

Abstract
Physical biochemical techniques are used to establish the structure, subunit stoichiometry, and assembly pathway of the primosome complex of the bacteriophage T4 DNA replication system. Analytical ultracentrifugation and fluorescence anisotropy methods show that the functional T4 primosome consists of six gp41 helicase subunits that assemble into a hexagon, driven by the binding of six NTPs (or six nonhydrolyzable GTPγS analogues) that are located at and stabilize the intersubunit interfaces, together with a single tightly bound gp61 primase subunit. Assembling the components of the primosome onto a model DNA replication fork is a multistep process, but equilibrium cannot be reached along all mixing pathways. Producing a functional complex requires that the helicase hexamer be assembled in the presence of the DNA replication fork construct prior to the addition of the primase to avoid the formation of metastable DNA-protein aggregates. The gp41 helicase hexamer binds weakly to fork DNA in the absence of primase, but forms a much more stable primosome complex that expresses full and functional helicase (and primase) activities when bound to a gp61 primase subunit at a helicase:primase subunit ratio of 6∶1. The presence of additional primase subunits does not change the molecular mass or helicase activity of the primosome, but significantly inhibits its primase activity. We develop both an assembly pathway and a minimal mechanistic model for the structure and function of the T4 primosome that are likely to be relevant to the assembly and function of the replication primosome subassemblies of higher organisms as well.
Keywords: DNA–protein complexes, macromolecular machines, duplex DNA unwinding, replication complex assembly
The DNA replication system of bacteriophage T4 contains eight different types of protein subunits, several present in multiple copies. Subsets of these components form three stable and functional protein complexes that can be assembled onto a model DNA replication fork in vitro to form an integrated T4 DNA replication complex that is capable of unwinding the parental DNA duplex and synthesizing new viral DNA with essentially in vivo rates and fidelity (1, 2). These replication subassemblies are: (i) the leading- and lagging-strand replication polymerases that catalyze the template-directed copying of the two parental-strands of the DNA genome at each cell division; (ii) the clamp-clamp loader complex that controls the processivity of the replication process by linking the polymerases to their respective template strands and also regulates the release and recycling of the lagging strand polymerase following the completion of each Okazaki fragment; and (iii) the helicase-primase (primosome) complex that unwinds the double-stranded genome ahead of the replication fork in its capacity as a helicase, while also performing template-directed synthesis of the RNA primers that reinitiate discontinuous lagging-strand DNA synthesis after each Okazaki fragment has been completed.
The subunit components and stoichiometries of the replication complexes of higher organisms closely resemble those of T4 (3, 4). Their helicases, in the presence of NTP, all form hexameric rings of identical subunits and interact with one or more primase subunits to form functional primosomes (5–7). Thus, the T4 DNA replication system and its constituent subassemblies comprise the simplest model system for studying many aspects of DNA replication in higher organisms (8). Furthermore, each of the three functional protein subassemblies can be loaded onto an appropriate DNA construct in vitro, permitting their mechanisms to be studied separately. DNA helicase activity is required to unwind the genomic DNA at the replication fork and to provide access for the DNA polymerases to their respective templates. This helicase function must be integrated with that of the primase, which serves as a low processivity RNA polymerase that carries out template-directed synthesis of RNA (5-mer) primers at approximately 1-kb intervals along the lagging strand DNA template. This functional integration of the helicase and the primase components of the T4 system alters both activities within the replication complex, and these interactions, together with the assembly pathway and subunit composition of the T4 primosome, form the main focus of this paper.
Biophysical studies of T4 primosome complexes (6, 7, 9, 10) have provided some information about their subunit stoichiometry, as well as insight into how these components work in conjunction with one another. However, two different sets of observations have come to different conclusions about the stoichiometry (and thus the mechanism) of the T4 primosome. In early work gel shift and protein–protein chemical cross-linking methodologies had shown that the gp61 primase subunit binds to the gp41 helicase hexamer at an approximately 1∶6 subunit stoichiometry (5, 7). In contrast, Benkovic and coworkers have interpreted more recent and indirect fluorescence anisotropy and scanning calorimetry experiments (10–12) to argue that the primase subunits may interact with gp41 in an approximately 6∶6 subunit stoichiometry. This difference is important because in a primosome containing one primase and six helicase subunits the primase must be shared, which makes very different functional and mechanistic demands on both helicase and primase mechanisms than does a 6∶6 ratio, as is found in the hexameric helicase of bacteriophage T7, where every helicase subunit carries its own primase domain (13). The replication complex of T7 differs in many important aspects from that of T4 or the replication complexes of higher organisms.
