

The observation was made that dehydroepiandrosterone (DHEA), as the unconjugated steroid, and its sulfate ester (DHEAS) are present in the brain of adult male rats (1). This finding was unforeseen because the rodent steroidogenic glands, including the adrenals, do not secrete significant amounts of DHEA (2). It led to the discovery of a steroid biosynthetic machinery in the nervous system, in charge of producing neurosteroids.
This term, neurosteroids, was proposed in 1981 (3). It applies to the steroids, the accumulation of which occurs in the nervous system independently, at least in part, of supply by the steroidogenic endocrine glands and which can be synthesized de novo in the nervous system from sterol precursors. The steroid precursors along their biosynthetic pathways can be formed in situ and assayed.
This definition applies to DHEA. To demonstrate that brain DHEA is independent of peripheral steroidogenic glands, endocrine manipulations were conducted in rats. Injections for 3 days of long-acting preparations of corticotropin (β1–24 ACTH), to stimulate adrenal steroidogenesis, or of dexamethasone to inhibit endogenous ACTH secretion, were not accompanied by clear-cut changes of brain DHEAS (1). Brain DHEAS was unchanged 1 day after castration, whereas testosterone completely disappeared from the brain. No obvious difference was observed when castrated adrenalectomized males were compared 15 days after operation with sham-operated controls (1, 4).
It thus was logical to assume that DHEA synthesis evolves in two steps
from cholesterol, as described for steroidogenic glands, principally
catalyzed by two different cytochrome P450 enzymes:
cholesterol
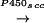 Pregnenolone (PREG)
Pregnenolone (PREG)
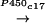 DHEA (Fig. 1).
DHEA (Fig. 1).
Figure 1.

Brain DHEA(S) metabolism. The biosynthesis of PREG and DHEA might proceed from cholesterol either through the classical pathway involving successively cytochromes P450scc and P450c17, or by an alternative pathway involving the intermediacy of sterol and/or steroid hydroperoxides. DHEA is reversibly converted to DHEAS and is the precursor of several metabolites, including sex steroid hormones.
Indeed PREG was found in the brain, at concentrations about 1 order of magnitude larger than those of DHEA, as might be expected from a precursor-to-product relationship (5). The concentrations of PREG and of its sulfate ester PREGS were of the same order of magnitude, and about 10-fold larger than in plasma. They were found at least in part independent of the activity of steroidogenic glands.
Solid evidence has been accrued for the biosynthesis of PREG in the nervous system (reviewed in ref. 6). Incubation of primary cultures of rat forebrain glial cells with a precursor to cholesterol led to the formation of cholesterol, PREG, progesterone, and 20α-dihydro-PREG, and incubation of rat oligodendrocyte mitochondria with cholesterol yielded pregnenolone (7, 8). There is only one cholesterol side-chain cleavage enzyme, named cytochrome P450scc, with strong structural similarity between rodent, bovine, and human species. The presence of immunoreactive P450scc protein in the rat and human brain has been established in the white matter and in primary cultures of the newborn rat forebrain glial cells (9). However, the abundance of P450scc mRNA is exceedingly low and could be demonstrated only by reverse transcription–PCR (RT-PCR) (10–12).
Major efforts were devoted to the elucidation of DHEA biosynthesis in the brain (13). However, incubations of [3H]PREG (and sulfated or acetyl ester derivatives) with brain slices, homogenates and microsomes, with primary cultures of mixed glial cells, or with astrocytes and neurons of rat and mouse embryos, never produced a radioactive metabolite with the chromatographic behavior of [3H]DHEA. Moreover, all attempts to demonstrate the P450c17 antigen immunohistochemically in the rat brain with antibodies to the enzyme purified from pig testis and in the guinea pig brain with specific antibodies to the enzyme from guinea pig adrenal were unsuccessful (14). Accordingly, Mellon and Deschepper (10) failed to detect the mRNA for P450c17 by RNase protection assays and RT-PCR. Only a transient expression of the mRNA for this enzyme during embryonic life was reported (15), however a conflicting report indicated its presence also in the adult rat brain (12). Thus, the pathway(s) by which DHEA biosynthesis occurs in the brain remains controversial.
