

Abstract
Advances in high-throughput mass spectrometry are making proteomics an increasingly important tool in genome annotation projects. Peptides detected in mass spectrometry experiments can be used to validate gene models and verify the translation of putative coding sequences (CDSs). Here, we have identified peptides that cover 35% of the genes annotated by the GENCODE consortium for the human genome as part of a comprehensive analysis of experimental spectra from two large publicly available mass spectrometry databases. We detected the translation to protein of “novel” and “putative” protein-coding transcripts as well as transcripts annotated as pseudogenes and nonsense-mediated decay targets.
We provide a detailed overview of the population of alternatively spliced protein isoforms that are detectable by peptide identification methods. We found that 150 genes expressed multiple alternative protein isoforms. This constitutes the largest set of reliably confirmed alternatively spliced proteins yet discovered. Three groups of genes were highly overrepresented. We detected alternative isoforms for 10 of the 25 possible heterogeneous nuclear ribonucleoproteins, proteins with a key role in the splicing process. Alternative isoforms generated from interchangeable homologous exons and from short indels were also significantly enriched, both in human experiments and in parallel analyses of mouse and Drosophila proteomics experiments. Our results show that a surprisingly high proportion (almost 25%) of the detected alternative isoforms are only subtly different from their constitutive counterparts.
Many of the alternative splicing events that give rise to these alternative isoforms are conserved in mouse. It was striking that very few of these conserved splicing events broke Pfam functional domains or would damage globular protein structures. This evidence of a strong bias toward subtle differences in CDS and likely conserved cellular function and structure is remarkable and strongly suggests that the translation of alternative transcripts may be subject to selective constraints.
Keywords: alternative splicing, shotgun proteomics, genome annotation, heterogeneous nuclear ribonucleoproteins, NAGNAG splicing, mutually exclusive exons
Introduction
Manual genome annotation projects can augment and refine gene models predicted by automatic annotation projects such as Ensembl (Hubbard et al. 2002). Manual determination of protein-coding genes and their structure is complicated and time consuming (Guigó et al. 2006) and relies on many sources of evidence. Data from mass spectrometry experiments have the potential to be a useful source of information since reliable proteomics data can confirm the coding potential of transcripts even where there is little supporting evidence. Tanner showed how proteomics can play a role in genome sequencing projects by validating the translation of 39,000 human exons (Tanner et al. 2007), and the Havana Group has also demonstrated the value of using peptide evidence to validate genome annotations by uncovering ten novel protein-coding genes and annotating several supposed pseudogenes as protein coding in mouse (Brosch et al. 2011).
Alternative splicing of mRNA can generate a wide range of mature RNA transcripts, and recent studies have estimated that practically all multiexon human genes (Wang et al. 2008; Pan 2008) are able to produce at least two differently spliced mRNA transcripts by alternative splicing of pre-mRNA. The rearrangement of exons from the primary transcripts to generate a range of splice variants will produce of protein isoforms with altered structure and biological function (Xing et al. 2006; Talavera et al. 2007; Tress et al. 2007). Alternative splicing has the potential to expand the cellular protein repertoire (Smith and Valcárcel 2000), as long as these alternative transcripts are translated into stable proteins.
The expression of many differently spliced mRNA transcripts is strongly supported by both cDNA and expressed sequence tag (EST) sequence evidence (Harrow et al. 2006) and by microarray data (Johnson et al. 2003). However, although there is considerable supporting evidence for the expression of alternative mature RNA transcripts, even when these transcripts are likely to encode protein sequences with unusual features (Yura et al. 2006; Tress et al. 2007; Melamud and Moult 2009), it has proved more difficult to demonstrate the existence of alternative protein isoforms at a global level. Much of the evidence for the translation of alternative splice variants as stable proteins has come from individual experiments (Guo et al. 2003; Bacart et al. 2010; Wang et al. 2011).
It should be possible to use proteomics technologies to confirm the translation to protein of alternatively spliced variants, but tandem mass spectroscopy (MS/MS) experiments are only able to identify a fraction of the peptide ions present in protease digests, and it remains a technical challenge to detect proteins that are expressed at low levels (Abu-Farha et al. 2009; Gstaiger et al. 2009). Given that it is already difficult to detect peptides for constitutive protein isoforms, it will be considerably more challenging to detect peptides that can uniquely identify alternative isoforms, especially if these are expressed at lower levels or in fewer tissues than their constitutive counterparts.
Recently, a handful of research groups have made use of evidence from proteomics experiments to demonstrate the expression of alternative isoforms in model organisms. Because of the problems inherent in searching for alternative isoforms, all these studies were either carried out using large-scale proteomics experiments or the pooled spectra from a large number of smaller proteomics experiments. Tanner (Tanner et al. 2007) published the first study to demonstrate the presence multiple alternative isoforms from proteomics experiments. The authors carried out their own experiments by interrogating a range of human tissues and identified peptides that mapped exclusively to 15 pairs of alternative isoforms from the search database.
Analysis of high-quality peptide catalogues from two extensive studies of the Drosophila melanogaster proteome demonstrated the presence of alternative isoforms for 130 genes (Tress, Bodenmiller, et al. 2008). This was the highest number of alternative isoforms detected prior to this study and demonstrated the potential usefulness of phopshoproteomics in search for alternative isoforms. A number of other studies have also detected low numbers of alternative isoforms in other species, such as Arabidopsis (Castellana et al. 2008; Severing et al. 2011), Aspergillus flavus (Chang et al. 2010), mouse (Brosch et al. 2011) and schistosome worms (DeMarco et al. 2010), and also via nonstandard peptide–proteomics mapping (Power et al. 2009; Ning and Nesvizhskii 2010).
In contrast to all the above experiments, the group of Ommen (Menon et al. 2009; Menon and Omenn 2010) found higher numbers of alternative isoforms from surprisingly few experiments. Unfortunately, the definition of alternative isoforms used in this research was not as rigorous as all the other experiments. The authors did not require evidence of the expression of two different isoforms; most of the “alternative” isoforms detected in these studies were assumed to be alternative based solely on their presence the isoform in the database ECgene (Lee et al. 2007). In ECgene, like most databases of this kind, the longest isoform is chosen as the constitutive isoform, and all others are assumed to be alternative. We have shown that the choice of the longest isoform as the constitutive isoform is not always the wisest (Tress, Wesselink, et al. 2008) and that a shorter isoform is likely to be the main variant in as many as 25% of human genes.
The number of alternative isoforms detected in these studies is surprisingly low, and this is certainly in part because of the low coverage of peptides from proteomics experiments and the difficulty of obtaining peptides that map to regions that differ between two alternative isoforms. Where the differences between two isoforms result in an indel, the most common type of alternative splicing event (Mudge et al. 2011), proteomics experiments must detect peptides that map to the exon junctions in both constitutive and alternative transcripts. The detection of alternative isoforms may also be complicated by the possibility that many of these isoforms are expressed infrequently, in very few tissues or have very short half-lives.
Here, we have used the data from peptide MS/MS experiments that is stored in two huge proteomics data repositories, the Global Proteome Machine (GPM) (Craig et al. 2004) and PeptideAtlas (Aebersold et al. 2006), to annotate the human genome. We mapped peptides to over 35% of the annotated protein-coding genes and detected the expression of novel transcripts, nonsense-mediated decay (NMD) targets and pseudogenes. We used the spectra to identify multiple splice isoforms for a record number of genes (150) at a 1% false discovery rate (FDR).
Many of the alternative isoforms that we detected were only subtly different from their constitutive counterparts. The most surprising result was that three groups of alternative isoforms were significantly overrepresented in this set, alternative isoforms that had homologous regions that were the product of mutually exclusively spliced exons (MEEs), alternative isoforms that differed by the insertion or deletion of a single amino acid residue (or a small number of residues), and alternative isoforms from heterogeneous nuclear ribonucleoproteins (hnRNPs). Not only were these three groups of genes significantly enriched in the set of genes that expressed alternative isoforms but also there was also more peptide evidence for their alternative isoforms. We confirmed that these findings were not just restricted to human proteomics experiments by carrying out the same analyses for mouse and Drosophila.
Materials and Methods
Spectra Data Sets and the Peptide Search Database
Our analysis was based on the peptide mass spectra from human proteomics experiments deposited in two publicly available proteomics databases. The GPM Organization set consisted of 5,809 mzXML (Craig et al. 2004) format spectra files and the PeptideAtlas set was 52,019 mzXML format spectra files. The spectra were sampled from a wide range of difference collection methods. The PeptideAtlas and GPM data files can be downloaded from the Tranche distributed file system (tranche.proteomecommons.org) and ftp://ftp.thegpm.org/data/msms/. The list of experiments used in this study can be found in the Supplementary Material online.
We analyzed the spectra from the experiment rather than the peptides that were detected in each experiment because each experiment used different peptide databases to assign peptides to the spectra, and the different peptide databases are not compatible.
The human peptides were identified by searching against release 3C of the GENCODE annotation of the human genome (Harrow et al. 2006). We used the GENCODE annotation because the GENCODE consortium produces high-quality reference gene sets through a combined manual, computational, and experimental strategy (Guigó et al. 2006). The GENCODE consortium (Harrow et al. 2006) is producing the reference gene set for the human genome as part of the ENCODE project (Birney et al. 2007). Release 3C is the initial merge of the GENCODE annotation data with the Ensembl annotations and corresponds to Ensembl version 56.
GENCODE release 3C contains 22,304 protein-coding genes and a total of 72,731 protein-coding transcripts. Of the polypeptide gene products, 9,788 are alternatively spliced only in the 3′ or 5′ untranslatable regions and will therefore have identical protein sequence to other isoforms from the same gene. These identical translated products cannot be distinguished with peptide data alone. A total of 12,842 protein-coding genes coded for more than one distinct gene product, 57.57% of all annotated genes.
The GENCODE annotation covers the whole human genome, but it is not yet final for all genes. Since we used the annotation from GENCODE, we could only detect peptides that were annotated in their manual annotation. That means that we cannot completely rule out the possibility that we were not catching single nucleotide polymorphisms and alternative variants that were not annotated by GENCODE.
Data Sets Used for the Mouse and Drosophila Comparison
The data set for mouse analysis was made up of the spectra from all mouse proteomics experiments that were deposited in PeptideAtlas (in total 3,509 mzXML format spectra files). We searched against the NCBIM37 release of the Ensembl mouse database (Church et al. 2009) using the X!Tandem search engine in the same way as for the human data (detailed below).
The Drosophila analysis was based on the peptides detected in two large-scale proteomics studies. The first experiment detected 32,729 nonoverlapping peptides from the D. melanogaster proteome and identified 6,980 genes unambiguously (Brunner et al. 2007). The second experiments (Bodenmiller et al. 2007) found 10,118 high-confidence phosphorylated peptides that mapped to 3,472 gene models. The detected peptides were mapped to the alternative variants annotated in FlyBase (Tweedie et al. 2009). Full details of the methods used in the analysis can be found in the original work (Tress, Bodenmiller, et al. 2008).
