

Abstract

A cascade of Pd-catalyzed N-to-C allyl transfer–intramolecular ketenimine–[2 + 2] cycloadditions of N-allyl ynamides is described. This tandem sequence is highly stereoselective and the [2 + 2] cycloaddition could be rendered in a crossed or fused manner depending on alkene substitutions, leading to bridged and fused bicycloimines.
Throughout our studies on palladium catalyzed N-to-C allyl transfers1 of N-allyl ynamides2–4 to ketenimines,5 we have anticipated the possibility of effecting an intramolecular ketenimine-[2 + 2] cycloaddition with tethered alkenes [Scheme 1]. Seminal work on cycloadditions involving ketenes6 and keteniminium ions have been studied extensively by Marko,7 Snider,8 Brady,9 and recently by Minehan,10 giving rise to cyclobutanones through fused–11 and/or crossed–[2 + 2]12 pathways. For our own designs, we imagined that ketenimino-Pd-π-allyl complexes prepared by N-to-C allyl transfers of N-allyl ynamides 1 could also participate in fused, or more rarely, crossed [2 + 2] cycloadditions to afford highly substituted bicycloimines 2 or 3 via intermediates 5 or 6.
Scheme 1.

Intramolecular Ketenimine–[2 + 2] Cycloadditions
During our pursuit of this endeavor, Tu13 demonstrated beautifully the feasibility of carrying out intramolecular crossed–[2 + 2] cycloadditions using ketenimines generated in situ by extrusion of N2 from N-tosyl azides in a retro [3 + 2] manner.2a,14 In their work, the resulting bicycloimines were immediately hydrolyzed to ketones, and the crossed cycloaddition was the exclusive pathway. Our findings deviate significantly from theirs, and we report herein our successful development of highly diastereoselective crossed and fused ketenimine–[2 + 2] cycloadditions from N-allyl ynamides.
We quickly discovered that, in fact, γ-branched N-allyl ynamide 7 featuring an oxygen tethered styryl moiety cleanly underwent the desired Pd-catalyzed rearrangement–intramolecular [2 + 2] cycloaddition sequence to give bridged bicycloimine 8 in 80% yield as a single diastereomer [Scheme 2].15 It is noteworthy that cycloadduct 9 from a fused–cycloaddition pathway was not observed.
Scheme 2.

Discovery of a Crossed Ketenimine–[2 + 2]
Unlike in Tu’s system,13 the directly resulting imine was isolable by silica gel column chromatography and also crystalline, allowing for unambiguous determination of its structure by single crystal X-ray analysis [Figure 1]. By orienting the alkene to engage the orthogonal imine π-system and the bulky c-hexyl group into a pseudo-equatorial position, diastereomeric transition states 10 and 10’ can be envisioned. The A1,3 strain between the c-hex group and imine disfavored 10’, leading to 8 as the exclusive product with the imine anti to the c-hexyl.
Figure 1.

X-ray of 8 and Diastereoselectivity Rationale
The substrate scope proved to be exceptional, tolerating an array of propargylic substitutents and tethered olefins [Table 1]. Styryl-tethered ynamide 11 led to bicycle 14 in near quantitative yield [entry 1]. By utilizing crotyl-tethered ynamides 12a–c, cycloadducts 15a–c were isolated in good yields, though a competing carbocyclization1a,16 involving the Pd-π-allyl moiety was also observed in 10–20% yield [entries 2–4, see Scheme 4].
Table 1.
Crossed Ketenimine–[2 + 2] Cycloadditionsa
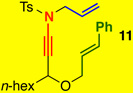
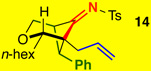
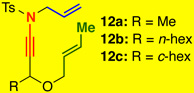
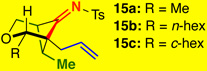
Reaction conditions: 5 mol % Pd(PPh3)4, Tol [conc = 0.1 M], 70 °C, 2 h.
Isolated yields.
≥20:1 dr by 1H NMR unless otherwise noted.
10–20% cyclopentenimine.
9:1 dr as measured by 1H NMR.
Scheme 4.

Crossed–[2 + 2] versus Carbocyclization
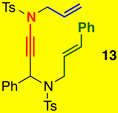
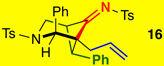
Gratifyingly, styryl-tethered ynamide 13 featuring an N-Ts linkage could also be used to afford 16 in quantitative yield as a 9:1 mixture of diastereomers [entry 9]. Interestingly, nOe of 1615 implied a switch of stereoselectivity. Subsequently, X-ray analysis showed that the once propargylic phenyl was indeed syn to the imine in 16 [Figure 2], opposite to the observed stereochemistry in the oxygen-tethered system employing a similarly sized propargylic c-hex moiety [see Figure 1].
Figure 2.

