

Abstract

Highly enantioselective aldol reactions of acetylphosphonates and activated carbonyl compounds was realized with cinchona alkaloid derived catalysts, in which the acetylphosphonate was directly used as an enolate precursor for the first time. The aldol product obtained was converted in situ to its corresponding ester or amide through methanolysis or aminolysis. The overall process may be viewed as formal highly enantioselective acetate or acetamide aldol reactions, which are very difficult to achieve directly with organocatalytic methods.
The aldol reaction1 is arguably one of the most important reactions in organic synthesis due to the fact that this reaction forms a β-hydroxycarbonyl moiety that may be found in many natural products.1b,2 Because a new carbon-carbon bond is formed with the concurrent creation of a stereogenic center in the aldol product, many asymmetric variants of this reaction have been developed in the past few decades.1,2 In recent years, significant advances have also been made on the organocatalytic direct aldol reactions.3 Despite these great progresses, the direct enantioselective acetate aldol reaction1b,4 still remains a very challenging task. Most of the reported asymmetric acetate aldol reactions have been achieved through diastereoselective reactions controlled by chiral auxiliaries or ligands involving the use of metal acetate enolates, such as those of tin,5 lithium,6 boron,7 or titanium.8 These are not catalytic methods and the reagents used in these reactions are often expensive and/or difficult to handle. Moreover, many of these methods also suffer from narrow substrate scopes and low stereoselectivities. Catalytic highly enantioselective acetate aldol reactions have been reported by Carreira,9 Denmark,10 Shibasaki,11 and List12 using Mukaiyama aldol reactions; however, preformed silyl ketene acetals have to be used in these reactions. On the other hand, to form an enolate or enamine from acetate directly with organocatalysts is very difficult. To our knowledge, there has been no report on an organocatalyzed direct acetate aldol reaction.
Previously, we demonstrated that α-ketophosphonates are excellent electrophiles in organocatalyzed asymmetric direct aldol reactions.13 Nevertheless, using enolizable α-ketophosphonates as nucleophiles in chiral amine derivative-catalyzed asymmetric direct aldol reactions has never been reported due to the fact that the labile and bulky phosphonate group in these compounds prevents the formation of the desired enamine intermediates. Most recently, we realized a base-catalyzed stereoselective aldol14 reaction of unactivated ketones.15 These reactions only involve the formation of the ketone enolate as the nucleophile in a complete noncovalent catalysis.14,15 Because no enamine formation is involved in the mechanisms, we envisioned that substrates with a labile and/or poor electrophilic ketone group may still be used in this reaction as nucleophiles. Since the α-hydrogen in acetylphosphonates (1) should be more acidic than that of an unactivated ketone, we hypothesized that a base-catalyzed enantioselective aldol reaction of acetylphosphonates should be feasible. Herein, we wish to report the first successful application of the enolizable acetylphosphonates (1) as a nucleophile in an organocatalyzed aldol reaction. More importantly, taking advantage of the lability of the α-ketophosphonate group,16 compound 1 was successfully employed as a surrogate of acetate or acetamide17 to achieve a convenient one-pot highly enantioselective formal acetate/acetamide aldol reaction.
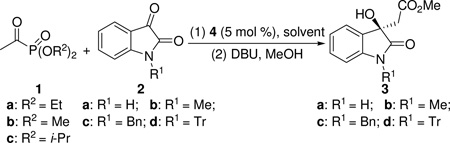
Isatin (2a, R1 = H) was chosen as the model substrate to test our hypothesis. On the basis of the results of our previous study,14 cinchona alkaloid derivatives18 were chosen as the catalysts (Figure 1). The results of the catalyst screening are summarized in Table 1. As the results show (Table 1), with the quinidine thiourea catalyst 4a, the desired aldol product was obtained when diethyl acetylphosphonate 1a and 2a were reacted in THF at rt for 4 h (Table 1, entry 1). Since the phosphonate group in α-ketophosphonates is a good leaving group,17 the original aldol product was directly converted to the corresponding methyl ester 3a in 68% yield via a nucleophilic acyl substitution reaction using MeOH in the presence of DBU (Table 1, entry 1).19 The ee value of this product was determined to be 53% (entry 1). The overall reaction may be regarded as a formal enantioselective acetate aldol reaction, which is difficult to achieve otherwise.
