

Abstract
Recent simultaneous progress in human and animal model genetics and the advent of microarray whole genome expression profiling have produced prodigious data sets on genetic loci, potential candidate genes, and differential gene expression related to alcoholism and ethanol behaviors. Validated target genes or gene networks functioning in alcoholism are still of meager proportions. Genetical genomics, which combines genetic analysis of both traditional phenotypes and whole genome expression data, offers a potential methodology for characterizing brain gene networks functioning in alcoholism. This chapter will describe concepts, approaches, and recent findings in the field of genetical genomics as it applies to alcohol research.
I. Introduction
Rapid progress in human and animal model genetics over the last 10–15 years has identified multiple genetic regions or candidate genes as associated with risk for alcoholism or ethanol-related behavioral responses. Simultaneously, the advent of high-throughput whole genome expression profiling with microarrays has produced prodigious data sets on differential gene expression related to alcoholism and ethanol behaviors. These expression profiling results, combined with modern bioinformatics tools, often produce insight into functional gene networks relevant to ethanol rather than simple lists of genes. Despite these parallel marked advances in our molecular databases relevant to alcoholism, there remains considerable difficulty in providing an “endgame” regarding identifying genes or gene networks that actually improve our ability to treat alcoholism or identify risk factors for becoming an alcoholic. Both genetics and genomics appear to have their individual weaknesses. Modern genetic approaches such as genome-wide association studies (GWAS) provide huge lists of questionably significant genes associated with alcoholism traits. Adequate statistical power to detect a large portion of the genetic variance underlying complex traits such as alcoholism seems difficult or impossible to achieve. Conversely, microarray studies provide dizzyingly complex lists of genes or gene network diagrams but most often without a causal link to ethanol behaviors or other complex phenotypes.
This chapter will describe a novel approach to solving the issues raised above. Genetical genomics, a term coined by Jansen and Nap (2001), combines the use of complex genetic studies on both behavior and gene expression. This represents a form of molecular triangulation whereby genetic variance in both gene expression and ethanol behaviors is used to define causal links between the two fields. The underlying premise for this approach is that gene expression per se can be treated as a quantitative trait just as with behavioral phenotypes, and that genetically driven alterations in gene expression might underlie much of the genetic contribution to complex traits. By using genetic linkage to anchor both gene expression networks and behavioral sub-phenotypes, the actual goal of defining an “alcoholism gene” or quantitative trait gene (QTG) becomes, at least in part, irrelevant. In short, it is “the network, stupid,” that becomes the focus of attention with this approach. We define such a focus with the operative term “quantitative trait gene network” (QTGN).
Numerous reviews and original papers concerning genetical genomics have been published over the last few years and many of these concern applications to ethanol behavioral genetics. This chapter will strive to describe the key elements and resources in each aspect of genetical genomics and summarize recent work relevant to ethanol responses and alcoholism. In particular, we will try to provide an overview of how genetical genomics in general can provide enormous power in ranking candidate genes underlying ethanol behavioral quantitative trait loci (QTLs). We will also introduce the concept of studying the genetics of ethanol-responsive gene expression networks (as opposed to basal gene expression) in mouse brain regions. This recent work from our own laboratory and others serves to provide novel insight for both the mechanisms and functional consequences associated with brain ethanol-responsive gene expression networks. It is our hope that the reader will be left with both a clear understanding of the goals and promise for ethanol genetical genomics studies, as well as the basic principles and shortcomings for this complex but exciting area of research.
II. Genetic and Genomic Approaches in Alcohol Research
A. Behavioral QTL Mapping and Identifying QTGs by Traditional Mapping Strategies
As the field of genetics moved beyond the challenge of identifying the genetic basis of binary phenotypes and began to shift its focus to complex traits, new tools were required. Like most human diseases, alcoholism is a complex trait, influenced by a combination of multiple genetic and environmental factors, and interactions thereof. The weapon of choice for identifying genes that influence complex diseases has become quantitative trait locus (QTL) mapping. A more complete description of QTL mapping will be found in the chapter by Buck and colleagues within this volume. Here we include a brief description of the concepts for reference to expression QTL mapping discussed below.
The strategy for QTL mapping a phenotype of interest is similar to that of classic linkage analysis, with a common goal of identifying alleles that co-segregate with a disease trait more frequently than would be expected by chance. Working with complex traits that typically vary in their manifestation across a continuous distribution, in contrast to the binary nature of monogenic traits, QTLs are discovered by simply identifying loci with alleles that consistently covary with a phenotype across a population. Genomic regions that show a sufficiently strong association with a phenotype are considered QTLs. The simplest, or most hopeful, interpretation of a mapped QTL is that the implicated region harbors a single gene affecting manifestation of the associated phenotype. However, it is quite possible that a QTL is driven by multiple genes, non-coding RNA species, epigenetic mechanisms (including environmental interactions), or a combination thereof.
While QTL mapping studies may be carried out in human populations, the inflated non-genetic variance contributed to a phenotype by each subject's unique environment and life experience introduces a tremendous amount of noise, making results difficult to interpret (Broman, 2009). These issues, plus the potential population stratification issues and tremendous costs associated with assembling sufficiently large human samples, explain why most QTL mapping studies are conducted with rodents, and primarily mice.
The mouse is a very attractive subject for dissecting complex traits by QTL analysis for a variety of reasons. Apart from its small size, relatively low maintenance cost, and short gestation period (Peters et al., 2007), the mouse provides an incredibly deep and ever-expanding arsenal of genetic tools. Laboratory mouse providers such as Jackson Laboratories and Charles Rivers Laboratories make available hundreds of inbred mice that vary greatly in genotype and phenotype. Perhaps the greatest advantage to using mice for QTL mapping regards the fact that the entire genome from multiple mouse strains has been fully sequenced with 17 strains currently being completed by the Wellcome Trust Sanger Institute (http://www.sanger.ac.uk/resources/mouse/genomes/).
Because these inbred animals are completely isogenic, meaning they carry two effectively identical haploid genomes, each generation of progeny is a genetic clone of its forebears. As such, these genetic lines are essentially immortal, an incredible boon for scientific reproducibility because an experiment conducted in laboratory “A” can be directly compared against the results published by laboratory “B” when the same inbred strain is used, even if laboratory B's results were published years earlier. However, some confounding interactions with the laboratory environment have been reported for mouse genetic studies (Crabbe et al., 1999). Nevertheless, the popularity of inbred mice has greatly encouraged interlab collaborations and more open data sharing.
Of course QTLs can only be mapped in the presence of genetic variation; therefore, QTL studies are usually conducted using derivatives of inbred strains. A typical experimental design might involve characterizing a panel of F2 progeny for a phenotype where the inbred parental strains differ significantly. QTL mapping could then commence after genotyping each of the F2 progeny. A special derivative of inbred strains, recombinant inbred (RI) strains, are produced much the same as an F2 panel but includes an additional phase of multiple generations of sibling inbreeding (Bailey, 1971). The result is a panel of new inbred strains, each one carrying a unique combination of the progenitor genomes.
Established RI panels carry several significant advantages over other gene mapping populations. Being inbred, each strain needs to be genotyped only once, provided that breeding strategies are designed to minimize genetic drift. In practice, this has meant that larger laboratories specializing in genotyping, such as the Wellcome Trust Sanger Institute and the Jackson Laboratory (see http://cgd.jax.org/cgdsnpdb/), have genotyped many inbred strains or RI panels and generously made the results publicly available. This exemplifies the true power of utilizing RI panels in QTL mapping studies; all acquired data is cumulative and directly relatable, regardless of where it originates. A bold new extension of the use of RI panels will soon be available with the generation of a new panel, termed the Collaborative Cross, containing up to 1000 inbred lines derived from eight progenitor inbred lines (Churchill et al., 2004). This enormous resource should allow the fine mapping of complex traits with detection of loci having even very low contributions to genetic variance of a trait. Furthermore, the use of multiple strains in developing the collaborative cross will enrich the genetic variation of the panel compared to traditional RI batteries that are derived from two progenitor strains.
At present, the most widely used RI panels for alcohol research have been the BXD and LXS batteries of RI lines. The BXD lines, derived from progenitor C57BL/6J (B6) and DBA2/J (D2), currently have over 80 inbred strains (Peirce et al., 2004). The LXS strains were derived from the ILS and ISS inbred strains that were originally derived by selective breeding for sensitivity to ethanol sedation (Badcock et al., 2004). A large collection of behavioral, anatomical and neurochemical phenotypes derived from the BXD and LXS RI lines is maintained at the GeneNetwork web resource (www.GeneNetwork.org), with many of the >2000 phenotypes being related to ethanol or drugs of abuse.
The molecular and genetic resources outlined above serve to greatly increase the power and resolution of QTL mapping for various behaviors or other traits of interest. The major barriers to mapping are the availability of a trait amenable to high-throughput study with low technical or environmental variance and showing significant genetic variance across the reference population used for QTL mapping. However, despite tremendous advances in genetic resources and identification of a large number of behavioral, anatomical, or neurochemical QTLs, validation of corresponding QTGs has greatly lagged. This difficulty stems largely from lack of sufficient recombination events in existing mouse panels to reduce haplotype block size for fine mapping. Additionally, small effect sizes and epistatic interactions complicate detection of a QTL as fine mapping efforts proceed. The effect size issue may be due in part to the existence of multiple QTGs underlying QTL detected by initial screens. Strategies such as derivation of congenic lines have been successful for fine-mapping a number of ethanol traits and for identifying Mpdz as one of the first QTGs mapped for a mammalian behavioral phenotype (Buck et al., 1999; Fehr et al., 2002; Shirley et al., 2004). However, such approaches take large investments in time, animals, and research expenditures. In many cases, even with the derivation of congenic lines, the investigator is left with a genetic interval comprising millions of base pairs and possibly hundreds of candidate genes. As described below, the use of whole genome expression profiling has provided a powerful approach for obviating some of the difficulties presented by traditional genetic QTL mapping approaches.