The Decoy Set
Peptide matching algorithms can generate incorrect peptide assignments, even at very low e values. False positive matches can be attributed to several error sources (Nesvizhskii et al. 2007). Many database search tools model the peptide spectra in a simplistic way, and this results in the failure to assign the peptide sequences to the correct spectra. Other common sources of error include the low quality of MS/MS spectra and deficiencies in scoring schemes.
In order to improve the performance of peptide matching techniques, the overall rate of false matches (FDR) must be estimated. We used a target/decoy strategy (Moore et al. 2002) to accomplish this task. It consists of the creation of a random decoy database that preserves the general composition of the target database but does not overlap with it. The matches obtained from the decoy database can be used to estimate the FDR of the target set, as they do not correspond to true peptides. The detected decoy peptides represent an estimate of the true peptides that are matched to the wrong spectra.
A decoy database was generated for the 62,943 unique transcripts in the GENCODE 3C release database. The decoy database was made by replacing each real sequence entry with a random sequence of the same length and the average amino acid composition for the entire database using a perl script from http://www.matrixscience.com/downloads/decoy.pl.gz. This approach for the generation of the decoy database is conservative as randomizing the sequences generates many more decoy peptides. The other option, reversing the peptide sequences, generates many identical peptides. The randomized GENCODE 3C database that we used included over 4 million unique random tryptic peptides compared with the 1.7 million unique tryptic peptides in the GENCODE 3C annotations.
In addition, we included in the search set a list of proteins commonly found in proteomics experiments from the GPM database. This is labeled as the Common Repository of Adventitious Proteins (cRAP). These peptides are detectable in most proteomics experiments but are considered contaminants.
Analysis of the Spectra
We obtained a list of peptide-spectrum matches (PSMs) by matching the spectra from PeptideAtlas and the GPM to the release 3C of the GENCODE annotation of the human genome using the X!Tandem peptide identification tool (Craig and Beavis 2004).
X!Tandem searches were performed in a Linux cluster based on an massive passing interface environment. Default parameters were used: tryptic cleavage specificity, mass tolerance of ± 20 ppm for precursor ions, ±0.4 Da for fragment ions, one missed cleavages permitted. If the same peptide was identified from multiple spectra from the same experiment in the analysis, we took just the PSM with the best e value.
The search was performed against the GENCODE 3C database, the cRAP database, and the random GENCODE database. The target–decoy database search was conducted on a concatenated target/decoy/cRAP database. Those PSM that matched proteins from the GENCODE 3C database were labeled as “target PSM.” The matched peptides that we deemed reliable were used to identify genes and splice isoforms from the GENCODE 3C annotation.
Most proteomics experiments use trypsin to cleave protein samples into fragments. If the protein has been efficiently cleaved, the peptides generated by the process should have an arginine (R) or lysine (K) amino acid residue at the C-terminal end (unless it is a C-terminal peptide) and the peptide should contain few missed cleavages (lysines or arginines). Peptides were tagged as “nontrypsin” if they did not terminate in an arginine or lysine (and were not C-terminal fragments) or if they contained more than one uncleaved lysine or arginine residue. We looked only at the peptides from the “trypsin” set in this study.
X!Tandem assigns an expectation value (e value) to each matched peptide. The e values are derived from the X!Tandem scoring scheme score distribution, and the peptide candidate selection is fully guided by the associated e values. X!Tandem detected a total of 950,684 target PSM at the default e value cutoff of 0.01. Of these 918,484 were labeled as cleaved by trypsin and 32,270 not cleaved by trypsin. The nontryptic peptides were discarded.
As previously mentioned, we selected one just one spectra match per experiment for each peptide to guarantee the independence in the initial set of spectra. However, each experiment is independent by definition so peptides can map to spectra from different experiments and each identification will have a different e value.
Calculating the FDR
The FDR is defined as the expected proportion of incorrect assignments within the set of accepted target peptides. The target and decoy PSM were used to estimate the proportion of incorrect assignments. In this target/decoy approach, the set of accepted peptides is represented by the target PSMs, while the incorrect assignments are estimated from the decoy matches.
The FDR was calculated from the e values from the X!Tandem searches. We only allowed a single PSM for each peptide in the calculation. If a target or decoy peptide was detected in more than one experiment, we selected the lowest e value and discarded the rest in the FDR calculation. At the 1% FDR cutoff threshold we chose, we identified a single decoy peptide for every hundred target peptides, and this decoy peptide represented the number of mismatched peptides. At the 1% FDR cutoff, we identified 75,474 unique tryptic peptides, and there were 918,484 target PSM with e values below the FDR threshold.
Identifying Alternatively Spliced Isoforms
Peptides were matched to the proteins in the GENCODE 3C release using a simple Perl script. Each of the more than 75,000 peptides mapped to at least one gene in the GENCODE annotation.
We distinguished alternative protein isoforms with discriminating peptides—those peptides that could be mapped to some but not all annotated transcripts from the same locus. We confirmed the presence of multiple alternative gene products only if two or more protein sequence distinct isoforms only if they could be unambiguously distinguished by the discriminating peptides.
Transcript Expression Levels
We estimated protein expression at the level of transcripts using the expression evidence in the HuGE Index database (Haverty et al. 2002). The database contains data for 19 different tissue types. All tissue samples are from normal human subjects, and the expression level is provided in arbitrary units that undergo the same normalization and scaling procedures. This means that the expression values are comparable across the whole gene set. The HuGE Index is a database that contains transcript expression data for more than 7,000 human genes.
In Silico Proteomics Analysis
We carried out a simulated experiment using in silico–generated peptides from the GENCODE 3C release. The in silico proteomics analysis was designed to determine which genes we would most expect to detect with alternative isoforms. The theoretical probability of detecting more than one protein isoform per gene is directly related to the differences in protein sequence between splice variants—the greater the differences between isoforms, the easier it should be to find PSM that map to distinct variants. The theoretical probability of detecting more than one protein isoform per gene can be approximated with a random simulation.
We generated peptides by a standard in silico trypsin lysis for all 22,304 GENCODE annotated genes. We drew just 14,000 peptides at random from the in silico trypsin digest (the number of peptides was calibrated so that we would identify approximately 8,000 genes, as in the main analysis) and we counted the number of times each gene would have been detected with at least two unambiguously identified splice isoforms. This experiment was repeated 1,000 times, and we gave each gene a score (the theoretical “probability of detection”) based on the percentage of simulations in which the drawn peptides identified at least two different splice isoforms for each gene.
Results
Peptides were detected by mapping the spectra from the individual human proteomics experiments in the PeptideAtlas and GPM databases to the gene products from the GENCODE 3C annotation of the human genome (Harrow et al. 2006). The GENCODE 3C gene set comprised a total of 22,304 genes that code for 72,731 protein sequences.
Peptide-spectrum matching was carried out using the search engine X!Tandem (Craig and Beavis 2004). Individual PSMs from different experiments were pooled for each peptide, and we used a target–decoy approach to determine the combined P values and corrected FDRs from the scores generated for each peptide by X!Tandem.
At corrected FDR of 1%, we detect 75,474 peptides, which in turn map unambiguously to 34.1% of the genes annotated by GENCODE (7,597 genes). We identify another 375 genes with peptides that map to more than one gene (there are many genes with identical or almost identical gene products in the annotation).
All the peptides detected in our analysis are included as evidence tracks in the GENCODE annotation and in the PROTEO Web site (http://proteo.bioinfo.cnio.es/).
Using Proteomics Evidence to Aid Genome Annotation
We have been able to add value to the GENCODE annotation of the human genome by validating “novel” and “putative” genes and coding sequences (CDSs). The GENCODE manual annotation of the human genome subdivides transcripts into three main categories based on the evidence supporting their annotation. “Known” CDSs can be mapped to known protein sequences from Swiss-Prot, “novel” CDSs have at least 60% of the length or the same domain structure as known CDSs, and “putative” CDSs share less than 60% length of the known CDSs. We detected peptides for seven genes that were annotated solely with putative or novel transcripts, while for the set of known genes that expressed multiple splice isoforms, we validated the expression of five putative isoforms and nine novel isoforms (fig. 1).
FIG. 1.

Annotation of putative and novel isoforms. In A, the GENCODE annotation for the COL6A3 locus viewed using the Ensembl genome browser. The large arrow picks out the “novel” transcript ENST00000472056 annotated for this gene. The transcript is missing three nonconsecutive exons (small arrows) at the 3′ end, and we detected 40 PSM that mapped uniquely to this isoform. In B, the GENCODE annotation for the APOA1 locus viewed using the Ensembl genome browser. The large arrow picks out the “putative” transcript ENST00000375329 annotated for this gene. The transcript codes for a different N-terminal, and we detected 56 PSM for this isoform.
The Ensembl automatic annotations that form part of the GENCODE 3C release also include a number of pseudogenes. We detected peptide evidence (two or more valid PSM) for the translation to protein of 15 of these pseudogenes. Several have since been reclassified, for example, MSTP9, which was reclassified as protein-coding gene as a result of the peptide evidence and from the identification of novel transcriptional evidence that supported locus-specific splice junctions that restored the full-length reading frame (Zheng et al. 2007). There was additional compelling evidence of coding potential to support the reannotation of ZNF66P as a protein-coding gene, and IGLC6 was confirmed as a polymorphic pseudogene with both protein-coding and pseudogenic alleles. Of the remaining 12, 10 have peptides that map to ORFs headed by an ATG, which suggests protein-coding potential, although all but one lack evidence of transcription that could be mapped unambiguously to the putative pseudogene locus.
Evidence for the Translation of NMD Isoforms
NMD is a cellular surveillance mechanism that degrades abnormal mRNA transcripts containing premature termination codons (BehmAnsmant et al. 2007; Isken and Maquat 2008; Chang et al. 2010). In mammals, NMD is triggered when translation terminates more than 50–55 nt ahead of a splicing-generated exon junction (Isken and Maquat 2007) and the protein Upf1 binds to the exon junction complex (Isken et al. 2008). The process is only partly understood, and recent work has suggested that there may be substantial differences between the NMD mechanisms in different organisms (BehmAnsmant et al. 2007; Brogna and Wen 2009). The proportion of alternative transcripts that will actually be targeted for degradation by NMD is not clear.
Of the 72,031 coding transcripts in the GENCODE 3C release, 3,939 are labeled as potential NMD targets because they have premature stop codons. If NMD were an efficient process, we would not expect to find evidence of the expression of NMD targets as proteins, but we detected the expression of four protein isoforms that were tagged as candidates for NMD degradation in the annotation. One example of an NMD-tagged isoform that we detected in the proteomics experiments can be found in the gene RBM6 (RNA-binding motif protein 6). The transcript ENST00000425608 skips an exon leading to a frame shift and a premature stop codon. The peptide “EYNTGNSNDPGQR” is generated from the region of the transcript sequence that crosses the exon junction at the skipped exon (fig. 2) and we detected a total of five PSMs for this peptide across different experiments.
