carriers of antithrombin, protein C, or protein S deficiency, 0.42% (0.12%-0.53%) for factor II G202010A, 0.25% (0.12%-0.53%) for factor V G1691A, and 0.10% (0.06%-0.17%) in relatives with no IT. Given the high absolute risk of VTE in relatives with protein C, protein S, and antithrombin deficiency, we suggest screening for these forms of hereditary thrombophilia in children with VTE and their relatives. Interventional studies are required to assess whether thromboembolism can be prevented in this high-risk population.
Introduction
Venous thromboembolism (VTE) is a multifactorial disease, to which both genetic and acquired risk factors contribute.1 Established genetic risk factors include protein C (PC), protein S (PS), and antithrombin (AT) deficiency as well as the factor V G1691A (FV) and prothrombin G20210A (FII) polymorphisms.2 Within the last 15 years, there has been a substantial increase in our understanding of both the pathogenetic mechanisms and the contribution of established and putative thrombophilic defects to the development of VTE.3 As a consequence, diagnostic evaluation for thrombophilia has become standard practice in many centers treating patients with VTE.
Although it is well established that inherited thrombophilia (IT) increases the risk of a first VTE event,4 mass screening for IT in healthy persons is not warranted, given an overall low incidence of VTE. Selected screening for IT among persons at heightened risk of VTE appears more reasonable. In addition, although it is known that a family history of VTE, particularly of early-onset during childhood, increases the risk for VTE, the extent to which this relationship is explained by IT has not been well established. It remains unclear whether asymptomatic relatives of patients with thrombophilia benefit from knowing their thrombophilic state.5 In one small pediatric series, Tormene et al observed no appreciable difference in risk of VTE among thrombophilic children of adults with a first VTE who did, versus did not, have thrombophilia.6 Whereas Calhoon et al7 showed a high risk of multitrait thrombophilia in children with a first- or second-degree family history of early VTE, the risk of VTE in such children was low during a median 22 months of prospective follow-up. However, focusing on adult relatives, Lijfering et al showed that VTE risk in family members differs by IT status.8
The aim of the present study, therefore, was to determine the influence of IT on relative and absolute risks of first symptomatic VTE in family members of children with a first episode of VTE.
Methods
Ethics
The present multicenter cohort study was performed in accordance with the ethical standards laid down in the updated version of the 1964 Declaration of Helsinki and was approved by the medical ethics committee of the University of Münster (Münster, Germany). Written informed consent was provided in all cases before study participation in accordance with the Declaration of Helsinki.
Study population and study design
From July 1996 to April 2010, 206 consecutively admitted term neonates and children younger than 18 years with newly diagnosed VTE and their 667 first- and second-degree relatives were enrolled in the study.
Inclusion criteria
Inclusion criteria for relatives of pediatric VTE cases were the following: (1) first- or second-degree relationship to the pediatric index case; (2) age younger than 18 years at the time of VTE diagnosis in the pediatric index case; and (3) objective confirmation of VTE in the pediatric index case by standard radiologic imaging methods, including compression sonography, venography, computed tomography (CT) venography, or magnetic resonance (MR) venography for venous thrombosis, and spiral CT angiography or lung perfusion scintigraphy for pulmonary embolism. Exclusion criteria consisted of: (1) lack of consent to participate, (2) nonpaternity as determined by personal interviews and DNA analysis in all cases, and (3) no access to blood samples for analysis on IT.
Clinical procedures
Diagnosis of VTE in pediatric index cases was confirmed by an experienced pediatric hematologist specialized in thromboembolic disease. Family members underwent baseline evaluation at enrollment by history and physical examination to evaluate evidence of previous or existing VTE and were seen subsequently on at least an annual basis with repeat history and physical examination. All reports of interim VTE in family members were confirmed by review of the medical records, including radiologic imaging studies. Clinical data collection included patient demographics and disease characteristics, clinical risk factors for VTE, clinical laboratory testing results, antithrombotic therapy regimens, and outcomes (including future VTE events).
Blood sample collection
Blood samples were collected from patients and relatives at the study center in Münster in the morning after a 12-hour fasting period (infants 4-6 hours); samples were drawn by peripheral venipuncture into plastic tubes containing one-tenth by volume of 3.8% trisodium citrate (Sarstedt) and were immediately placed on melting ice. The blood samples from patients were collected 6 to 12 months after the acute thrombotic event, and at least 6 weeks after discontinuation of anticoagulation therapy. Platelet-poor plasma was prepared by double centrifugation at 3000g at 4°C for 20 minutes, aliquoted in polystyrene long-term freezer storage tubes, stored at −70°C, and thawed immediately before assay. DNA extraction was performed by a spin column procedure (QIAGEN) as previously described.