Given the importance of the bacteriophage T4 replication complex as a model system, we have undertaken additional and careful studies by several methodologies to further examine the issues of subunit stoichiometry and assembly of the T4 primosome. The results increase our understanding of the molecular interactions that regulate primosome assembly and its function as a helicase and a primase, as well as providing insight into interactions between the primosome and the other subcomplexes of the T4 DNA replication system and, by extension, our understanding of helicase-primase complexes in higher organisms.
Results
Assembly of the T4 helicase hexamer depends on the concentrations of gp41 subunits and NTP ligands. Analytical ultracentrifugation was used to characterize the equilibria involved in the assembly of the T4 gp41 helicase hexamer. Sedimentation velocity profiles of gp41 at a subunit concentration of 1.5 μM in the absence of NTP ligands showed only one significant peak with an s20,w of approximately 4.5 S (Fig. 1A; see also Fig. S1). Assuming average hydration, this is close to the sedimentation coefficient expected for these relatively spherical gp41 subunits (14), and confirms earlier findings that gp41 protein exists in solution primarily as a monomer at these protein concentrations in the absence of NTP (6). We confirmed also that the addition of GTPγS (a nonhydrolyzable analogue of GTP) to a solution of gp41 monomers at this concentration drives this replication helicase into a stable hexameric state. We note that two additional components appeared in the c(s) versus s20,w distribution plots as the GTPγS concentration was increased (Fig. 1 B and C). These components fall at s20,w values between that of the monomer (approximately 4.5 S) and the approximately 10.5 S peak that corresponds to the sedimentation of the hexamer form that dominates at high GTPγS concentrations (Fig. 1 C and D and Fig. S1F).* We suggest that these intermediate peaks represent hexamer assembly intermediates, with the approximately 6 S peak corresponding to gp41 dimers at approximately 8.2 S to gp41 tetramers. These assignments are consistent with hydrodynamic modeling calculations (see discussion of hydrodynamic calculations in SI Text). These intermediate peaks decreased in size as the concentration of GTPγS was increased over the experimentally accessible concentration range, as expected for an equilibrium system.† At a 20-fold molar excess of GTPγS over protein subunits most of the complexes formed physiologically relevant hexamers (Fig. 1D and Fig. S1 D and F) stabilized by NTP ligands binding at the gp41 subunit-subunit interfaces. At 3 μM concentrations of gp41 subunits an approximately 60-μM concentration of GTPγS was required to drive the assembly of gp41 hexamers essentially to completion under the solution conditions used here and in the absence of gp61 and DNA.
Fig. 1.

Oligomerization of T4 helicase (gp41) subunits as a function of gp41 and GTPγS concentrations. Sedimentation velocity profiles [(c)s versus s20,w distribution plots] at: (A) 1.5 μM gp41 (monomers) with no GTPγS; (B) 3 μM gp41 and 5 μM GTPγS; (C) 3 μM gp41 and 30 μM GTPγS; and (D) 3 μM gp41 and 60 μM GTPγS. The approximately 4.5 S peak corresponds to gp41 monomers and the peak at approximately 10.5 S to gp41 hexamers. The peaks at intervening s20,w values represent intermediate oligomers of gp41 subunits and show a progressive shift into hexamers as the concentration of GTPγS is increased. (Additional plots showing complete sedimentation profiles and residual values for the (c)s versus s20,w distribution plots of B and D are presented in Fig. S1.)
The Order of Addition of Components is Important in Assembling Stable T4 Primosomes.
Analytical ultracentrifugation and steady state fluorescence experiments showed that gp41 hexamers do not form stable complexes with ssDNA or DNA fork constructs at the concentrations tested (see SI Text and Fig. S2 A and B). Nevertheless, the helicase hexamer does exist as a stable component within the T4 replication system at the DNA replication fork. We had previously suggested that T4 gp61 primase might play this stabilizing role (7). However, undefined primase-DNA aggregates are formed when T4 primase, which sediments as a monomer with an s20,w of approximately 2.6 S in isolation (Fig. S3A), is mixed with ssDNA or DNA fork constructs (Fig. S3 B and C). Fluorescence anisotropy experiments in which increasing concentrations of gp61 were added to ssDNA confirmed the appearance of undefined primase-DNA aggregates (Fig. S3D). To assemble the components of the T4 replication system and determine the ratios of individual subunits in the final complex requires that we establish an assembly pathway that produces a stable complex and avoids the formation of metastable aggregates. To this end, a series of experiments were performed in which various order-of-addition protocols were tested and the concentrations of individual components were varied to define optimal (and equilibrium) mixing conditions for assembling stable and active primosomes on DNA fork constructs. The details of some of these assembly pathway experiments are presented in SI Text.