The biosynthesis of PREG also occurs in the rat retina, where it is the most abundant steroid. Although it has been suggested by Guarneri et al. (16), the presence and activity of P450c17 in retina has not been conclusively demonstrated. We failed also to demonstrate the direct conversion of radioactive cholesterol or sesterpene to DHEA (17, 18). However, Prasad et al. (19) have been able to generate PREG and DHEA from organic solvent extracts of rat brain by reaction with various reagents. They have suggested the intermediate formation of cholesterol 17,20-cycloperoxide or 17-hydroperoxide in an hypothetical biochemical pathway from cholesterol to DHEA. Their results are supported by a recent report of Cascio et al. (20). Previous reports had indicated that C6 rat glioma cells in culture biosynthesize both PREG and DHEA, despite the complete lack of expression of P450c17. Adding FeSO4 to the culture medium increased the synthesis of both neurosteroids, even in presence of specific inhibitors of P450scc and/or P450c17. These results were interpreted as caused by the fragmentation of in situ-formed tertiary hydroperoxides (“hydroperoxide pathway”). Namely, the precursor of brain DHEA might be a steroid where both C-17 and C-20 are oxygenated. The enzyme(s) responsible for these conversions are unknown.
The description of DHEA as a neurosteroid has triggered a vast body of pharmacological studies showing that DHEA and DHEAS are neuroactive steroids. Their neurotrophic effects first were reported by Roberts et al. (21). When cultures of dissociated brain cells of mouse embryos were cultured with either DHEA or DHEAS, prominent increases were found in the numbers of neurofilament-positive neurons and glial fibrillary acid protein-positive astrocytes, with extensions of the processes of both types of cells.
The work of Compagnone and Mellon (22) in this issue of the Proceedings is a remarkable contribution to the neurotrophic function of DHEA and DHEAS. They have postulated that the developmentally regulated, region-specific expression of P450c17 in the rat embryo might be involved in brain development. Because P450c17 is expressed in neurons of the cortical subplate, a region involved in guiding thalamic fibers to their cortical targets, Compagnone and Mellon have considered the eventual role of DHEA and DHEAS in regulating motility and/or growth of cortico-thalamic projections. For that purpose, they have used cultures of neocortical neurons of embryonic day 16.5 rats. They have made the observation that DHEA and DHEAS not only were both active, but that their effects were different. DHEA selectively increased the length of neurites containing the axonal marker Tau-1, and the incidence of varicosities and basket-like process formations, whereas DHEAS selectively increased the length of neurites containing the dendritic marker MAP-2. Moreover, these effects were dose-related and observed at subnanomolar concentrations of either DHEA or DHEAS, within the range reported in the adult and newborn rat brain (23).
Compagnone and Mellon have attempted to define the mechanism by which DHEA and DHEAS exert their neurotrophic effects. They have observed that the noncompetitive N-methyl-d-aspartate (NMDA) receptor antagonist MK801 (dizocilpine) blocked axonal growth stimulated by DHEA, but did not block dendritic growth stimulated by DHEAS. The involvement of NMDA receptors was further substantiated by the study of calcium uptake in cultured neocortical neurons. Subnanomolar concentrations of DHEA increased intracellular Ca2+ concentration ([Ca2+]i), whereas DHEAS was ineffective. NMDA receptor antagonists decreased the response of [Ca2+]i to DHEA.
Indeed, DHEA is a naturally occurring excitatory neuroactive steroid. DHEAS has been reported to increase neuronal excitability (firing rate) when directly applied to septal-preoptic neurons (24). Both DHEAS and PREGS behave as γ-aminobutyric acid type A (GABAA) receptor antagonists at low micromolar concentrations, although their properties are not identical, PREGS displays mixed GABA agonistic/antagonistic features, whereas DHEAS behaves as a mere antagonist (25, 26). DHEA mimicks the effects of DHEAS, although less potently.
In the nanomolar range, PREGS is also a potent positive allosteric modulator at the NMDA receptor, whereas DHEAS is weakly active (27). DHEA was not previously reported to be directly active at NMDA receptors. However DHEAS, in the high nanomolar range, might indirectly stimulate NMDA receptors via a modulation of σ1 receptors (28). DHEA was not active on these receptors when applied to hippocampal slices (28); it was shown to be active in vivo after intravenous administration, but in that case its conversion to another active molecule could not be excluded (29). DHEAS also attenuated dizocilpine-induced learning impairment in mice via σ1 receptors, because its enhancing effect was antagonized by co-administration of the σ1 antagonist BMY-14802 (30).
Therefore, although several reports indicate a direct or indirect involvement of exogenous DHEA in the potentiation of NMDA receptors, they disagree on the nature of the active moiety (DHEA or DHEAS) and its active concentration (subnanomolar or submicromolar).