Laboratory testing
Standard laboratory techniques were used to investigate the FV G1691A and FII G20210A polymorphisms, AT activity (intra-assay coefficient of variation, 3.1%; run-to-run coefficient of variation, 6.5%), PC activity (intra-assay coefficient of variation, 1.6%; run-to-run coefficient of variation, 5.0%), and free PS antigen (intra-assay coefficient of variation, 2.2%; run-to-run coefficient of variation, 4.2%), as per the manufacturer's instructions. For all plasma-based assays, a clotting abnormality was regarded as a defect only if the level was outside 2 SDs of the mean for age-dependent normative values. Confirming criteria for hereditary nature of AT, PC, and PS deficiency consisted of reproducibility of the abnormality in a second plasma sample (3-6 months after initial testing), and either identification of a causative gene mutation or detection of the defect in at least 1 first-degree relative. PC and PS values were performed in the absence of vitamin K antagonist administration.
Statistical analysis
We assessed the influence of genetic risk factors on VTE. As genetic factors do not change over time, we defined the exposure time to IT as the period from birth until the study end point or the age at which the event occurred. We calculated the absolute risk of VTE in relatives as incidence rates per 100 patient-years (%). If concomitant thrombophilic defects occurred in one relative, this person was not counted for the single thrombophilic defects but assigned to a group of combined defects. We calculated 95% CIs of the incidence based on a Poisson distribution. Event-free survival for “first thromboembolic event” was analyzed by survival analysis. We used a Cox regression model adjusting for age, sex, and body mass index (BMI) to calculate relative risks, as hazard ratios (HRs), with 95% CI for HR calculated using the Wald method. We built 2 different models: one using IT as an independent variable and the other using type of IT as independent variables. Because of small numbers, we combined PC, PS, and AT deficiency as a single type of IT. In the first approach, exposure time was defined as the period from birth until the first thromboembolic event or the end of the study. To assess the effect of pubertal status on incidence rates, we used age as a proxy for pubertal status and defined for that analysis the exposure time from the age of 15 years on. Relatives younger than 15 years or with a thromboembolic event before the age of 15 years subsequently were excluded from this analysis. We calculated BMI SD scores for patients younger than 18 years according to sex-specific reference values for German children and adolescents.9,10 This score indicates how many SDs the BMI is above the age- and sex-specific mean of German children and adolescents.
Continuous variables were expressed as median (range), and categorical data as counts and percentage. Differences between groups were assessed using a Mann-Whitney U test for continuous data and χ2 test for categorical data. A 2-tailed P value < .05 was used to indicate statistical significance. All data were analyzed using SPSS statistical package (PASW Statistics Version 18 for Windows).
Results
Clinical characteristics
The final population for analysis consisted of 533 (79.9%) first- and second-degree family members (Figure 1) of 206 index patients 18 years of age or younger. In our cohort, the median age of first VTE was 4 years (range, 0-18 years) in index patients and 30 years (range, 0-58 years) in relatives. Relatives with IT did not differ significantly from those without with respect to sex, age, and BMI. The median BMI (adults) was 26 kg/m2, and the median BMI SD score (≤ 18 years) was 0 in both groups. A total of 18.3% of all relatives reported to smoke regularly, and 7.9% of women were on oral contraceptives at the time they were included in the study (Table 1).
Figure 1.