Figure 1.

Catalyst Screened in the Aldol Reaction [Nap = naphthyl]
Table 1.
Catalyst Screen and Reaction Condition Optimizationa
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | 4 | R1 | R2 | solvent | time (h) |
yield (%)b |
ee (%)c |
| 1 | 4a | H | Et | THF | 4 | 68 | 53 |
| 2 | 4a | H | Me | THF | 4 | 39 | 62 |
| 3 | 4a | H | i-Pr | THF | 4 | 91 | 53 |
| 4 | 4b | H | i-Pr | THF | 2 | 87 | 75 |
| 5 | 4c | H | i-Pr | THF | 2 | 84 | 72 |
| 6 | 4d | H | i-Pr | THF | 4 | 88 | 50d |
| 7 | 4e | H | i-Pr | THF | 2 | 63 | 23d |
| 8 | 4f | H | i-Pr | THF | 2 | 83 | 53 |
| 9 | 4g | H | i-Pr | THF | 2 | trace | nde |
| 10 | 4h | H | i-Pr | THF | 2 | 44 | 47 |
| 11 | 4i | H | i-Pr | THF | 2 | 90 | 65 |
| 12 | 4j | H | i-Pr | THF | 2 | 91 | 73 |
| 13 | 4k | H | i-Pr | THF | 4 | trace | nde |
| 14 | 4l | H | i-Pr | THF | 4 | trace | nde |
| 15f | 4b | H | i-Pr | THF | 2 | 88 | 79 |
| 16g | 4b | H | i-Pr | THF | 4 | 90 | 81 |
| 17g | 4j | H | i-Pr | THF | 6 | 88 | 86 |
| 18g | 4b | Me | i-Pr | THF | 4 | 88 | 85 |
| 19g | 4b | Bn | i-Pr | THF | 4 | 88 | 87 |
| 20g | 4b | Tr | i-Pr | THF | 6 | 80 | 94 |
| 21g | 4j | Me | i-Pr | THF | 4 | 85 | 84 |
| 22g | 4j | Bn | i-Pr | THF | 4 | 84 | 86 |
| 23g | 4j | Tr | i-Pr | THF | 6 | 83 | 95 |
Unless otherwise specified, all aldol reactions were conducted with acetylphosphonate (1, 0.50 mmol), isatin (2, 0.10 mmol), and catalyst 4 (0.0050 mmol, 5 mol %) in the indicated solvent (1.0 mL) at rt under a nitrogen atmosphere. After the aldol reaction was completed, the reaction mixture was treated with DBU (0.10 mmol) and MeOH (1.0 mL) at rt for 15 min to convert the original reaction product to compound 3 in situ.
Yield of isolated compound 3 after column chromatography.
Determined by HPLC analysis using a ChiralCel OD-H column. For the assignment of the absolute configuration of the major enantiomer, please see text.
The S-enantiomer was obtained as the major product.
Not determined.
The aldol reaction was carried out at 0 °C.
The aldol reaction was carried out at −15 °C.
The effects of the ester group (R2) of the acetylphosphonate were then investigated. It was found that, although dimethyl acetylphosphonate 1b led to a slightly higher ee value of the product (62% ee, entry 2), the yield was much poorer (39%). In contrast, diisopropyl acetylphosphonate 1c led to an excellent yield (91%) of the expected product 3a with the same ee value (entry 3) as that of diethyl acetylphosphonate (entry 1). Therefore, 1c was adopted as the model substrate for catalyst screening. As the results in Table 1 show, except for catalysts 4g and 4h, all the other cinchona alkaloid catalysts led to the formation of 3a in good to high yields (entries 4–12). High ee values were achieved for the quinidine thiourea catalysts 4b (75% ee, entry 4) and 4c (72% ee, entry 5), and 9-O-(1-naphthylmethyl)cupreidine (4j, 73% ee, entry 12). In contrast, proline-based catalysts 4k and 4l failed to generate the desired product from acetylphosphonate 1c (entries 13–14). These results clearly demonstrate the advantages of the enolate mechanism over the enamine mechanism for such bulky and labile ketone substrates. A solvent study with catalyst 4b revealed that THF is the best solvent for this reaction.20 Lower reaction temperature was found to be beneficial for improving the enantioselectivity of this reaction (entries 15–16), and the ee value of the product 3a was improved to 81% when the reaction was conducted at −15 °C (entry 16). Under these optimized conditions, catalyst 4j yielded 3a in 88% yield and 86% ee in 6 h (entry 17). Intrigued by our previous finding that the substituent on the isatin nitrogen atom (R1) has a dramatic effect on the enantioselectivity,14 we also studied N-substituted isatins in this aldol reaction. It is interesting to find that higher ee values of the products were obtained when the size of the N-substituent was increased. For examples, with catalyst 4b, N-methyl (2b), N-benzyl (2c), and N-trityl (2d) isatins lead to the corresponding products 3b, 3c, and 3d in 85%, 87%, and 94% ee, respectively (entries 18–20). A similar trend was also observed for catalyst 4j (entries 21–23).