We tested a possible assembly route involving initial mixing of helicase and primase subunits in the presence of GTPγS, followed by the addition of DNA. This procedure did not result in stable primosome-DNA complexes (Fig. S4). The addition of gp41 to mixtures of DNA, gp61, and GTPγS also resulted in aggregation and precipitation (Fig. S5), showing that this order-of-addition pathway leads to the formation of metastable aggregates rather than stable and equilibrated DNA-bound primosome complexes. Finally, we tried adding gp61 to an initial mixture of DNA and hexameric helicase molecules. Using this assembly protocol we were able to show that adding GTPγS to a solution containing the DNA construct, followed by the addition of gp41 subunits, resulted in the formation of unstable DNA-gp41 helicase hexamer complexes that could then be stabilized by adding primase. This order-of-addition pathway led to clear and reproducible sedimentation velocity boundaries (Fig. 2 A–D) and to well-defined stoichiometric complexes in fluorescence anisotropy titrations (Fig. 2E).
Fig. 2.

Association states of ssDNA-gp41-gp61 complexes in the presence of GTP or GTPγS show that the helicase∶primase subunit ratio in the T4 primosome is 6∶1. Sedimentation velocity c(s) versus s20,w distribution plots for: (A) 500 nM dT45, 3 μM gp41, 30 μM GTPγS and 500 nM gp61 (ratio of gp41∶gp61 subunits is 6∶1); (B) 3 μM dT45, 3 μM gp41, 60 μM GTPγS and 3 μM gp61 (ratio of gp41∶gp61 subunits is 1∶1); and (C) 3 μM dT45, 3 μM gp41, 60 μM GTPγS and 4.3 μM gp61 (ratio of gp41∶gp61 subunits is 2∶3). All plots show three peaks with s20,w values of approximately 4.5 S (gp41 monomers), approximately 2.6 S (gp61 monomers) and approximately 12.1 S (DNA-primosome complex). (D) Primosome-DNA s20,w values as a function of increasing concentrations of gp61 subunits per gp41 hexamer. (E) Steady state fluorescence anisotropy monitoring the titration of gp61 into a solution containing 3 μM 5′-Oregon green-labeled ssDNA, 3 μM gp41 (monomers), and 60 μM GTPγS. The anisotropy data show a stoichiometric break point at approximately 500 nM gp61, corresponding to a T4 primosome complex containing gp41 and gp61 subunits (and DNA constructs) in a 6∶1∶1 molar ratio.
A Stable Primosome Complex is Formed with DNA at a 6∶1 Helicase-Primase Subunit Stoichiometry.
To achieve maximal processivity and activity, a functional helicase must bind tightly to the DNA fork construct that serves as an unwinding substrate. Sedimentation velocity experiments showed that the addition of even small amounts of gp61 increased the stability of complexes between gp41 hexamers and DNA (Fig. S6). Titration of gp61 into preformed gp41-GTPγS-DNA complexes, monitored by fluorescence anisotropy (Fig. 2E), confirmed this result. Given that gp61 can interact with both gp41 and DNA, it was necessary to determine the subunit stoichiometry at which these components interact to form a stable and active T4 helicase-primase (primosome) complex.
As summarized above, two sets of experiments in the T4 DNA replication system literature have previously dealt with the subunit stoichiometry of these complexes and came to different conclusions. Using physical methods, Dong, et al. (7) showed that the gp41∶gp61 subunit ratio in these complexes was approximately 6∶1, consistent with an earlier approximately 5∶1 subunit ratio measured in functional studies by Richardson and Nossal (5). In contrast, Benkovic and coworkers (10–12) interpreted their scanning calorimetry, fluorescence anisotropy, and electron microscopy studies on T4 helicase-primase complexes to suggest that the relevant subunit ratio might be 6∶6. We note that these workers formed their primase-DNA complexes by direct mixing and that their fluorescence anisotropy titrations (figure 1 in ref. 11) did not reach stable plateaus. Rather, their published titration curves closely resemble those of Fig. S3D, which we have shown to reflect the formation of undefined DNA-primase aggregates.
Further assembly experiments were performed to test these contrasting stoichiometry conclusions and define the correct subunit ratio for the functional T4 primosome subassembly. As stated above, our successful approach involved forming weakly associating DNA-gp41-GTPγS complexes and then adding gp61. This procedure permits the system to equilibrate without forming metastable primase-DNA aggregates. Experiments were performed in which the concentration of input gp61 subunits was varied relative to the concentration of the DNA-gp41-GTPγS components present (Fig. 2). Components mixed at an input (subunit or molecule) ratio of 6∶1∶1 (gp41∶gp61∶DNA) showed a major sedimentation peak at s20,w = 12.1 S, and smaller peaks at 2.6 S and 4.5 S (Fig. 2A). These smaller peaks sediment with the s20,w values characteristic of gp41 and gp61 monomers (Fig. 1A and Fig. S3A). We suggest that the 12.1 S peak corresponds to an equilibrium ternary (DNA-gp41-gp61) primosome complex that contains one primase subunit per gp41 hexamer. The s20,w of this component is substantially greater than the 10.5 S value measured for the GTPγS-stabilized gp41 hexamer in the absence of primase (Fig. 1D).‡
Varying the Input Ratios of gp41 and gp61 Subunits.