FIG. 2.
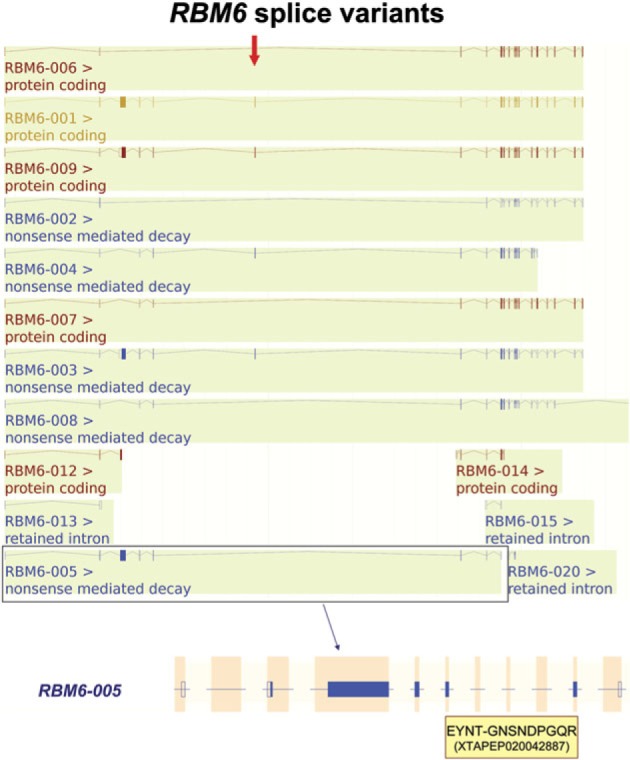
NMD target RBM6-005. The GENCODE annotation for the RBM6 locus viewed using the Ensembl genome browser showing the peptide detected for NMD-targeted isoform RBM6-005 (ENST00000425068). GENCODE currently annotates 22 variants for this gene, not all are shown for clarity. Transcript RBM6-005 skips the exon marked with the arrow that leads to a frame shift and a premature stop codon. Transcript RBM6-005 is highlighted below and aligned with the detected peptide fragment (in the shaded box). The dash (–) in the peptide sequence marks where the peptide spans the exon junction.
Although evidence for the expression of NMD transcripts has been amply demonstrated (Barberan-Soler et al. 2009; Filichkin et al. 2009), we believe that this is one of the first times that NMD-tagged protein isoforms have been detected in proteomics experiments. The HAVANA group did not detect the expression of any NMD candidates for the mouse proteome (Brosch et al. 2011). Although a recent paper (de Lima Morais and Harrison 2010) was able to map peptides from the PRIDE database (Vizcaíno et al. 2009) to a staggering 205 human NMD targets, this incredible number is almost certainly not correct—many, if not all, of the candidate NMD transcripts listed in the study are, in fact, annotated as protein coding in Ensembl.
Mouse Proteomics Analysis Identifies More Novel Isoforms
We performed exactly the same experiment for mouse using the X!Tandem search engine, the Ensembl mouse database and the spectra from PeptideAtlas mouse experiments. We did not include the mouse spectra from the GPM, and the mouse protein database is annotated with fewer alternative isoforms, so we detected fewer peptides than for human. The peptides mapped to just 4,956 genes.
We checked the annotations for the transcripts for which we detected as alternative isoforms, and even in this subset, we were able to validate the translation to protein of nine mouse transcripts labeled as novel and two transcripts labeled as putative. The relatively high proportion of uncharacterized transcripts is almost certainly because the mouse proteome has been less analyzed than the human genome at this point and suggests that proteomics ought to be an even more powerful tool for annotation for less well-studied genomes.
Drosophila Proteomics Experiments Detect Further NMD Targets
We also analyzed the peptides detected in two large proteomics experiments with Drosophila (Brunner et al. 2007; Bodenmiller et al. 2007). The peptides detected in these two large-scale experiments mapped uniquely to 8,166 Drosophila genes.
Although there is no experimental evidence to show that the four human NMD targets are targeted for NMD, we were able to detect experimentally characterized NMD targets in the analysis of the Drosophila proteomics experiments. Hansen (Hansen et al. 2009) identified NMD targets by performing knockdowns of Upf1 and Upf2, proteins that are a crucial part of the NMD process, and recording those variants that were upregulated by Upf1 or Upf2 knockdowns. The study provided a high-confidence set of more than 60 NMD targets that were upregulated by the loss of Upf1 or Upf2.
We were able to distinguish four isoforms coded by transcripts that were upregulated by Upf1 or Upf2 knockdowns with the peptides detected in the Drosophila proteomics experiments. Three of these NMD targets were upregulated in Upf1 knockdowns and the fourth was upregulated by the loss of Upf2.
Alternative Splice Isoforms
We used the PSM from the analysis to look for the expression of multiple alternative isoforms from the same gene. In order to validate the expression of alternative isoforms, peptides had to distinguish at least two protein sequence distinct alternative splice isoforms (see the example in fig. 3). We detected evidence of the expression of two or more alternative splice isoforms for total of 150 genes and we detected evidence for the expression of three or more distinct gene products for 13 genes. The number of genes detected with evidence of the expression of at least two alternative gene products is higher than any previous comparable study. The set of genes for which we detected the presence of at least two sequence distinct protein isoforms is referred to throughout this work as the AI Detected genes.
FIG. 3.

PSMD4 splice isoforms. (A) The four annotated isoforms of PSMD4 aligned. Shaded areas represent the peptides found in the experiments. The strength of shading denotes the total number of unique peptide-spectra matches encountered for each peptide. Just two of the isoforms were detected in the proteomics studies—they differed by a three-residue indel. (B) The detected peptides are shown mapped onto the Protein Data Bank (PDB) (Berman et al. 2000) structure 2X5N, which is 50% identical to the N-terminal of PSMD4, and onto 1YX4, the structure of the central section of the PSMD4 isoforms. Most of the structure of 1YX4 is disordered (the image is just one of the 15 NMR models of 1YX4), and the three extra residues detected for isoform ENST00000368881 would be inserted in a disordered region, as indicated by the arrow.
Among the genes for which we detected the translation of alternative peptides were several well-known examples of alternative splicing. We found detected isoforms p16INK4a and p14ARF from CDKN2A (Norman and Sharpless 2005). We also found alternative isoforms for Lamin A/C (LMNA). Lamin A is the major component of nuclear lamins along with thymopoietin (TMPO), another gene for which we detected two well-studied isoforms, LAP2-alpha and LAP2-beta. Protein 4.1 (EPB41), another nuclear envelope protein for which we detected two isoforms, has been shown to be intimately involved with lamin A/C and LAP2-alpha (Meyer et al. 2011). Indeed, we detect alternative isoforms for five of the main components of the nucleoskeleton, lamin A/C, LAP2, Protein 4.1, titin, and alpha-II spectrin (Simon et al. 2011).
We found evidence of the expression of distinct protein isoforms for genes CUX1, NEBL, and MACF1. These three genes generate isoforms that share part of their protein sequence but then diverge considerably over the rest of the sequence and in each case have well characterized but distinct functional domains in the diverged region of their sequences. CUX1 generates two isoforms, CDP and CASP, with different C-terminals (Lievens et al. 1997). The C-terminal section of CDP contains three copies of the DNA-binding CUT domain; in CASP, thought to have a role in vesicle transport (Gillingham et al. 2002), these are replaced by a single CASP_C domain. MACF1 (microtubule-actin cross-linking factor 1) generates two isoforms that differ at the N-terminus. The N-terminal region in isoform 1 has two CH domains, while isoform 4 contains multiple plectin domains. NEBL (nebulette) also differs in the N-terminus and has one isoform with multiple nebulette repeats and another where these are replaced by a LIMS domain.
We also detected two isoforms for ketohexokinese (KHK), a protein whose alternative isoforms have been studied to such an extent that structures for both isoforms have been crystallized (fig. 4). There are only subtle differences between the two structures for these isoforms and although these differences do affect the active site, both isoforms bind the substrate fructose (Trinh et al. 2009).
FIG. 4.

The active site of the two isoforms detected for ketohexokinase (KHK). The structures of the two KHK isoforms detected in the experiments are known. The structure of the fructose-binding site of the “peripheral” isoform (3NBV) is shown superimposed on the fructose-binding site of the “central” isoform (2HQQ). The closest residues to the fructose substrate for each isoform are shown in stick form. The binding residues are identical and are oriented in an almost identical form, with the exception of those residues in the region coded for by the homologous exon. These are the two beta sheets on the right of the image. Here, the residues in contact with the fructose are different in the two isoforms, alanine and tyrosine in the central isoform and leucine and isoleucine in the peripheral. Despite the differences, the substituted residues have similar hydrophobicity, and both isoforms were crystallized with bound fructose (not shown) and have conserved catalytic residues (López et al. 2007).
Alternative Splicing in the Mouse and Drosophila Analyses
The annotations in the NCBIM37 release of the Ensembl mouse database contain fewer annotated isoforms than the equivalent human database, in part because the Ensembl mouse merge contains substantially less manual annotation. In addition, we searched against only PeptideAtlas spectra for mouse, so we would expect to find fewer genes with expressed alternative isoforms. We detected alternative isoforms for just 49 genes. Strikingly, 18 of the genes were orthologs of human genes that were found to express pairs of isoforms in the AI Detected set. Of these 18 genes, 16 expressed pairs of alternative isoforms with the same alternative splicing event as the human equivalent.
For Drosophila, we detected the presence of alternative isoforms for 130 of the 8,166 genes identified in two large-scale Drosophila proteomics experiments (Tress, Bodenmiller, et al. 2008). Although we detected alternative isoforms for 11 Drosophila genes that were orthologs of genes from the human AI Detected set, none of the alternative isoforms detected for Drosophila were generated from splicing events orthologous to those detected in human or mouse.
In fact, alternative splicing events are rarely conserved between human and Drosophila. The Flybase annotation of Drosophila records similar splicing events for just 3 of the 150 genes in the AI Detected set. The gene squid has an insert in the identical position as in HNRNPAB, but the insert is shorter in the human isoforms. The same is true of the indel in zipper that is in the same position as an indel found in human orthologs MYH10 and MYH11. Finally, the Drosophila and human tropomyosin genes both have four sets of exons that can be swapped in a homologous fashion, although curiously, only one set of the four sets of homologous exons is in the same sequence position in both species. The other three homologous exons do not overlap between the two species (supplementary fig. 1, Supplementary Material online).