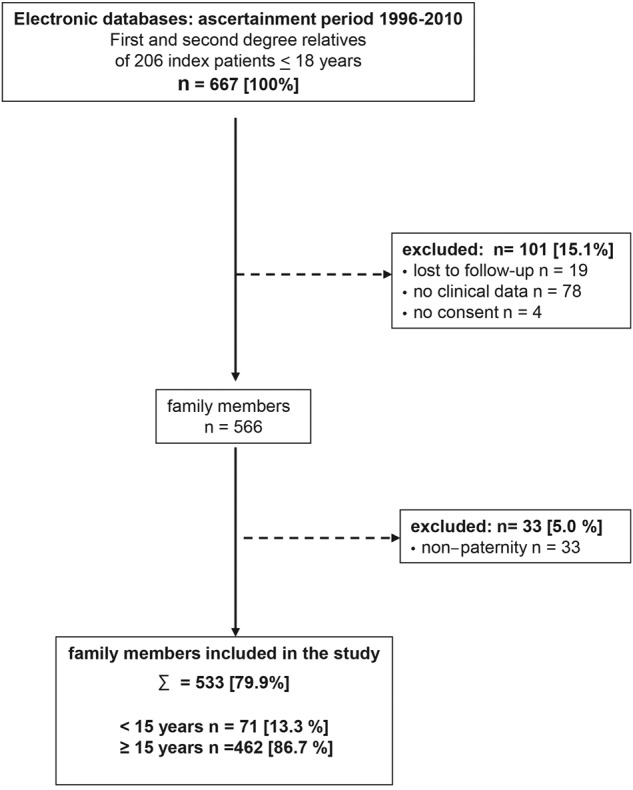
Flow chart of study population.
Table 1.
Characteristics of relatives with and without IT
| IT (n = 146) | No IT (n = 387) | Total (n = 533) | P | |
|---|---|---|---|---|
| Sex, % male | 69 (47.3) | 191 (49.5) | 260 (48.9) | .7 |
| BMI, kg/m2 | 26 (18-43) | 26 (17-40) | 26 (17-43) | .6 |
| BMI, SD score | 0 (−1.2-3.0) | 0 (−1.5-3.0) | 0 (−1.5-3.0) | .8 |
| HRT/oral contraceptives, % female | 6.3 | 8.5 | 7.9 | .3 |
| Smoking, % | 21.4 | 17.2 | 18.3 | .3 |
| Median exposure time, y (range) | 33 (0-60) | 38 (0-73) | 37 (0-73) | .1 |
| Exposure time, person-years | 4711 | 13 567 | 18 278 |
Categorical variables are presented as percentages. Continuous variables are presented as median (range). χ2 test for comparison of categorical data, Mann-Whitney test for comparison of continuous data.
HRT indicates hormone replacement therapy; and IT, inherited thrombophilia.
VTE occurred in 47 relatives; 6 of those 47 relatives were younger than 18 years at the event time. In both, index patients as well as in relatives with VTE, the majority of events were triggered by transient risk factors (70% in index patients, 64% in relatives) that did not qualify for a primary thromboprophylaxis according to standard medical care.11,12 In our cohort of relatives with VTE, transient risk factors were diverse and included immobilization/surgery (8 of 30), hormonal replacement therapy/use of oral contraceptive or pregnancy (5 of 30), and obesity (10 of 30).
IT carrier rate in relatives
Table 2 gives the prevalence of IT in relatives dependent on VTE outcome. A total of 146 of 533 relatives (27%) had at least 1 IT. Among these 146, 93% had single defects, of which heterozygous FV G1691A was the most prevalent (57%). The frequency of each IT (considering homozygous and heterozygous together) was higher than expected (relative to the population norm). Homozygous carrier status was low, with 3 relatives each for FV or FII, and none for PC, PS, or AT.
Table 2.
Distribution of IT in relatives with and without VTE
| IT | No VTE (n = 486), no. (%) | VTE (n = 47), no. (%) | VTE per IT status, no. (%) |
|---|---|---|---|
| No IT | 373 (76.7) | 14 (29.8) | 14/387 (3.6) |
| Any IT | 113 (23.3) | 33 (70.2) | 33/146 (22.6) |
| Type of IT | |||
| FV G1691A | 76 | 12 | 12/88 (13.6) |
| Single | 74 | 7 | 7/81 (8.6) |
| + FII G20210A | 2 | 0 | 0 |
| + AT/PC/PS deficiency | 0 | 5 | 5/5 (100.0) |
| FII G202010A | 35 | 5 | 5/40 (12.5) |
| Single | 32 | 5 | 5/37 (13.5) |
| + FV G1691A | 2 | 0 | 0 |
| + AT/PC/PS deficiency | 1 | 0 | 0 |
| AT/PC/PS deficiency | 5 | 18 | 18/23 (78.2) |
| Single | 4 | 13 | 13/17 (76.5) |
| + FV G1691A | 0 | 5 | 5/5 (100.0) |
| + FII G20210A | 1 | 0 | 0 |
IT indicates inherited thrombophilia; and VTE, venous thromboembolism.