B. Genome-Wide Analysis of mRNA
Through an unbiased, parallel examination of mRNA expression from virtually the entire genome, DNA microarrays identify genes or gene networks associated with a given phenotype (drug response, disease, developmental stage, etc.). Gene networks provided by such studies can provide novel information regarding the mechanisms underlying a disease state. Thus, microarrays provide an alternative or complementary approach to genetic mapping for the non-biased identification of disease-related genes. As described below, a large number of studies have indeed applied microrarray analysis to the study of alcoholism or responses to ethanol in animal models. In this section we provide a brief overview of mRNA expression microarrays as this is important to understanding the limitations of this technology for QTG identification. Many reviews have been written regarding the use of microarrays and provide a rich source for more detailed information (Brown and Botstein, 1999; Kerns and Miles, 2008; Lyons, 2003; Miles, 2001). In addition to mRNA expression analysis, a large number of microarray platforms now exist for conducting assays of microRNA abundance, mRNA splicing (exon arrays), transcription factor binding (ChIP on chip assays), and DNA methylation. Additionally, recent technological advances in DNA sequencing are enabling a whole new approach to transcriptome profiling. Such approaches, termed NextGen RNASeq, rely on ultrafast sequencing to provide a transcript count across the entire genome. This approach, not discussed further in this chapter, offers the advantage of profiling all splice variants of transcripts as well as not being susceptible to hybridization artifacts on microarrays due to single nucleotide polymorphisms (SNP effects; see below).
Multiple platforms now exist for whole genome–wide mRNA expression profiling. However, the existing literature mainly utilizes oligonucleotide microarrays produced by photolithography (Chee et al., 1996) or spotted cDNA microarrays (Schena et al., 1995). Recent work has turned to the use of oligonucleotides (50–70 nucleotides (nt)) either spotted on glass slides or immobilized on beads as preferable to the initial “Brown arrays” that used cDNA spotting. Thus, most investigators currently use either short (25 nt), high-density oligonucleotide arrays (www.affymetrix.com), bead-based oligonucleotide arrays (www.illumina.com), or “home-grown” spotted oligonucleotide arrays.
All microarray platforms depend on similar underlying molecular fundamentals. Fluorescent labeled probes are generated from RNA isolated from a desired tissue or cell type. Such “target” molecules may be either RNA or DNA based, depending on the particular microarray platform. A “target” gene of interest, through the basic principle of complementary base pairing of nucleotides, adheres to small “probes” attached to a solid-phase support such as a glass slide, bead, or nylon membrane. The probes are generally DNA oligonucleotides 25–70 nt in length as mentioned above. Following hybridization and washing, fluorescent signals are detected and these correlate directly to specific mRNA abundance.
The experimental design for microarray experiments is relatively straightforward, but at each individual stage of sample processing, considerable attention to detail is needed to minimize batch effects. Batch effects are non-biological changes in gene expression occurring from a systematic grouping of samples throughout the protocol. Supervised randomization techniques are needed at every step in the protocol to ensure that changes in gene expression are due to independent variables of the experiment and not from misleading factors such as different lots of reagents. A number of methods exist to “correct” batch effects (Alter et al., 2000; Benito et al., 2004; Johnson et al., 2007); however, in our opinion, it is best to avoid these problems as much as possible in the initial experimental design. As discussed below, such batch effect issues are very important considerations when performing very large expression profiling experiments such as those used across RI panels.
The analysis of microarray data is an extremely detailed topic beyond the scope of this chapter. Investigators should be aware that there are multiple algorithms available for analyzing virtually all microarray platforms. Major factors to be considered include the particular approach used for background subtraction, normalization, and probe summarization. Following the initial calculation of expression values for individual probes or genes, a variety of statistical approaches are used for detection of expression differences across independent variables. Given the huge multiple testing confound of microarray analyses, statistical approaches utilizing a false-discovery rate calculation rather than a family-wise error rate analysis are generally most appropriate (Tusher et al., 2001). Perhaps the most challenging and potentially rewarding aspect of analyzing microarray data comes from the bioinformatic approaches that are needed to identify and interpret gene network information contained within microarray results (Kerns and Miles, 2008). This important topic is discussed in Section IV and represents one of the most powerful aspects of applying microarray technology to identification of ethanol-responsive gene expression or identification of QTGs for ethanol traits.
Another critical factor affecting microarray expression results concerns the genetic background of both the array design itself and the mRNA under study. This is an important factor since most microarrays in use today depend on hybridization of probes to relatively short oligonucleotide targets. The stability of such hybrids is very sensitive to secondary structure as can be introduced by probe/target mismatching caused by sequence polymorphism differences between the probe and target. For example, SNPs have been shown to produce false-positive strain-specific (B6 mice vs. D2 mice) differences in expression for up to 10–20% of the probes on Affymetrix oligonucleotide arrays (Walter et al., 2007). Any microarray experiment detailing expression differences across strains or individuals with differing genetic backgrounds has to take such SNP effects into account when interpreting microarray results. Such SNP effects can generate devastating false-positive results in genetical genomics studies as discussed in Section V.A.
C. Genomic Analysis of Alcoholism and Ethanol Behavioral Responses
Alcoholism is a complex disease influenced by both genetic and environmental factors. The genetic predisposition for the risk of developing alcoholism has been well documented through a host of publications on the familial origin of substance abuse (Hill et al., 2008; Merikangas et al., 1998; Prescott et al., 1999). Human and animal model genetic studies, however, have to date only identified a few well-documented genes contributing to the genetic risk for alcoholism or genetic variation in behavioral responses to ethanol. Although the genes involved in the development or progression of this complex trait are largely unknown, genomic studies are currently elucidating gene expression patterns relevant to the neurobiology of this disease. Understanding the molecular genetic basis of an alcoholic phenotype requires defining the genes underlying the predisposition for abuse, as well as the genes altered through acute and chronic ethanol exposure. These sets of genes likely overlap but are not identical. Microarray studies in human or animal models of ethanol responses offer the promise of “ranking/identifying” candidate genes for genetic variance in risk for alcoholism or ethanol behavioral responses. These genomic studies also provide a mechanism for identifying the larger network of genes that contribute to the overall behavioral and toxic responses to ethanol in both humans and animal models.
Microarray studies on ethanol can be subdivided into human versus animal model, acute versus chronic, basal versus treated, and whole brain versus brain regional. In general, for the animal model studies, two different experimental approaches have been used. The first entails profiling of “basal gene expression” across two or more lines of animals that are known to have important differences in ethanol drinking or other behavioral responses to ethanol. The second approach looks at responses to acute or chronic ethanol exposure or ethanol withdrawal. The two approaches have been merged in a limited number of studies where ethanol responses across different lines of animals are also studied. As a general conclusion, all of these approaches have produced significant findings, sometimes with very large lists of genes being identified.
Microarray studies have been done on naïve inbred strains of mice differing in one or more ethanol behavioral phenotypes to define gene networks contributing to such behavioral differences. For example, whole-brain expression profiling of ILS and ISS mice, bred for their initial sensitivity to a sedative-hypnotic dose of ethanol, shows differences in their expression of approximately 81 cDNA clones (Xu et al., 2001). The candidate genes from this study suggested the involvement of several gene ontologies including glycolysis through differential expression of pyruvate dehydrogenase E1-a subunit, a key enzyme in the conversion of pyruvate to acetyl coenzyme A (acetyl-CoA). Follow-up reverse transcriptase polymerase chain reaction (RT-PCR) of ILS and ISS samples suggested potential splice variants for unknown cDNA clones that may contribute to ILS and ISS phenotypic differences.
Mulligan et al. (2006) recently defined a set of genes involved in the predisposition for abuse through a meta-analysis of whole-brain samples of 13 ethanolnaïve inbred strains of mice that are known to differ in voluntary ethanol drinking behavior. Their results pointed to a diverse array of molecular pathways with those genes of largest effect size related to cellular homeostasis and neuronal function. However, expression differences suggested the potential involvement of thousands of genes for a wide array of functional categories across distinct genotypes of alcohol “preferring” and “non-preferring” genotypes. The subtle complexities of functional neuroanatomical differences in the brain may further complicate these genotypic disparities between alcohol phenotypes because of the regional variation in gene expression across heterogeneous populations of inbred mice. Complex behavioral traits are likely the result of numerous genomic differences encompassing integrative processes across multiple brain regions.
A number of laboratories including our own are interested in brain region–specific differences contributing to ethanol behavioral phenotypes. Genotype-specific differences have been demonstrated within individual brain sections using microarray analysis across five distinct brain regions for eight different strains of inbred mice from priority group A of the Mouse Phenome Project (Bogue and Grubb, 2004; Letwin et al., 2006). Correlation of these expression profiling results in treatment-naïve mice identified a subset of genes solely within the ventral striatum related to the N-methyl-d-aspartate (NMDA)/glutamate signaling pathway for distance traveled following 1.5 g/kg ethanol, suggesting a brain region–specific expression network associated with this behavioral feature. This correlation of basal gene expression with an ethanol behavioral phenotype across multiple inbred mouse lines suggested the potential power of combining genomic profiling with genetic analysis of ethanol behaviors.
Human and animal studies continue to demonstrate that the acute behavioral responses to alcohol may serve as a functional barometer indicative of the risk for long-term drinking behavior (Metten et al., 1998b; Schuckit, 1994). Progression toward the full onset of alcoholism, exemplified by abuse and dependence, may thus be affected by acute ethanol sensitivity and concurrent acute ethanol-provoked signaling events. Such acute signaling events are reflected in downstream changes in gene expression. Expression patterns responding to acute ethanol might thus serve as surrogate measures of signal transduction events underlying experience-dependent plasticity that leads to long-term behavioral responses to ethanol and other drugs of abuse (Miles, 1995; Nestler and Aghajanian, 1997). Thus, several microarray studies have profiled expression responses to acute ethanol so as to define the full extent of the ethanol transcriptome.
B6 and D2 are two inbred strains of mice commercially available from the Jackson Laboratory (http://www.jax.org/) that differ in a number of ethanol behavioral phenotypes (McClearn, 1959). For example, D2 mice demonstrate greater acute locomotor responses, withdrawal-induced seizures, and reduced ethanol preference/consumption compared to B6 mice (Belknap et al., 1993; Goldstein, 1973; Metten et al., 1998a; Phillips et al., 1994). An initial global whole-brain expression profiling experiment of B6 and D2 mice acutely treated with 6 g/kg ethanol revealed a small number of genes responding to ethanol and related to cell signaling, gene regulation, and homeostasis/stress response (Treadwell and Singh, 2004). Several of their ethanol-responsive genes resided within known ethanol behavioral QTLs; however, only 16 genes were identified with a differential ethanol response between B6 and D2 mice. The authors attributed this low extent of differential expression between the two strains to heterogeneity of the brain tissue in conducting genomic comparisons on whole brain.