Many of the Alternative Isoforms We Identify Are from Highly Expressed Genes
In order to find out why we detected alternative isoforms for the 150 genes of the AI Detected set and not others, we investigated whether the genes for which we detected multiple alternative isoforms were particularly highly expressed or whether there were any features that might make the alternative isoforms easier to detect.
If proteins are present in the cell in high quantities, it ought to be easier to detect alternative splice isoforms even if they are present in relatively low proportions—in theory, the more copies of a protein, the greater the likelihood that one of them is an alternative variant. We looked at expression at the transcript level using the HuGE Index database (Haverty et al. 2002). The HuGE Index database contained expression data for 2,512 genes from the Background set and 109 genes from the AI Detected subset. For each gene, we calculated the arithmetic mean of each of the tissue samples and calculated a global mean expression score from the sample means.
We also estimated the expression at protein level by counting the number of valid experimental PSM that matched uniquely to each gene (proteins that are present in many cell types should have more experimental PSM).
Differences between annotated isoforms are another factor that could influence in the likelihood of detecting distinct isoforms. The a priori probability of detecting more than one protein isoform per gene is related to the differences in protein sequence between splice variants and to other factors such as the length of the proteins. Clearly, if only a handful of detected peptides map to a gene, there is more chance of discriminating between two isoforms that have entirely different amino acid sequences than two isoforms that differ only by a single inserted residue. Here, we measured features that could be related to the theoretical possibility of detecting of splice isoforms, such as mean protein length, the number of annotated variants, and the number of exons. We also performed an in silico proteomics analysis (see Materials and Methods) to estimate the theoretical probability of detecting two different splice isoforms for a gene.
We compared the features for the 150 genes with alternative isoforms (the “AI Detected” genes) against two larger sets of genes; those genes with multiple splice variants for which we detected peptides in the proteomics experiments (the “Background” genes) and those genes annotated with multiple variants for which there was no evidence of protein expression at all (the “Not Detected” genes).
The results are shown in table 1. The genes in the Background set were slightly longer than those in the Not Detected set, and they had slightly more variants and exons per gene. The biggest difference between the two sets could be found in the mean expression intensity from the HuGE Index database. The mean for the Background set was over twice as high as that for the 7,329 genes from the Not Detected set (190.8–77.2). As might be expected proteins detected in the proteomics experiments were more highly expressed than those that were not detected.
Table 1.
Features of Genes Detected with Alternative Isoforms Compared with Those from the Background Sets.
| Mean CDS Length | Total Variants | Total Exons | Expression | PSM | Detection Probability | |
|---|---|---|---|---|---|---|
| Not expressed | 450.9 | 4.19 | 11.8 | 77.21 | — | 15.89 |
| Background | 488.82 | 4.52 | 14.64 | 190.82 | 106.96 | 18.2 |
| AI detected | 602.38 | 7.63 | 20 | 323.71 | 754.66 | 32.57 |
The differences between the Background set and the AI Detected set were even more noticeable. The average protein sequence was 110 residues longer for the genes in the AI Detected set. The AI Detected genes also had more exons on average than the Background set and more annotated variants per gene. There were also striking differences in the mean theoretical probability of the detection of alternative isoforms between those genes in the AI Detected set (33.0%) and the Background set (18.2%). The genes in the AI Detected also set had markedly higher levels of transcript expression and had more than seven times as many peptide matches per gene than the proteins detected in the Background set (table 1).
The genes in the AI Detected set are expressed at higher levels and detected much more frequently in cells, so it should be considerably easier to detect alternative isoforms for these genes than for those in the Background set. This strongly suggests that the alternative isoforms that we are detecting are the low hanging fruit, those variants that are easiest to detect.
The AI Detected Set Is Enriched in Cytoskeleton and RNA-Binding Genes
Many groups have sought to discover whether there is a relationship between alternative splicing and protein function (Modrek et al. 2001; Pan et al. 2004; Omenn et al. 2005; Hansen et al. 2009). We used functional annotation clustering tools from the DAVID resource (Huang et al. 2008) to determine whether there were functional annotations that were significantly enriched in the AI Detected set.
The details of the investigation can be seen in the Supplementary Material online. In short, although Gene Ontology (GO) functional terms (Ashburner et al. 2000) related to actin and cytoskeletal binding were highly enriched in the AI Detected set (33 genes) and in mouse and Drosophila, we found that the overrepresentation of these genes was likely to be related to their greater size and higher levels of expression (fig. 5). However, the AI Detected set was also enriched with genes annotated as RNA binding, and these genes did turn out to be worthy of further investigation.
FIG. 5.

Functional bias and the genes with alternative isoforms. The genes from the AI Detected set were clustered into four groups based on their functional annotations. In part B, the percentage of genes in three functional clusters and the remaining genes (Miscellaneous). Mean values for the features from table 1 were calculated for the genes from each of the groups. These values were normalized by the scores from the AI Detected set and are plotted in A. The cytoskeleton subset has a much higher number of exons and longer sequence, and their alternative isoforms have a much higher probability of detection in proteomics experiments. Peptides for the “immune response” genes are detected in much higher numbers in the proteomics experiments (the number of PSM). We detected substantially fewer PSM for the RNA-binding subset than for the rest of the AI Detected set.
Within the genes tagged as RNA binding by DAVID, one family of genes stood out. We detected alternative isoforms for 10 hnRNPs, genes that are implicated in the regulation of alternative splicing (Martinez-Contreras et al. 2007; Venables et al. 2008). There are 26 hnRNP genes annotated with sequence distinct protein isoforms in the GENCODE 3C release, and we detected peptides for 23 of them. If we assume that it is equally possible to detect a pair of alternative isoforms for any gene from the Background set, we would only have expected to detect alternative isoforms for 0.62 of an hnRNP gene in the AI Detected set. We calculated the odds ratio for proportion of the hnRNP genes in the AI Detected set comparing the proportion of isoforms we detected for the hnRNP genes against the proportion of isoforms we would expect to detect in the AI Detected set. The odds ratio was 17.1, which translates to a P value of 0.
There is also support for these results from a previous identification of human alternative isoforms (Tanner et al. 2007). Of the 15 genes detected with pairs of alternative isoforms that were recorded in the search databases, two were hnRNP genes (HNRNPU and HNRNPK).
Many More Alternate Isoforms Than Expected Are Only Subtly Different
Another eight of the pairs of alternative isoforms detected in the Tanner paper were single residue indels. Insertions of a single amino acid residue are usually the result of “NAGNAG” alternative splicing (Hiller et al. 2006). These events can occur when two acceptors with the pattern “NAG” are separated by 3 nt (or a multiple of 3 nt) and are not especially common (Mudge et al. 2011). We detected 11 genes that expressed alternative isoforms differing only by the addition (or deletion) of just a single residue. Ten of these pairs of alternative isoforms are annotated in TASSDB (Hiller et al. 2007) as being generated from NAGNAG acceptors or GYNGYN donors. One example is VDAC3, a mitochondrial porin voltage-dependent anion-selective channel (Reina et al. 2010) that has a single extra methionine residue as a result of an inserted 3-nt exon (Decker and Craigen 2000). Besides these 11 genes, another six pairs of alternative isoforms are generated from insertions/deletions of two, three, or four residues; five of these indels are also recorded in TassDB. We also detected five genes with alternative isoforms generated by NAGNAG splicing and by other splicing events (CUX1, IMMT, HNRNPR, HNRNPK, and RBM26).
The single residue indels are clearly enriched in the AI Detected set. There are 1,070 pairs of alternative isoforms with a single residue indel among the genes in the Background set for which we detected peptides. In total, there are 73,579 alignments between sequence distinct isoforms in the Background set. Assuming that the detection of any pair of alternative isoforms is equally possible, we would expect to detect just 2.18 genes with alternative isoforms generated from single residue indels in the AI Detected set.
The odds ratio between the proportion of isoforms detected with single residue indels in the AI Detected set and the proportion of isoforms with single residue indels in the Background set was 5.36, which again translated to a P value of 0.
Another set of alternative isoforms we detected were those that were generated from homologous equivalent (HE) exon substitutions. Here, we defined HE exons as exons that are homologous to the exon that they replace. Many of these HE isoforms are generated from MEEs. However, the definition of MEEs is much stricter than HE exons, MEE requires that the two MEEs are never seen in the same transcript, and many HE isoforms that we detected do not fit the strict criteria for mutually exclusive splicing, often because there is some (albeit infrequent) evidence for the inclusion of both homologous exons in the same transcript (e.g., this is the case with TPM1). MEE is a form of alternative splicing that is very rare (Tress et al. 2007; Wang and Burge 2008).
We detected alternative isoforms generated from homologous exons for 19 genes, including TPM1 for which we identified at least four different splice isoforms. The gene has nine variants annotated by GENCODE that differ in their 5′ and 3′ coding exons and in also in two sets of internal exons. The 3′ exons and the internal exons are homologous in sequence and are spliced in a mutually exclusive manner. In total, we detected 572 discriminating PSM for 28 unique peptides that mapped to TPM1 variants. We detected PSMs for each alternative exon in all four of the alternatively spliced regions, suggesting that all nine annotated isoforms may be translated. However, since there were no peptides that spanned across two alternatively spliced regions, we could only guarantee the presence of four protein isoforms.
The alternative splicing of HE exons is of interest from an evolutionary point of view because swapping homologous exons could allow subtle modifications of function. This stands in contrast to the vast majority of annotated alternative isoforms, which are generated from large indels or nonhomologous substitutions and that are likely to generate isoforms with functions that are radically different from the main function of the gene.
One example from the proteomics experiments is ketohexokinse, an enzyme that catalyzes the phosphorylation of fructose. We detect two peptides that map to two isoforms for this gene. These isoforms differ by a single interchangeable and homologous exon. The region of the protein structure covered by this binding site includes substrate-binding residues and a catalytic residue (fig. 4). Several of the fructose-binding residues are different between the two isoforms, but both isoforms bind fructose (Trinh et al. 2009), and predictions by the functional residue prediction server, firestar (López et al. 2007), suggest that the affected catalytic residue is likely to still be functional.
A total for the number of genes with alternative transcripts that can be generated from HE is hard to come by, in part because it is hard to define automatically what constitutes a homologous exon. We chose to use a definition based on protein sequence homology. A pair of proteins from the same gene was defined as generated by HE if the two homologous regions were adjacent to each other in the primary mRNA transcripts, and if they generated an amino acid sequence of at least eight residues. The alignment of the amino acid sequences of the exons also had to have at least 30% sequence identity, and a maximum of 20% gaps were allowed. For the shortest translated exon sequences, these limitations were stricter, 40% identity and 0% gaps in the alignments. These limits excluded 4 of the 19 genes we had defined (by eye) as having isoforms generated from HE, leaving us with just 15.