This 6∶1 helicase∶primase subunit assignment was further tested in sedimentation experiments by varying the subunit input ratios. In one set of experiments the input gp61 concentration was increased either sixfold or ninefold, resulting in a 6∶6 (Fig. 2B) or 6∶9 (Fig. 2C) helicase∶primase input subunit ratios. The formation of stable primosome complexes containing a gp41 helicase hexamer and more than one primase subunit should increase the observed s20,w values for the ternary complexes at the higher gp61 input levels. We found that a s20,w of 12.1 ± 0.5 S was maintained at all three input ratios, suggesting that larger stable primosome complexes containing more primase subunits per helicase hexamer do not form (Fig. 2D). These experiments also showed that the gp61 subunits present beyond those needed to form the 6∶1 complex sedimented at approximately 2.2 S, close to the s20,w for free gp61 monomers, while the primosome peak remained at approximately 12.1 S. Similar experiments were performed with solutions containing input gp41 concentrations in excess of the 6∶1 helicase∶primase subunit ratio (see SI Text and Fig. S6). Here, the excess gp41 appeared as gp41 oligomer peaks that sedimented at approximately 10.5 S or less, while the largest (presumably primosome) peak continued to sediment at 12.1 S.
Fluorescence anisotropy experiments in which gp61 was titrated into a solution containing premixed DNA-gp41-GTPγS solutions at a total gp41 subunit concentration of 3 μM (Fig. 2E) were also performed. The addition of gp61 resulted in a steep increase in anisotropy and a clear breakpoint at approximately 500 nM, again confirming a 6∶1 association stoichiometry for the T4 DNA-primosome complex. In contrast, the addition of gp41 to a preformed complex of gp61 and DNA under the same solution conditions showed an initial drop in anisotropy and then an increase, again suggesting that undefined and concentration-dependent aggregation occurs under these experimental conditions (Fig. S4B). We conclude that the assembly pathway described here and a 6∶1 gp41∶gp61 subunit ratio lead to a physically stable and discrete T4 primosome-DNA complex.
To further test this conclusion we also performed hydrodynamic modeling calculations (15, 16) in which the complexes were represented by constellations of appropriately sized spherical beads in varying geometries, and values of s20,w were calculated and compared with our experimental measurements of s20,w. The calculations are summarized in Fig. S7 and show that, while these hydrodynamic calculations cannot help us to discriminate between reasonable primosome structures containing from one to three primase subunits per gp41 hexamer, complexes containing six primase subunits per helicase hexamer are clearly ruled out by this approach as well.
Functional Unwinding and Priming Assays Confirm an Approximately 6∶1 gp41∶gp61 Subunit Stoichiometry for the T4 Primosome.
Finally, we ask whether complexes assembled according to the above protocols and subunit stoichiometries are also optimal in functional assays. Previous experiments in which Richardson and Nossal (5) examined the effects of primase on the helicase activity demonstrated that the T4 primosome unwound a helicase assay substrate construct (formed from M13mp2 ssDNA bound to a 51-bp complementary strand with a 15-nt long noncomplementary 3′ tail) much more efficiently than did gp41 helicase hexamer alone. Furthermore, these experiments showed that helicase unwinding activity plotted as a function of input gp61 reached a plateau at a helicase∶primase subunit ratio of approximately 6∶1 (5). These results are included in Fig. 3 to compare with new experiments that we carried out to examine the physiological priming activity of the T4 primosome complex, using the RNA priming assay described in Materials and Methods and SI Text.
Fig. 3.

The primase and helicase activities of the T4 primosome are optimal at an approximately 6∶1 gp41∶gp61 subunit ratio. RNA primer synthesis activity (filled circles) measures the amount of [α32P]rCTP incorporated into RNA primer pentamers after a reaction time of 20 min (see Materials and Methods). Increasing concentrations of gp61 subunits were titrated into a reaction mix containing a fixed concentration of gp41 helicase hexamers. The priming activity reached a maximum at a gp61 concentration ratio somewhat in excess of one primase subunit per gp41 helicase hexamer and then decreased progressively as more gp61 was added (see text). The helicase data were taken from Fig. 4 of Richardson and Nossal (5), and show that the unwinding activity of either 50 pmols (open squares) or 100 pmols (open circles) of T4 gp41 hexamer on a helicase substrate consisting of a 3-tailed ssDNA M13 template complexed with a 51 bp duplex construct reached a plateau value at a gp61 subunit concentration ratio of approximately one gp61 subunit per gp41 hexamer.