Incidence of first VTE
In our cohort, the life-long cumulative incidence of VTE in the group of relatives of index patients with PC, PS, or AT deficiency was very high (ie, 76.5%). In carriers of FV G1691A or FII G20210A, cumulative incidence of VTE was substantially lower with 8.6% and 13.5%, respectively. In relatives without IT, life-long cumulative incidence of VTE was only 3.6% (Table 2). There were no significant differences of the median age, sex, or BMI between these groups (data not shown).
Annual incidences of VTE dependent on IT are shown in Table 3. Annual incidence in patients with PS, PC, or AT deficiency was significantly increased compared with those with no IT (2.82%; 95% confidence interval [CI], 1.63%-4.80%; vs 0.1%; 95% CI, 0.06%-0.17%; P < .001). Annual incidences in carriers of FII G20210A and FV G1691A were not significantly increased compared with those without IT.
Table 3.
Absolute and relative risks for VTE in relatives dependent on IT
| Type of IT | Exposure time, y | Event, n | Annual incidence, % (95% CI) | Hazard ratio,* % (95% CI) | P |
|---|---|---|---|---|---|
| AT/PC/PS | 463 | 13 | 2.82 (1.63-4.80) | 25.7 (12.2-54.2) | < .001 |
| FV G1691A | 2783 | 7 | 0.25 (0.12-0.53) | 1.7 (0.7-4.4) | .24 |
| FII G20210A | 1192 | 5 | 0.42 (0.17-1.01) | 2.6 (0.9-7.6) | .09 |
| Combined defects | 258 | 6 | 2.33 (1.04-5.19) | 19.6 (7.7-49.6) | < .01 |
| No IT | 13 567 | 14 | 0.10 (0.06-0.17) | 1 | 1 |
| Total | 18 278 | 47 | 0.26 (0.19-0.34) |
IT indicates inherited thrombophilia; and VTE, venous thromboembolism.
Adjusted for age and sex.
Impact of IT on risk of first VTE
To evaluate the influence of IT on first VTE in relatives, we built a Cox regression model comparing presence versus absence of IT in these relatives. In our cohort, the relative risk of a first VTE was significantly higher in relatives with IT than in those without (HR = 7.6; 95% CI, 4.0-14.5). To analyze whether this effect was true for all types of IT, we analyzed separately the effect on VTE of (1) FV; (2) FII; and (3) PC, PS, or AT deficiency; and (4) combined IT. In this model, PC, PS, and AT deficiency had a strong influence on VTE risk (HR = 25.7; 95% CI, 12.2-54.2; P < .001). Carriers of FV G1691A had only a slightly higher risk of developing VTE than those without any IT (Table 3). Restricting the analysis to relatives 15 years of age or older did not substantially alter the results. All models included sex, age, and BMI as covariates to correct for potential confounding factors (Figure 2). In our cohort of relatives, IT status was not associated with the occurrence of spontaneous versus triggered VTE (spontaneous VTE in relatives with no IT 40% vs relatives with IT 34%, P = .75).
Figure 2.

VTE event-free survival dependent on IT. Freedom from VTE over time is shown for relatives of pediatric index patients stratified for different types of IT.
Impact of age
Age is one of the strongest risk factors for the development of thrombosis, and prepubertal events are very rare. In a second step, we therefore restricted the analysis to postpubertal children. We used 15 years as a proxy for pubertal status and excluded observation years in relatives 14 years of age or younger. Annual incidences increased to 4.89% (95% CI, 2.74-7.56, P < .001) for PC, PS, and AT deficiency (Figure 3).
Figure 3.
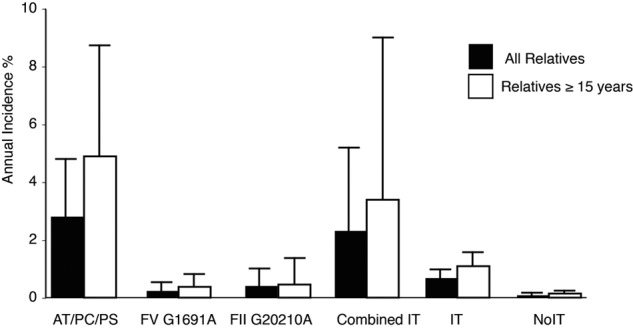
Annual incidence of VTE in all relatives and restricted to relatives 15 years of age or older. Bars represent the annual incidence of VTE stratified for different forms of IT, any IT, or no IT. Error bars represent respective 95% CIs of the estimates.