Furthermore, all of these studies do not bring any information about the physiological role of endogenous DHEA and DHEAS. Such information rests primarily on the availability of sensitive and specific methods for the assay of neurosteroids in relevant brain structures. Indeed the radioimmunoassays, which are still by far the most often used technique for the measurement of neurosteroids, have a far too low sensitivity and an eventual lack of specificity to allow the rigorous measurement of DHEA and DHEAS in the developing neocortex, and therefore to validate their potential physiological role. For this purpose, GC negative ion chemical ionization-mass fragmentography recently has been introduced, with still only preliminary results (31).
To date, there are only two reports showing a correlation between endogenous neurosteroid levels in defined nervous structures and corresponding function: the first one deals with progesterone synthesis in Schwann cells and its role in peripheral nerve regeneration after a cryolesion (32), the second one shows a striking positive correlation between the concentration of PREGS in the hippocampus of 2-year-old rats and their spatial memory performance (33). DHEAS is abundant in monkey and human brain (23) and might regulate memory performance in humans as PREGS does in rodents.
In conclusion, DHEA, formerly believed to be only an intermediary steroid in the biosynthetic pathway of sex steroid hormones (androgens and estrogens), has documented previously unsuspected activities apparently of its own, although its conversion to active metabolites cannot be formally excluded. The “receptor(s)” responsible for the direct action of DHEA(S) are still unknown. In humans, the concentrations of DHEA(S) are very large in plasma and in brain, and even more so in the fetus, because of the presence of the so-called fetal zones in the adrenals, which regress after birth. The only documented function of fetal DHEAS is as a precursor of placental estrogens (34). An organizational role in the developing central nervous system is suggested by the report of Compagnone and Mellon (22).
References
- 1.Corpéchot C, Robel P, Axelson M, Sjövall J, Baulieu E E. Proc Natl Acad Sci USA. 1981;78:4704–4707. doi: 10.1073/pnas.78.8.4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vinson G P, Whitehouse B J, Godard C. J Steroid Biochem. 1978;9:677–683. doi: 10.1016/0022-4731(78)90181-4. [DOI] [PubMed] [Google Scholar]
- 3.Baulieu E E. In: Steroid Hormone Regulation of the Brain. Fuxe K, Gustafsson J A, editors. Oxford: Pergamon; 1981. pp. 3–14. [Google Scholar]
- 4.Young J, Corpéchot C, Perché F, Haug M, Baulieu E E, Robel P. Endocrine. 1994;2:505–509. [Google Scholar]
- 5.Corpéchot C, Synguelakis M, Talha S, Axelson M, Sjövall J, Vihko R, Baulieu E E, Robel P. Brain Res. 1983;270:119–125. doi: 10.1016/0006-8993(83)90797-7. [DOI] [PubMed] [Google Scholar]
- 6.Robel P, Baulieu E E. Trends Endocrinol Metab. 1994;5:1–8. doi: 10.1016/1043-2760(94)90114-7. [DOI] [PubMed] [Google Scholar]
- 7.Hu Z Y, Jung-Testas I, Robel P, Baulieu E E. Proc Natl Acad Sci USA. 1987;84:8215–8219. doi: 10.1073/pnas.84.23.8215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jung-Testas I, Hu Z Y, Baulieu E E, Robel P. Endocrinology. 1989;125:2083–2091. doi: 10.1210/endo-125-4-2083. [DOI] [PubMed] [Google Scholar]
- 9.Le Goascogne C, Robel P, Gouézou M, Sananès N, Baulieu E E, Waterman M. Science. 1987;237:1212–1215. doi: 10.1126/science.3306919. [DOI] [PubMed] [Google Scholar]
- 10.Mellon S, Deschepper C F. Brain Res. 1993;629:283–292. doi: 10.1016/0006-8993(93)91332-m. [DOI] [PubMed] [Google Scholar]
