

The ribosomal RNA of Escherichia coli contains 24 methylated residues. A set of 22 methyltransferases responsible for the modification of 23 residues has been described previously. The authors report the identification of the 24th enzyme that modifies the 23S rRNA, at nucleotide A2030.
Keywords: rRNA, modification, methylation, yhiR, N6-methyl adenosine
Abstract
The ribosomal RNA (rRNA) of Escherichia coli contains 24 methylated residues. A set of 22 methyltransferases responsible for modification of 23 residues has been described previously. Herein we report the identification of the yhiR gene as encoding the enzyme that modifies the 23S rRNA nucleotide A2030, the last methylated rRNA nucleotide whose modification enzyme was not known. YhiR prefers protein-free 23S rRNA to ribonucleoprotein particles containing only part of the 50S subunit proteins and does not methylate the assembled 50S subunit. We suggest renaming the yhiR gene to rlmJ according to the rRNA methyltransferase nomenclature. The phenotype of yhiR knockout gene is very mild under various growth conditions and at the stationary phase, except for a small growth advantage at anaerobic conditions. Only minor changes in the total E. coli proteome could be observed in a cell devoid of the 23S rRNA nucleotide A2030 methylation.
INTRODUCTION
Ribosomal RNAs (rRNAs) contain modified nucleotides distributed within their functional centers. In the rRNA of Escherichia coli, 36 modified nucleotides are represented by 10 pseudouridines, one dihydrouridine, and one unknown modification, while the rest of the 24 modified nucleotides are methylated ones (Sergiev et al. 2011). After the introduction of a convenient gene inactivation method based on the lambda Red system (Datsenko and Wanner 2000), a comprehensive collection of E. coli gene knockout strains was created (Baba et al. 2006). This methodological advance accelerated the discovery of genes responsible for rRNA modification. By early 2012 (Basturea et al. 2012), only for one methylated rRNA residue, m6A2030 of the 23S rRNA (Branlant et al. 1981), did the methyltransferase remain unknown.
The nucleotide m6A2030 is buried inside the large ribosomal subunit close to the peptidyltransferase center (PTC) and on the way between the PTC and the elongation factor binding site (Fig. 1; Schuwirth et al. 2005; Sergiev et al. 2005a,b). It forms a strong stacked contact with the U571 residue of the 23S rRNA, thus connecting structural elements of domains II and V located at half-23S rRNA length distance in the primary structure. Curiously, in the archaea Haloarcula marismortui, the equivalent of A2030 is unmethylated, but the residue located at a position equal to U571 is methylated (Ban et al. 2000; Kirpekar et al. 2005). Thus, the methyl group that enhances interdomain interactions occupies the same position in the tertiary structure of the large ribosomal subunit, while being attached to completely different nucleotide residues.
FIGURE 1.

Location of the methylated residue m6A2030 of the 23S rRNA in the 50S subunit of E. coli. Shown is a structure of the E. coli 50S ribosomal subunit as viewed from the 30S side. Modified nucleotide m6A2030 is shown as dark gray van der Waals sphere model. (Inset) A closer view of the interacting m6A2030 and U571 nucleotides.
In this work, we demonstrate that the yhiR gene of E. coli is solely responsible for the modification of A2030 of the 23S rRNA. The identification of the yhiR target nucleotide was followed by a comprehensive proteome study of a strain devoid of yhiR. The expression of a very small subset of genes was shown to be affected by yhiR inactivation, while the majority of the proteome remained independent of the A2030 modification.
RESULTS
YhiR methylates 23S rRNA in vitro
The recombinant protein YhiR, containing a His6 tag on its C terminus, was expressed and purified from an E. coli strain AG1 carrying the pCA24YhiR plasmid (Kitagawa et al. 2005). The resulting protein was pure, according to the SDS-PAGE, and was thus used to modify the potential substrates. The substrates were prepared from the strain JW3466 lacking the yhiR gene on the chromosome (termed hereafter as ΔyhiR) (Baba et al. 2006). The cell lysate was fractionated into a ribosome and S100 by ultracentrifugation. The ribosomes were split into subunits. Each of the ribosomal subunit fractions, as well as the S100 fraction, were phenol-deproteinized. Thus, five putative substrate groups were generated: large and small ribosomal subunits, 16S rRNA, 23S + 5S rRNA, and the entire small RNA fraction. All the substrate fractions were mixed with recombinant YhiR protein and radioactive S-adenosyl –L-[methyl-3H]methionine ([3H]SAM). The reactions were then analyzed for tritium incorporation. The results showed that the deproteinized 23S rRNA is modified by YhiR in vitro. Assembled 50S or 30S subunits could be modified only marginally (Fig. 2).
FIGURE 2.

In vitro modification of potential YhiR substrates prepared from the ΔyhiR strain. Methylation of the 50S subunits, NH4Cl/ethanol split particles, LiCl split particles, and deproteinized 23S rRNA prepared from the ΔyhiR strain.
To test whether partially deproteinized 50S subunits are better substrates for YhiR, we prepared LiCl and NH4Cl/ethanol split particles. NH4Cl/ethanol split particles lack proteins L1, L5, L6, L7, L10, L11, L16, L25, L31, and L33 (Nierhaus 1990). The treatment of the 50S subunit with 3.5M LiCl is known to remove L9, L14, L15, L18, L19, L24, L27, L28, L30, and L32 proteins (Nierhaus 1990) in addition to those removed by NH4Cl/ethanol, retaining only 23S rRNA and proteins L2, L3, L4, L13, L17, L20, L21, L22, L23, L29, and L34. Although the split particles do not exactly match in vivo assembly intermediates, they could be used as assembly intermediate mimics (Nierhaus 1990). In vitro the modification of LiCl and NH4Cl/ethanol split particles (Fig. 2) revealed that their modification efficiency is intermediate between the 23S rRNA and the assembled 50S subunits. Modification of NH4Cl/ethanol split particles was somewhat better than that of particles split with LiCl despite the fact that the former possess more ribosomal proteins. Either a small residual amount of LiCl was inhibiting for modification or partial deproteinization with LiCl was more damaging to the structure of 23S rRNA. Nascent 23S rRNA seems to be the natural substrate for YhiR. This result is in agreement with an earlier study (Siibak and Remme 2010) indicating that modification of the 23S rRNA nucleotide A2030 occurs early in the 50S subunit assembly.