As previously shown (17), the addition of gp61 to gp41, together with DNA substrates containing suitable priming template sequences, results in the synthesis of pentameric RNA primers on the template DNA. Here (Fig. 3), we demonstrate that the titration of gp61 subunits into a primase assay system containing a fixed concentration of gp41 helicase hexamers, ssDNA and excess concentrations of all four canonical NTPs resulted in a progressive increase in the rate of formation of pentameric RNA primers, and that this primase activity reached a maximum value at an input primase to helicase subunit ratio of 1 to 1.5 gp61 subunits per helicase hexamer (grey zone in Fig. 3). The addition of more primase subunits resulted in a progressive decrease in the rate of primer formation, showing that the addition of primase beyond an approximately 6∶1 helicase∶primase subunit ratio inhibits RNA primer synthesis. At a 6∶6 molar ratio of gp41∶gp61 subunits this inhibition was essentially complete (Fig. 3). We suggest that this inhibition may reflect the occlusion of priming sequences as the excess gp61 subunits form metastable ssDNA-primase aggregates with the ssDNA template.
Discussion
Overview.
The following results relevant to the structure, function, subunit stoichiometry and assembly pathway of the T4 primosome complex have been established or confirmed. (i) The T4 primosome comprises six helicase (gp41) and one primase (gp61) subunits, additional primase subunits are not bound, and a primosome complex with this 6∶1 subunit stoichiometry is stable and fully active as both a helicase and a primase. (ii) The helicase hexamer of the primosome assembles from free gp41 monomers via dimer and tetramer intermediates in the presence of hydrolyzable (GTP) or non-hydrolyzable (GTPγS) ribonucleotide triphosphates. (iii) Primosome assembly is an equilibrium process and intermediates can be detected by ultracentrifugation in the presence of limited concentrations of helicase or primase subunits or GTPγS. (iv) An NTP-saturated primosome complex with a 6∶1 helicase∶primase subunit composition binds stably to ssDNA or to a DNA replication fork, while an NTP-saturated gp41 hexameric helicase alone binds much more weakly.
We have defined a primosome assembly (order-of-addition) pathway that leads to a stable equilibrium complex with model DNA replication forks. This pathway is shown in Fig. 4. We have also shown that the use of other assembly protocols, such as adding gp61 to a forked DNA construct in the absence of stable gp41 hexamers, leads to aggregation and metastable complexes. We suggest that this may explain some earlier fluorescence anisotropy and titration calorimetry data that seemed to suggest helicase-primase subunit stoichiometries approaching 6∶6 (10–12). Clearly even in equilibrium situations one cannot assume that a stable and functional macromolecular complex has been formed just because the components are mixed in correct proportions at concentrations above the relevant dissociation constants; rather it must be demonstrated directly that the mixing protocol does not result in the formation of undefined complexes that are trapped in metastable states. In vivo this pathway problem is probably avoided by specific or nonspecific chaperones that help macromolecular complexes attain conformational and compositional equilibrium.
Fig. 4.

Assembly pathway for the T4 DNA replication primosome. The primosome components are: gp41 subunits (blue ovals), gp61 subunit (red ellipse), GTP or GTPγS (yellow rectangles), and GDP (violet rectangle). The gp61 primase subunits are shown as elongated ellipsoids to reflect their higher frictional ratios compared with the relatively globular helicase subunits. The potential assembly pathways on the Right or Left of the figure lead to the formation of metastable aggregates, while the central pathway leads to a well-defined equilibrium complex with 6∶1 helicase to primase subunit stoichiometry, as shown by the included titration plots. For further details see text.
Significance of a 6∶1 Helicase∶Primase Subunit Stoichiometry.
The operation of the primosome with a single primase subunit puts important constraints on mechanistic models for helicase and primase functions, both for T4 and for the replication complexes of higher organisms that seem also to function with significantly fewer primase than helicase subunits. The primase monomer is smaller than a helicase subunit, making it unlikely that more than one or two helicase subunits can be in contact with the primase subunit at any one time. Our demonstration here that the primase subunit plays a central role in binding the primosome to the replication fork suggests, for any model in which all the subunits of the gp41 hexamer play active roles, that the primase is likely to be repositioned on the helicase hexamer at every iteration of the helicase cycle that moves the primosome complex progressively into the replication fork.
A Simple Mechanistic Model for the T4 Primosome Helicase.