- 11.Sanne J L, Krueger K E. Gene. 1995;165:327–328. doi: 10.1016/0378-1119(95)00536-f. [DOI] [PubMed] [Google Scholar]
- 12.Strömsted M, Waterman M R. Mol Brain Res. 1995;34:75–88. doi: 10.1016/0169-328x(95)00140-n. [DOI] [PubMed] [Google Scholar]
- 13.Robel P, Akwa Y, Corpéchot C, Hu Z Y, Jung-Testas I, Kabbadj K, Le Goascogne C, Morfin R, Vourc’h C, Young J, Baulieu E E. In: Brain Endocrinology. Motta M, editor. New York: Raven; 1991. pp. 105–132. [Google Scholar]
- 14.Le Goascogne C, Sananès N, Gouézou M, Takemori S, Kominami S, Baulieu E E, Robel P. J Reprod Fertil. 1991;93:609–622. doi: 10.1530/jrf.0.0930609. [DOI] [PubMed] [Google Scholar]
- 15.Compagnone N A, Bulfone A, Rubenstein J L R, Mellon S H. Endocrinology. 1995;136:5212–5223. doi: 10.1210/endo.136.11.7588260. [DOI] [PubMed] [Google Scholar]
- 16.Guarneri P, Guarneri R, Cascio C, Pavasant P, Piccoli F, Papadopoulos V. J Neurochem. 1994;63:86–96. doi: 10.1046/j.1471-4159.1994.63010086.x. [DOI] [PubMed] [Google Scholar]
- 17.Jungman R A. Biochim Biophys Acta. 1968;164:110–123. doi: 10.1016/0005-2760(68)90077-5. [DOI] [PubMed] [Google Scholar]
- 18.Tait A D, Hodge L C. J Steroid Biochem. 1985;22:237–242. doi: 10.1016/0022-4731(85)90118-9. [DOI] [PubMed] [Google Scholar]
- 19.Prasad V V K, Vegesna S R, Welch M, Lieberman S. Proc Natl Acad Sci USA. 1994;91:3220–3223. doi: 10.1073/pnas.91.8.3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cascio C, Prasad V V K, Lin Y Y, Lieberman S, Papadopoulos V. Proc Natl Acad Sci USA. 1998;95:2862–2867. doi: 10.1073/pnas.95.6.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts E, Bologa L, Flood J F, Smith G E. Brain Res. 1987;406:357–362. doi: 10.1016/0006-8993(87)90807-9. [DOI] [PubMed] [Google Scholar]
- 22.Compagnone N A, Mellon S H. Proc Natl Acad Sci USA. 1998;95:4678–4683. doi: 10.1073/pnas.95.8.4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baulieu E E, Robel P, Vatier O, Haug M, Le Goascogne C, Bourreau E. Receptor-Receptor Interactions. Basingstoke, U.K.: MacMillan; 1987. pp. 89–104. [Google Scholar]
- 24.Carette B, Poulain P. Neurosci Lett. 1984;45:205–210. doi: 10.1016/0304-3940(84)90100-9. [DOI] [PubMed] [Google Scholar]
- 25.Majewska M D, Demirgören S, Spivak C E, London E D. Brain Res. 1990;526:143–146. doi: 10.1016/0006-8993(90)90261-9. [DOI] [PubMed] [Google Scholar]
- 26.Majewska M D. Prog Neurobiol. 1992;38:379–395. doi: 10.1016/0301-0082(92)90025-a. [DOI] [PubMed] [Google Scholar]
- 27.Wu F S, Gibbs T T, Farb D H. Mol Pharmacol. 1991;40:333–336. [PubMed] [Google Scholar]
- 28.Monnet F P, Mahe V, Robel P, Baulieu E E. Proc Natl Acad Sci USA. 1995;92:3774–3778. doi: 10.1073/pnas.92.9.3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Debonnel G, Bergeron R, de Montigny C. J Endocrinol. 1996;150:S33–S42. [PubMed] [Google Scholar]
- 30.Maurice T, Junien J L, Privat A. Brain Res. 1997;83:159–164. doi: 10.1016/s0166-4328(97)86061-5. [DOI] [PubMed] [Google Scholar]
- 31.Uzunov D P, Cooper T B, Costa E, Guidotti A. Proc Natl Acad Sci USA. 1996;93:12599–12604. doi: 10.1073/pnas.93.22.12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koenig H, Schumacher M, Ferzaz B, Do-Thi A, Ressouches A, Guennoun R, Jung-Testas I, Robel P, Akwa Y, Baulieu E E. Science. 1995;268:1500–1503. doi: 10.1126/science.7770777. [DOI] [PubMed] [Google Scholar]
- 33.Vallée M, Mayo W, Darnaudéry M, Corpéchot C, Young J, Koehl M, Le Moal M, Baulieu E E, Robel P, Simon H. Proc Natl Acad Sci USA. 1997;94:14865–14870. doi: 10.1073/pnas.94.26.14865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siiteri P K, MacDonald P C. Steroids. 1963;2:713–730. [Google Scholar]