YhiR is responsible for N6-methylation of nucleotide A2030 of the 23S rRNA
Only the 23S rRNA methylated nucleotide m6A2030 still lacked the identified methyltransferase responsible for its modification (Basturea et al. 2012). To prove that the YhiR protein is responsible for the modification of the nucleotide A2030 of the 23S rRNA, we purified the 23S rRNA from the wild-type strain, the ΔyhiR strain, and the ΔyhiR strain, transformed with the plasmid that encodes the YhiR protein. Additionally, we purified the 23S rRNA and the 50S subunits from the ΔyhiR strain, treated these samples with recombinant YhiR and SAM, and analyzed the 23S rRNA extracted from the YhiR-treated samples. All 23S rRNA samples were hybridized with the oligodeoxyribonucleotide, complementary to its 2017- to 2053-nt region, and were treated with the mixture of RNase A and T2 to cleave all 23S rRNA parts outside the hybridization area. After the inactivation of RNases and the removal of nucleotides, the sample was treated with DNase I to remove the DNA oligonucleotide and was purified by gel electrophoresis: The 23S rRNA fragment was hydrolyzed with the RNase A and subjected to matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (Fig. 3). One fragment, 2029GAAGAUp, was found to be methylated in the wild-type strain, as expected (Fig. 3). The site of modification in this fragment was previously shown to be Gm6AAGAU (Branlant et al. 1981). The inactivation of the yhiR gene resulted in the complete loss of A2030 modification (Fig. 3) The expression of the yhiR gene from the plasmid restored the A2030 modification. Thus, the inactivation of the yhiR gene, but not any other possible changes caused by yhiR gene substitution with the kanamycin-resistance cassette on a chromosome, resulted in the loss of the 23S rRNA nucleotide A2030 modification. The analysis of the 23S rRNA from the ΔyhiR strain treated with recombinant YhiR and SAM in vitro revealed that YhiR by itself, without any additional proteins, is able to modify 23S rRNA (Fig. 3). It is obvious that the assembled 50S subunit is not a substrate for the YhiR enzyme, according to the previous data obtained with the help of rRNA isotope labeling with [3H]SAM (Fig. 2). An increased background level of 30S and 50S subunit modification in the assay with [3H]SAM may indicate some nonspecific methyltransferase activity of YhiR in vitro at high enzyme concentrations, similar to those observed for RluC and RluD activity in vitro at high enzyme concentrations (Huang et al. 1998). The results of mass spectrometry show explicitly that the YhiR protein alone is necessary and sufficient for the 23S rRNA methylation at the nucleotide A2030.
FIGURE 3.

MALDI MS analysis of the RNase A digest of the 23S rRNA fragment's 2017–2053 nt. The part of spectrum around mass/charge (m/z) 1990–2050 is shown. MALDI spectra correspond to fragments of the 23S from the (A) wild-type strain; (B) ΔyhiR strain; (C) ΔyhiR strain, transformed with pCA24YhiR; (D) 50S subunits from the ΔyhiR strain, treated with SAM and recombinant YhiR in vitro; and (E) deproteinized 23S rRNA from the ΔyhiR strain, treated with SAM and recombinant YhiR in vitro. Oligonucleotides corresponding to peaks are indicated.
Phenotype of the yhiR gene knockout
We examined the way in which the modified nucleotide m6A2030 of 23S rRNA influences E. coli cell fitness under different growth conditions. The E. coli doubling time was measured at 30°C, 37°C, and 42°C in LB-rich media and at 37°C and 42°C in M9 poor media (Table 1). The comparison of the growth of the wild-type and ΔyhiR strains revealed no significant difference. Additionally, growth rates were monitored at anaerobic conditions at LB media, 37°C (Table 1; Fig. 4B). Although the growth rates at the early log phase were identical (see Table 1), the wild-type strain stopped growth at a smaller optical density than the ΔyhiR strain (Fig. 4B).
TABLE 1.
Proteins whose quantity was affected by yhiR gene inactivation
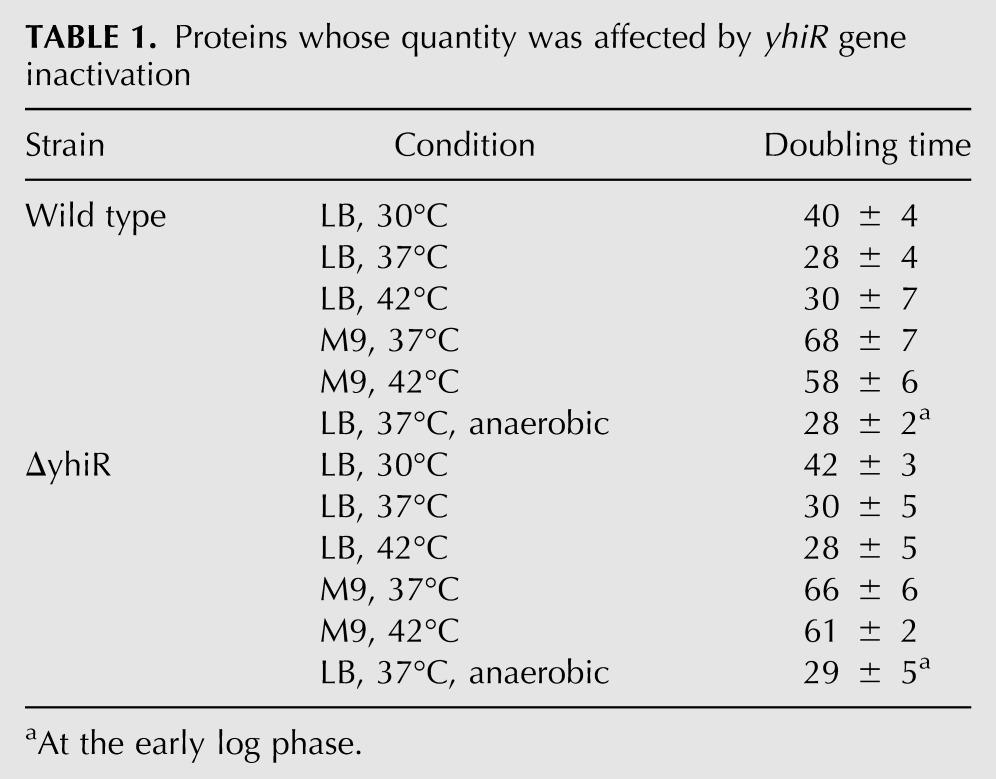
FIGURE 4.

(A) Cell titer drop in the logarithmic scale after incubation of bacterial cultures over time. Data points and approximation curves for the wild-type strain are shown in black, while those for the ΔyhiR strain are shown in gray. Squares correspond to incubation in LB at 42°C. Stars correspond to incubation in LB at 37°C. Diamonds correspond to incubation in LB at 30°C. Circles correspond to incubation in M9 at 42°C. Crosses correspond to incubation in M9 at 37°C. Triangles correspond to incubation in M9 at 30°C. Additionally, incubation conditions are indicated on the right side of the figure at the ends of the corresponding approximation curves. (B) Growth of the wild-type strain (black curve) and the ΔyhiR strain (gray curve) at anaerobic conditions. Error bars, SDs from an average of five independently grown cultures.