According to the definition, the genes in the Background set have just 195 pairs of alternative isoforms that are generated from homologous exons. Based on these figures, we expect to detect approximately 0.4 of a gene with alternative isoforms generated from homologous exons for in the AI Detected set. The odds ratio between the proportion of alternative isoforms generated by HE splicing in the AI Detected set and the proportion of alternative isoforms generated by HE splicing in the Background set was 41.18. Once again, the proportion of alternative isoforms with this form of splicing was highly significant.
Would We Expect These Genes to Be Enriched in the AI Detected Set?
Clearly, these three sets of genes were significantly overrepresented in the AI Detected set. However, it may be that (like the actin-binding genes), it is simply easier to detect alternative isoforms for the HE, NAGNAG, and hnRNP genes. To test this, we used the features previously calculated for each gene, such as the mean number of exons per gene, the theoretical probability of detecting multiple alternative isoforms and the numbers of PSM detected for each gene.
We divided the genes in the AI Detected set into eight groups by type of alternative splicing event. The eight groups were those genes with pairs of expressed alternative isoforms that were known to be generated from NAGNAG splicing or had inserts of four residues or less (17 genes), genes with isoforms that differed by larger indels (48), those genes with isoforms that differed by HE substitutions (19), genes with isoforms that differed by C-terminal substitutions (27), genes with isoforms that differed by N-terminal substitutions (6), those genes with evidence for the expression of two completely unrelated proteins (7), those genes with isoforms that differed by more than one type of alternative splicing event (16), and the 10 hnRNP genes (the only group not defined by their splicing event, almost all of these were small indels).
We calculated the mean score for each of the features in table 1 for each individual group and then normalized these scores against the mean values for the whole AI Detected set. The results are shown in table 2.
Table 2.
Normalized Features of Eight Separate Groups of Genes Detected with Alternative Isoforms.
| Mean CDS Length | Mean Variants | Mean Exons | PSM | Detection Probability | |
|---|---|---|---|---|---|
| Homologous exons | 0.54 | 0.821 | 0.642 | 0.848 | 0.856 |
| NAGNAG | 0.784 | 0.764 | 0.726 | 0.495 | 0.660 |
| HNRNP | 0.595 | 0.826 | 0.610 | 0.670 | 0.362 |
| C-substitution | 0.836 | 1.073 | 0.776 | 0.391 | 1.126 |
| N-substitution | 1.907 | 1.289 | 1.367 | 2.959 | 1.423 |
| Indels | 1.284 | 1.087 | 1.277 | 1.557 | 0.824 |
| Complex | 1.907 | 1.260 | 2.267 | 1.102 | 1.752 |
| Different proteins | 0.480 | 0.768 | 0.600 | 0.166 | 1.783 |
Note.—Values that are lower than AI Detected set are shown in italics.
The hnRNP genes and the genes that generate isoforms from NAGNAG splicing and homologous exons have fewer exons and variants than average, and their protein sequences are shorter. On average, we detected fewer peptides for these genes than the others, and there is a lower theoretical probability of detecting alternative isoforms.
Based on all these features, we would actually expect it to be less easy to detect alternative isoforms for these genes than for the other genes in the AI Detected set.
Subtly Different Alternative Isoforms Are Also Supported by More Discriminating PSM
The peptides we detected came from a large number of separate experiments. Although we have no way of measuring the level of expression of the individual peptides in each experiment, we can count the number of times each peptide is detected over all the experiments. Here, we are counting PSMs, the numbers of experiments in which a peptide was detected by X!Tandem. Although this is not a direct substitution for expression levels, it does give an idea of the ubiquity of each peptide.
It was also possible to match the discriminating experimental PSM to each splice isoform. Here, we only counted PSM that discriminated between different isoforms (as in fig. 3). We nominated the isoform with the most discriminating PSM as the main isoform for each gene.
On average, we mapped 515 discriminating PSM to the main isoform for each gene, but only 8 PSM to the isoform with the second highest number of discriminating PSM. Clearly, we found considerably more experimental evidence for the main isoform in the AI Detected set.
The fact that the main isoform for each gene is detected in substantially more experiments certainly suggests that most alternative isoforms are translated only rarely, although the low coverage of proteomics studies means that such comparisons are provisional. Despite this, there were a number of alternative isoforms for which we did detect high numbers of discriminating PSM. These alternative isoforms may possibly have a more prominent functional role in the cell.
We calculated an “alternative PSM score” (AP Score) for each alternative isoform in order to identify highly expressed alternative isoforms. The AP Score was calculated as follows. We took the PSM that discriminated between the first two isoforms and from this calculated the percentage of the PSM that mapped to the alternative isoform.
We calculated the AP Scores for all genes in the AI Detected set and the mean AP Scores for each of the eight subsets. The results (fig. 6) show that genes with alternative isoforms generated from homologous exons, the hnRNP genes, and the genes with alternative isoforms generated from NAGNAG indels, have much higher AP Scores than the other groups. Not only are the hnRNP genes and genes generated from homologous exons or NAGNAG splicing significantly enriched in the AI Detected set, but their alternative isoforms are also supported by a larger proportion of PSM.
FIG. 6.

Alternative isoforms generated from three subsets are supported by more peptide evidence. The percentage of PSM that distinguish alternative isoforms (the AP Score) contrasted with the theoretical likelihood of detecting splice isoforms (detectability) calculated for each of the eight subsets from the AI Detected set. Detectability is calculated from the number of PSM detected for the each gene and the theoretical probability of detection, both normalized by the equivalent values from the Background set. The higher the detectability, the more likely we are to detect PSM that match to alternative isoforms. The AP Score is surprisingly high for three of the groups (the hnRNP genes, the genes with isoforms generated from NAGNAG splicing, and the genes with alternative isoforms generated from homologous exons).
Splice Events in Mouse and Drosophila Show Similar Patterns
As already mentioned, 18 of the 49 genes we detected in the mouse analysis were orthologs of human genes in the AI Detected set, including 3 of the 4 tropomyosin genes (the mouse ortholog of TPM4 is not annotated with alternative isoforms, so we cannot detect any). Of the 49 mouse genes with multiple splice isoforms, 10 had alternative isoforms generated from interchangeable homologous exons and 6 of these genes were generated with the same event as their human orthologs. seven pairs of isoforms differed by the insertion of a single residue as a result of NAGNAG splicing. We detected alternative isoforms with the same splicing event for a single NAGNAG gene. The proportions of these two types of splice isoforms are similar to (but higher than) those found in human (see fig. 7). We also detected alternative isoforms for a heterogenous nuclear ribonucleoprotein, Hnrnpd, the ortholog of HNRNPD in human, which were generated by a splicing event that was conserved between mouse and human.
FIG. 7.

Alternative splice isoforms detected for human, mouse, and Drosophila. The proportion of genes with detected alternative isoforms that come from hnRNP genes, that have alternative isoforms generated from homologous exons, and that have alternative isoforms generated by NAGNAG splicing (or small indels), in different proteomics experiments for Drosophila, human, and mouse.
It was also possible to compare the proportion of discriminating PSM that mapped to each isoform for the mouse set. Again, the AP Score was substantially higher for the genes that generated their splice isoforms through NAGNAG splicing (27.2%) or homologous isoforms (39.4%) than for the remaining genes (4.2%).
Alternative isoforms for 19 of the 130 genes detected with alternative isoforms in Drosophila were generated from homologous exons. Five genes expressed isoforms that were different by a single residue insertion. We detected only one hnRNP gene that expressed multiple alternative isoforms (bancal, a remote homologue of the HNRNPK gene from the AI Detected set), but at the time of the proteomics experiments, there were only six Drosophila hnRNP genes annotated with alternative isoforms. The proportion of genes that generated splice isoforms through homologous exons is similar to that of mouse and human, though we detected smaller numbers of genes that expressed isoforms different by a single inserted residue (see fig. 7), probably in part because there are considerably fewer NAGNAG acceptors annotated in Drosophila than in human (Hiller et al. 2004). In contrast to the human set, we detected fewer alternative isoforms with indels and more pairs of isoforms that differed in the C-terminal and N-terminal regions.
One further result that stood out was that isoforms with C-terminal substitutions were significantly enriched in GO terms related with DNA-dependent transcription and regulation of transcription. Four of these genes (lola, mod(mdg4), tramtrack, and fruitless) generate multiple isoforms with distinct unique zinc finger domains. The generation of alternative isoforms that have a range of distinct but homologous functional domains is interesting because this type of complex exon organization appears to be particularly rare in human and mouse.
The Cell Favors Intact Pfam Domains
It is striking that 24% of the alternative isoforms we detected would probably only have slightly modified structure and function in relation to the constitutive variant. The bias toward isoforms with homologous equivalently spliced exons and with small indels is not the only indication that there may be some sort of selection process at the level of the expression of splice isoforms. One crucial advantage that these isoforms have over other annotated isoforms is that there is less potential for toxic effects on the cell. If this pattern were reflected in the rest of the set, we would also expect to find less evidence for the expression of alternative isoforms with damaged domains, both structural and functional.
First, we determined whether Pfam A functional domains were more conserved for the proteins detected in the proteomics experiments than for those proteins that were not detected. Pfam functional domains are hand-curated functional motifs, and it has been shown that they are more conserved than would be expected in annotated alternative splice variants (Kriventseva et al. 2003; Tress et al. 2007; Severing et al. 2009).
Using the program Pfamscan (Finn et al. 2009), we classed the translated transcripts as having damaged, slightly damaged, and whole domains based on their alignments with the Pfam hidden Markov models. Isoforms with Pfam domains were classed as “damaged” if one or more Pfam A domain in each isoform was damaged. Isoforms were only classified as “whole” if all Pfam domains were undamaged according to Pfamscan. There is a high background level of variants with damaged domains, and many genes had all their annotated variants classified as damaged because protein sequences do not always contain whole Pfam domains. Despite this noise, there were clear differences between the isoforms that we detected and those that we did not detect (fig. 8). The biggest differences between the detected and undetected isoforms was in the Background set—45.2% of the detected isoforms had damaged Pfam domains, this rose to 72.3% for the isoforms that were not detected, reinforcing the idea that there may be some sort of selective pressure toward conserving functional domains at the level of translation.
FIG. 8.

Pfam functional domain damage in isoforms generated from different sets of genes. The y axis shows the percentage of isoforms with Pfam domains defined as damaged. The x axis shows the five sets of isoforms tested. The isoforms detected in proteomics experiments for the genes in the Background set, the isoforms not detected in proteomics experiments for the genes in the Background set, the main isoform detected for each gene in the AI Detected set (the isoform that most PSM mapped to), the alternative isoforms detected for each gene in the AI Detected set, and the isoforms not detected for each gene in the AI Detected set.
We can see similar differences between detected and undetected isoforms for the AI Detected genes. The main isoforms (those for which we detected the most PSM) had the lowest proportion of isoforms with at least one damaged Pfam A domain (44.0%), the alternative isoforms we detected would have a slightly higher proportion of damaged Pfam domains (49.6%), whereas the isoforms that we did not detect for the genes in the AI Detected set had the highest proportion of damaged Pfam domains (59.1%).