Our laboratory has previously characterized basal and acute ethanol-responsive neurogenomic differences within the ventral tegmental area (VTA), nucleus accumbens (NAC), and prefrontal cortex (PFC) between D2 and B6 mice (Kerns et al., 2005). Extensive multivariate analysis of VTA, NAC, and PFC uncovered basal and ethanol-evoked region-specific differences in gene expression patterns. Acute ethanol induced coordinated changes in gene expression patterns that suggested an overall functional involvement with neuroplasticity. Region-specific functional relationships were determined for retinoic acid signaling (VTA), Bdnf signaling and neuropeptide expression (NAC), and glucocorticoid signaling and myelination (PFC). A subset of differentially expressed genes mapped to QTLs that have been previously implicated in acute ethanol behaviors. This overlap of ethanol-responsive genes and genes contained within support intervals for behavioral QTLs provided a glimpse of the power that microarrays might have for prioritizing candidate QTGs. Overall, this study strongly suggested that the activity of divergent brain region–specific gene expression networks, and their cognate signaling mechanisms, might play an important role in the different ethanol behaviors seen with D2 and B6 mice.
The studies from Kerns et al. (2005) also defined myelin gene expression results in PFC that were particularly interesting. A set of genes, all relating to myelin function, showed divergent basal and ethanol-responsive effects between the two strains. Ethanol regulation of myelin gene expression is further supported by genomic analysis of postmortem human brain tissue, which demonstrates a coordinate regulation of myelin gene expression in the PFC (Iwamoto et al., 2004; Lewohl et al., 2000; Mayfield et al., 2002). Similar results, demonstrating white matter abnormalities have been obtained at the protein level (Lewohl et al., 2005) as well as with neuropathology and neuroimaging studies (Kril and Harper, 1989; Rosenbloom et al., 2003). Microarray evidence of myelin gene dysregulation has also been identified in studies on schizophrenia and cocaine addiction, suggesting that PFC myelin gene expression may be sensitive to dopaminergic signaling (Sokolov, 2007).
A number of other functional categories, such as glutamate signaling, protein trafficking, and cyclic adenosine monophosphate (cAMP) signaling, have also been revealed through genomic studies of postmortem alcoholic brain tissue (Mayfield et al., 2002; Sokolov, 2007). These changes reflect a large cohort of cell signaling mechanisms cooperating in a highly orchestrated series of central nervous system (CNS) plastic events occurring with prolonged ethanol exposure. The cAMP signaling pathway, for example, has been implicated in a large number of experimental models relevant to alcohol abuse (for review, please see Diamond and Gordon, 1997). A potential weakness of these human autopsy microarray studies on alcoholism, however, is that they only represent the final end point of the disorder, and not necessarily the neuroadaptations occurring over time which lead to the development of abuse and dependence. The possibility of other confounding environmental or population stratification factors affecting the microarray results also cannot be discounted.
Chronic ethanol exposure in animal models perhaps represents the most valid and yet challenging experimental design for understanding the neurobiology of alcoholism. Animal models, although lacking many of the functional criteria for clinical diagnoses of alcoholism, are able to mimic different phenotypic components of the disorder. Brain region–specific differences in gene expression have been detected in the NAC shell and central nucleus of the amygdala of alcohol-preferring rats (P rats) following an 8-week model of alcohol binge–like behavior (McBride et al., 2010). Interestingly, there was little overlap in gene expression results between this study and a previous analysis from the same research group of the NAC within the same strain following ethanol operant self-administration, suggesting that differences in behavioral procedures and the neural circuits they activate may contribute to deviations in expression-profiling results (McBride et al., 2010; Rodd et al., 2008).
A particularly attractive model for progressive levels of high ethanol intake involves chronic intermittent exposure to ethanol vapor. This model produces chronic intermittent episodes of ethanol withdrawal and has been used by multiple investigators to produce high levels of ethanol intake in mice or rats. Rimondini et al. performed expression profiling on cingulate cortex and amygdala in rats that had been exposed to the chronic intermittent ethanol exposure (Rimondini et al., 2002). These investigators found striking alterations in expression for genes involved with glutamate neurotransmission, synaptic plasticity, and mitogen-activated protein kinase (MAPK) signaling in the animals exposed to intermittent ethanol. Most intriguingly, these expression changes were detected 3 weeks after withdrawal from the last ethanol vapor exposure. These genomic responses could thus represent important molecular adaptations underlying the dramatic increases in ethanol intake seen with this experimental model. However, these studies used only a single time point and only two microarrays per treatment/brain region group, and thus may be plagued by type I and type II errors.
Ethanol withdrawal–induced changes in gene expression have been demonstrated in a model of acute and chronic withdrawal using expression profiling of hippocampi for both B6 and D2 mice (Daniels and Buck, 2002). These two strains exhibit significant differences in ethanol withdrawal behavioral responses. Consistent with other reports of ethanol's effects on gene expression, this study showed strain- and treatment-specific changes, some of which were consistent with the behavioral phenotypes under study. In a similar study on protein kinase C gamma (PKCγ) null mutants that do not develop tolerance to the sedative-hypnotic effects of ethanol following chronic ethanol exposure (Bowers et al., 1999), Bowers et al. showed by microarray analysis of striatal and cerebellar tissue that a variety of functional categories could be associated with tolerance; however, both studies used a liberal statistical approach to their analysis and confirmed only two candidate genes (Bowers et al., 2006; Smith et al., 2006).
Overall, the results to date on expression profiling in alcholism or animal models of ethanol behavioral responses provide unique insight into the genomic response to ethanol. The data may include gene networks contributing to the predisposition for alcoholism, as well as the acute and chronic CNS plastic events associated with the development of the disease. However, validation of these multifaceted gene networks and their direct relationship to behavioral phenotypes remains a considerable challenge. Most of these expression results could be likened to defining a correlation coefficient for a line drawn through two points—treated versus untreated, sensitive strain versus insensitive, or alcoholic versus control. Defining a causal connection between gene expression patterns and behavior requires a much more powerful experimental design.
III. Genetic Analysis of Gene Expression
A. Genetical Genomics: Using Gene Expression as a Quantitative Trait
QTL mapping is, of course, not limited to dissecting classical phenotypes. Any quantitative trait influenced by genetic factors is amenable to QTL analysis, including transcript, protein, or metabolite abundance. Analyzing the genetic regulation of such molecular phenotypes offers a much closer look at the cellular biochemical processes driving the variation in standard quantitative traits (Schadt et al., 2003). By extending these analyses to include high-throughput molecular phenotypes, such as genome-wide microarrays, it becomes possible to map out entire molecular networks or signaling pathways that underlie complex traits.
Several reports of microarray expression data discussed in Section II were done across two or more lines of mice, allowing low-resolution genetic correlation analysis of gene expression traits and cross-correlation with behavioral (or other) phenotypes. Applying robust genetic approaches to a multitude of molecular phenotypes was perhaps first conducted in a study of a subset of the maize proteome (Damerval et al., 1994). Two-dimensional polyacrylamide gels were used to separate 72 proteins and measure their relative abundance levels across a population of 60 F2 individuals. Looking for associations between these measurements and a panel of 100 genetic markers, the authors identified QTLs significantly influencing the abundance of over half of the analyzed proteins. Furthermore, this study effectively demonstrated the potential of this approach to provide unprecedented insight into structural complexities of quantitative trait regulation, by not only determining the number of QTLs influencing a given trait but also characterizing the dominance effects and epistatic interactions between QTLs and uncovering genetic regulators driving the co-expression between proteins with highly similar expression patterns.
Years later, this strategy of performing genetic linkage analysis on genome-wide molecular profiles was formalized and deemed “genetical genomics,” a term coined by Jansen and Nap (2001). The proposed methodology outlined by that paper primarily focused on gene expression microarrays, and predicted that mapping expression QTL (or eQTL) would make it possible to construct gene networks and elucidate the roles of genes in metabolic and regulatory pathways. The authors also asserted that eQTL mapping could greatly benefit the search for causative candidate genes underlying classical QTL for disease traits.
Less than a year after Jansen's paper, the first study to carry out QTL analysis across genome-wide gene expression microarrays was published using an experimental cross between two strains of Saccharomyces cerevisiae (Brem et al., 2002). Results from this landmark paper shed a great deal of light on the genetic basis of gene expression in a complex organism. Of the 6215 genes measured, 1528 were differentially expressed between the progenitor strains and 570 showed significant linkage to at least one locus. Importantly, the authors noted that the power to detect eQTL for a gene is a direct function of the number of loci regulating that gene and the relative contribution made by each locus. By comparing results derived from empirical computer simulations to their observed data, they determined the majority of differentially expressed genes were likely being regulated by at least five eQTLs.
B. Identifying Gene Networks and Regulatory Mechanisms by Genetical Genomics
Strictly speaking, because microarrays measure steady-state mRNA abundance, eQTLs concern a gene's mRNA level and not the rate of transcription per se. There are also two distinct types of eQTLs, classified based on their physical location relative to that of the linked gene. An eQTL regulating a gene that is genetically unlinked, for example by being located on an entirely different chromosome, is considered to be a trans-eQTL. Interestingly, Brem et al. (2002) identified several loci regulating the expression of many genes simultaneously: over 40% of identified eQTLs mapped to just 8 loci. They noted that the collection of genes mapping to a shared eQTL, sometimes referred to as a trans-band, appeared to be functionally related. For example, over half of the genes that comprise the largest trans-band are mitochondrial ribosomal proteins. This suggests that functionally related genes may share common genetic regulators. While there are many possible explanations for such an observation, it is easy to imagine that a transcription factor with a polymorphic DNA binding region could exert such drastic downstream effects.
The complement to a trans-eQTL is a cis-eQTL, which indicates a gene is regulated by a local sequence polymorphism. An obvious example of this would be an SNP located within a gene's promoter sequence, affecting its ability to initiate transcription. However, an apparent cis-eQTL may actually represent a gene being regulated in trans by a neighboring gene carrying a functional polymorphism. It is not possible to distinguish between such mechanisms without performing follow-up molecular assays (Doss et al., 2005). For a more complete discussion of such nuan ces, an excellent review has been written by Rockman and Kruglyak (2006). Williams and colleagues have also contributed multiple primary publications and reviews on the general area of expression genetics (Chesler et al., 2003, 2004), as well as providing the GeneNetwork resource (www.genenetwork.org) for pursuing such studies (see discussion below).