In addition, we checked the ability of the wild-type strain to compete for growth with the ΔyhiR strain. Both rich LB and minimal M9 media were used for growth competition assays at 30°C, 37°C, and 42°C. Equal numbers of cells of the kanamycin-sensitive parental strain and kanamycin-resistant ΔyhiR strain (A600 0.001) were mixed and grown for 24 h. The growth cycles were repeated so that at the end of each cycle 1/1000th of the media containing the mixture of the cells was used to re-inoculate fresh media with 1000-fold dilution. The cells amplify 1000-fold with each growth cycle, which corresponds to about 10 doublings. After each growth cycle, titers of the wild-type and ΔyhiR strains were measured. Under all conditions tested, no growth disadvantage could be revealed for the ΔyhiR strain, as the proportions of ΔyhiR to wild-type cells fluctuated randomly at all growth cycles (data not shown).
The only known phenotype of the ΔyhiR strain described in the scientific literature is its inability to grow using DNA as a sole nutrition source (Palchevskiy and Finkel 2006) and decreased survival in the stationary phase (Palchevskiy and Finkel 2006). Here we decided to test the survival of the ΔyhiR strain in the stationary phase at 25°C, 37°C, and 42°C in rich LB and poor M9 media (see Fig. 4A). No significant difference in the survival of the wild-type and ΔyhiR strains was revealed under all conditions tested.
Lack of modification at the 23S rRNA nucleotide: A2030 does not cause the accumulation of assembly intermediates
The deficiency in several rRNA modification enzymes is known to affect the ribosome assembly (Hager et al. 2002; Connolly et al. 2008; Mangat and Brown 2008; Xu et al. 2008) and lead to the accumulation of assembly intermediates in vivo (Hager et al. 2002; Gutgsell et al. 2005), although the latter could sometimes be an indirect consequence of a functional defect of an unmodified ribosome in a specific genetic context (O'Connor and Gregory 2011). The accumulation of such intermediates could be visualized by sucrose gradient centrifugation. We analyzed the optical density profiles after sucrose gradient centrifugation for the lysates of the wild-type and the ΔyhiR strains. Both profiles were identical at high and low magnesium ion concentrations, and thus it is apparent that YhiR does not affect the rate of ribosome subunit assembly (see Supplemental Fig. S1).
Proteome analysis of the yhiR knockout strain
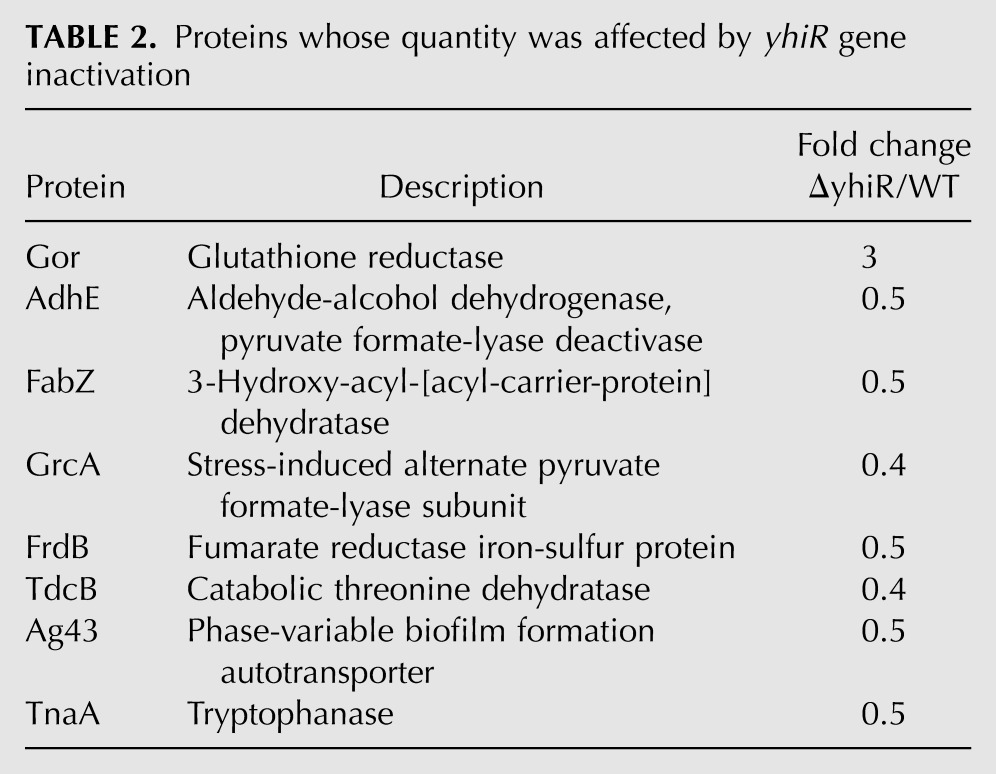
To reveal fine differences in the translation efficiencies of cellular mRNA caused by the A2030 modification, we compared the proteomes of wild-type cells and cells lacking the yhiR gene (see Supplemental Fig. S2). We found only a very limited number of proteins whose abundance was dependent on the modification of A2030 (Table 2). The only protein in this list whose synthesis is regulated by a well-known mechanism related to translation is tryptophanase (TnaA) (Stewart and Yanofsky 1986; Konan and Yanofsky 1997).
TABLE 2.
Proteins whose quantity was affected by yhiR gene inactivation
To check if the lack of A2030 modification represses tnaA gene expression by interfering with tnaC stop peptide stalling, we constructed a reporter plasmid coding for the Cerulean fluorescent protein (CER) (Rizzo et al. 2004) under the control of a T5 phage promoter and a regulatory region similar to those of the tnaA gene. This region involved tnaC open reading frame coding for a peptide stalling in the ribosomal exit tunnel in the presence of tryptophan. In addition to the CER, the plasmid codes for Red fluorescent protein (RFP) (Merzlyak et al. 2007) under the control of a similar T5 phage promoter served as internal control for gene expression (Osterman et al. 2012). Similarly, we constructed a reporter plasmid coding for CER under the control of the secA regulatory region containing the secM gene (Nakatogawa and Ito 2002). The latter gene codes for the peptide stalled in the ribosomal tunnel in the case of decreased efficiency in the protein export machinery.
We checked whether CER reporters, placed under the control of tnaA and secA regulatory regions that encoded tnaC and secM stop peptides, were differentially expressed in the wild-type and ΔyhiR strains. Unfortunately, no significant differences in the expression level were found (data not shown).
Another functionally distinct group of proteins whose abundance depends on the 23S rRNA nucleotide A2030 modification is the group of proteins whose function is related to iron ions (GrcA, AdhE, FrdB) and anaerobic growth (AdhE, FrdB, TdcB) (Keseler et al. 2011). All these groups of genes are down-regulated in the ΔyhiR strain relative to the wild-type strain, according to the comparative proteome analysis.
DISCUSSION
In this work, we identified the yhiR gene as the coding for the last unknown rRNA methyltransferase, responsible for the formation of m6A2030. The results of the experiments in vivo and in vitro demonstrated clearly that YhiR is necessary and sufficient for the methylation of the nucleotide A2030 of 23S rRNA. We hereby suggest renaming the yhiR gene to rlmJ according to the rRNA methyltransferase nomenclature. The substrate preference in vitro was shown to be deproteinized 23S rRNA; thus, it is likely that the in vivo substrates are early assembly intermediates or an immediately transcribed 23S rRNA. No other modification substrates for YhiR could be detected. A complete modification of the nucleotide A2030 could be observed in the wild-type strain. The challenging question of how enzymes like YhiR could complete the modification of rRNA before the incorporation of ribosomal proteins into the subunit remains open.