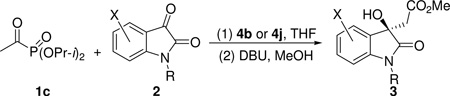
Once the reaction conditions were optimized, the scope of this reaction was then established using N-tritylisatins. The results are collected in Table 2. As is evident from the data, high yields and good to excellent ee values of the expected formal acetate aldol products were obtained for a variety of substituted N-tritylisatins using either catalyst 4b or 4j. Interestingly, these two catalysts were found to be complementary in terms of enantioselectivity for substituted isatins. For example, catalyst 4b is very sensitive towards the substituent at the 4-position of isatin and leads to the formation of the opposite enantiomers as the major products in very poor ee values (Table 2, entries 2–3). The exact reason for this inversion is not clear at the moment, but most likely it is due to steric effects.21 In contrast, substituents on any other locations of the ring do not show much effect (entries 1, 4–14). On the other hand, catalyst 4j is much less sensitive towards the substituents on the isatin ring and high ee values of products were obtained for the 4-substituted isatins (data in parentheses, entries 2–3). Nevertheless, it normally produces slightly lower ee values of the products than catalyst 4b does when there is a substituent on the 5, 6, or 7-positions (entries 4–14). It should be pointed out that, when isatin has a substituent at the 7-position, it is impossible to protect it with the large trityl group and, therefore, N-benzyl protected substrates were used instead (entries 12–14), and very good results were obtained as well. The absolute configuration of the major enantiomer formed in this reaction was assigned as R by X-ray crystallographic analysis of a crystal of compound 3o. On the basis of this result, transition state models are proposed to account for the stereochemical outcome of this reaction.21
Table 2.
Substrate Scope of the Direct Aldol Reactiona
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | X | R | 3 | time (h) | yield (%)b | ee (%)c | |
| 1 | H | Tr | 3d | 6.0 (6.0) | 80 (83) | 94 (95) | |
| 2 | 4-Cl | Tr | 3e | 5.5 (4.0) | 81 (87) | 8d (96) | |
| 3 | 4-Br | Tr | 3f | 5.0 (4.5) | 70 (92) | 21d (96) | |
| 4 | 5-Me | Tr | 3g | 4.0 (4.5) | 92 (94) | 98 (95) | |
| 5 | 5-MeO | Tr | 3h | 4.0 (4.0) | 90 (88) | 94 (94) | |
| 6 | 5-F | Tr | 3i | 3.0 (4.0) | 88 (93) | 95 (90) | |
| 7 | 5-Cl | Tr | 3j | 4.0 (3.5) | 84 (86) | 95 (90) | |
| 8 | 5-Br | Tr | 3k | 4.0 (3.0) | 89 (92) | 93 (89) | |
| 9 | 5-I | Tr | 3l | 3.0 (2.0) | 91 (93) | 94 (89) | |
| 10 | 5-NO2 | Tr | 3m | 4.5 (4.5) | 88 (85) | 84 (72) | |
| 11 | 6-Br | Tr | 3ne | 4.0 (3.0) | 92 (94) | 97 (94) | |
| 12 | 7-Br | Bn | 3oe | 3.0 (3.0) | 91 (92) | 93 (88) | |
| 13 | 5,7-Br2 | Bn | 3pe | 1.0 (1.5) | 85 (88) | 93 (82) | |
| 14 | 5,7-Me2 | Bn | 3qe | 5.0 (5.0) | 82 (80) | 94 (92) | |
Unless otherwise specified, all aldol reactions were conducted with diisopropyl acetylphosphonate (1c, 0.50 mmol), isatin (2, 0.1 mmol), and catalyst 4b or 4j (0.0050 mmol, 5 mol %) in THF (1.0 mL) at −15 °C under a nitrogen atmosphere. After the aldol reaction was completed, the reaction mixture was treated with DBU (0.10 mmol) and MeOH (1.0 mL) at rt for 15 min to convert the original reaction product to compound 3 in situ. Data in parentheses are those of catalyst 4j.