Switch in Diastereoselectivity with N-Ts Tether
It is noteworthy that such a selectivity switch was not observed in Tu’s study.13 The switch in diastereoselectivity for the crossed cycloaddition with N-Ts tethered ynamides is likely a result of a gauche interaction between the phenyl and the N-sulfonyl moiety as shown in 17’ [Figure 2]. Instead, the cycloaddition favored chair-flipped 17 with the phenyl pseudo-axial, explaining the formation of 16 with the imine and phenyl syn as the major diastereomer.
In Table 1, we revealed that a competing carbocyclization was operational to give cyclopentenimines in 10–20% yield with several of the γ-branched ynamides. Upon attempting to carry out cycloadditions with tethered cis alkenes, this reaction dichotomy was exemplified [Scheme 3]. As anticipated, ynamide 18 bearing a tethered trans-olefin afforded the desired crossed cycloadduct 20 in ≥95% yield. However, the cis-olefin tethered analogue 21 [10:1 cis:trans] gave cyclopentenimine 24 in 55% yield with only a trace amount of cycloadduct 20 observed, which likely arose from the trans impurity in the starting ynamide and not from reaction of the cis alkene. Clearly, the cis olefin geometry would have disfavored the cycloaddition transition state 22, and instead the carbocyclization through 23 ensued.
Scheme 3.

A Dichotomy Based on Olefin Geometry
Furthermore, when t-Bu-substituted ynamide 24 was subjected to the reaction conditions, the desired cycloadduct 25 was isolated in only 22% yield, however cyclopentenimine 26 was obtained in 57% yield [Scheme 4]. This further illustrates the necessity for the cycloaddition to occur through the highly-organized transition state 28, as disfavorable steric interactions clearly favor carbocyclization through 29.
Next, we wished to assess how an unsubstituted allyl group serving as the cycloaddition partner would behave under the reaction conditions, as Pd-catalyzed deallylation was also possible [Scheme 5]. Additionally and notably, Tu found unsubstituted alkenes to be unreactive in their system.13 Interestingly, when ynamide 30 was heated to 70 °C with 5 mol % Pd(PPh3)4, a 1:1 mixture of cycloadduct 31 and cyclopentenimine 32 was isolated in 61% yield, arising from competing fused–[2 + 2] cycloaddition and carbocyclization. Fortunately, fused cycloadduct 31 crystallized cleanly from the mixture, allowing us to confirm its structure by X-ray analysis. The cycloaddition was highly diastereoselective, giving 31 with the imine syn to the c-hexyl as a single diastereomer through 33 to minimize A1,2 strain suffered in 33’.
Scheme 5.

Discovery of a Fused–[2 + 2] Cycloaddition
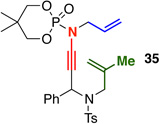
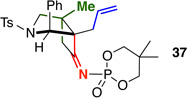
Similar to what has been well documented for ketene–[2 + 2] cycloadditions,6 we found that tethered internally substituted alkenes also favored formation of fused cycloadducts in our system [Table 2]. Ynamides 34a–d featuring a variety of propargylic substituents gave fused cycloadducts 36a–d in good yields with excellent diastereoselectivity. Further supporting the switch to a fused cycloaddition pathway, the imine carbon NMR signal for the fused cycloadducts was consistently 3–5 ppm upfield from the imine signal in the related bridged systems [194–195 ppm vs. 198–199 ppm]. The relative stereochemistry of 36a and 37, derived from the phosphoryl-substituted ynamide 35,17 were assigned by nOe analysis.15
Table 2.
Fused Ketenimine–[2 + 2] Cycloadditions
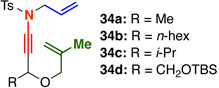