Another intriguing question is the functional role of the modified nucleotide m6A2030 of the 23S rRNA. Orthologs of YhiR can be found in α, β, and γ proteobacteria and in several distant species (Dehal et al. 2010; Szklarczyk et al. 2011). The latter cases are most likely a result of either horizontal gene transfer or misidentification of false orthologs. Despite a rather narrow distribution of orthologous genes, an interesting observation could be made. In archaea H. marismortui, the 23S rRNA nucleotide C2071, which occupies a position equivalent to A2030 of E. coli, is unmethylated. However, the nucleotide A628 of H. marismortui 23S rRNA (the E. coli equivalent is U571), which forms a conserved contact with C2071 (Ban et al. 2000; Schuwirth et al. 2005), is methylated (Kirpekar et al. 2005). This is an intriguing example of a methyl group location in the same spatial position in such diverse species, while the nature and sequence context of the modified residues differ so drastically.
A possible role of the modification might be stabilization of the local tertiary structure (Sergiev et al. 2011). We were unable, however, to observe any disadvantages caused by the lack of methylation at any growth conditions, including growth at an elevated temperature. Since the destabilization of folding should be manifested at elevated temperatures, we think that such destabilization is unlikely for 50S subunits unmodified at A2030. A high-throughput screening of the total E. coli knockout collection for growth at unfavorable conditions does not reveal any significant difference in the wild-type and ΔyhiR strains growth at any temperature up to 45°C (Nichols et al. 2011).
Some rRNA methyltransferases are known to play the role of switch proteins in the ribosome assembly. The inactivation of genes coding for such enzymes causes the accumulation of assembly intermediates (Hager et al. 2002; Gutgsell et al. 2005; Connolly et al. 2008; Xu et al. 2008; Nichols et al. 2011) and cold sensitivity (Connolly et al. 2008; Xu et al. 2008; Nichols et al. 2011). The inactivation of the yhiR gene does not lead to either of these phenotypes. No assembly intermediates could be detected by the sucrose density centrifugation, and no growth retardation relative to the wild type could be observed in our experiments at 30°C and in the high-throughput screening experiments at several temperatures down to 16°C (Nichols et al. 2011). We can conclude that it is unlikely that YhiR activity is necessary for ribosome assembly.
The only phenotype associated with the yhiR gene knockout described in the literature is the inability to grow using pure DNA as the only source of nutrition and decreased survival in the stationary phase (Palchevskiy and Finkel 2006). We did not observe a decrease in the stationary phase survival for a ΔyhiR strain relative to the wild-type strain for up to 10 d of incubation. The overall survival in the stationary phase depended on the temperature of incubation in the way that high temperatures decreased the survival. Within the 10-d incubation at 42°C, the titer of survived cells dropped two orders of magnitude, and even at such harsh conditions, we could not detect the difference in survival between the wild-type and the ΔyhiR strains. It might be that the difference in the stationary state survival could be detected at longer incubation rounds, as it occurred in the original experiment by Palchevskiy and Finkel (2006).
Comparative proteome analysis of the wild-type strain and the ΔyhiR strain revealed several proteins whose abundance differed. Among them, only the tnaA gene is known to be regulated by a translation-dependent mechanism (Stewart and Yanofsky 1986; Konan and Yanofsky 1997). To confirm the results of the comparative proteome analysis by an independent method, we created two reporter constructions based on dual CER/RFP fluorescent proteins (Osterman et al. 2012). The latter served as the internal control, while the CER gene expression was controlled by either the tnaA (Stewart and Yanofsky 1986; Konan and Yanofsky 1997) or the secA (Nakatogawa and Ito 2002) regulatory region. These two genes are regulated by ribosome stalling on the preceding tnaC and secM reading frames. No differences in the activity of the tnaA or secA regulatory regions were found between the wild-type and ΔyhiR strains. Thus, we think it is unlikely that the 23S rRNA nucleotide A2030 modification influences the stalling of tnaC and secM stop peptides in the ribosomal tunnel.
Another group of proteins whose quantity changed in the strain devoid of the yhiR gene is involved in anaerobic growth and mechanisms with Fe2+ ion participation (Keseler et al. 2011). This group of proteins demonstrates decreased abundance in the ΔyhiR strain. Surprisingly, we observed not an inhibition but an advantage of ΔyhiR strain growth at anaerobic conditions (Fig. 4B). We can hypothesize that rRNA methylation at A2030 could be involved in the regulation of gene expression at oxygen limitation. Although we do not have a straightforward explanation of the mechanism of how rRNA modification could influence Fe2+ utilization, a study of Vecerek et al. (2007) proposes a translation-related mechanism for the regulation of intracellular Fe2+ concentration. We can hypothesize that 23S rRNA nucleotide A2030 modification could influence the efficiency of this regulatory mechanism. Definitely, the modification of A2030 is beneficial for E. coli vitality at certain conditions, since bacteria maintain a gene for this modification. Obviously, the variety of environmental conditions of natural habitats where bacteria are found exceeds the limits testable in a laboratory.