Yield of isolated compound 3 after column chromatography.
Unless otherwise noted, ee values were determined by HPLC analyses using a ChiralCel OD-H column. The absolute configuration of the major enantiomer of compound 3o was assigned by X-ray crystallographic analysis. The configuration of the rest compounds was assigned on the basis of the reaction mechanism.
The S-enantiomer was obtained as the major product.
Determined by HPLC analysis using a ChiralPak AD-H column.
When diisopropyl propionylphosphonate (1d) was used as the substrate, the corresponding aldol product 3r was obtained in a dr of 80:20 with a 94% ee for the major diastereomer (Scheme 1, equation A). The absolute stereochemistry of compound 3r was determined on the basis of the X-ray crystallographic analysis of its crystals.21
Scheme 1.

Additional Substrate Scope Studies of the Base-Catalyzed Aldol Reactions
Besides isatins, phenylglyoxal hydrates (5) may also be used as the substrates in this reaction, and the corresponding β-hydroxy esters 6 were obtained in good ee values (87% and 84% for the 4-H- and 4-MeO-subsituted 6, respectively) after converting the original aldol products through methanolysis (Scheme 1, equation B).
Since the phosphonate group may be easily replaced by nucleophiles, we also attempted the in situ aminolysis of the original aldol product (Scheme 1, equation C) and the corresponding β-hydroxyamide 7 was obtained in 90% yield and 96% ee. Similarly, a β,γ-alkynyl-α-keto ester 8 gave directly the cyclized product 9 in 76% ee after the aldol-aminolysis sequence. This reaction provides a one-pot synthesis of optically enriched 3-hydoxy-2,5-pyrrolidinediones (Scheme 1, equation D). These results may be regarded as formal acetamide aldol reactions, which again are very difficult to achieve directly.
The N-trityl protecting group that is necessary for achieving high enantioselectivity in this reaction may be easily removed by using a reported procedure22 to give the deprotected product in high yield with complete retention of the stereochemistry (Scheme 2).22
Scheme 2.

Deprotection of the Trityl Group
To further demonstrate the utility of our method, we also developed an efficient two-step synthesis of compound 11 from the aldol product 3d (Scheme 3). Compound 11, which is the half fragment used in the total synthesis of natural products Madindoline A and B,23 was obtained in 53% overall yield and 94% ee.
Scheme 3.

Synthesis of the Half Fragment of Madindoline A and B
Reaction conditions: A) LiAlH4 / THF, 0 °C to rt, 4 h, 64%; B) TFA, Et3SiH / CH2Cl2, 0 °C, 4 h, 83%
In conclusion, we have developed the first highly enantioselective aldol reaction of acetylphosphonates and activated carbonyl compounds, such as, isatins, phenylglyoxals, and α-ketoesters, using cinchona alkaloid derived catalysts, in which acetylphosphonate was used as a nucleophile. Through an in-situ methanolysis or aminolysis of the phosphonate group of the original aldol products obtained in this reaction, the corresponding esters or amide were obtained. The overall reaction may be viewed as formal highly enantioselective acetate or acetamide aldol reactions, which are very difficult to achieve directly with organocatalytic methods.
Supplementary Material
Acknowledgment
The generous financial support of this research from the National Science Foundation (Grant No. CHE 0909954), the NIH-NIGMS (Grant-No. SC1GM0828718), and the Welch Foundation (Grant No. AX-1593) is gratefully acknowledged. The authors also thank Dr. Hadi Arman (UTSA) for his assistance in the X-ray crystallographic analyses of the reaction products.