Discussion
VTE is a serious disease that can lead to long-term morbidity or death.13 Therefore, major goals include (1) identification of populations/subgroups at risk and (2) prevention of VTE in these persons.14
In family members of pediatric patients who were carriers of IT compared with those without IT, the risk of VTE was significantly increased. In particular, we demonstrate a markedly high risk to develop VTE in relatives with PC, PS, or AT deficiency. The risk is approximately 20-fold higher than those in family members without any inherited trait. Absolute risk estimates of a relative's PC, PS, or AT deficiency from our cohort exceeded those of relatives of adult patients with the same thrombophilic defect and comparable age group in a large retrospective study and results from other prospective studies.15–17 As expected, restricting the analysis to patients older than 14 years resulted in increased incidence rates. As age is an important risk factor for VTE,18 we suggest that the development of VTE at a young age indicates an especially high intrinsic thrombosis risk. In a recent study in adult patients, the risk to develop VTE in relatives was associated with the age of first VTE irrespective of IT.19 The low incidence rates of VTE observed in children of adult index patients again underscore the influence of age in index patients as well as in relatives when defining high-risk groups.6
In our cohort, the familial risk of VTE was not increased among carriers of FII of FV mutations. Analyzing all patients with FII or FV mutations as well showed an increased incidence rate of VTE in relatives compared with persons with none of the tested IT. The risks appeared to be the result of concomitance of other thrombophilia defects, especially PC, PS, or AT deficiency. After exclusion of combined defects, the effect diminished and was no longer statistically significant. These results are in accordance with the findings from a large retrospective study in adult patients and their first-degree relatives,8 which showed only mildly increased relative risks for the so-called “low-risk thrombophilic” defects FII, FV, and elevated FVIII after exclusion of PC, PS, or AT deficiencies. Furthermore, this pattern of results points to the fact that studies identifying FV or FII or other low-risk mutations as risk factor for VTE need to be treated with caution; cumulative effects of multiple thrombophilia defects have to be taken into account.
The FV G1691A mutation is the most frequent genetic test performed in the United States.17 For FII or FV mutations, absolute risk estimates were only slightly elevated compared with patients with none of the tested IT in our study, and differences were not significant in the present cohort. Our results are in accordance with other studies that showed only slightly elevated risks associated with single FII or FV polymorphisms. As absolute risks are clearly outweighed by the risk of serious bleeding in long-term anticoagulation and are mainly influenced by concomitant thrombophilic defects, we argue against screening for FII G20210A or FV G1691A mutation Leiden exclusively. Further studies, especially on the interaction of FII and FV with other risk factors, are needed to define patients in whom screening for those factors may be most beneficial.
It is generally accepted that unselected population screening for thrombophilia is inappropriate; in contrast, testing for heritable thrombophilia in patients with VTE may allow case finding of affected asymptomatic family members.20 The benefit of screening depends on (1) the prevalence of the thrombophilic defect, (2) the incidence of VTE in positively screened persons, and (3) the safety and effectiveness of preventive measures. PC, PS, or AT deficiency are rare in the general population and therefore account only for a small percentage of the overall thrombosis risk, even though they are associated with a high thrombotic risk. Our cohort of index patients was selected for VTE before the age of 18 years, thus representing a high-risk group. The annual incidence of VTE of 4.9% identifies relatives of children with VTE and PC, PS, or AT deficiency as a population with high VTE risk. Putative preventive measures are either long-term anticoagulation with vitamin K antagonists or intermittent anticoagulation during risk periods. The risk of major bleeding for patients treated with vitamin K antagonist has been reported as 2% to 3% in an unselected population and 0.5% in PC, PS, or AT-deficient patients with VTE and therewith is less than the annual risk of developing VTE in that subgroup.19,20 However, as 64% of VTE cases in our cohort occurred in association with transient clinical triggers, a risk-adapted intermittent anticoagulation regimen (eg, prophylactic administration of low molecular heparin during high-risk periods and the avoidance of triggers, such as hormonal contraception) may be preferable.