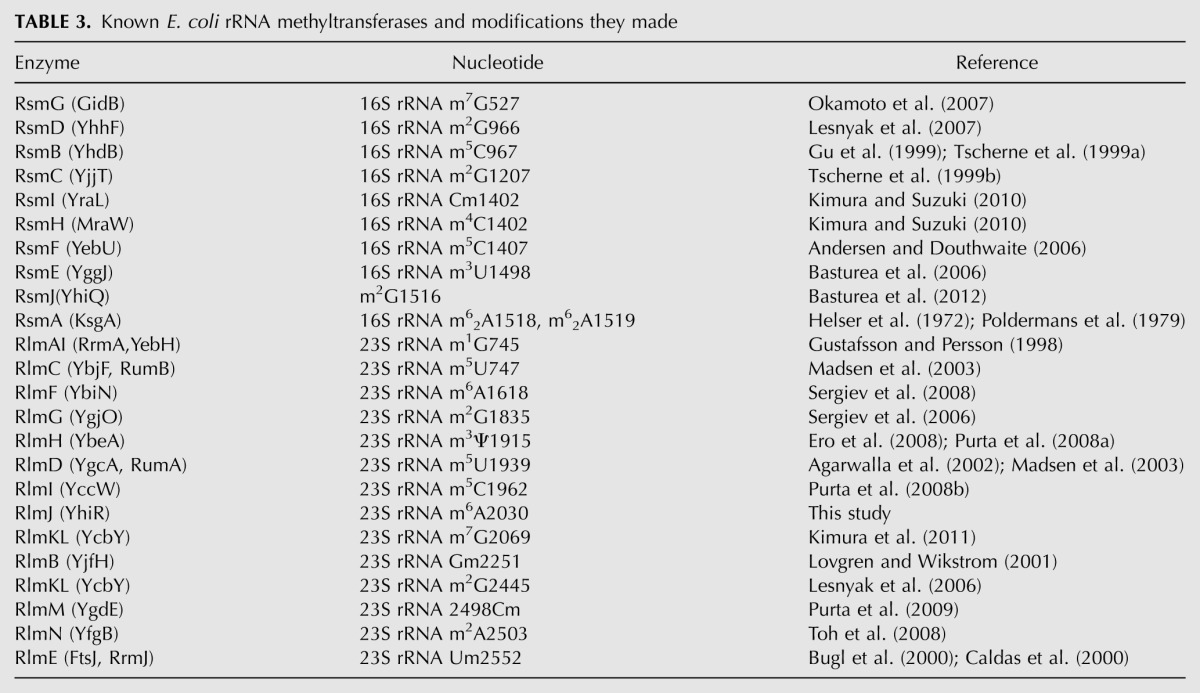
In the present report, we have completed the list of E. coli rRNA methyltransferases (Table 3). The majority, i.e., 18 out of the rRNA methyltransferases described (including YhiR), are monospecific in both ways; i.e., a single methyltransferase corresponds to a single modified nucleotide. Two methyltransferases modify two rRNA residues each. KsgA makes two identical modifications, m26A1518 and m26A1519 (Helser et al. 1972; Poldermans et al. 1979), while RlmKL is responsible for the formation of two differently modified nucleotides, m7G2069 (Kimura et al. 2011) and m2G2445 (Lesnyak et al. 2006). The two rRNA methyltransferases, RsmI and RsmH (Kimura and Suzuki 2010), introduce two chemically distinct modifications into the same C1402 nucleotide of the 23S rRNA. One methyltransferase, RlmH (Ero et al. 2008; Purta et al. 2008a), modifies pseudouridine residue. The result of this work is that no more known E. coli rRNA methylation sites are left with a nonidentified methyltransferase gene. However, several rRNA modification enzymes remained undiscovered. It is still unknown which genes are responsible for the formation of D2449 and oh5C2501 (Havelund et al. 2011). Also, it is likely that some yet-unknown modification sites, perhaps active only under certain conditions, still could be found in the ribosome (Sergiev et al. 2012). An even larger and probably much more interesting field is now open for study, i.e., the functional role of rRNA modification.
TABLE 3.
Known E. coli rRNA methyltransferases and modifications they made
MATERIALS AND METHODS
Strains and plasmids
The BW25141 parental strain (Datsenko and Wanner 2000) and the JW2559 strain, carrying a kanR cassette inserted at the place of the yhiR gene (Baba et al. 2006), as well as the pCA24yhiR plasmid (Kitagawa et al. 2005), were kindly provided by Dr. H. Mori.
Protein preparation
The recombinant N-terminal hexahistidine tagged YhiR protein was prepared from M15 E. coli cells, harboring the pCA24N plasmid with the yhiR gene cloned under control of the T5lac promoter (Kitagawa et al. 2005). The cells were grown in 500 mL LB media at 37°C until A600 0.4 and induced by IPTG, 1 mM final concentration (f.c.). After the induction, the cells were grown for an additional 3.5 h and were harvested, washed, and then lysed by sonication in buffer 1 (20 mM Hepes K at pH 7.5, 1 mM Mg(OAc)2, 200 mM NH4OAc). Protein purification on 1 mL Ni-NTA agarose (Qiagen) was done in the same buffer with 10 mM imidazole added. After washing four times in buffer 1 containing 20 mM imidazole, the protein was eluted by increasing imidazole concentration up to 500 mM. The YhiR purity was proven by SDS-PAGE.
Substrate preparation
To prepare YhiR putative substrates, wild-type cells and cells of the JW2559 strain were grown to A600 0.8 in LB media at 37°C. The cells were harvested by centrifugation, washed by buffer 2 (20 mM Hepes K at pH 7.5, 1 mM Mg(OAc)2, 200 mM NH4OAc, 4 mM 2-mercaptoethanol), and lysed by sonication in the same buffer. The cell debris was pelleted by 40-min centrifugation at 15000 rpm in a JA20 rotor. The lysates were subjected to 2-h ultracentrifugation in a MLA-80 rotor at 80,000 rpm with a further resuspension of the ribosomal pellet in buffer 2. The ribosomes and S100 were used to prepare ribosomal subunits, ribosomal core particles, rRNA, and total tRNA. The ribosomal subunits were prepared by 20-h ultracentrifugation in 10%–30% sucrose gradient in buffer 2 in a SW32Ti rotor at 18000 rpm. The 30S and 50S subunits were fractionated according to A260 optical density and then were harvested in a Ti70 rotor at 30000 rpm for 14 h.
To get core particles P37, the NH4Cl/EtOH-split procedure (Nierhaus 1990) was used. 70 A260 units of 50S subunits were diluted in a 1 mL buffer containing 10 mM imidazole-HCl (pH 7.4), 1.5 M NH4Cl, 20 mM MgCl2, and 1 mM 2-mercaptoethanol to f.c. 35 A260 units/mL and then were warmed up to 37°C. Prewarmed ethanol (0.5 mL) was added twice and shaken gently for 5 min after each addition. The core particles were harvested at 16,000g for 30 min. The pellet was resuspended in buffer containing 20 mM Tris-HCl (pH 7.4), 4 mM Mg(OAc)2, 400 mM NH4Cl, 0.2 mM EDTA, and 5 mM 2-mercaptoethanol, and resulting core particles were dialyzed in the same buffer.
To obtain 3.5 core particles, a LiCl-split procedure was used (Nierhaus 1990). Eighty A260 units were diluted in 4.5 mL of buffer containing 10 mM Tris-HCl (pH 7.6 at 0°C), 10 mM Mg(OAc)2, and 3.5 LiCl. The mixture was incubated for 5 h at 0°C and shaken gently every hour. The core particles were pelleted by centrifugation at 40,000 rpm in a MLA-80 rotor for 5 h. The pellet was resuspended in 200 μL of storage buffer: 20 mM Tris-HCl (pH 7.4), 20 mM Mg(OAc)2, 400 mM NH4Cl, 1 mM EDTA, and 5 mM 2-mercaptoethanol.
Naked RNAs were prepared from S100 and ribosomal subunits by a two-step phenol extraction in buffer 300 mM NaOAc, 0.5% SDS, and 5 mM EDTA, followed by ethanol precipitation.
In vitro methylation
For in vitro methylation, 100 pmol of ribosomal subunits, rRNA, or equal volume ratio of total tRNA was mixed with 2 mkCi [3H]SAM (GE Healthcare) and 10 pmol of recombinant YhiR protein in 100 μL volume of buffer 2. After 30 min of incubation at 37°C, the reaction was quenched by an equal volume of 300 mM NaOAc, 0.5% SDS, and 5 mM EDTA, and RNA was extracted twice by phenol. Ethanol precipitation was performed twice to remove the residual [3H]SAM. The radioactivity of tritium incorporated into RNA was measured by scintillation counting.