Alternative Isoforms Conserved in Mouse Do Not Have Damaged Functional Domains
We investigated the conservation of the alternative isoforms in mouse by comparing the human and mouse annotations in Ensembl for the 150 genes in the AI Detected set. We were able to find mouse orthologs for 145 of the 150 genes. Over half of the genes (75 genes) are annotated with the same splice event (or events) as those we detected for the human alternative isoforms. As a comparison, the Havana group found that just 28% of the human splicing events annotated in 1% of the human genome studied in depth in the ENCODE Pilot Project (Birney et al. 2007) were conserved in mouse (Mudge et al. 2011). There were 25 genes annotated with a single protein-coding isoform (possibly in part because the annotation of the mouse genome is lagging behind the human annotation) and the alternative splicing events we detected for the other 45 genes were not annotated in the Ensembl mouse annotation.
There are clear differences between the genes in the conserved and “nonconserved” subsets. Twenty-six of the 70 splicing events (37.1%) that generated the alternative isoforms in the nonconserved set would break Pfam A functional domains, while Pfam domains would be broken in only 3 of the 75 events in the conserved set (4%). Indeed in each of the three cases where the Pfam domain as defined would be broken, there is evidence that the loss of protein sequence would not necessarily cause catastrophic damage to the structure or function of the protein. In one case (CALD1), the Pfam domain is almost certainly not well defined (it is a domain of more than 700 residues defined by just two sequences with a large low complexity region in the center, just where the splicing event falls). In a second case, EBP41, the splicing event would damage a well-defined Pfam domain but falls in a part of the Pfam domain that is also annotated as “low complexity,” and in the third case (PEA15) the peptide that covers the alternative splicing event (an indel) overlaps the indel by just a single residue, leaving open the possibility that the peptide may actually represent a single nucleotide polymorphism or unannotated NAGNAG splicing.
The evidence from structure mapping is even clearer. We used alignments between the isoforms and protein structures to observe the likely effect of alternative splicing on 3D structure. For 22 of 70 nonconserved alternative splicing events (31.4%), the splicing event would result in clear damage to protein 3D structure, while for the 75 genes with conserved splicing events, 3D structure would be affected in only a single case (PEA15 again).
Once again we could turn to the PSM evidence from the experiments to see to what extent the alternative isoforms were detected. We were able to show that alternative isoforms that came from genes with conserved splicing events were detected much more frequently and that alternative spicing events that would break Pfam domain or damage 3D structure were supported by very little PSM evidence (see table 3).
Table 3.
AP Scores for Alternative Isoforms with Conserved or Nonconserved Splicing Events.
| Intact Pfam domains | Lost Pfam domains | Breached Pfam domains | Damaged 3D structure | |
|---|---|---|---|---|
| Event conserved in mouse | 18.659 | 23.191 | 8.823 | 3.846 |
| Event not conserved | 8.194 | 1.497 | 0.5 | 0.431 |
The genes in the conserved set have an AP Score (the mean percentage of discriminating PSMs that map uniquely to the alternative isoforms) of over 19.44% for their alternative isoforms, whereas the AP Score for the isoforms with splicing events that were not conserved is just over 2%. Within this set, the isoforms with splicing events that broke Pfam domains had a mean AP Score of 0.5%, and those with splicing events that would damage 3D structure had a mean AP Score of just 0.43%. Notably, both these scores are below the FDR threshold for the proteomics experiments.
Perhaps the most curious result concerns those alternative isoforms where the splicing event would result in the loss or gain of entire protein domains. There are 13 of these genes in the conserved set and 17 in the nonconserved set. Although there is plenty of evidence for the expression of alternative isoforms for the genes from the conserved set (an AP Score of 23.2%), there is practically no evidence for the translation to protein of the alternative isoforms in the nonconserved set (1.5% AP Score).
For the 70 genes in the nonconserved set, it is not clear whether the event is not conserved in mouse or whether the event has yet to be annotated. Among the 33 genes with conserved Pfam domains, there are 19 genes that are likely candidates for the annotation of further splice variants. These have an AP Score greater than 10% and more than 2 PSM that map to their alternative isoforms. These 19 genes include 3 hnRNP genes, 3 genes with NAGNAG splicing, 4 genes generated by homologous exons, and both sorbin and SH3 domain-containing genes (SORBS1 and SORBS2).
Discussion
We have performed a large-scale analysis of protein expression in the human, mouse, and Drosophila proteomes. At a corrected FDR of 1%, we detected evidence of the expression of peptides for 7,972 human genes. This is an exceptionally high coverage of the human genome (it is 35% of the genes annotated by GENCODE) and more than the unprecedented 7,000 human genes reported by Wisniewski using a state of the art peptide detection method (Wiśniewski et al. 2009). However, given that the spectra that we analyzed came from a much larger number of different experiments, it is also perhaps a surprisingly low coverage of the genome. To a certain extent, it demonstrates the limitations of what can be detected with current shotgun proteomics techniques.
Here, we have emphasized the usefulness of proteomics data for genome annotation, detecting evidence for the expression of isoforms that are annotated as putative and novel, and have shown that a small number of NMD-targeted transcripts and pseudogenes are expressed as protein isoforms in sufficient quantities to be detected in proteomics experiments. This suggests that pseudogenes and NMD may be subject to leaky control processes, like many others, or that we do not yet totally understand the mechanisms behind NMD regulation and pseudogene transcription.
Alternative Splicing at the Protein Level
Microarray data (Black 2000) and cDNA and EST sequences (Smith and Valcárcel 2000) have demonstrated the widespread expression of alternatively spliced transcripts. Here, we show that many of these alternative transcripts are translated in quantities high enough to be recorded in proteomics experiments. In total, we detected 150 genes with evidence of the translation of distinct protein isoforms, a number that is somewhat higher than previous comparable studies.
Evidence for large-scale alternative splicing at the level of transcripts has lead many to suggest that the role of alternative splicing is to expand functional complexity in the cell (Modrek and Lee 2003). If this were true, we might expect to detect the expression of a substantial range of alternative splice isoforms because we are interrogating spectra from so many different proteomics experiments.
However, studies suggest that the majority of the proteins generated from annotated alternative splicing events would have very different structure and function (Talavera et al. 2007; Tress et al. 2007), and this has lead several groups to suggest that the role of alternative splicing in the expansion of functional diversity is limited (Tress et al. 2007; Severing et al. 2009) or may be the result of a noisy splicing process (Melamud and Moult 2009). If this were true, we would expect to find little evidence of alternative splicing (since the cell is unlikely to tolerate large numbers of nonfunctional proteins).
In fact, we discover evidence to suggest that the reality may lie somewhere between these two opposing theories. We detected surprisingly low numbers of alternative splice isoforms—we recovered only a small fraction of the potential alternative isoforms—something that might be expected if many of the isoforms generated by alternative splicing were nonfunctional and potentially damaging. Against that a number of the alternative isoforms were detected at levels that suggested that they may play cellular roles, and many of these more highly expressed alternative isoforms were either well documented in the literature, had splicing events that were conserved in mouse, or were generated from the sort of small changes in sequence that suggest subtle changes in cellular function. The results from numerous proteomics experiments suggest that although many alternative variants are expressed as transcripts, only a percentage of these are translated into measurable quantities of stable proteins in the cell.
Conservation of Splice Events in Mouse Sheds Light on the Evolution of Functional Alternative Isoforms
Alternative splicing events were conserved in human and mouse for 50% of the AI Detected genes. The alternative isoforms detected for these genes were supported by much higher proportion of the discriminating PSMs than those from genes without conserved splicing events. In fact, proportionally, the evidence for alternative isoforms with conserved splicing events was almost 10-fold higher than for those isoforms with nonconserved splicing events.
It has been shown that annotated splice variants would produce fewer isoforms with damaged Pfam domains than expected by chance (Kriventseva et al. 2003; Tress et al. 2007). Here, we have gone one step further by demonstrating that the subset of annotated AIs that can be detected in proteomics experiments are even less likely to have split Pfam domains. Indeed for the 75 alternative isoforms detected in proteomics experiments and conserved in mouse, Pfam domains (and protein structural domains) are almost entirely conserved. This suggests that conservation of functional and structural domain integrity plays a crucial role in the development of conserved and functional alternative splice isoforms.
Heterogeneous Nuclear Ribonucleoproteins
The hnRNP genes were notably enriched in the study. We found evidence of alternative splicing for 10 of the 26 hnRNP genes annotated with splice variants by GENCODE and a much higher proportion of the discriminating PSM mapped to their alternative isoforms. The enrichment of the hnRNP genes was also notable in the results of Tanner (Tanner et al. 2007).
The fact that we detect much more evidence than expected for the translation of alternative hnRNP isoforms is especially interesting given that the hnRNP genes are directly related with the generation of alternative mRNA transcripts (Dreyfuss et al. 2002; Matlin et al. 2005; Martinez-Contreras et al. 2007; Venables et al. 2008). The genes associate with pre-mRNA in the nucleus and have an effect on RNA processing and the selection of exons.
It is not clear what role alternative isoforms of hnRNP proteins might play in the cell, though a study on the hnRNP A and B proteins showed that alternative splicing affected the binding affinity for their targets (Han et al. 2010), and it has recently been shown that the alternative isoform we detected for HNRNPK is present only in the nucleus and will be missing a serine that is usually phosphorylated (Kimura et al. 2010).
If hnRNP are involved in the regulation of alternative splicing, it might be expected that we see a relation between the genes that we detect with alternative isoforms and hnRNPs. PTBP1 is the best studied of the hnRNP that we detected with alternative isoforms. We detected two isoforms that differed by an indel of 26 residues in the long linker between the second and third RNA-binding motifs. The indel does not affect the RNA-binding motifs, so the two isoforms should still have the same RNA-binding specificity. PTBP1 has been shown to be involved in exon selection for the HE transcripts of TPM1 (Mulligan et al. 1992) and PKM2 (Clower et al. 2010; David et al. 2010) and to have an effect on the expression of transcripts from ACTN1 (Southby et al. 1999) and RTN4 (Venables et al. 2008). Xue (Xue et al. 2009) detected binding sites for PTBP1 around the HE exons of PKM2 and TPM2 and showed that the shorter (alternative) isoform of PTBP1 is less effective at regulating exon inclusion. The authors list 54 genes that have exhibited PTBP1-dependent exon skipping and we detect alternative isoforms for 9 of them, ACTN1, GANAB, LMNA, MPRIP, PKM2, PTBP1, TPM1, TPM2, and TPM3.