C. Combining Phenotypic and Expression Genetics to Identify Genes or Gene Networks Linked to Complex Traits
The first mammalian transcriptome genetical genomics study of a given complex trait used 111 F2 mice placed on high-fat diets to induce an obesity-like phenotype (Schadt et al., 2003). Microarray analysis of liver tissue from these animals identified 280 genes that changed significantly with the high-fat diet. Similar to the yeast transcriptome, eQTL anlalysis of the mouse liver identified several large trans-bands that exert enormous control over downstream genes. In fact, 25% of the high-fat diet-responsive genes were regulated primarily by a single eQTL, a so-called “eQTL hotspot.” However, this paper is notable for recognizing the potential in intersecting genetical genomics with classical phenotypic QTL, as a way to greatly assist in identifying causative candidate genes underlying disease traits. The authors discovered two high-priority candidates for their obesity phenotype by screening for genes with cis-eQTL that overlapped with the QTL for fat pad mass, the phenotype used as a marker for obesity. Their hypothesis is consistent with a number of recent studies suggesting that genetic variation in the expression of QTGs rather than genetic variation in the function of proteins coded for by the QTGs might be important to the biology of complex traits.
The increased genetic resolution afforded by the rapid evolution of genotyping technology has made it possible to obtain continuously denser genetic maps and associate disease traits with correspondingly smaller regions. Additional information provided by eQTL mapping greatly benefits the process of prioritizing candidate genes localized to the disease-associated region by screening for genes whose expression patterns are regulated by local polymorphisms and strongly correlate with the disease trait (see Fig. 1). This will typically yield a small handful of interesting candidate genes. However, the final steps necessary to definitively identify the susceptibility gene in a linked region are still tremendously difficult and remain largely elusive. The experiments necessary to validate a gene's role in disease are expensive and time consuming enough that even a relatively small list of genes may be too large to evaluate each individually. As such, more information is required to further evaluate and prioritize genes that are, empirically, equally strong candidates.
FIG. 1.

Overall schematic for identifying a QTG or QTGN for ethanol and alcoholism. Inbred and RI lines (or other animal models) undergo a series of congruent steps for identifying a gene or gene network underlying a specific phenotype relevant to ethanol or risk for the development of alcoholism. Microarray analysis with and without ethanol is used to identify differentially expressed gene expression patterns and ethanol-responsive gene networks. QTL mapping is conducted to narrow genomic regions contributing to an ethanol response. Combining gene expression data and nucleotide information from microarray analysis and genotyping assists in identifying pertinent cis- or trans-eQTLs. Multilayered bioinformatic analysis of all the data, as well as that of public data and literature resources, is used to identify relevant QTG(s) or QTGN(s).
Constructing molecular networks from high-throughput molecular data associated with a disease trait could very well provide the additional context necessary to distinguish the causal gene from a list of positional candidates by providing a more comprehensive view of the involved biological pathways and how they are affected by the genetic polymorphism driving the linkage with an implicated QTL (Chen and Charness, 2008). Studies of gene co-expression networks have already revealed much about the structural architecture of gene regulation by demonstrating that gene networks conform to a scale-free toplogy (van Noort et al., 2004). Scale-free networks are characterized by the power distribution they are named for, with many sparsely connected nodes and a few that are highly interconnected, and also provide accurate models for protein and metabolite interaction networks, the neural networks of Caenorhabditis elegans, and even the World Wide Web (Junker and Schreiber, 2008). Armed with this knowledge, we can begin to ask interesting questions, such as: does the topology of gene networks change in response to repeated drug use or can better drug targets be discovered by identifying genes that represent major hubs of communication within a network?
Network analysis of microarray expression data is a rapidly progressing field of study, fueled by a seemingly exponential increase in the rate of microarray data being accumulated and a surge in the number of computational biologists cultivating it. Novel methods for constructing and analyzing gene expression networks are being released continuously, using increasingly sophisticated methods (Baldwin et al., 2005; Horvath and Dong, 2008). However, gene co-expression networks can be constructed by calculating a simple Pearson product moment for all pairwise gene expression data across a segregating population (Fig. 3C). Identifying clusters of densely intercorrelated genes has proven to be an effective method for identifying gene product interactions that are evolutionarily conserved across multiple species and may participate in common biological pathways or form a protein complex (Stuart et al., 2003). Identifying coregulated genes from microarray expression across segregating populations has been used to identify novel protein interactions (Scott et al., 2005).
FIG. 3.

Ethanol-responsive eQTL for Gabrb2. PFC S-score microarray data described in Fig. 2 was searched for Gabrb2 responses within GeneNetwork. (A) QTL tracing for GABA-A receptor subunit beta 2 (Gabrb2). The transcript abundance of this gene is being regulated in trans by loci on chromosomes 7, 13, and proximal 17. (B) The change in Gabrb2's expression induced by acute ethanol (1.8 g/kg) is significantly correlated with anxiety as measured by elevated plus maze (Yang et al., 2008). (C) Gabrb2 gene coexpression network. The ethanol response of all plotted genes correlate significantly with that of Gabrb2 (p-value ≤ 5 × 10−9).
Robert Williams and colleagues have greatly facilitated the process of identifying eQTLs, correlating the eQTL with phenotypic QTL and generating coexpression networks through the development of the comprehensive GeneNetwork web resource (www.genenetwork.org). This site includes databases of phenotypes and many different microarray expression data sets from a variety of species, tissues, and brain regions. This allows an exhaustive investigation of links between genetic variation, gene expression, and phenotypes. The data displayed in Fig. 3A–C are examples of output from GeneNetwork analyses.
D. Genetical Genomics Analysis for Ethanol QTG Mapping
As discussed in Section II, identifying QTGs underlying ethanol behavioral QTL has been frustratingly slow despite the mapping and confirmation of many QTLs over the last 15 years. Applying expression profiling to ethanol behaviors or mouse/rat lines selected for ethanol phenotypes, followed by superimposing these results on behavioral QTL data, has been one approach used to prioritize candidate QTGs (Fig. 1). This essentially is an extension of the analysis of ethanol-related microarray data discussed in Section II.C. As an example, Mulligan et al. reported a meta-analysis of basal microarray expression data derived from several inbred lines divergent in ethanol drinking behaviors. This resulted in a large set of ~3800 genes theorized to be involved in ethanol preference. To narrow this gene set and also provide candidate QTGs for previously mapped QTLs on ethanol preference drinking, Mulligan et al. superimposed the meta-analysis data set on genes contained within the support interval of a Chr 9 ethanol preference QTL (Belknap and Atkins, 2001; Mulligan et al., 2006). Additionally, by using eQTL data from GeneNetwork, a subset of high-priority candidates was identified among genes within the support interval having strong correlations with the ethanol drinking phenotype across BXD lines (and having a significant cis-eQTL) and also being positive in the meta-analysis for ethanol preference–related genes. This combination of approaches resulted in a testable set of fewer than six candidate genes rather than the hundreds of genes that resulted from the behavioral genetics study alone.
A similar approach combining known QTLs, gene expression data, and selective breeding strategies in mice was used to identify potential QTGs and signaling pathways involved in acute functional tolerance to ethanol (Tabakoff et al., 2003). A congenic specific approach has also been implemented for a QTL on rat Chr 4 associated with alcohol preference using selectively inbred rats denoted alcohol-preferring (iP) and alcohol-non-preferring (iNP) (Carr et al., 2007). Using congenic approaches largely decreases differential expression between strains because they are essentially isogenic at all loci except for the QTL region of interest, thus allowing for a method of QTG identification restricted to a chromosomal region mapped for an ethanol behavioral feature (e.g., preference). The study on iP and iNP rats identified ~34 genes with expression differences correlating with the QTL region, and residing within the Chr 4 QTL influencing ethanol preference, with suggested involvement in PKC signaling pathways. All of the studies described above used ethanol-naïve samples for either whole brain or a single measure from an average of results across multiple brain regions. Therefore, the results may be limited to genes involved in the predisposition for the ethanol phenotype in question rather than direct differences in ethanol-related signaling events and may fail to delineate brain region–specific QTGs of these complex traits.
Kerns et al. (also discussed in Section II.C) performed microarray studies across multiple brain regions in ethanol-treated versus control mice and thus identified expression patterns regulated by ethanol as well as strain-specific expression differences (Kerns et al., 2005). These results suggested that expression analysis across genetically different strains tends to produce similar expression patterns across multiple brain regions in contrast to the more regionally selective expression changes caused by ethanol treatment. Thus, whole-brain expression profiling might be adequate for detection of at least some important QTGs showing eQTL across multiple brain regions. These authors also superimposed these brain regional ethanol-responsive expression results with behavioral and eQTL data from GeneNetwork. The net result of this layered analysis was the identification of several candidate QTGs or QTGNs for previously identified behavioral QTLs. For example, brain-derived neurotrophic factor (Bdnf) was identified as a possible candidate gene for a Chr 2 ethanol locomotor activation QTL (Actre3) on the basis of ethanol-responsive expression changes in the NAC. Importantly, multiple genes related to Bdnf signaling were also shown to be ethanol responsive within the same brain region. Bdnf signaling could thus be considered a QTGN for the Actre3 QTL. This illustrates an additional power of merging expression studies and behavioral genetics because it suggests that identifying a QTG may become irrelevant if a QTGN of defined function can be mapped to the QTL. Thus, superimposing trans-eQTL bands with behavioral QTL might pose a more powerful approach than just looking for cis-eQTL within a QTL support interval.
Direct genetic analysis of ethanol-responsive gene expression has not been reported on a scale larger than several inbred lines at a time. However, our laboratory has recently completed analysis of ethanol-responsive gene expression across the PFC, NAC, and VTA for over 30 BXD strains (see Virginia Commonwealth University (VCU) databases in GeneNetwork). Analysis of this enormous data set is ongoing (Wolen and Miles, in preparation) but it is already evident that this eQTL analysis of ethanol-responsive genes is uncovering previously unappreciated aspects of brain gene regulation by ethanol. As has been seen for some eQTL mapping of basal gene expression, we have identified a very limited set of genetic loci influencing the ethanol responsiveness for hundreds of genes (Fig. 2). This suggests that a limited number of signaling mechanisms may underlie the bulk of genomic responses to acute ethanol. This has important implications for strategies to prevent or reverse ethanol-related neuroplasticity related to alcoholism.
FIG. 2.