To obtain rRNA fragments that contain the nucleotide A2030 and that are appropriate for MALDI analysis, a special digestion method was chosen. We modified the method described by Andersen and Douthwaite (2006) in the following way: 120 pmol of 23S/5S rRNA were diluted in a hybridization buffer containing 75 mM HEPES and 150 mM KCl together with 600 pmol of oligonucleotide, complementary to the 2017- to 2053-nt region with additional five noncomplementary nucleotides at both ends (attatCGGGTACACTGCATCTTCACAGCGAGTTCAtaaag). Thirty microliters of this mixture was heated for 5 min at 90°C and then slowly cooled to 37°C. Hybridized probes were treated with a mixture of 0.15 μg RNase A, 0.01 unit RNase T2, and 1.5 units RNase T1 at 37°C for 1 h. The reaction was extracted twice with phenol/chloroform with further ethanol precipitation. To get rid of DNA oligonucleotide, the precipitate was diluted in 50 μL of the buffer for DNase I and treated with 5 units of DNase I (Fermentas) for 30 min at 37°C. The reaction was phenol-extracted and precipitated by adding LiClO4 to f.c. of 0.5 M and 5 volumes of acetone. The resulting RNA fragments were diluted in 10 μL of RNA Loading Dye (Fermentas) and separated in a 12% polyacrylamide gel containing 7 M urea. Visualization of the bands was made by ethidium bromide staining. An appropriate band was retrieved by elution at 0°C in 0.5 M LiClO4 and precipitated with 4 volumes of acetone.
The RNA fragment was additionally digested by 0.5 μg of RNase A in 4 μL of 10 mM ammonium citrate. The reaction was stopped by adding 0.8 μL 0f 0.5M HCl at room temperature for 30 min with further drying under vacuum. The digested RNA fragments were diluted in 2 μL of H2O and were analyzed by MALDI MS.
Examination of cell growth and survival characteristics
Growth rate measurements and growth competition experiments were carried out for 10 d, as previously described for YfiC methyltransferase (Golovina et al. 2009). Cell titers were calculated.
To examine cell survival in the stationary phase in different conditions, both wild-type cells and the cells of the JW2559 strain were inoculated in LB or M9 media at 37°C, and such fresh cultures were kept at 25°C, 37°C, or 42°C for 1–10 d. Cell titers were also calculated. Growth at anaerobic conditions was monitored in the sealed titer plate without any air left. Monitoring was done with the help of a Perkin Elmer Victor X5 plate reader.
Proteomics
Two-dimensional gel electrophoresis of total proteins was carried out as follows. The cell pellets were disrupted by sonication for 5 min at 4°C in ice water and centrifuged (10 min at 15,000g) to discard insoluble particles. The samples (10 μL) were dissolved in buffer: 8 M urea, 2 M thiourea, 4% CHAPS, and 30 mM TrisHCl (pH 8.5). The protein concentration was determined using Quick start Bradford dye reagent (Bio-Rad). Fifty micrograms of the sample proteins was labeled with 400 pmol of either Cy3 or Cy5 CyDye DIGE Fluor minimal dyes (Amersham Biosciences) according to the manufacturer's instructions. After the equalization of Cy3 and Cy5 labeled proteins, the samples were mixed, and 100 mM DTT (Bio-Rad) and 2% (v/v) Ampholine 3-10 (Bio-Rad) were added. Isoelectric focusing (IEF) was performed using tube gels (20 cm × 1.5 mm) containing carrier ampholytes and applying a voltage gradient in an IEF chamber. After IEF, the ejected tube gels were incubated in equilibration buffer (125 mM TrisHCl, 40% [w/v] glycerol, 3% [w/v] SDS, 65 mM DTT at pH 6.8) for 10 min. The tube gels were placed onto polyacrylamide gels (9%–16%) of 1.5-mm thickness, 20 × 18 cm (Protean II Multi-Cell, Bio-Rad), and fixed using 1.0% (w/v) agarose containing 0.01% (w/v) bromphenol blue. The electrophoresis was carried out for 12–14 h, and then the gels were scanned using a Typhoon Trio Imager at 200-dpi resolutions (Amersham Biosciences). Only afterward were the gels fixed and silver stained as described (Shevchenko et al. 1996). Image analysis was performed using an Imagemaster DIGE (GE Healthcare). Two independent experiments were performed for each experimental setup. Spot quantities were normalized to remove nonexpression-related variations in spot intensity. At least a twofold change in spot normalized fluorescence between the two sets of proteins was recognized as the significant difference in their expression levels.
Plasmids and dual fluorescent proteins reporter assay
pRFPCER, a dual fluorescent protein reporter made in our laboratory (Osterman et al. 2012), was used as host vector for construction reporter plasmids. pRFPCER was digested with NdeI and SacII restriction enzymes, and the obtained linearized vector was ligated with digested (NdeI and SacII) PCR products of 5′ UTR regions of the secM and tnaA genes, obtained with following primers:
TnaA F 5′-CCGCGGTGTCTTGCGAGGATAAGTG,
TnaA R 5′-CATATGATAATCCTTCATTTATTTTAATTACAG,
SecM F 5′-CCGCGGATCGGGATGGCAATAACGTG, and
SecM R 5′-CATATGTAGCATAATAAAATCTCAAACGC.
Chemically competent cells made from BW25113 and ΔyhiR were transformed with TnaA and SecM reporter plasmids, and the Cerulean/RFP fluorescent ratio was measured as previously described (Osterman et al. 2012).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Dr. H. Mori, NIG, Japan, for providing us with the knockout strain JW3466 and a plasmid, encoding yhiR. We thank Ekaterina Malashko and Alex Lebedeff for improvement of the English of the manuscript. This work was supported by grants from the Russian Foundation for Basic Research 10-04-01345-a, 11-04-01314-a, 11-04-01018-a, 11-04-12060-ofi, 11-04-91337-nnio_a; the Russian Ministry of Science 16.512.11.2108; Federal Agency for Science and Innovations 02.740.11.0706; and Moscow State University Development Program PNR 5.13.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.034207.112.