Several aspects warrant consideration. First, our risk estimates exceed those of other studies that did not select based on the presence of a pediatric index case. Second, we only examined for currently established risk factors; given that VTE is a multifactorial disease, additional risk factors probably contribute to differences between studies and may change risk estimates. However, as annual incidence of VTE in relatives with none of the tested IT was low at 0.1%, the likelihood of a strong contribution from nonidentified risk factors is low. In addition, because nonidentified risk factors are likely to be distributed randomly across groups, those risk factors would not materially alter relative risk estimates. Third, selection bias is a potential concern in any observational study. Severity of disease or IT carrier status may have influenced referral to our hospital and may result in an overestimation of absolute risks of recurrence in family members. However, because VTE is a rare and serious disease in childhood, the majority of patients are transferred to academic hospitals; hence, our study population is probably representative of the general pediatric VTE population. Furthermore, we tested consecutive patients and their families to minimize selection bias. Fourth, as the families included in the study were mainly of white ethnicity and were ascertained exclusively in Germany, the results presented here cannot be assumed to be directly applicable to other ethnicities or populations. Lastly, the strength of our conclusions is limited by the size of our study population. This restriction applies in particular to the analysis of rare variants, the influence of combined or homozygous IT. Numbers were too small to assess the impact of PC, PS, and AT deficiency separately. For the same reason, we could not investigate the impact of these severe or combined types of thrombophilia on the risk to develop spontaneous versus triggered VTE. Notwithstanding these limitations, to the best of our knowledge, the study presented here is the largest study on relatives of pediatric index patients and the first to systematically investigating absolute risks of relatives of pediatric patients with VTE.
In conclusion, in this multicenter cohort study of the risk of VTE in relatives of pediatric index cases with VTE, risk of VTE was markedly increased in relatives with, versus without, PC, PS, or AT deficiency. By contrast, risk of VTE was not significantly increased among those with common mild thrombophilic defects. Given the high annual incidence of VTE in those with PC, PS, and AT deficiency, we suggest that screening for IT in relatives of pediatric VTE index cases with PC, PS, and AT deficiency be performed, and that (based on the findings) appropriate prophylactic measures are taken. Future studies need to investigate the safety and efficacy of thromboprophylactic strategies in this setting.
Acknowledgments
This work was supported by Förderverein Schlaganfall und Thrombosen im Kindesalter eV and Interdisziplinäres Zentrum für Klinische Forschung (CRA01-09), University of Münster. N.A.G. was supported in part by the National Institutes of Health, National Heart, Lung, and Blood Institute (Career Development Award 5K23HL084055).
Footnotes
There is an Inside Blood commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.H. and U.N.-G. designed the study, analyzed the data, and wrote the paper; and R.J., C.H., D.M., N.A.G., M. Stoll, M. Stach, R.M., and A.K. had full access to the data, took part in the design, execution, data analysis, and discussion, and wrote the report.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Susanne Holzhauer, Department of Pediatric Hematology/Oncology, Charité, Augustenburger Platz 1, 13353, Berlin, Germany; e-mail: susanne.holzhauer@charite.de.
References
- 1.Rosendaal FR. Venous thrombosis: a multicausal disease. Lancet. 1999;353(9159):1167–1173. doi: 10.1016/s0140-6736(98)10266-0. [DOI] [PubMed] [Google Scholar]
- 2.Middeldorp S, van Hylckama Vlieg A. Does thrombophilia testing help in the clinical management of patients? Br J Haematol. 2008;143(3):321–335. doi: 10.1111/j.1365-2141.2008.07339.x. [DOI] [PubMed] [Google Scholar]
- 3.Wu O, Greer IA. Is screening for thrombophilia cost-effective? Curr Opin Hematol. 2007;14(5):500–503. doi: 10.1097/MOH.0b013e32825f5318. [DOI] [PubMed] [Google Scholar]