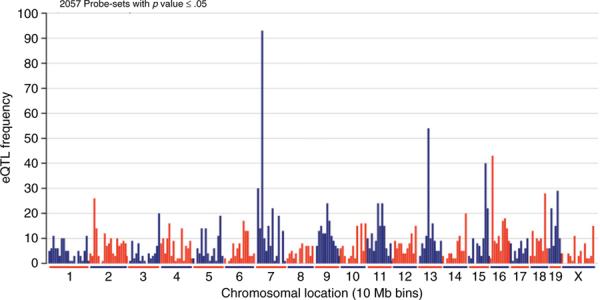
Ethanol-responsive eQTL from the PFC of BXD RI lines. Mice were treated with saline or ethanol (1.8 g/kg × 4 h) and then brain region RNA isolated for microarray analysis. S-score analysis was used to identify significant ethanol-regulated gene expression (Kerns et al., 2003). There are 2057 probe sets from the BXD PFC saline vs. ethanol S-score expression data with a significant eQTL, using a genome-wide corrected p-value ≤ .05 as the significance threshold. Each bar indicates the number eQTL located within a 10-megabase region of the genome. There are large ethanol responsive trans-bands on chromosomes 7, 13, 15, and 16.
Our ethanol-responsive expression genetics analysis has already identified potentially important individual gene networks responding to ethanol in a coordinated manner. Figure 3A illustrates ethanol-responsive eQTL for the Gabrb2 gene coding for a subunit of GABA-A receptors, an important molecular target for ethanol action. The interval map in Fig. 3A shows several trans-eQTLs for ethanol regulation of Gabrb2 but no significant cis-eQTLs at the site of the Gabrb2 gene (Chr 11). The Chr 7 trans eQTL is also the site of a suggestive QTL for several basal anxiety phenotypes in the GeneNetwork database. Figure 3B shows the correlation between one of these basal anxiety phenotypes and ethanol-regulated Gabrb2 expression. Importantly, Gabrb2 showed strong correlation with ethanol regulation of an entire network of genes (Fig. 3C). As expected, these genes showed Chr 7 ethanol-responsive eQTL responses similar to Gabrb2 (not shown), and thus this group of genes represents a potential ethanol-responsive QTGN with relevance to anxiety phenotypes.
Putative ethanol behavioral QTLs and QTGs identified in animal models may not be directly paralleled in humans; however, associations do exist between the two approaches. The multiple PDZ domain protein (Mpdz) was identified as a QTG for ethanol-induced withdrawal activity through a succession of studies involving F2 intercrosses, RI line, and interval-specific congenic strains (Buck et al., 1997, 2002; Fehr et al., 2002; Shirley et al., 2004). Mpdz was the only gene among five to demonstrate genotype-dependent differences in coding sequence and gene expression which co-segregated with withdrawal severity in mice. Mpdz also showed regional specific regulation by ethanol and genotype-specific expression differences in microarray studies by Kerns et al. (2005). In humans MPDZ does not demonstrate an association with alcohol-induced withdrawal seizures, but haplotype and single-SNP association analyses do suggest a possible association with alcohol dependence (Karpyak et al., 2009). Disconnects existing between human and animal research may merely represent our lack of understanding for cross-species comparisons or demonstrate the need for more relevant animal models and phenotypes (Crabbe, 2008). Nevertheless, the identification of ethanol-related QTGs or QTGNs through genetical genomics continues to emerge as a powerful approach for the discovery of fundamental processes central to the risk, progression, and full onset of alcoholism and may help resolve some discrepancies between human and animal studies because of the focus on gene networks rather than an individual gene.
IV. Recognizing Biological Function in Genetical Genomics Studies
Interpreting results with such complexity as those from DNA microarrays is a daunting task that has spawned entirely new bioinformatic approaches for assigning biological function to coordinated changes in gene expression. The process of using genetical genomics does serve to organize and focus the often bewildering mass of data derived from microarray studies. Bioinformatic efforts, however, can further reduce the data to manageable sizes and provide a phenotypic or functional “trace” to a particular gene network or trans-eQTL band. Associating a presumed biological function to a gene network for a given behavioral phenotype might lead to new hypothesis generation regarding underlying molecular mechanisms and, eventually, avenues for intervention. Such an approach is obviously the hope of using genetical genomics and rigorous bioinformatics analysis with studies on ethanol phenotypes.
A. Over-Representation Analysis
The first step in organizing microarray data is often to determine whether a given list of genes (for example, a trans-band) has some coherent function. This is derived by essentially interrogating the biological function of each gene in the list and asking whether any particular “functional group” is over-represented beyond what would be expected by chance alone. Such analysis requires databases of genes categorized in some fashion. This can be done with a number of different categorization schemes: overall biological function (gene ontology), biochemical pathways, protein–protein binding partners, genes containing common transcription factors or miRNA binding sites, and a host of other such methods. The gene ontology consortium (http://www.geneontology.org/) is a collection of investigators working in a collaborative effort to assign function to gene products based on three broad categories—cellular component, biological process, and molecular function—which are further segmented into numerous functional subcategories. A number of tools and browsers are available to determine if a particular data set is enriched for a functional category. For a list of available tools, please see http://www.geneontology.org/GO.tools.shtml. The Database for Annotation, Visualization and Integrated Discovery (DAVID) (Dennis et al., 2003; Huang et al., 2009) is likely the most popular method for assessing gene ontologies with greater than 1600 citations currently among the literature. Hosted by the Laboratory of Immunopathogenesis and Bioinformatics (LIB), SAIC-Frederic, Inc., DAVID is a free online resource for conducting functional over-representation analysis.
The Web-based tool ToppGene (Chen et al., 2009) is a similar bioinformatics program for conducting over-representation analyses but contains several additional databases for querying than does DAVID. ToppGene, for example, will determine whether there are particular miRNA or transcription factor binding sites that are enriched within a given gene set. A very important additional database utilized by ToppGene concerns sets of experimentally derived gene lists available through the Broad Institute Gene Set Enrichment Analysis (GSEA) database (http://www.broadinstitute.org/gsea/index.jsp). ToppGene also has additional tools for identifying and prioritizing candidate genes for further study when compared to a training set of genes known to be associated with a given phenotype of interest.
B. Literature Association Analysis
A published literature association analysis is an additional tool often used for querying functional information in genomics data. These approaches often use expert user–curated databases derived from natural language processing interrogation of the biomedical literature or other sources of gene/gene interaction data. The Ingenuity Pathway Analysis (www.ingenuity.com) is a popular commercial bioinformatics program for interpreting the network structure of data sets based on curated literature associations, biological function, and cell signaling mechanisms. The network structure of gene lists can reveal innovative associations between genes outside of predetermined functional categories. Other commercial programs such as Bibliosphere (Genomatrix Software, Munich, Germany) and GeneGo (www.genego.com) and free programs such as Chilibot (Chen and Sharp, 2004) are available for “literature mining” and drawing network diagrams among gene sets. Unfortunately, all of these programs will draw some sort of network given enough input genes. Therefore, care must be taken in over-interpreting the results of such network associations and verifying the implied literature co-citations. Deriving the same or similar network structure from different bioinformatics resources or through combinations of literature association, functional group over-representation, and expression correlation is a reassuring indication of some meaningful functional correlation to the gene set being interrogated.
C. Gene Set Correlation and Ontology Analysis
Finally, comparing a given genetical genomics–derived gene set or network with other microarray or functional gene sets can often provide valuable information about the biological function of the new gene list. As mentioned previously, GeneNetwork (www.genenetwork.org) is a collaborative Web-based resource equipped with tools and features for studying gene/gene and exploring genetic correlates to neurobehavioral phenotypes (Chesler et al., 2003, 2004). The Web site is home to a growing collection of gene expression and phenotypic data from a variety of species and brain regions, with a host of links to external resources for tracing the interrelationships of a gene among multiple Web-based resources. GeneNetwork also offers a number of correlation and mapping strategies for assessing associations among multiple genes and QTLs. GeneNetwork aims to make the study of complex traits through the use of systems genetics widely available to the scientific community. A powerful tool that can be integrated with GeneNetwork or used on gene sets from a wide variety of sources is the Ontological Discovery Environment (ODE; http://ontologicaldiscovery.org). This public analysis platform provides an ever-growing list of gene sets derived from gene ontology categories, mouse genome informatics definitions, or experimentally derived genomic data sets (Baker et al., 2009). Users can input their own data sets and look for intersection with other gene sets having previously defined ontologies. In this regard, ODE has features of both ToppGene and GSEA but allows users great flexibility in organizing their own gene sets among defined user groups.
As genome-wide research continues to produce ever enlarging data sets from genetic analysis of gene expression and phenotypic traits, there is considerable need for interactive bioinformatics resources that can quickly aid in characterizing biological function underlying the whole genome analyses. Here, we highlighted just a fraction of the bioinformatics tools available to researchers for extracting meaningful information from these complex data sets. Such investigations provide a hypothesis-generating framework for leveraging genetical genomics data on the neurobiology of alcoholism and other complex diseases.
V. Current Problems and Future Solutions
A. SNP Effects Causing False Positive cis-eQTL
While the potential of genetical genomics to provide insight into complex diseases continues to grow as high-throughput technologies improve and become more commonplace, the field is not without problems. We have discussed the value added by genetical genomics in the pursuit of causal disease genes by identifying cis-eQTL coincident with QTL for a disease trait. However, an important caveat is that putative cis-eQTL can suffer from a high false-positive rate. A thorough examination of this issue was performed using microarray expression data from the liver of B6xD2 F2 mice, and revealed a significant bias in the additive effects of cis-eQTL in the direction of the B6 allele (Doss et al., 2005). That is, the B6 allele was associated with higher transcript abundance more frequently than would be expected by chance, suggesting that undetected polymorphisms within microarray probe target binding regions were affecting hybridization. This hypothesis is well supported by facts; as the microarray probe sequences were designed against the B6 genome, any undetected probe polymorphisms would bias measurement in favor of the B6 allele. This issue was discussed briefly in Section II.C because such “SNP effects” also affect traditional expression profiling studies comparing expression across a few strains.
In order to obtain an estimate of the proportion of true versus spurious cis-eQTL in their data, Doss et al. (2005) performed a cis–trans-test in a B6xD2 F1 population for a semi-randomly selected subset of probe sets with putative cis-eQTL, and found 35% (10/28) to be potentially false positives. This emphasizes the importance of validating a cis-eQTL before resources are spent attempting to confirm a gene's role in a complex disease. However, even before performing a cis–trans-test, spurious cis-eQTL can often be flagged by applying a statistical screen that eliminates deviating probes and repeats the QTL analysis with the remaining probes (Alberts et al., 2007). If the significant association between a probe set and a locus is lost after excluding the deviating probes, the corresponding gene's cis-linkage should be considered highly suspect. Conversely, probe target SNPs may also diminish the significance of a potentially genuine cis-eQTL by reversing the expression/allele relationship for a subset of probes (see Fig. 4).