REFERENCES
- Agarwalla S, Kealey JT, Santi DV, Stroud RM 2002. Characterization of the 23 S ribosomal RNA m5U1939 methyltransferase from Escherichia coli. J Biol Chem 277: 8835–8840 [DOI] [PubMed] [Google Scholar]
- Andersen NM, Douthwaite S 2006. YebU is a m5C methyltransferase specific for 16 S rRNA nucleotide 1407. J Mol Biol 359: 777–786 [DOI] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol Syst Biol 2: 2006.0008 doi: 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban N, Nissen P, Hansen J, Moore PB, Steitz TA 2000. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science 289: 905–920 [DOI] [PubMed] [Google Scholar]
- Basturea GN, Rudd KE, Deutscher MP 2006. Identification and characterization of RsmE, the founding member of a new RNA base methyltransferase family. RNA 12: 426–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basturea GN, Dague DR, Deutscher MP, Rudd KE 2012. YhiQ is RsmJ, the methyltransferase responsible for methylation of G1516 in 16S rRNA of E. coli. J Mol Biol 415: 16–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branlant C, Krol A, Machatt MA, Pouyet J, Ebel JP, Edwards K, Kossel H 1981. Primary and secondary structures of Escherichia coli MRE 600 23S ribosomal RNA. Comparison with models of secondary structure for maize chloroplast 23S rRNA and for large portions of mouse and human 16S mitochondrial rRNAs. Nucleic Acids Res 9: 4303–4324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugl H, Fauman EB, Staker BL, Zheng F, Kushner SR, Saper MA, Bardwell JC, Jakob U 2000. RNA methylation under heat shock control. Mol Cell 6: 349–360 [DOI] [PubMed] [Google Scholar]
- Caldas T, Binet E, Bouloc P, Costa A, Desgres J, Richarme G 2000. The FtsJ/RrmJ heat shock protein of Escherichia coli is a 23 S ribosomal RNA methyltransferase. J Biol Chem 275: 16414–16419 [DOI] [PubMed] [Google Scholar]
- Connolly K, Rife JP, Culver G 2008. Mechanistic insight into the ribosome biogenesis functions of the ancient protein KsgA. Mol Microbiol 70: 1062–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci 97: 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehal PS, Joachimiak MP, Price MN, Bates JT, Baumohl JK, Chivian D, Friedland GD, Huang KH, Keller K, Novichkov PS, et al. 2010. MicrobesOnline: An integrated portal for comparative and functional genomics. Nucleic Acids Res 38: D396–D400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ero R, Peil L, Liiv A, Remme J 2008. Identification of pseudouridine methyltransferase in Escherichia coli. RNA 14: 2223–2233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovina AY, Sergiev PV, Golovin AV, Serebryakova MV, Demina I, Govorun VM, Dontsova OA 2009. The yfiC gene of E. coli encodes an adenine-N6 methyltransferase that specifically modifies A37 of tRNA1Val(cmo5UAC). RNA 15: 1134–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu XR, Gustafsson C, Ku J, Yu M, Santi DV 1999. Identification of the 16S rRNA m5C967 methyltransferase from Escherichia coli. Biochemistry 38: 4053–4057 [DOI] [PubMed] [Google Scholar]
- Gustafsson C, Persson BC 1998. Identification of the rrmA gene encoding the 23S rRNA m1G745 methyltransferase in Escherichia coli and characterization of an m1G745-deficient mutant. J Bacteriol 180: 359–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutgsell NS, Deutscher MP, Ofengand J 2005. The pseudouridine synthase RluD is required for normal ribosome assembly and function in Escherichia coli. RNA 11: 1141–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager J, Staker BL, Bugl H, Jakob U 2002. Active site in RrmJ, a heat shock-induced methyltransferase. J Biol Chem 277: 41978–41986 [DOI] [PubMed] [Google Scholar]
- Havelund JF, Giessing AM, Hansen T, Rasmussen A, Scott LG, Kirpekar F 2011. Identification of 5-hydroxycytidine at position 2501 concludes characterization of modified nucleotides in E. coli 23S rRNA. J Mol Biol 411: 529–536 [DOI] [PubMed] [Google Scholar]
- Helser TL, Davies JE, Dahlberg JE 1972. Mechanism of kasugamycin resistance in Escherichia coli. Nat New Biol 235: 6–9 [DOI] [PubMed] [Google Scholar]
- Huang L, Ku J, Pookanjanatavip M, Gu X, Wang D, Greene PJ, Santi DV 1998. Identification of two Escherichia coli pseudouridine synthases that show multisite specificity for 23S RNA. Biochemistry 37: 15951–15957 [DOI] [PubMed] [Google Scholar]
- Keseler IM, Collado-Vides J, Santos-Zavaleta A, Peralta-Gil M, Gama-Castro S, Muniz-Rascado L, Bonavides-Martinez C, Paley S, Krummenacker M, Altman T, et al. 2011. EcoCyc: A comprehensive database of Escherichia coli biology. Nucleic Acids Res 39: D583–D590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Suzuki T 2010. Fine-tuning of the ribosomal decoding center by conserved methyl-modifications in the Escherichia coli 16S rRNA. Nucleic Acids Res 38: 1341–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Ikeuchi Y, Kitahara K, Sakaguchi Y, Suzuki T 2011. Base methylations in the double-stranded RNA by a fused methyltransferase bearing unwinding activity. Nucleic Acids Res 40: 4071–4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirpekar F, Hansen LH, Rasmussen A, Poehlsgaard J, Vester B 2005. The archaeon Haloarcula marismortui has few modifications in the central parts of its 23S ribosomal RNA. J Mol Biol 348: 563–573 [DOI] [PubMed] [Google Scholar]
- Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H 2005. Complete set of ORF clones of Escherichia coli ASKA library (A complete Set of E. coli K-12 ORF Archive): Unique resources for biological research. DNA Res 12: 291–299 [DOI] [PubMed] [Google Scholar]
- Konan KV, Yanofsky C 1997. Regulation of the Escherichia coli tna operon: Nascent leader peptide control at the tnaC stop codon. J Bacteriol 179: 1774–1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnyak DV, Sergiev PV, Bogdanov AA, Dontsova OA 2006. Identification of Escherichia coli m2G methyltransferases: I. The ycbY gene encodes a methyltransferase specific for G2445 of the 23 S rRNA. J Mol Biol 364: 20–25 [DOI] [PubMed] [Google Scholar]
- Lesnyak DV, Osipiuk J, Skarina T, Sergiev PV, Bogdanov AA, Edwards A, Savchenko A, Joachimiak A, Dontsova OA 2007. Methyltransferase that modifies guanine 966 of the 16 S rRNA: Functional identification and tertiary structure. J Biol Chem 282: 5880–5887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovgren JM, Wikstrom PM 2001. The rlmB gene is essential for formation of Gm2251 in 23S rRNA but not for ribosome maturation in Escherichia coli. J Bacteriol 183: 6957–6960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen CT, Mengel-Jorgensen J, Kirpekar F, Douthwaite S 2003. Identifying the methyltransferases for m5U747 and m5U1939 in 23S rRNA using MALDI mass spectrometry. Nucleic Acids Res 31: 4738–4746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangat CS, Brown ED 2008. Ribosome biogenesis; the KsgA protein throws a methyl-mediated switch in ribosome assembly. Mol Microbiol 70: 1051–1053 [DOI] [PubMed] [Google Scholar]