A conceptually simple (and speculative) model for primosome function at the replication fork, which is based on the literature and the issues of subunit stoichiometry and interaction that we have defined here and is also compatible with what we know about hexameric replication helicases that react with limited numbers of primase subunits in higher organisms, is outlined in Fig. S8. It has the following features. (i) The primosome is positioned at the replication fork with the lagging DNA template strand passing through the center of the helicase hexamer and the leading template strand bound to the outside of approximately two gp41 subunits. This is compatible with a ssDNA binding site size for the T4 helicase of approximately 20 nts (18) and is also consistent with the binding site size that has been demonstrated for the closely related hexameric dnaB helicase of E. coli (19). (ii) The primase subunit binds to two ssDNA-bound gp41 subunits at the replication fork (20), bringing it into effective contact with the NTP ligand that lies between these subunits. This primase binding proximity could then specifically “activate” the hydrolysis of the NTP located at this position, while not perturbing the NTP ligands that lie between and stabilize other gp41 subunit interfaces and are not directly adjacent to the primase subunit.§ The primase subunit likely binds leading strand ssDNA at the fork and functional arguments show that in the complete replication complex the primase must also lie sufficiently close to the lagging strand to permit it to use this ssDNA as templating sequences for RNA synthesis. This primer synthesis could take place as the separated ssDNA of the lagging strand enters the central hole in the helicase hexamer. (iii) The activated NTP of the primosome helicase then hydrolyzes to NDP and Pi, transiently releasing the primase subunit from the lagging strand ssDNA and also specifically weakening the interaction of the approximately 2 gp41 subunits with the fork junction. This could permit the helicase hexamer to rotate by one subunit relative to the fork and also to “move into” the dsDNA of the fork by one nucleotide position as a consequence of local fork “breathing” (21), followed by the rebinding of the next pair of gp41 subunits at the replication fork and the concomitant rebinding of the primase to the fork DNA, but now translocated around the hexamer helicase ring by one gp41 subunit position. The primase subunit could achieve this reorientation by dissociating and rebinding, or it could remain bound to one of the ssDNA strands and/or gp41 subunits and “pivot” into its new position without dissociation.
This model is consistent with a step-wise and sequential hydrolysis of the primosome-bound NTPs around the helicase ring. We emphasize that this mechanism requires a “mobile” primase and is thus fundamentally different from one that might apply to a T7 DNA helicase-primase complex in which each subunit of the T7 helicase-primase complex carries both a helicase and a primase domain. In a recent single molecule study Sun et al. (22) have also proposed a mechanism for the T7 helicase in which the NTPs bound at the interface fire sequentially and “cooperatively” as the helicase moves into the replication fork. We note that the model outlined above is basically a Brownian ratchet, with the primase playing the role of the ratchet. Other proposed mechanisms for hexameric helicases, developed largely on the basis of crystal structures, are conceptually closer to power-stroke models (23, 24). Elsewhere (DJ, SEW and PHvH; manuscript under review) we have further developed and refined the molecular details of this mechanistic model on the basis of near UV spectral measurements with site-specifically placed DNA analogue base probes that can directly monitor dsDNA base pair opening and closing events and the stacking and unstacking of vicinal bases within the replication fork as the T4 primosome helicase advances.
Materials and Methods
Materials.
Unless stated otherwise, all experiments were performed at 20 °C in buffer containing 20 mM HEPES (pH 7.5), 150 mM K(OAc), 10 mM Mg(OAc)2, 0.1 mM EDTA and 1 mM DTT. Purine nucleoside triphosphates and their non-hydrolysable analogues were purchased from Sigma-Aldrich. [γ-32P] GTP and [γ-32P] ATP were obtained from NEN (Boston, MA). Unlabeled and 2-AP labeled DNA oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). DNA oligonucleotides 5′-labeled with Oregon Green 488 were from Operon (Huntsville, AL). Oligonucleotide concentrations were determined by absorbance at 260 nm using extinction coefficients furnished by the manufacturer. Forked DNA constructs were annealed by heating equimolar concentrations of DNA primer and template strands at 90 °C for 5 min, followed by gradual (2 h) cooling to 25 °C. The DNA constructs used in this study are shown in Table S1. The T4-coded DNA replication helicase (gp41) and replication primase (gp61) were prepared and concentrations determined as previously described (see 6, 20 and SI Text).
Primase Assay.
Primase assays were performed in a reaction buffer containing 33 mM Tris-OAc, 125 mM KOAc, 6 mM Mg(OAc)2, 0.5 mM DTT and 200 μM each of all four canonical rNTPs, in addition to 3.5 mM ATP and 100 nM [α-32P]rCTP. Unless otherwise indicated, the reaction mixtures contained 800 nM gp41 and 50–900 nM gp61 (concentrations as protein subunits) and 100 nM of ssDNA construct (Table S1) in a 10-μl reaction volume. Reaction mixtures were assembled on ice and preincubated at 37 °C for 2 min. Reactions were started by the addition of the proteins components, incubated at 37 °C for 30 min and then quenched by adding 10 μl of 95% deionized formamide containing 20 mM EDTA, 0.25% xylene cyanol and 0.25% bromophenol blue. The quenched reactions were subjected to electrophoresis on 22% polyacrylamide 8 M urea gels (19∶1 acrylamide∶bis-acrylamide) at 40 watts for 90 min and the dried gels were quantitated using an AMBIS scanner.