FIG. 4.

False eQTL analysis. Probe-level log2 intensities for Kruppel-like factor 5 (Klf5) measured in NAC tissue from 36 BXD RI strains. Line color indicates strain genotype at the locus underlying Klf5's significant cis-eQTL. There are consistently higher levels of Klf5 transcript in strains carrying the D2 allele, with the exception of probe 4, where the trend is completely reversed. This suggests that an SNP effect may be hindering probe 4's ability to properly hybridize; thus it is contaminating the probe set's expression summary value and should be excluded. The plot was generated using code adapted from the affyGG package for R (Alberts et al., 2008).
Even after applying the above methods and excluding genes with dubious cis-linkage, the QTL region for a classical phenotype may still harbor a daunting number of candidate genes with high-confidence cis-eQTL. When the QTL region is characterized by strong linkage disequilibrium, many of the local genes' expression patterns will be tightly correlated with each other as well as the linked phenotype, making it very difficult to discern which gene (or genes) is actually contributing to the phenotypic variation. If the region also contains a number of trans-eQTLs, it may be possible to tease apart the effects of colocalized genes using a partial correlation analysis to determine which local gene most strongly correlates with the trans-band after removing the genetic variance contributed by the underlying locus (Liu et al., 2008; Mozhui et al., 2008). Others have proposed a structural equation modeling approach to distinguish between a string of cis-eQTLs by identifying the genetic model that best fits the data (Schadt et al., 2005).
B. Batch Effects Generating Spurious Linkage or Correlation
Another important issue to consider is systematic batch effects that can introduce considerable bias into microarray data. In studies seeking to differentiate between two or more classes of samples by identifying unique expression patterns, these hidden factors may cause expression variation that dilutes differences driven by the variable of interest, or masks them completely (Johnson et al., 2007; Leek and Storey, 2007). In microarray studies across genetic mapping panels, batch effects can produce a biologically meaningless correlation between samples. If alleles for a particular SNP happen to segregate among the samples in a manner consistent with the spurious correlation, this may produce what appears to be major trans-band, potentially driven by an important genetic regulator (Kang et al., 2008). Such an exciting result is difficult to ignore, and substantial resources could be spent chasing a technical artifact masquerading as an important discovery, causing potentially irrevocable heartbreak to the involved investigators. While there are available novel statistical methods for mapping expression QTLs designed to correct for inter-sample correlations and avoid generating potentially spurious trans-bands, these methods are currently unable to distinguish between artifactual and biologically genuine trans-bands. This makes the use of such statistical filter prone to high false-negative rates insofar as trans-eQTL detection is concerned (Kang et al., 2008).
Of course batch effects are best dealt with by avoiding them as much as possible, using carefully executed randomization schemes during the stages of producing microrarray data. In our own laboratory we conduct careful experimental design studies before initiating genetical genomics or behavioral genetics studies so as to balance and randomize grouping of strains as much as possible. An optimal design to prevent batch effects regarding strain grouping during phenotypic (e.g., behavioral) testing might be to test as many strains as possible at one time, even if only a single or few animals from each strain are tested in a given batch. The process is then repeated until the desired number of animals is tested per strain but with randomization of strains tested at one time if all strains cannot be tested together. We perform supervised re-randomization of samples at each stage of the microarray analysis: RNA extraction, probe preparation, and chip hybridization. While it probably is not possible to resolve all potentially confounding factors (Churchill, 2002), their influence can be mitigated in a properly designed microarray experiment.
C. Defining Causality For Genes or Gene Networks in Behavioral Responses to Ethanol
Establishing causality among genes or gene networks in ethanol behavioral phenotypes is an important feature in discerning their prospective role in the neurobiology of alcoholism. The process is extremely difficult and potentially nearly impossible for genes having small effects and multiple epistatic interactions in regard to their final influence on a given behavior. Other chapters in this volume also cover this topic, so we will only discuss these issues briefly so as to frame the “endgame” for genetical genomics studies. It should be emphasized that such validation studies are crucial because combining expression and phenotypic genetics, although capable of detecting functionally relevant correlations between gene expression and phenotype, still only represents a correlation study and does not represent causality per se.
Gene targeting techniques have advanced our understanding of how specific genes may influence behavioral responses to ethanol and modulate risk for alcoholism. Multiple reports exist where genetically engineered transgenic, null knockout, and conditional knockout mice have determined the role(s) that individual genes play in the context of different ethanol behavioral models. For example, transgenic overexpression of the serotonin receptor 5-HT3 and the neuropeptide Y (Npy) decreases ethanol preference and consumption (Engel et al., 1998; Thiele et al., 1998). Interestingly, Npy knockout mice show increased ethanol preference and low sensitivity to high doses of ethanol, clearly suggesting that expression of Npy may have an important role on ethanol behaviors (Thiele et al., 1998). However, such transgenic or knockout approaches suffer several potential weaknesses related mainly to underlying physiological factors and experimental techniques. Transgenes can often integrate at a random chromosomal region not associated with the gene of interest, and creating a null mutation (knockout) can cause developmental compensation that may contribute to the observed phenotype. Yet these animals are still powerful models for investigating ethanol traits as evidenced by the number of genetically modified mice that have been established and evaluated for the role of the manipulated gene in acute and chronic ethanol behavioral phenotypes (for review, Crabbe see et al., 2006). Cre/loxP or TetR-based transactivators avoid some of the weaknesses of traditional knockout approaches and confirm that a given phenotype is due to the specific gene mutation and not caused by developmental compensation in an adult animal model (Lewandoski, 2001). For example, PKCε knockout mice exhibit decreased ethanol preference and acute ethanol sensitivity (Hodge et al., 1999). This is not a developmental effect because conditional expression under control of the tet operator sequences coupled to a minimal cytomegalovirus (CMV) promoter subsequently rescued acute ethanol sensitivity and ethanol preference in PKCε knockout mice (Choi et al., 2002).
Viral vector–mediated transfection techniques are a complementary approach to traditional genetic mouse models for investigating the behavioral responses of alcohol and other drugs of abuse. Viruses (e.g., adenovirus and adeno-associated virus) and virions (i.e., herpes simplex virus and lentivirus) infect mammalian cells by binding to specific cell surface receptor elements. Packaging a gene of interest inside a viral vector allows for site-specific injection into a targeted brain region with spatiotemporal controlled expression. There are now numerous examples whereby viral vector gene delivery has been used to validate a causal role of given candidate genes for ethanol or other drug abuse–related behaviors. For example, viral-mediated gene transfer of RGS9-2, a regulator of G-protein signaling, into the NAC of RGS9 knockout mice reversed the increased sensitivity to morphine seen in knockout mice (Zachariou et al., 2003). Additionally, viral-mediated downregulation of BDNF in the dorsal lateral striatum, and not the dorsal medial striatum, increases ethanol self-administration in rats (Jeanblanc et al., 2009).
D. Sharing of Resources
High-throughput methods of analyses, such as microarrays and sequencing technologies, have created the need for public repositories to keep pace with the sharing of data sets generated from individual experiments. In recent years, the sharing of such high-throughput data has become a condition for publication and funding. The National Center for Biotechnology Information (NCBI) and the European Bioinformatics Institute (EBI) have respectively created Gene Expression Omnibus (GEO)(Barrett et al., 2009; Edgar et al., 2002) and ArrayExpress (Parkinson et al., 2009) to assist in the sharing of data sets from numerous gene expression studies, as well as some sequencing and epigenetic information. The repositories seek to remain flexible to the constantly evolving nature of genomics, while conforming to the standards of the Minimum Information About a Microarray Experiment (MIAME) (Brazma et al., 2001), which provides colleagues enough information to replicate experiments. Collectively, the two repositories have greater than 20,000 experiments logged to date with tools available for searching and downloading data sets for subsequent analyses. The logistics of quality control and intellectual property rights are obviously of concern and a topic of considerable debate, but the precedent of data sharing in this post-genomic era continues to drive the field forward.
In the case of alcoholism research, genetic/genomic investigations are identifying a large number of genes that may contribute to a variety of ethanol phenotypic traits. Research scientists have created independent gene expression information warehouses equipped with simple tools for understanding the genetical genomics of complex traits. As already discussed, GeneNetwork has a large collection of behavioral, genetic marker, and expression data relevant to alcohol research and available for online analysis. PhenoGen Informatics (Bhave et al., 2007) is a Web-based tool for investigating complex traits originally developed for sharing information among the Integrative Neuroscience Initiative on Alcoholsim (INIA). PhenoGen awards investigators a place to store data, conduct common methods of analyses, research candidate genes of interest, and investigate their relationships to QTLs. Ontological Discovery Environment (ODE) (Baker et al., 2009) is another example of a gene list repository and Web-based tool created through the INIA consortium, which also allows investigators to draw associations between gene networks and phenotypic traits. Undoubtedly the creation of repositories such as PhenoGen and ODE, which are combined with methods for integrating and analyzing differential transcript expression across copious data sets, will make genomic investigations more accessible to the scientific community. Additionally, the Ethanol-Related Gene Resource (ERGR, http://bioinfo.vipbg.vcu.edu/ERGR/) is a public database integrating published ethanol-related genetic and genomic information across multiple species and platforms (Guo et al., 2009). The existence of publicly and privately shared data sets envisions disease-specific management libraries that allow investigators to quickly identify how a given gene or network of genes respond under different experimental parameters and across species.
Acknowledgments
The authors thank Nate Bruce and Paul Vorster for their contributions in generating some of the microarray data discussed in this chapter, and Robert Williams at University of Tennessee Health Sciences Center for his assistance with data entry and analysis within GeneNetwork. This work was supported in part by grants from the National Institute on Drug Abuse (5T32DA007027, SPF), National Institute of Mental Health (MH-20030, ARW), and the National Institute on Alcohol Abuse and Alcoholism (F31AA018615 to SPF; U01AA016662, U01AA016667, P20AA017828, and R01AA014717 to MFM).