Footnotes
Supporting Information Available Full experimental procedures, transition state models, compound characterization data, ORTEP drawings and cif files of compounds 3o and 3r, and copy of NMR spectra and HPLC chromatograms. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews, see: Mukaiyama T. Org. React. 1982;28:203. Mahrwald R, editor. Modern Aldol Reactions. Weinheim, Germany: Wiley-VCH; 2004.
- 2.For review, see: Norcross RD, Paterson I. Chem. Rev. 1995;95:2041.
- 3.For a reviews, see: List B. Acc. Chem. Res. 2004;37:548. doi: 10.1021/ar0300571. Notz W, Tanaka F, Barbas CF., III Acc. Chem. Res. 2004;37:580. doi: 10.1021/ar0300468. List B. Tetrahedron. 2002;58:5573. Pellissier H. Tetrahedron. 2007;63:9267.
- 4.For reviews, see: Braun M. Angew. Chem. Int. Ed. 1987;26:24. Palomo C, Oiarbide M, García JM. Chem. Eur. J. 2002;8:36. doi: 10.1002/1521-3765(20020104)8:1<36::aid-chem36>3.0.co;2-l. Ariza X, Garcia J, Romea P, Urpí F. Synthesis. 2011:2175.
- 5.For selected examples, see: Nagao Y, Yamada S, Kumagai T, Ochiai M, Inoue T, Fujita E. J. Chem. Soc. Chem. Commun. 1985:1418. Nagao Y, Hagiwara Y, Kumagai T, Ochiai M, Inoue T, Hashimoto K, Fujita E. J. Org. Chem. 1986;51:2391.
- 6.(a) Braun M, Devant R. Tetrahedron Lett. 1984;25:5031. [Google Scholar]; (b) Saito S, Hatanaka K, Kano T, Yamamoto H. Angew. Chem. Int. Ed. 1998;37:3378. doi: 10.1002/(SICI)1521-3773(19981231)37:24<3378::AID-ANIE3378>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]; (c) Song JJ, Xu J, Tan Z, Reeves JT, Grinberg N, Lee H, Kuzmich K, Feng X, Yee NK, Senanayake CH. Org. Process Res. Dev. 2007;11:534. [Google Scholar]
- 7.(a) Yan TH, Hung AW, Lee HC, Chang CS. J. Org. Chem. 1994;59:8187. [Google Scholar]; (b) Zhang YC, Sammakia T. Org. Lett. 2004;6:23. doi: 10.1021/ol036020y. [DOI] [PubMed] [Google Scholar]; (c) Zhang YC, Phillips AJ, Sammakia T. Org. Lett. 2004;6:3139. doi: 10.1021/ol048810t. [DOI] [PubMed] [Google Scholar]
- 8.(a) Yan TH, Hung AW, Lee HC, Chang CS, Liu WH. J. Org. Chem. 1995;60:3301. [Google Scholar]; (b) Gonzalez A, Aiguade J, Urpi F, Vilarrasa J. Tetrahedron Lett. 1996;37:8949. [Google Scholar]; (c) Guz NR, Phillips AJ. Org. Lett. 2002;4:2253. doi: 10.1021/ol026108w. [DOI] [PubMed] [Google Scholar]; (d) Crimmins MlT, Shamszad M. Org. Lett. 2007;9:149. doi: 10.1021/ol062688b. [DOI] [PubMed] [Google Scholar]
- 9.Carreira EM, Singer RA, Lee W. J. Am. Chem. Soc. 1994;116:8837. [Google Scholar]
- 10.(a) Denmark SE, Wynn T, Buetner GL. J. Am. Chem. Soc. 2002;124:13405. doi: 10.1021/ja0282947. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Fan Y. J. Am. Chem. Soc. 2002;124:4233. doi: 10.1021/ja025670e. [DOI] [PubMed] [Google Scholar]
- 11.Oisaki K, Zhao D, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2006;128:7164. doi: 10.1021/ja061815w. [DOI] [PubMed] [Google Scholar]
- 12.García-García P, Lay F, García-García P, Rabalakos C, List B. Angew. Chem. Int. Ed. 2009;48:4363. doi: 10.1002/anie.200901768. [DOI] [PubMed] [Google Scholar]
- 13.(a) Samanta S, Zhao C-G. J. Am. Chem. Soc. 2006;128:7442. doi: 10.1021/ja062091r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dodda R, Zhao C-G. Org. Lett. 2006;8:4911. doi: 10.1021/ol062005s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mandal T, Samanta S, Zhao C-G. Org. Lett. 2007;9:943. doi: 10.1021/ol070209i. 16. [DOI] [PubMed] [Google Scholar]; (d) Samanta S, Perera S, Zhao C-G. J. Org. Chem. 2010;75:1101. doi: 10.1021/jo9022099. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Perera S, Naganaboina VK, Wang L, Zhang B, Guo Q, Rout L, Zhao C-G. Adv. Synth. Catal. 2011;353:1729. doi: 10.1002/adsc.201000835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo Q, Bhanushali M, Zhao C-G . Angew. Chem. Int. Ed. 2010;49:9460. doi: 10.1002/anie.201004161. See also reference 15b for a similar study.