- Merzlyak EM, Goedhart J, Shcherbo D, Bulina ME, Shcheglov AS, Fradkov AF, Gaintzeva A, Lukyanov KA, Lukyanov S, Gadella TW, et al. 2007. Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat Methods 4: 555–557 [DOI] [PubMed] [Google Scholar]
- Nakatogawa H, Ito K 2002. The ribosomal exit tunnel functions as a discriminating gate. Cell 108: 629–636 [DOI] [PubMed] [Google Scholar]
- Nichols RJ, Sen S, Choo YJ, Beltrao P, Zietek M, Chaba R, Lee S, Kazmierczak KM, Lee KJ, Wong A, et al. 2011. Phenotypic landscape of a bacterial cell. Cell 144: 143–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nierhaus K. 1990. Reconstitution of ribosomes. In Ribosomes and protein synthesis: A practical approach (ed. G Spedding), pp. 161–189. Oxford University Press, Oxford. [Google Scholar]
- O'Connor M, Gregory ST 2011. Inactivation of the RluD pseudouridine synthase has minimal effects on growth and ribosome function in wild-type Escherichia coli and Salmonella enterica. J Bacteriol 193: 154–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto S, Tamaru A, Nakajima C, Nishimura K, Tanaka Y, Tokuyama S, Suzuki Y, Ochi K 2007. Loss of a conserved 7-methylguanosine modification in 16S rRNA confers low-level streptomycin resistance in bacteria. Mol Microbiol 63: 1096–1106 [DOI] [PubMed] [Google Scholar]
- Osterman IA, Prokhorova IV, Sysoev VO, Boykova YV, Efremenkova OV, Svetlov MS, Kolb VA, Bogdanov AA, Sergiev PV, Dontsova OA 2012. Attenuation-based dual-fluorescent-protein reporter for screening translation inhibitors. Antimicrob Agents Chemother 56: 1774–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palchevskiy V, Finkel SE 2006. Escherichia coli competence gene homologs are essential for competitive fitness and the use of DNA as a nutrient. J Bacteriol 188: 3902–3910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poldermans B, Roza L, Van Knippenberg PH 1979. Studies on the function of two adjacent N6,N6-dimethyladenosines near the 3′ end of 16 S ribosomal RNA of Escherichia coli. III. Purification and properties of the methylating enzyme and methylase-30 S interactions. J Biol Chem 254: 9094–9100 [PubMed] [Google Scholar]
- Purta E, Kaminska KH, Kasprzak JM, Bujnicki JM, Douthwaite S 2008a. YbeA is the m3Ψ methyltransferase RlmH that targets nucleotide 1915 in 23S rRNA. RNA 14: 2234–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purta E, O'Connor M, Bujnicki JM, Douthwaite S 2008b. YccW is the m5C methyltransferase specific for 23S rRNA nucleotide 1962. J Mol Biol 383: 641–651 [DOI] [PubMed] [Google Scholar]
- Purta E, O'Connor M, Bujnicki JM, Douthwaite S 2009. YgdE is the 2′-O-ribose methyltransferase RlmM specific for nucleotide C2498 in bacterial 23S rRNA. Mol Microbiol 72: 1147–1158 [DOI] [PubMed] [Google Scholar]
- Rizzo MA, Springer GH, Granada B, Piston DW 2004. An improved cyan fluorescent protein variant useful for FRET. Nat Biotechnol 22: 445–449 [DOI] [PubMed] [Google Scholar]
- Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JH 2005. Structures of the bacterial ribosome at 3.5 A resolution. Science 310: 827–834 [DOI] [PubMed] [Google Scholar]
- Sergiev PV, Bogdanov AA, Dontsova OA 2005a. How can elongation factors EF-G and EF-Tu discriminate the functional state of the ribosome using the same binding site? FEBS Lett 579: 5439–5442 [DOI] [PubMed] [Google Scholar]
- Sergiev PV, Lesnyak DV, Burakovsky DE, Kiparisov SV, Leonov AA, Bogdanov AA, Brimacombe R, Dontsova OA 2005b. Alteration in location of a conserved GTPase-associated center of the ribosome induced by mutagenesis influences the structure of peptidyltransferase center and activity of elongation factor G. J Biol Chem 280: 31882–31889 [DOI] [PubMed] [Google Scholar]
- Sergiev PV, Lesnyak DV, Bogdanov AA, Dontsova OA 2006. Identification of Escherichia coli m2G methyltransferases: II. The ygjO gene encodes a methyltransferase specific for G1835 of the 23 S rRNA. J Mol Biol 364: 26–31 [DOI] [PubMed] [Google Scholar]
- Sergiev PV, Serebryakova MV, Bogdanov AA, Dontsova OA 2008. The ybiN gene of Escherichia coli encodes adenine-N6 methyltransferase specific for modification of A1618 of 23 S ribosomal RNA, a methylated residue located close to the ribosomal exit tunnel. J Mol Biol 375: 291–300 [DOI] [PubMed] [Google Scholar]
- Sergiev PV, Golovina AY, Prokhorova IV, Sergeeva OV, Osterman IA, Nesterchuk MV, Burakovskii DE, Bogdanov AA, Dontsova OA 2011. Modifications of ribosomal RNA: From enzymes to function. In Ribosomes: Structure, function, and dynamics (ed. MV Rodnina et al.), pp. 97–110. Springer, New York [Google Scholar]
- Sergiev PV, Golovina AY, Sergeeva OV, Osterman IA, Nesterchuk MV, Bogdanov AA, Dontsova OA 2012. How much can we learn about the function of bacterial rRNA modification by mining large-scale experimental datasets? Nucleic Acids Res 40: 5694–5705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68: 850–858 [DOI] [PubMed] [Google Scholar]
- Siibak T, Remme J 2010. Subribosomal particle analysis reveals the stages of bacterial ribosome assembly at which rRNA nucleotides are modified. RNA 16: 2023–2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart V, Yanofsky C 1986. Role of leader peptide synthesis in tryptophanase operon expression in Escherichia coli K-12. J Bacteriol 167: 383–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, et al. 2011. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 39: D561–D568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh SM, Xiong L, Bae T, Mankin AS 2008. The methyltransferase YfgB/RlmN is responsible for modification of adenosine 2503 in 23S rRNA. RNA 14: 98–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tscherne JS, Nurse K, Popienick P, Michel H, Sochacki M, Ofengand J 1999a. Purification, cloning, and characterization of the 16S RNA m5C967 methyltransferase from Escherichia coli. Biochemistry 38: 1884–1892 [DOI] [PubMed] [Google Scholar]
- Tscherne JS, Nurse K, Popienick P, Ofengand J 1999b. Purification, cloning, and characterization of the 16 S RNA m2G1207 methyltransferase from Escherichia coli. J Biol Chem 274: 924–929 [DOI] [PubMed] [Google Scholar]
- Vecerek B, Moll I, Blasi U 2007. Control of Fur synthesis by the non-coding RNA RyhB and iron-responsive decoding. EMBO J 26: 965–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, O'Farrell HC, Rife JP, Culver GM 2008. A conserved rRNA methyltransferase regulates ribosome biogenesis. Nat Struct Mol Biol 15: 534–536 [DOI] [PubMed] [Google Scholar]