Analytical Ultracentrifugation.
Sedimentation velocity experiments were performed in a Beckman Optima XL-I Analytical Ultracentrifuge. Protein and nucleic acid samples were dialyzed extensively against reference buffer [20 mM HEPES (pH 7.5), 150 mM K(OAc), 10 mM Mg(OAc)2] prior to centrifugation and protein and nucleic acid concentrations were re-measured after dialysis. Sedimentation runs were performed and analyzed by standard techniques; see SI Text and Fig. S1.
Steady State Fluorescence and Anisotropy Experiments.
Fluorescence experiments were performed with a Jobin-Yvon Fluorolog or Fluoromax spectrofluorimeter. The excitation wavelength for Oregon Green labeled samples was 494 nm and emission was monitored at 520 nm. When 2-AP served as the fluorescent probe the excitation wavelength was 315 nm and emission spectra were recorded from 330 to 400 nm. Binding curves were fit to Eq. 1:
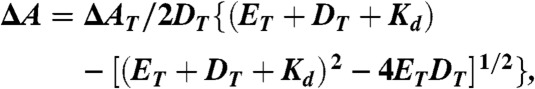 |
[1] |
where ΔA is the change in anisotropy at each point in the titration, ΔAT is the total anisotropy change, DT is the total DNA concentration, ET is the total gp61 concentration and Kd is the dissociation constant.
Supplementary Material
ACKNOWLEDGMENTS.
We are grateful to our laboratory colleagues (especially Walt Baase and Sandra Grieve) for many helpful discussions of this work. These studies were supported by NIH Grant GM-15792 to P.H.v.H., who is also an American Cancer Society Research Professor of Chemistry.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1210040109/-/DCSupplemental.
*We note that these c(s) versus S distribution plots show a nonzero point (and a peak) at each sedimentation coefficient value at which a resolvable boundary is centered in a sedimentation velocity run. Points are taken at 0.1 S intervals. The height of each peak represents the concentration of the corresponding component, but we note that spreading due to homogenous (Gaussian) diffusion has been removed from these representations and therefore the apparent width of each peak (when it contains more than one point) is due only to the presence of minor apparently independently sedimenting components detected by the program. For further details of the steps involved in the analysis of such sedimentation velocity experiments see SI Text, Fig. S1.
†The GTPγS concentrations used were ≤ 60 μM; at higher concentrations the optical density of this ligand at 280 nm overwhelmed the optical density of the protein components.
‡The estimated standard error of the determination of these sedimentation coefficients between different runs is approximately ± 0.6 S, and the standard error for the position of a peak on an individual c(s) versus s20,w plot is (by definition) ± 0.1 S, because that is the spacing at which we took points in the experiments.
References
- 1.Alberts BM. Prokaryotic DNA replication mechanisms. Philos Trans R Soc Lond B Biol Sci. 1987;317:395–420. doi: 10.1098/rstb.1987.0068. [DOI] [PubMed] [Google Scholar]
- 2.Nossal NG. In: Molecular Biology of Bacteriophage T4. Karam JD, editor. Washington, DC: American Society for Microbiology; 1994. editor-in-chief. [Google Scholar]
- 3.Mueser TC, Hinerman JM, Devos JM, Boyer RA, Williams KJ. Structural analysis of bacteriophage T4 DNA replication: A review in the Virology Journal series on bacteriophage T4 and its relatives. Virol J. 2010;7:359. doi: 10.1186/1743-422X-7-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson A, O’Donnell M. Cellular DNA replicases: Components and dynamics at the replication fork. Annu Rev Biochem. 2005;74:283–315. doi: 10.1146/annurev.biochem.73.011303.073859. [DOI] [PubMed] [Google Scholar]
- 5.Richardson RW, Nossal NG. Characterization of the bacteriophage T4 gene 41 DNA helicase. J Biol Chem. 1989;264:4725–4731. [PubMed] [Google Scholar]
- 6.Dong F, Gogol EP, von Hippel PH. The phage T4-coded DNA replication helicase (gp41) forms a hexamer upon activation by nucleoside triphosphate. J Biol Chem. 1995;270:7462–7473. doi: 10.1074/jbc.270.13.7462. [DOI] [PubMed] [Google Scholar]
- 7.Dong F, von Hippel PH. The ATP-activated hexameric helicase of bacteriophage T4 (gp41) forms a stable primosome with a single subunit of T4-coded primase (gp61) J Biol Chem. 1996;271:19625–19631. doi: 10.1074/jbc.271.32.19625. [DOI] [PubMed] [Google Scholar]
- 8.Sinha NK, Morris CF, Alberts BM. Efficient in vitro replication of double-stranded DNA templates by a purified T4 bacteriophage replication system. J Biol Chem. 1980;255:4290–4293. [PubMed] [Google Scholar]