Translation of Alternative Isoforms May Be Subject to Regulation
We detected peptides for an unexpectedly large number of alternative isoforms that differed by the insertion or deletion of just a single residue and also for a surprisingly large number of alternative isoforms generated from homologous exons. Almost half the alternative peptides detected by Tanner (Tanner et al. 2007) had just a single residue indel, and these sets of alternative isoforms were also detected in surprisingly high numbers in mouse and Drosophila proteomics experiments. Indeed, the proportions of both types of splicing events were higher in mouse than in human experiments. There was also substantially more peptide support for the alternative isoforms we detected for these genes.
These splice events would give rise to alternative splice isoforms with just small differences in protein sequence and structure. Crucially, these changes are also unlikely to lead to protein aggregation. These sort subtle changes are more likely to produce the sort minor modulations in function that could allow an alternative gene product to be incorporated into the general cellular processes. This bias toward alternative isoforms with subtle changes in protein structure and function is a strong indication that there may be significant selective constraints on the translation of protein isoform.
Supplementary Material
Supplementary files 1–4 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
This work was supported by the ENCODE Project (Grant Number: U54 HG0004555) and by grants from Consolider E-Science (CSD2007-00050), the Instituto de Salud Carlos III (ISCIII) COMBIOMED (RD07/0067/0014), the Spanish Ministry of Science and Innovation (BIO2007-66855), and the ENFIN Network of Excellence (LSHG-CT-2005-518254). Technical assistance was provided by the Spanish Bioinformatics Institute (INB), a platform of the ISCII. The authors would like to thank the members of the Structural Computational Biology Group and in particular Ramon Díaz Uriarte for his statistical input.
References
- Abu-Farha M, Elisma F, Zhou H, Tian R, Zhou H, Asmer MS, Figeys D. Proteomics: from technology developments to biological applications. Anal Chem. 2009;81:4585–4599. doi: 10.1021/ac900735j. [DOI] [PubMed] [Google Scholar]
- Aebersold R, Desiere F, Deutsch E, King N, Nesvizhskii A, Mallick P, Eng J, Chen S, Eddes J, Loevenich S. The PeptideAtlas project. Nucleic Acids Res. 2006;34:D655–D658. doi: 10.1093/nar/gkj040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball C, Blake J, et al. (19 co-authors) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacart J, Leloire A, Levoye A, Froguel P, Jockers R, Couturier C. Evidence for leptin receptor isoforms heteromerization at the cell surface. FEBS Lett. 2010;584:2213–2217. doi: 10.1016/j.febslet.2010.03.033. [DOI] [PubMed] [Google Scholar]
- Barberan-Soler S, Lambert N, Zahler A. Global analysis of alternative splicing uncovers developmental regulation of nonsense-mediated decay in C. elegans. RNA. 2009;15:1652–1660. doi: 10.1261/rna.1711109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BehmAnsmant I, Kashima I, Rehwinkel J, Sauliere J, Wittkopp N, Izaurralde E. mRNA quality control: an ancient machinery recognizes and degrades mRNAs with nonsense codons. FEBS Lett. 2007;581:2845–2853. doi: 10.1016/j.febslet.2007.05.027. [DOI] [PubMed] [Google Scholar]
- Berman H, Westbrook J, Feng Z, Gilliland G, Bhat T, Weissig H, Shindyalov I, Bourne P. The Protein Data Bank. Nucl. Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E, Stamatoyannopoulos J, Dutta A, et al. (311 co-authors) Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black D. Protein diversity from alternative splicing: a challenge for bioinformatics and post-genome biology. Cell. 2000;103:367–370. doi: 10.1016/s0092-8674(00)00128-8. [DOI] [PubMed] [Google Scholar]
- Bodenmiller B, Malmstrom J, Gerrits B, et al. (13 co-authors) PhosphoPep—a phosphoproteome resource for systems biology research in Drosophila Kc167 cells. Mol Syst Biol. 2007;3:139. doi: 10.1038/msb4100182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogna S, Wen J. Nonsense-mediated mRNA decay (NMD) mechanisms. Nat Struct Mol Biol. 2009;16:107–113. doi: 10.1038/nsmb.1550. [DOI] [PubMed] [Google Scholar]
- Brosch M, Saunders GI, Frankish A, et al. (11 co-authors) Shotgun proteomics aids discovery of novel protein-coding genes, alternative splicing, and “resurrected” pseudogenes in the mouse genome. Genome Res. 2011;21:756–767. doi: 10.1101/gr.114272.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner E, Ahrens CH, Mohanty S, et al. (20 co-authors) A high-quality catalog of the Drosophila melanogaster proteome. Nat Biotechnol. 2007;25:576–583. doi: 10.1038/nbt1300. [DOI] [PubMed] [Google Scholar]
- Castellana NE, Payne SH, Shen Z, Stanke M, Bafna V, Briggs SP. Discovery and revision of Arabidopsis genes by proteogenomics. Proc Natl Acad Sci U S A. 2008;105:21034–21038. doi: 10.1073/pnas.0811066106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K, Georgianna D, Heber S, Payne G, Muddiman D. Detection of alternative splice variants at the proteome level in Aspergillus flavus. J Proteome Res. 2010;9:1209–1217. doi: 10.1021/pr900602d. [DOI] [PubMed] [Google Scholar]
- Church D, Goodstadt L, Hillier L, et al. (30 co-authors) Lineage-specific biology revealed by a finished genome assembly of the mouse. PLoS Biol. 2009;7:e1000112. doi: 10.1371/journal.pbio.1000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clower C, Chatterjee D, Wang Z, Cantley L, Vander Heiden M, Krainer A. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci U S A. 2010;107:1894–1899. doi: 10.1073/pnas.0914845107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig R, Beavis R. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- Craig R, Cortens J, Beavis R. Open source system for analyzing, validating, and storing protein identification data. J Proteome Res. 2004;3:1234–1242. doi: 10.1021/pr049882h. [DOI] [PubMed] [Google Scholar]
- David C, Chen M, Assanah M, Canoll P, Manley J. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364–368. doi: 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker W, Craigen W. The tissue-specific, alternatively spliced single ATG exon of the type 3 voltage-dependent anion channel gene does not create a truncated protein isoform in vivo. Mol Genet Metab. 2000;70:69–74. doi: 10.1006/mgme.2000.2987. [DOI] [PubMed] [Google Scholar]
- de Lima Morais DA, Harrison PM. Large-scale evidence for conservation of NMD candidature across mammals. PLoS One. 2010;5:e11695. doi: 10.1371/journal.pone.0011695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMarco R, Mathieson W, Manuel S, Dillon G, Curwen R, Ashton P, Ivens A, Berriman M, Verjovski-Almeida S, Wilson R. Protein variation in blood-dwelling schistosome worms generated by differential splicing of micro-exon gene transcripts. Genome Res. 2010;20:1112–1121. doi: 10.1101/gr.100099.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfuss G, Kim V, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol. 2002;3:195–205. doi: 10.1038/nrm760. [DOI] [PubMed] [Google Scholar]
- Filichkin S, Priest H, Givan S, Shen R, Bryant D, Fox S, Wong W, Mockler T. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 2009;20:45–58. doi: 10.1101/gr.093302.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R, Mistry J, Tate J, et al. (13 co-authors) The Pfam protein families database. Nucleic Acids Res. 2009;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingham AK, Pfeifer AC, Munro S. CASP, the alternatively spliced product of the gene encoding the CCAAT-displacement protein transcription factor, is a Golgi membrane protein related to giantin. Mol Biol Cell. 2002;13:3761–3774. doi: 10.1091/mbc.E02-06-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gstaiger M, Aebersold R, Gstaiger M, Aebersold R. Applying mass spectrometry-based proteomics to genetics, genomics and network biology. Nat Rev Genet. 2009;10:617. doi: 10.1038/nrg2633. [DOI] [PubMed] [Google Scholar]
- Guigó R, Flicek P, Abril J, et al. (17 co-authors) EGASP: the human ENCODE Genome Annotation Assessment Project. Genome Biol. 2006;7(Suppl 1):S2.1–S2.31. doi: 10.1186/gb-2006-7-s1-s2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Xu D, Kim D, Xu Y. Improving the performance of DomainParser for structural domain partition using neural network. Nucleic Acids Res. 2003;31:944–952. doi: 10.1093/nar/gkg189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Kassahn K, Skarshewski A, Ragan M, Rothnagel J, Smith R. Functional implications of the emergence of alternative splicing in hnRNP A/B transcripts. RNA. 2010;16:1760–1768. doi: 10.1261/rna.2142810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen K, Lareau L, Blanchette M, et al. (11 co-authors) Genome-wide identification of alternative splice forms down-regulated by nonsense-mediated mRNA decay in Drosophila. PLoS Genet. 2009;5:e1000525. doi: 10.1371/journal.pgen.1000525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Denoeud F, et al. (14 co-authors) GENCODE: producing a reference annotation for ENCODE. Genome Biol. 2006;7:S4. doi: 10.1186/gb-2006-7-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haverty P, Weng Z, Best N, Auerbach K, Hsiao L, Jensen R, Gullans S. HugeIndex: a database with visualization tools for high-density oligonucleotide array data from normal human tissues. Nucleic Acids Res. 2002;30:214–217. doi: 10.1093/nar/30.1.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller M, Huse K, Szafranski K, Jahn N, Hampe J, Schreiber S, Backofen R, Platzer M. Widespread occurrence of alternative splicing at NAGNAG acceptors contributes to proteome plasticity. Nat Genet. 2004;36:1255–1257. doi: 10.1038/ng1469. [DOI] [PubMed] [Google Scholar]
- Hiller M, Nikolajewa S, Huse K, Szafranski K, Rosenstiel P, Schuster S, Backofen R, Platzer M. TassDB: a database of alternative tandem splice sites. Nucleic Acids Res. 2007;35:D188–D192. doi: 10.1093/nar/gkl762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller M, Szafranski K, Backofen R, Platzer M. Alternative splicing at NAGNAG acceptors: simply noise or noise and more? PLoS Genet. 2006;2:e207. doi: 10.1371/journal.pgen.0020207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA, Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2008;4:44. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Hubbard T, Barker D, Birney E, et al. (34 co-authors) The Ensembl genome database project. Nucleic Acids Res. 2002;30:38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isken O, Kim Y, Hosoda N, Mayeur G, Hershey J, Maquat L. Upf1 phosphorylation triggers translational repression during nonsense-mediated mRNA decay. Cell. 2008;133:314–327. doi: 10.1016/j.cell.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isken O, Maquat L. Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev. 2007;21:1833–1856. doi: 10.1101/gad.1566807. [DOI] [PubMed] [Google Scholar]