Reaction conditions: 5 mol % Pd(PPh3)4, Tol [conc = 0.1 M], 70 °C, 2 h.
Isolated yields.
≥20:1 dr by 1H NMR unless otherwise noted.
10–20% cyclopentenimine.
9:1 dr as measured by 1H NMR.
We have showcased here a highly diastereoselective cascade of Pd-catalyzed N-to-C allyl transfer–intramolecular–[2 + 2] cycloadditions to afford highly substituted bicycloimines from N-allyl ynamides. The alkene substitution pattern played an imminent role in favoring either the fused or crossed cycloaddition pathway, leading to fused or bridged cycloadducts. Also uncovered is a competing carbocyclization pathway when hindered alkenes or sterically-demanding propargylic substitutents were employed, giving rise to α,β-unsaturated cyclopentenimines. Applications and a further mechanistic understanding these unique cycloaddition manifolds are currently underway.
Supplementary Material
Acknowledgement
We thank NIH [GM066055] for funding. KAD thanks the American Chemical Society for a Division of Medical Chemistry Predoctoral Fellowship. We thank Dr. Victor G. Young of the University of Minnesota for providing X-ray structural analysis.
Footnotes
Supporting Information Available: Experimental procedures as well as NMR spectra, and characterizations are available for all new compounds and free of charge via Internet http://pubs.acs.org.
References
- 1.(a) Zhang Y, DeKorver KA, Lohse AG, Zhang YS, Huang J, Hsung RP. Org. Lett. 2009;11:899. doi: 10.1021/ol802844z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) DeKorver KA, Hsung RP, Lohse AG, Zhang Y. Org. Lett. 2010;12:1840. doi: 10.1021/ol100446p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) DeKorver KA, Johnson WL, Zhang Y-S, Zhang Y, Lohse AG, Hsung RP. J. Org. Chem. 2011;76:5092. doi: 10.1021/jo200780x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For current leading reviews on ynamides, see DeKorver KA, Li H, Lohse AG, Hayashi R, Lu Z, Zhang Y, Hsung RP. Chem. Rev. 2010;110:5064. doi: 10.1021/cr100003s. Evano G, Coste A, Jouvin K. Angew. Chem. Int. Ed. 2010;49:2840. doi: 10.1002/anie.200905817.
- 3.For leading reviews on the synthesis of ynamides, see: Tracey MR, Hsung RP, Antoline JA, Kurtz KCM, Shen L, Slafer BW, Zhang Y. In: Science of Synthesis, Houben-Weyl Methods of Molecular Transformations. Weinreb Steve M., editor. Chapter 21.4. Stuttgart, Germany: Georg Thieme Verlag KG; 2005. Mulder JA, Kurtz KCM, Hsung RP. Synlett. 2003:1379.
- 4.For ynamide papers published since February 2012, see: Saito N, Ichimaru T, Sato Y. Org. Lett. 2012;14:1914. doi: 10.1021/ol300571b. Kerr DJ, Miletic M, Chaplin JH, White JM, Flynn BL. Org. Lett. 2012;14:1732. doi: 10.1021/ol300316a. Garcia P, Evanno Y, George P, Sevrin M, Ricci G, Malacria M, Aubert C, Gandon V. Chem. Eur. J. 2012;18:4337. doi: 10.1002/chem.201103906. Jin X, Yamaguchi K, Mizuno N. Chem. Commun. 2012;48:4974. doi: 10.1039/c2cc31159c. Tchabanenko K, Sloan C, Bunetel YM, Mullen P. Org. Biomol. Chem. 2012;10:4215. doi: 10.1039/c2ob07007c.
- 5.For reviews on the chemistry of ketenimines, see: Krow GR. Angew. Chem. Int. Ed. Engl. 1971;10:435. Gambaryan NP. Usp. Khim. 1976;45:1251. Dondoni A. Heterocycles. 1980;14:1547. Barker MW, McHenry WE. In: The Chemistry of Ketenes, Allenes and Related Compounds. Patai S, editor. Part 2. Chichester, UK: Wiley-Interscience; 1980. pp. 701–720. Alajarin M, Vidal A, Tovar F. Targets Heterocycl. Syst. 2000;4:293.
- 6.For a review, see: B. B. Snider BB. Chem. Rev. 1988;88:793.
- 7.Marko I, Ronsmans B, Hesbain-Frisque A-M, Dumas S, Ghosez L. J. Am. Chem. Soc. 1985;107:2192. [Google Scholar]
- 8.For leading references, see: Snider BB, Hui RAHF, Kulkarni YS. J. Am. Chem. Soc. 1985;107:2194. Lee SY, Kulkarni YS, Burbaum BW, Johnston MI, Snider BB. J. Org. Chem. 1988;53:1848.
- 9.Brady WT, Giang YF. J. Org. Chem. 1985;50:5177. [Google Scholar]
- 10.Tran V, Minehan T. Org. Lett. 2011;13:6588. doi: 10.1021/ol202989c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For more examples of fused–[2 + 2] ketene cycloadditions, see: Snider BB, Hui RAHF, Kulkarni YS. J. Am. Chem. Soc. 1985;107:2194. Zhang W, Collins MR, Mahmood L, Dowd P. Tetrahedron Lett. 1995;36:2729.
- 12.For recent crossed ketene–[2 +2]’s, see: McCaleb KL, Halcomb RL. Org. Lett. 2000;2:2631. doi: 10.1021/ol000145a. Bélanger B, Léevesque F, Pâquet J, Barbe G. J. Org. Chem. 2005;70:291. doi: 10.1021/jo0483466.
- 13.Li B-S, Yang B-M, Wang S-H, Zhang Y-Q, Cao X-P, Tu Y-Q. Chem. Sci. 2012;3:1975. [Google Scholar]
- 14.For a recent review, see: Meldal M, Tornoe CW. Chem. Rev. 2008;108:2952. doi: 10.1021/cr0783479.
- 15.See Supporting Information.
- 16.DeKorver KA, Wang X-N, Walton MC, Hsung RP. Org. Lett. 2012;14:1768. doi: 10.1021/ol300366e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeKorver KA, Walton MC, North TD, Hsung RP. Org. Lett. 2011;13:4862. doi: 10.1021/ol201947b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.