- 15.For related examples, see: Paradowska J, Rogozinńska M, Mlynarski J. Tetrahedron Lett. 2009;50:1639. Allu S, Molleti N, Panem R, Singh VK. Tetrahedron Lett. 2011;52:4080. Li L, Klauber EG, Seidel D. J. Am. Chem. Soc. 2008;130:12248. doi: 10.1021/ja804838y. Misaki T, Takimoto G, Sugimura T. J. Am. Chem. Soc. 2010;132:6286. doi: 10.1021/ja101216x. Liu G-G, Zhao H, Lan Y-B, Wu B, Huang X-F, Chen J, Tao J-C, Wang X-W. Tetrahedron. 2012;68:3843.
- 16.For examples, see: Maeda H, Takahashi K, Ohmori H. Tetrahedron. 1998;54:12233. Afarinkia K, Twist AJ, Yu H-w. J. Organomet. Chem. 2005;690:2688. Afarinkia K, Twist AJ, Yu H-w. J. Org. Chem. 2004;69:6500. doi: 10.1021/jo049133j.
- 17.For an example of using acylphosphonate as a surrogate of esters and amides in an enantioselective Michael addition reaction, see: Jiang H, Paixao MW, Monge D, Jorgensen KA. J. Am. Chem. Soc. 2010;132:2775. doi: 10.1021/ja9097803.
- 18.These catalysts were prepared according to the reported procedures, see: Vakulya B, Varga S, Csámpai A, Soós T. Org. Chem. 2005;7:1967. doi: 10.1021/ol050431s. Wu W, Min L, Zhu L, Lee C-S. Adv. Synth. Catal. 2011;353:1135. Amere M, Lasne M-C, Rouden J. Org. Chem. 2007;9:2621. doi: 10.1021/ol070712v. Wang H-F, Cui H-F, Chai Z, Li P, Zheng C-W, Yang Y-Q, Zhao G. Chem. Eur. J. 2009;15:13299–13303. doi: 10.1002/chem.200902303. Denis J-B, Masson G, Retailleau P, Zhu J. Angew. Chem. Int. Ed. 2011;50:5356. doi: 10.1002/anie.201100706.
- 19.This also facilitates the isolation and purification of the reaction product.
- 20.The product was obtained in 74% ee in dioxane, while lower ee values were obtained in DME, ether, CH2Cl2, EtOAC, and MeCN. Only trace amount of the product was obtained when MeOH was used.
- 21.For details, please see the Supporting Information.
- 22.Vedejs E, Klapars A, Warner DL, Weiss AH. J. Org. Chem. 2001;66:7542. doi: 10.1021/jo0106243. [DOI] [PubMed] [Google Scholar]
- 23.For examples, see: Itoh T, Ishikawa H, Hayashi Y. Org. Lett. 2009;11:3854. doi: 10.1021/ol901432a. Hirose T, Sunazuka T, Shirahata T, Yamamoto D, Harigaya Y, Kuwajima I, Omura S. Org. Lett. 2002;4:501. doi: 10.1021/ol017058i. Wan L, Tius MA. Org. Lett. 2007;9:647. doi: 10.1021/ol062919e. Sunazuka T, Hirose T, Shirahata T, Harigaya Y, Hayashi M, Komiyama K, Omura S, Smith AB., III J. Am. Chem. Soc. 2000;122:2122.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.