- Isken O, Maquat L. The multiple lives of NMD factors: balancing roles in gene and genome regulation. Nat Rev Genet. 2008;9:699–712. doi: 10.1038/nrg2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J, Castle J, Garrett-Engele P, Kan Z, Loerch P, Armour C, Santos R, Schadt E, Stoughton R, Shoemaker D. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2003;302:2141–2144. doi: 10.1126/science.1090100. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Nagata K, Suzuki N, Yokoyama R, Yamanaka Y, Kitamura H, Hirano H, Ohara O. Characterization of multiple alternative forms of heterogeneous nuclear ribonucleoprotein K by phosphate-affinity electrophoresis. Proteomics. 2010;10:3884–3895. doi: 10.1002/pmic.201000349. [DOI] [PubMed] [Google Scholar]
- Kriventseva E, Koch I, Apweiler R, Vingron M, Bork P, Gelfand M, Sunyaev S. Increase of functional diversity by alternative splicing. Trends Genet. 2003;19:124–128. doi: 10.1016/S0168-9525(03)00023-4. [DOI] [PubMed] [Google Scholar]
- Lee Y, Lee Y, Kim B, Shin Y, Nam S, Kim P, Kim N, Chung W, Kim J, Lee S. ECgene: an alternative splicing database update. Nucleic Acids Res. 2007;35:D99–D103. doi: 10.1093/nar/gkl992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lievens PM, Tufarelli C, Donady JJ, Stagg A, Neufeld EJ. CASP, a novel, highly conserved alternative-splicing product of the CDP/cut/cux gene, lacks cut-repeat and homeo DNA-binding domains, and interacts with full-length CDP in vitro. Gene. 1997;197:73–81. doi: 10.1016/s0378-1119(97)00243-6. [DOI] [PubMed] [Google Scholar]
- López G, Valencia A, Tress M. firestar—prediction of functionally important residues using structural templates and alignment reliability. Nucleic Acids Res. 2007;35:W573–W577. doi: 10.1093/nar/gkm297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Contreras R, Cloutier P, Shkreta L, Fisette J, Revil T, Chabot B. hnRNP proteins and splicing control. Adv Exp Med Biol. 2007;623:123–147. doi: 10.1007/978-0-387-77374-2_8. [DOI] [PubMed] [Google Scholar]
- Matlin A, Clark F, Smith C. Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol. 2005;6:386–398. doi: 10.1038/nrm1645. [DOI] [PubMed] [Google Scholar]
- Melamud E, Moult J. Stochastic noise in splicing machinery. Nucleic Acids Res. 2009;37:4873–4886. doi: 10.1093/nar/gkp471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon R, Omenn G. Proteomic characterization of novel alternative splice variant proteins in human epidermal growth factor receptor 2/neu-induced breast cancers. Cancer Res. 2010;70:3440–3449. doi: 10.1158/0008-5472.CAN-09-2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon R, Zhang Q, Zhang Y, Fermin D, Bardeesy N, DePinho R, Lu C, Hanash S, Omenn G, States D. Identification of novel alternative splice isoforms of circulating proteins in a mouse model of human pancreatic cancer. Cancer Res. 2009;69:300–309. doi: 10.1158/0008-5472.CAN-08-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer AJ, Almendrala DK, Go MM, Krauss SW. Structural protein 4.1R is integrally involved in nuclear envelope protein localization, centrosome—nucleus association and transcriptional signaling. J Cell Sci. 2011;124:1433–1444. doi: 10.1242/jcs.077883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrek B, Lee C. Alternative splicing in the human, mouse and rat genomes is associated with an increased frequency of exon creation and/or loss. Nat Genet. 2003;34:177–180. doi: 10.1038/ng1159. [DOI] [PubMed] [Google Scholar]
- Modrek B, Resch A, Grasso C, Lee C. Genome-wide detection of alternative splicing in expressed sequences of human genes. Nucleic Acids Res. 2001;29:2850–2859. doi: 10.1093/nar/29.13.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore R, Young M, Lee T. Qscore: an algorithm for evaluating SEQUEST database search results. J Am Soc Mass Spectrom. 2002;13:378–386. doi: 10.1016/S1044-0305(02)00352-5. [DOI] [PubMed] [Google Scholar]
- Mudge JM, Frankish A, Fernandez-Banet J, Alioto T, Derrien T, Howald C, Reymond A, Guigo R, Hubbard T, Harrow J. The origins, evolution and functional potential of alternative splicing in vertebrates. Mol Biol Evol. 2011;28:2949–2959. doi: 10.1093/molbev/msr127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan GJ, Guo W, Wormsley S, Helfman DM. Polypyrimidine tract binding protein interacts with sequences involved in alternative splicing of beta-tropomyosin pre-mRNA. J Biol Chem. 1992;267:25480–25487. [PubMed] [Google Scholar]
- Nesvizhskii A, Vitek O, Aebersold R. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat Methods. 2007;4:787–797. doi: 10.1038/nmeth1088. [DOI] [PubMed] [Google Scholar]
- Ning K, Nesvizhskii A. The utility of mass spectrometry-based proteomic data for validation of novel alternative splice forms reconstructed from RNA-Seq data: a preliminary assessment. BMC Bioinformatics. 2010;11:S14. doi: 10.1186/1471-2105-11-S11-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman N, Sharpless NE. INK4a/ARF: a multifunctional tumor suppressor locus. Mutat Res. 2005;576:22–38. doi: 10.1016/j.mrfmmm.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Omenn G, States D, Adamski M, et al. (39 co-authors) Overview of the HUPO Plasma Proteome Project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database. Proteomics. 2005;5:3226–3245. doi: 10.1002/pmic.200500358. [DOI] [PubMed] [Google Scholar]
- Pan Q. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Revealing global regulatory features of mammalian alternative splicing using a quantitative microarray platform. Mol Cell. 2004;16:929–941. doi: 10.1016/j.molcel.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Power K, McRedmond J, de Stefani A, Gallagher W, Gaora PO. High-throughput proteomics detection of novel splice isoforms in human platelets. PLoS One. 2009;4:e5001. doi: 10.1371/journal.pone.0005001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina S, Palermo V, Guarnera A, Guarino F, Messina A, Mazzoni C, De Pinto V. Swapping of the N-terminus of VDAC1 with VDAC3 restores full activity of the channel and confers anti-aging features to the cell. FEBS Lett. 2010;584:2837–2844. doi: 10.1016/j.febslet.2010.04.066. [DOI] [PubMed] [Google Scholar]
- Severing E, van Dijk A, Stiekema W, van Ham R. Comparative analysis indicates that alternative splicing in plants has a limited role in functional expansion of the proteome. BMC Genomics. 2009;10:154. doi: 10.1186/1471-2164-10-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severing E, van Dijk A, van Ham R. Assessing the contribution of alternative splicing to proteome diversity in Arabidopsis thaliana using proteomics data. BMC Plant Biol. 2011;11:82. doi: 10.1186/1471-2229-11-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DN, Wilson KL, Simon DN, Wilson KL. The nucleoskeleton as a genome-associated dynamic ‘network of networks’. Nat Rev Mol Cell Biol. 2011;12:695. doi: 10.1038/nrm3207. [DOI] [PubMed] [Google Scholar]
- Smith CW, Valcárcel J. Alternative pre-mRNA splicing: the logic of combinatorial control. Trends Biochem Sci. 2000;25:381–388. doi: 10.1016/s0968-0004(00)01604-2. [DOI] [PubMed] [Google Scholar]
- Southby J, Gooding C, Smith C. Polypyrimidine tract binding protein functions as a repressor to regulate alternative splicing of alpha-actinin mutally exclusive exons. Mol Cell Biol. 1999;19:2699–2711. doi: 10.1128/mcb.19.4.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera D, Vogel C, Orozco M, Teichmann SA, de la Cruz X. The (in)dependence of alternative splicing and gene duplication. PLoS Comput Biol. 2007;3:e33. doi: 10.1371/journal.pcbi.0030033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner S, Shen Z, Ng J, Florea L, Guigó R, Briggs S, Bafna V. Improving gene annotation using peptide mass spectrometry. Genome Res. 2007;17:231–239. doi: 10.1101/gr.5646507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tress M, Bodenmiller B, Aebersold R, Valencia A. Proteomics studies confirm the presence of alternative protein isoforms on a large scale. Genome Biol. 2008;9:R162. doi: 10.1186/gb-2008-9-11-r162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tress M, Martelli P, Frankish A, et al. (45 co-authors) The implications of alternative splicing in the ENCODE protein complement. Proc Natl Acad Sci U S A. 2007;104:5495–5500. doi: 10.1073/pnas.0700800104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tress M, Wesselink J, Frankish A, et al. (11 co-authors) Determination and validation of principal gene products. Bioinformatics. 2008;24:11–17. doi: 10.1093/bioinformatics/btm547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh CH, Asipu A, Bonthron DT, Phillips SE. Structures of alternatively spliced isoforms of human ketohexokinase. Acta Crystallogr D Biol Crystallogr. 2009;65:201–211. doi: 10.1107/S0907444908041115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweedie S, Ashburner M, Falls K, et al. (11 co-authors) FlyBase: enhancing Drosophila Gene Ontology annotations. Nucleic Acids Res. 2009;37:D555–D559. doi: 10.1093/nar/gkn788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables J, Koh C, Froehlich U, et al. (16 co-authors) Multiple and specific mRNA processing targets for the major human hnRNP proteins. Mol Cell Biol. 2008;28:6033–6043. doi: 10.1128/MCB.00726-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J, Côté R, Reisinger F, Foster J, Mueller M, Rameseder J, Hermjakob H, Martens L. A guide to the Proteomics Identifications Database proteomics data repository. Proteomics. 2009;9:4276–4283. doi: 10.1002/pmic.200900402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore S, Schroth G, Burge C. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Meng L, Hu H, Zhang Y, Zhao C, Li Q, Shi F, Wang X, Lin A. Oct-4B isoform is differentially expressed in breast cancer cells: hypermethylation of regulatory elements of Oct-4A suggests an alternative promoter and transcriptional start site for Oct-4B transcription. Biosci Rep. 2011;31:109–115. doi: 10.1042/BSR20100033. [DOI] [PubMed] [Google Scholar]
- Wang Z, Burge C. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–813. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiśniewski J, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Xing Y, Lee C, Xing Y, Lee C. Alternative splicing and RNA selection pressure—evolutionary consequences for eukaryotic genomes. Nat Rev Genet. 2006;7:499. doi: 10.1038/nrg1896. [DOI] [PubMed] [Google Scholar]
- Xue Y, Zhou Y, Wu T, et al. (11 co-authors) Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol Cell. 2009;36:996–1006. doi: 10.1016/j.molcel.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yura K, Shionyu M, Hagino K, et al. (12 co-authors) Alternative splicing in human transcriptome: functional and structural influence on proteins. Gene. 2006;380:63–71. doi: 10.1016/j.gene.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Zheng D, Frankish A, Baertsch R, et al. (15 co-authors) Pseudogenes in the ENCODE regions: consensus annotation, analysis of transcription, and evolution. Genome Res. 2007;17:839–851. doi: 10.1101/gr.5586307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.