- 4.Young G, Albisetti M, Bonduel M, et al. Impact of inherited thrombophilia on venous thromboembolism in children: a systematic review and meta-analysis of observational studies. Circulation. 2008;118(13):1373–1382. doi: 10.1161/CIRCULATIONAHA.108.789008. [DOI] [PubMed] [Google Scholar]
- 5.Keeling D. Thrombophilia screening or screaming. J Thromb Haemost. 2010;8(6):1191–1192. doi: 10.1111/j.1538-7836.2010.03871.x. [DOI] [PubMed] [Google Scholar]
- 6.Tormene D, Pagnan A, Prandoni P, Simioni P. Screening for thrombophilia in children: a puzzling decision with unclear implications. J Thromb Haemost. 2004;2(7):1193–1194. doi: 10.1111/j.1538-7836.2004.00755.x. [DOI] [PubMed] [Google Scholar]
- 7.Calhoon MJ, Ross CN, Pounder E, Cassidy D, Manco-Johnson MJ, Goldenberg NA. High prevalence of thrombophilic traits in children with family history of thromboembolism. J Pediatr. 2010;157(3):485–489. doi: 10.1016/j.jpeds.2010.03.031. [DOI] [PubMed] [Google Scholar]
- 8.Lijfering WM, Brouwer JL, Veeger NJ, et al. Selective testing for thrombophilia in patients with first venous thrombosis: results from a retrospective family cohort study on absolute thrombotic risk for currently known thrombophilic defects in 2479 relatives. Blood. 2009;113(21):5314–5322. doi: 10.1182/blood-2008-10-184879. [DOI] [PubMed] [Google Scholar]
- 9.Kromeyer-Hausschild K, Wabitsch M, Kunze D, et al. [Percentiles for body mass index for children and adolescents considering various samples from the German Population]. Monatsschr Kinderheilkd. 2001;149:807–818. [Google Scholar]
- 10.Cole TJ, Green PJ. Smoothing reference centile curves: the LMS method and penalized likelihood. Stat Med. 1992;11(10):1305–1319. doi: 10.1002/sim.4780111005. [DOI] [PubMed] [Google Scholar]
- 11.Michelson AD, Bovill E, Monagle P, Andrew M. Antithrombotic therapy in children. Chest. 1998;114(5 Suppl):748S–69S. doi: 10.1378/chest.114.5_supplement.748s. [DOI] [PubMed] [Google Scholar]
- 12.Haas S. Prevention of venous thromboembolism: recommendations based on the International Consensus and the American College of Chest Physicians Sixth Consensus Conference on Antithrombotic Therapy. Clin Appl Thromb Hemost. 2001;7(3):171–177. doi: 10.1177/107602960100700301. [DOI] [PubMed] [Google Scholar]
- 13.Clagett GP, Anderson FA, Jr, Heit J, Levine MN, Wheeler HB. Prevention of venous thromboembolism. Chest. 1995;108(4 Suppl):312S–334S. doi: 10.1378/chest.108.4_supplement.312s. [DOI] [PubMed] [Google Scholar]
- 14.Coppola A, Tufano A, Cerbone AM, Di Minno G. Inherited thrombophilia: implications for prevention and treatment of venous thromboembolism. Semin Thromb Hemost. 2009;35(7):683–694. doi: 10.1055/s-0029-1242722. [DOI] [PubMed] [Google Scholar]
- 15.Sanson BJ, Simioni P, Tormene D, et al. The incidence of venous thromboembolism in asymptomatic carriers of a deficiency of antithrombin, protein C, or protein S: a prospective cohort study. Blood. 1999;94(11):3702–3706. [PubMed] [Google Scholar]
- 16.Segal JB, Brotman DJ, Necochea AJ, et al. Predictive value of factor V Leiden and prothrombin G20210A in adults with venous thromboembolism and in family members of those with a mutation: a systematic review. JAMA. 2009;301(23):2472–2485. doi: 10.1001/jama.2009.853. [DOI] [PubMed] [Google Scholar]
- 17.Mahmoodi BK, Brouwer JL, Ten Kate MK, et al. A prospective cohort study on the absolute risks of venous thromboembolism and predictive value of screening asymptomatic relatives of patients with hereditary deficiencies of protein S, protein C or antithrombin. J Thromb Haemost. 2010;8(6):1193–1200. doi: 10.1111/j.1538-7836.2010.03840.x. [DOI] [PubMed] [Google Scholar]
- 18.Couturaud F, Kearon C. Predictors of thrombosis in relatives of patients with venous thromboembolism. Curr Opin Pulm Med. 2010;16(5):453–458. doi: 10.1097/MCP.0b013e32833bde6b. [DOI] [PubMed] [Google Scholar]
- 19.Couturaud F, Leroyer C, Julian JA, et al. Factors that predict risk of thrombosis in relatives of patients with unprovoked venous thromboembolism. Chest. 2009;136(6):1537–1545. doi: 10.1378/chest.09-0757. [DOI] [PubMed] [Google Scholar]
- 20.Cohn DM, Roshani S, Middeldorp S. Thrombophilia and venous thromboembolism: implications for testing. Semin Thromb Hemost. 2007;33(6):573–581. doi: 10.1055/s-2007-985753. [DOI] [PubMed] [Google Scholar]