References
- Alberts R, et al. Sequence polymorphisms cause many false cis eQTLs. PLoS ONE. 2007;2:e622. doi: 10.1371/journal.pone.0000622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberts R, et al. affyGG: Computational protocols for genetical genomics with Affymetrix arrays. Bioinformatics. 2008;24:433–434. doi: 10.1093/bioinformatics/btm614. [DOI] [PubMed] [Google Scholar]
- Alter O, et al. Singular value decomposition for genome-wide expression data processing and modeling. Proc. Natl. Acad. Sci. USA. 2000;97:10101–10106. doi: 10.1073/pnas.97.18.10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badcock JC, et al. Speed of processing and individual differences in IQ in schizophrenia: General or specific cognitive deficits? Cogn. Neuropsychiatry. 2004;9:233–247. doi: 10.1080/13546800344000228. [DOI] [PubMed] [Google Scholar]
- Bailey DW. Recombinant-inbred strains. An aid to finding identity, linkage, and function of histocompatibility and other genes. Transplantation. 1971;11:325–327. doi: 10.1097/00007890-197103000-00013. [DOI] [PubMed] [Google Scholar]
- Baker EJ, et al. Ontological discovery environment: A system for integrating gene-phenotype associations. Genomics. 2009;94:377–387. doi: 10.1016/j.ygeno.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin NE, et al. Computational, integrative, and comparative methods for the elucidation of genetic coexpression networks. J. Biomed. Biotechnol. 2005;2005:172–180. doi: 10.1155/JBB.2005.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T, et al. NCBI GEO: Archive for high-throughput functional genomic data. Nucleic Acids Res. 2009;37:D885–890. doi: 10.1093/nar/gkn764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belknap JK, Atkins AL. The replicability of QTLs for murine alcohol preference drinking behavior across eight independent studies. Mamm. Genome. 2001;12:893–899. doi: 10.1007/s00335-001-2074-2. [DOI] [PubMed] [Google Scholar]
- Belknap JK, et al. Quantitative trait loci (QTL) applications to substances of abuse: Physical dependence studies with nitrous oxide and ethanol in BXD mice. Behav. Genet. 1993;23:213–222. doi: 10.1007/BF01067426. [DOI] [PubMed] [Google Scholar]
- Benito M, et al. Adjustment of systematic microarray data biases. Bioinformatics. 2004;20:105–114. doi: 10.1093/bioinformatics/btg385. [DOI] [PubMed] [Google Scholar]
- Bhave SV, et al. The PhenoGen informatics website: Tools for analyses of complex traits. BMC Genet. 2007;8:59. doi: 10.1186/1471-2156-8-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogue MA, Grubb SC. The mouse phenome project. Genetica. 2004;122:71–74. doi: 10.1007/s10709-004-1438-4. [DOI] [PubMed] [Google Scholar]
- Bowers BJ, et al. Decreased ethanol sensitivity and tolerance development in gamma-protein kinase C null mutant mice is dependent on genetic background. Alcohol Clin. Exp. Res. 1999;23:387–397. [PubMed] [Google Scholar]
- Bowers BJ, et al. Microarray analysis identifies cerebellar genes sensitive to chronic ethanol treatment in PKCgamma mice. Alcohol. 2006;40:19–33. doi: 10.1016/j.alcohol.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazma A, et al. Minimum information about a microarray experiment (MIAME)-toward standards for microarray data. Nat. Genet. 2001;29:365–371. doi: 10.1038/ng1201-365. [DOI] [PubMed] [Google Scholar]
- Brem RB, et al. Genetic dissection of transcriptional regulation in budding yeast. Science. 2002;296:752–755. doi: 10.1126/science.1069516. [DOI] [PubMed] [Google Scholar]
- Broman K. A Guide to QTL Mapping with R/QTL. Springer; New York, NY: 2009. [Google Scholar]
- Brown PO, Botstein D. Exploring the new world of the genome with DNA microarrays. Nat. Genet. 1999;21:33–37. doi: 10.1038/4462. [DOI] [PubMed] [Google Scholar]
- Buck K, et al. Quantitative trait loci affecting risk for pentobarbital withdrawal map near alcohol withdrawal loci on mouse chromosomes 1, 4, and 11. Mamm. Genome. 1999;10:431–437. doi: 10.1007/s003359901018. [DOI] [PubMed] [Google Scholar]
- Buck KJ, et al. Quantitative trait loci involved in genetic predisposition to acute alcohol withdrawal in mice. J. Neurosci. 1997;17:3946–3955. doi: 10.1523/JNEUROSCI.17-10-03946.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck KJ, et al. Mapping murine loci for physical dependence on ethanol. Psychopharmacology (Berl.) 2002;160:398–407. doi: 10.1007/s00213-001-0988-8. [DOI] [PubMed] [Google Scholar]
- Carr LG, et al. Identification of candidate genes for alcohol preference by expression profiling of congenic rat strains. Alcohol. Clin. Exp. Res. 2007;31:1089–1098. doi: 10.1111/j.1530-0277.2007.00397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chee M, et al. Accessing genetic information with high-density DNA arrays. Science. 1996;274:610–614. doi: 10.1126/science.274.5287.610. [DOI] [PubMed] [Google Scholar]
- Chen H, Sharp BM. Content-rich biological network constructed by mining PubMed abstracts. BMC Bioinformatics. 2004;5:147. doi: 10.1186/1471-2105-5-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, et al. ToppGene suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37:W305–311. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Charness ME. Ethanol inhibits neuronal differentiation by disrupting activity-dependent neuroprotective protein signaling. Proc. Natl. Acad. Sci. USA. 2008;105:19962–19967. doi: 10.1073/pnas.0807758105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler EJ, et al. WebQTL: Rapid exploratory analysis of gene expression and genetic networks for brain and behavior. Nat. Neurosci. 2004;7:485–486. doi: 10.1038/nn0504-485. [DOI] [PubMed] [Google Scholar]
- Chesler EJ, et al. Genetic correlates of gene expression in recombinant inbred strains: A relational model system to explore neurobehavioral phenotypes. Neuroinformatics. 2003;1:343–357. doi: 10.1385/NI:1:4:343. [DOI] [PubMed] [Google Scholar]
- Choi DS, et al. Conditional rescue of protein kinase C epsilon regulates ethanol preference and hypnotic sensitivity in adult mice. J. Neurosci. 2002;22:9905–9911. doi: 10.1523/JNEUROSCI.22-22-09905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill GA. Fundamentals of experimental design for cDNA microarrays. Nat. Genet. 2002;32(Suppl.):490–495. doi: 10.1038/ng1031. [DOI] [PubMed] [Google Scholar]
- Churchill GA, et al. The collaborative cross, a community resource for the genetic analysis of complex traits. Nat. Genet. 2004;36:1133–1137. doi: 10.1038/ng1104-1133. [DOI] [PubMed] [Google Scholar]
- Crabbe JC. Review. Neurogenetic studies of alcohol addiction. Philos. Trans. R Soc. Lond. B. Biol. Sci. 2008;363:3201–3211. doi: 10.1098/rstb.2008.0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe JC, et al. Genetics of mouse behavior: Interactions with laboratory environment. Science. 1999;284:1670–1672. doi: 10.1126/science.284.5420.1670. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, et al. Alcohol-related genes: Contributions from studies with genetically engineered mice. Addict. Biol. 2006;11:195–269. doi: 10.1111/j.1369-1600.2006.00038.x. [DOI] [PubMed] [Google Scholar]
- Damerval C, et al. Quantitative trait loci underlying gene product variation: A novel perspective for analyzing regulation of genome expression. Genetics. 1994;137:289–301. doi: 10.1093/genetics/137.1.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels GM, Buck KJ. Expression profiling identifies strain-specific changes associated with ethanol withdrawal in mice. Genes Brain Behav. 2002;1:35–45. doi: 10.1046/j.1601-1848.2001.00008.x. [DOI] [PubMed] [Google Scholar]
- Dennis G, et al. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4(5):P3. [PubMed] [Google Scholar]
- Diamond I, Gordon AS. Cellular and molecular neuroscience of alcoholism. Physiol Rev. 1997;77:1–20. doi: 10.1152/physrev.1997.77.1.1. [DOI] [PubMed] [Google Scholar]
- Doss S, et al. cis-Acting expression quantitative trait loci in mice. Genome Res. 2005;15:681–691. doi: 10.1101/gr.3216905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, et al. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel SR, et al. 5-HT3 receptor over-expression decreases ethanol self administration in transgenic mice. Psychopharmacology (Berl.) 1998;140:243–248. doi: 10.1007/s002130050763. [DOI] [PubMed] [Google Scholar]
- Fehr C, et al. Congenic mapping of alcohol and pentobarbital withdrawal liability loci to a <1 centimorgan interval of murine chromosome 4: Identification of Mpdz as a candidate. J. gene. Neurosci. 2002;22:3730–3738. doi: 10.1523/JNEUROSCI.22-09-03730.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DB. Letter: Inherited differences in intensity of alcohol withdrawal reactions in mice. Nature. 1973;245:154–156. doi: 10.1038/245154a0. [DOI] [PubMed] [Google Scholar]
- Guo AY, et al. ERGR: An ethanol-related gene resource. Nucleic Acids Res. 2009;37:D840–845. doi: 10.1093/nar/gkn816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SY, et al. Psychopathology in offspring from multiplex alcohol dependence families with and without parental alcohol dependence: A prospective study during childhood and adolescence. Psychiatry Res. 2008;160:155–166. doi: 10.1016/j.psychres.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge CW, et al. Supersensitivity to allosteric GABA(A) receptor modulators and alcohol in mice lacking PKCepsilon. Nat. Neurosci. 1999;2:997–1002. doi: 10.1038/14795. [DOI] [PubMed] [Google Scholar]
- Horvath S, Dong J. Geometric interpretation of gene coexpression network analysis. PLoS Comput. Biol. 2008;4:e1000117. doi: 10.1371/journal.pcbi.1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, et al. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Iwamoto K, et al. Decreased expression of NEFH and PCP4/PEP19 in the prefrontal cortex of alcoholics. Neurosci. Res. 2004;49:379–385. doi: 10.1016/j.neures.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Jansen RC, Nap JP. Genetical genomics: The added value from segregation. Trends Genet. 2001;17:388–391. doi: 10.1016/s0168-9525(01)02310-1. [DOI] [PubMed] [Google Scholar]
- Jeanblanc J, et al. Endogenous BDNF in the dorsolateral striatum gates alcohol drinking. J. Neurosci. 2009;29:13494–13502. doi: 10.1523/JNEUROSCI.2243-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, et al. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- Junker BH, Schreiber F. Analysis of Biological Networks. Wiley; New York, NY: 2008. [Google Scholar]