- 9.Dong F, Weitzel SE, von Hippel PH. A coupled complex of T4 DNA replication helicase (gp41) and polymerase (gp43) can perform rapid and processive DNA strand-displacement synthesis. Proc Natl Acad Sci USA. 1996;93:14456–14461. doi: 10.1073/pnas.93.25.14456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valentine AM, Ishmael FT, Shier VK, Benkovic SJ. A zinc ribbon protein in DNA replication: Primer synthesis and macromolecular interactions by the bacteriophage T4 primase. Biochemistry. 2001;40:15074–15085. doi: 10.1021/bi0108554. [DOI] [PubMed] [Google Scholar]
- 11.Yang J, Xi J, Zhuang Z, Benkovic SJ. The oligomeric T4 primase is the functional form during replication. J Biol Chem. 2005;280:25416–25423. doi: 10.1074/jbc.M501847200. [DOI] [PubMed] [Google Scholar]
- 12.Norcum MT, et al. Architecture of the bacteriophage T4 primosome: Electron microscopy studies of helicase (gp41) and primase (gp61) Proc Natl Acad Sci USA. 2005;102:3623–3626. doi: 10.1073/pnas.0500713102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tabor S, Richardson CC. Template recognition sequence for RNA primer synthesis by gene 4 protein of bacteriophage T7. Proc Natl Acad Sci USA. 1981;78:205–209. doi: 10.1073/pnas.78.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erickson HP. Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol Proced Online. 2009;11:32–51. doi: 10.1007/s12575-009-9008-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bloomfield V, Dalton WO, Van Holde KE. Frictional coefficients of multisubunit structures. I. Theory. Biopolymers. 1967;5:135–148. doi: 10.1002/bip.1967.360050202. [DOI] [PubMed] [Google Scholar]
- 16.Carrasco B, Garcia de la Torre J. Hydrodynamic properties of rigid particles: Comparison of different modeling and computational procedures. Biophys J. 1999;76:3044–3057. doi: 10.1016/S0006-3495(99)77457-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu CC, Alberts BM. Pentaribonucleotides of mixed sequence are synthesized and efficiently prime de novo DNA chain starts in the T4 bacteriophage DNA replication system. Proc Natl Acad Sci USA. 1980;77:5698–5702. doi: 10.1073/pnas.77.10.5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Young MC, Kuhl SB, von Hippel PH. Kinetic theory of ATP-driven translocases on one-dimensional polymer lattices. J Mol Biol. 1994;235:1436–1446. doi: 10.1006/jmbi.1994.1099. [DOI] [PubMed] [Google Scholar]
- 19.Bujalowski W, Jezewska MJ. Interactions of Escherichia coli primary replicative helicase DnaB protein with single-stranded DNA. The nucleic acid does not wrap around the protein hexamer. Biochemistry. 1995;34:8513–8519. doi: 10.1021/bi00027a001. [DOI] [PubMed] [Google Scholar]
- 20.Jing DH, Dong F, Latham GJ, von Hippel PH. Interactions of bacteriophage T4-coded primase (gp61) with the T4 replication helicase (gp41) and DNA in primosome formation. J Biol Chem. 1999;274:27287–27298. doi: 10.1074/jbc.274.38.27287. [DOI] [PubMed] [Google Scholar]
- 21.Jose D, Datta K, Johnson NP, von Hippel PH. Spectroscopic studies of position-specific DNA ‘breathing’ fluctuations at replication forks and primer-template junctions. Proc Natl Acad Sci USA. 2009;106:4231–4236. doi: 10.1073/pnas.0900803106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun B, et al. ATP-induced helicase slippage reveals highly coordinated subunits. Nature. 2011;478:132–135. doi: 10.1038/nature10409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Enemark EJ, Joshua-Tor L. Mechanism of DNA translocation in a replicative hexameric helicase. Nature. 2006;442:270–275. doi: 10.1038/nature04943. [DOI] [PubMed] [Google Scholar]
- 24.Thomsen ND, Berger JM. Running in reverse: The structural basis for translocation polarity in hexameric helicases. Cell. 2009;139:523–534. doi: 10.1016/j.cell.2009.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pietroni P, von Hippel PH. Multiple ATP binding is required to stabilize the ‘activated’ (clamp open) clamp loader of the T4 DNA replication complex. J Biol Chem. 2008;283:28338–28353. doi: 10.1074/jbc.M804371200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelch BA, Makino DL, O’Donnell M, Kuriyan J. How a DNA polymerase clamp loader opens a sliding clamp. Science. 2011;334:1675–1680. doi: 10.1126/science.1211884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.