- Kang HM, et al. Accurate discovery of expression quantitative trait loci under confounding from spurious and genuine regulatory hotspots. Genetics. 2008;180:1909–1925. doi: 10.1534/genetics.108.094201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpyak VM, et al. Sequence variations of the human MPDZ gene and association with alcoholism in subjects with European ancestry. Alcohol. Clin. Exp. Res. 2009;33:712–721. doi: 10.1111/j.1530-0277.2008.00888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerns RT, Miles MF. Microarray analysis of ethanol-induced changes in gene expression. Methods Mol. Biol. 2008;447:395–410. doi: 10.1007/978-1-59745-242-7_26. [DOI] [PubMed] [Google Scholar]
- Kerns RT, et al. Application of the S-score algorithm for analysis of oligonucleotide microarrays. Methods. 2003;31:274–281. doi: 10.1016/s1046-2023(03)00156-7. [DOI] [PubMed] [Google Scholar]
- Kerns RT, et al. Ethanol-responsive brain region expression networks: Implications for behavioral responses to acute ethanol in DBA/2J versus C57BL/6J mice. J. Neurosci. 2005;25:2255–2266. doi: 10.1523/JNEUROSCI.4372-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kril JJ, Harper CG. Neuronal counts from four cortical regions of alcoholic brains. Acta Neuropathol. 1989;79:200–204. doi: 10.1007/BF00294379. [DOI] [PubMed] [Google Scholar]
- Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3:1724–1735. doi: 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letwin NE, et al. Combined application of behavior genetics and microarray analysis to identify regional expression themes and gene–behavior associations. J. Neurosci. 2006;26:5277–5287. doi: 10.1523/JNEUROSCI.4602-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandoski M. Conditional control of gene expression in the mouse. Nat. Rev. Genet. 2001;2:743–755. doi: 10.1038/35093537. [DOI] [PubMed] [Google Scholar]
- Lewohl JM, et al. Gene expression in human alcoholism: Microarray analysis of frontal cortex. Alcohol. Clin. Exp. Res. 2000;24:1873–1882. [PubMed] [Google Scholar]
- Lewohl JM, et al. Expression of MBP, PLP, MAG, CNP, and GFAP in the human alcoholic brain. Alcohol. Clin. Exp. Res. 2005;29:1698–1705. doi: 10.1097/01.alc.0000179406.98868.59. [DOI] [PubMed] [Google Scholar]
- Liu B, et al. Gene network inference via structural equation modeling in genetical genomics experiments. Genetics. 2008;178:1763–1776. doi: 10.1534/genetics.107.080069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons P. Advances in spotted microarray resources for expression profiling. Brief. Funct. Genomic. Proteomic. 2003;2:21–30. doi: 10.1093/bfgp/2.1.21. [DOI] [PubMed] [Google Scholar]
- Mayfield RD, et al. Patterns of gene expression are altered in the frontal and motor cortices of human alcoholics. J. Neurochem. 2002;81:802–813. doi: 10.1046/j.1471-4159.2002.00860.x. [DOI] [PubMed] [Google Scholar]
- McBride WJ, et al. Changes in gene expression in regions of the extended amygdala of alcohol-preferring rats after binge-like alcohol drinking. Alcohol. 2010;44:171–183. doi: 10.1016/j.alcohol.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClearn GE, Rodgers DA. Differences in alcohol preference among inbred strains of mice. Q. J. Stud. Alcohol. 1959;20:691–695. [Google Scholar]
- Merikangas KR, et al. Familial transmission of substance use disorders. Arch Gen. Psychiatry. 1998;55:973–979. doi: 10.1001/archpsyc.55.11.973. [DOI] [PubMed] [Google Scholar]
- Metten P, et al. Drug withdrawal convulsions and susceptibility to convulsants after short-term selective breeding for acute ethanol withdrawal. Behav. Brain Res. 1998a;95:113–122. doi: 10.1016/s0166-4328(97)00216-7. [DOI] [PubMed] [Google Scholar]
- Metten P, et al. High genetic susceptibility to ethanol withdrawal predicts low ethanol consumption. Mamm. Genome. 1998b;9:983–990. doi: 10.1007/s003359900911. [DOI] [PubMed] [Google Scholar]
- Miles MF. Alcohol's effects on gene expression. Alcohol Health Res. World. 1995;19:237–243. [PMC free article] [PubMed] [Google Scholar]
- Miles MF. Microarrays: Lost in a storm of data? Nat. Rev. Neurosci. 2001;2:441–443. doi: 10.1038/35077582. [DOI] [PubMed] [Google Scholar]
- Mozhui K, et al. Dissection of a QTL hotspot on mouse distal chromosome 1 that modulates neurobehavioral phenotypes and gene expression. PLoS Genet. 2008;4:e1000260. doi: 10.1371/journal.pgen.1000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan MK, et al. Toward understanding the genetics of alcohol drinking through transcriptome meta-analysis. Proc. Natl. Acad. Sci. USA. 2006;103:6368–6373. doi: 10.1073/pnas.0510188103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Science. 1997;278:58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- Parkinson H, et al. ArrayExpress update—From an archive of functional genomics experiments to the atlas of gene expression. Nucleic Acids Res. 2009;37:D868–872. doi: 10.1093/nar/gkn889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peirce JL, et al. A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC Genet. 2004;5:7. doi: 10.1186/1471-2156-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters LL, et al. The mouse as a model for human biology: A resource guide for complex trait analysis. Nat. Rev. Genet. 2007;8:58–69. doi: 10.1038/nrg2025. [DOI] [PubMed] [Google Scholar]
- Phillips TJ, et al. Behavioral sensitization to drug stimulant effects in C57BL/6J and DBA/2J inbred mice. Behav. Neurosci. 1994;108:789–803. doi: 10.1037//0735-7044.108.4.789. [DOI] [PubMed] [Google Scholar]
- Prescott CA, et al. Sex differences in the sources of genetic liability to alcohol abuse and dependence in a population-based sample of US twins. Alcohol Clin. Exp. Res. 1999;23:1136–1144. doi: 10.1111/j.1530-0277.1999.tb04270.x. [DOI] [PubMed] [Google Scholar]
- Rimondini R, et al. Long-lasting increase in voluntary ethanol consumption and transcriptional regulation in the rat brain after intermittent exposure to alcohol. FASEB J. 2002;16:27–35. doi: 10.1096/fj.01-0593com. [DOI] [PubMed] [Google Scholar]
- Rockman MV, Kruglyak L. Genetics of global gene expression. Nat. Rev. Genet. 2006;7:862–872. doi: 10.1038/nrg1964. [DOI] [PubMed] [Google Scholar]
- Rodd ZA, et al. Differential gene expression in the nucleus accumbens with ethanol self-administration in inbred alcohol-preferring rats. Pharmacol. Biochem. Behav. 2008;89:481–498. doi: 10.1016/j.pbb.2008.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom M, et al. Using magnetic resonance imaging and diffusion tensor imaging to assess brain damage in alcoholics. Alcohol Res. Health. 2003;27:146–152. [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nat. Genet. 2005;37:710–717. doi: 10.1038/ng1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, et al. Genetics of gene expression surveyed in maize, mouse and man. Nature. 2003;422:297–302. doi: 10.1038/nature01434. [DOI] [PubMed] [Google Scholar]
- Schena M, et al. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Schuckit MA. Low level of response to alcohol as a predictor of future alcoholism. Am. J. Psychiatry. 1994;151:184–189. doi: 10.1176/ajp.151.2.184. [DOI] [PubMed] [Google Scholar]
- Scott RE, et al. P2P-R expression is genetically coregulated with components of the translation machinery and with PUM2, a translational repressor that associates with the P2P-R mRNA. J. Cell Physiol. 2005;204:99–105. doi: 10.1002/jcp.20263. [DOI] [PubMed] [Google Scholar]
- Shirley RL, et al. Mpdz is a quantitative trait gene for drug withdrawal seizures. Nat. Neurosci. 2004;7:699–700. doi: 10.1038/nn1271. [DOI] [PubMed] [Google Scholar]
- Smith AM, et al. Microarray analysis of the effects of a gamma-protein kinase C null mutation on gene expression in striatum: A role for transthyretin in mutant phenotypes. Behav. Genet. 2006;36:869–881. doi: 10.1007/s10519-006-9083-6. [DOI] [PubMed] [Google Scholar]
- Sokolov BP. Oligodendroglial abnormalities in schizophrenia, mood disorders and substance abuse. Comorbidity, shared traits, or molecular phenocopies? Int. J. Neuropsychopharmacol. 2007;10:547–555. doi: 10.1017/S1461145706007322. [DOI] [PubMed] [Google Scholar]
- Stuart JM, et al. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302:249–255. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- Tabakoff B, et al. Selective breeding, quantitative trait locus analysis, and gene arrays identify candidate genes for complex drug-related behaviors. J. Neurosci. 2003;23:4491–4498. doi: 10.1523/JNEUROSCI.23-11-04491.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele TE, et al. Ethanol consumption and resistance are inversely related to neuropeptide Y levels. Nature. 1998;396:366–369. doi: 10.1038/24614. [DOI] [PubMed] [Google Scholar]
- Treadwell JA, Singh SM. Microarray analysis of mouse brain gene expression following acute ethanol treatment. Neurochem. Res. 2004;29:357–369. doi: 10.1023/b:nere.0000013738.06437.a6. [DOI] [PubMed] [Google Scholar]
- Tusher VG, et al. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Noort V, et al. The yeast coexpression network has a small-world, scale-free architecture and can be explained by a simple model. EMBO Rep. 2004;5:280–284. doi: 10.1038/sj.embor.7400090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter NA, et al. SNPs matter: Impact on detection of differential expression. Nat. Methods. 2007;4:679–680. doi: 10.1038/nmeth0907-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, et al. Comparison of global brain gene expression profiles between inbred long-sleep and inbred short-sleep mice by high-density gene array hybridization. Alcohol Clin. Exp. Res. 2001;25:810–818. [PubMed] [Google Scholar]
- Yang RJ, et al. Variation in mouse basolateral amygdala volume is associated with differences in stress reactivity and fear learning. Neuropsychopharmacology. 2008;33:2595–2604. doi: 10.1038/sj.npp.1301665. [DOI] [PubMed] [Google Scholar]
- Zachariou V, et al. Essential role for RGS9 in opiate action. Proc. Natl. Acad. Sci. USA. 2003;100:13656–13661. doi: 10.1073/pnas.2232594100. [DOI] [PMC free article] [PubMed] [Google Scholar]