

Abstract
Tuberculosis, one of the leading causes of death worldwide, stimulates inflammatory responses with beneficial and pathologic consequences. The regulation and nature of an optimal inflammatory response to Mycobacterium tuberculosis (MTb) remains poorly understood in humans. Insight into mechanisms of negative regulation of the Toll-like Receptor (TLR)-mediated innate immune response to MTb could provide significant breakthroughs in the design of new vaccines and drugs. We hypothesized that TOLLIP and its common variants negatively regulate TLR signaling in human monocytes and are associated with susceptibility to tuberculosis. Using shRNA knockdown of TOLLIP in peripheral blood human monocytes, we found that TOLLIP suppresses TNF and IL-6 production after stimulation with TLR2 and TLR4 ligands. In contrast, secretion of the anti-inflammatory cytokine IL-10 was induced by TOLLIP. We also discovered 2 common polymorphisms that are associated with either decreased levels of mRNA expression (rs3750920) or increased IL-6 production (rs5743899) in a sample of 56 healthy volunteers. Furthermore, in a case-population study in Vietnam with 760 cord-blood samples and 671 TB case patients, we found that SNPs rs3750920 and rs5743899 were associated with susceptibility to tuberculosis (p=7.03×10−16, 6.97×10−7, respectively). Together, these data demonstrate that TOLLIP has an anti-inflammatory effect on TLR signaling in humans and that TOLLIP deficiency is associated with an increased risk of TB. To our knowledge, these data also show the first associations of TOLLIP polymorphisms with any infectious disease. These data also implicate an unexpected mechanism of negative regulation of TLR signaling in human TB pathogenesis.
Introduction
Tuberculosis is one of the leading causes of infectious death worldwide and yet its immune pathogenesis remains incompletely understood (1, 2). The innate immune system is critical for the initial host defense against mycobacteria (3). Several lines of evidence suggest that TLR1, TLR2, TLR4, TLR6, TLR8, and TLR9 are important for host defense against Mycobacterium tuberculosis (MTb) (4–11). For example, in vitro studies in murine macrophages demonstrate that TLR1, TLR2, TLR4, TLR6, TLR9 and MyD88 regulate Mtb-induced cytokine secretion (5, 12–21). The mycobacterial cell wall contains a number of pro-inflammatory TLR2 ligands, including lipoproteins, mycolylarabinogalactan-peptidoglycan complex [(mAGP), the cell wall core structure], lipids, and LAM (5, 6, 12, 13, 22). Despite the multiple lines of evidence demonstrating a critical role for TLR mediation of MTb recognition in vitro, the in vivo significance of individual TLRs has been more difficult to consistently demonstrate. Although some studies demonstrated increased susceptibility to MTb in MyD88−/−, and Tlr2−/−, and Tlr2/9−/− mice, these results have not been uniformly observed under all experimental conditions (10, 18, 21, 23–26). Furthermore, the effects appear to be strongest when multiple TLRs are impaired. Together, these in vivo studies suggest an important though not absolute role for the TLR pathway in mediating host protection to in vivo murine MTb infection.
Through the TLR pathway, proinflammatory (IL-6, TNF, and IL-12) and anti-inflammatory (IL-10) cytokines are secreted from macrophages stimulated with MTb (27, 28). The interplay between proinflammatory and anti-inflammatory cytokines is important in determining the quality and quantity of both the innate and adaptive immune response to pathogens. The signaling mechanisms that specifically modulate IL-10 production after TLR stimulation are unclear, and understanding how IL-10 secretion is modulated by TLR regulators has important implications in MTb pathogenesis and vaccine design. IL-10 −/− mice develop severe colitis in response to gut microbiota (29), making extrapolation of the role of IL-10 in humans difficult. Several investigators reported that IL-10−/− mice have enhanced MTb clearance (30), but others have reported no such differences (31, 32). Human studies of the role of IL-10 in tuberculosis have shown that IL-10 levels are increased in lung tissue of patients with pulmonary tuberculosis (33). Furthermore, neutralization of IL-10 enhances IFN-gamma and T cell proliferation in vitro (34). Human genetic studies examining whether polymorphisms in the IL-10 gene are associated with susceptibility to TB are inconclusive (35–37). Together, these studies suggest that the role of IL-10 in the pathogenesis of tuberculosis remains unclear and further study is required.
Negative regulators have been discovered at nearly every step of the TLR signaling cascade (38–40) and include Toll-interacting protein (TOLLIP, also called IL-1RAcPIP), A20 (41), Single Ig IL-1-receptor (SIGIRR, also called TIR8) (42), and interleukin-1 receptor-associated kinase 3 (IRAK-3, also called IRAK-M) (43). TOLLIP is a 274 amino acid protein with highly conserved C2 (amino acids 54–186, similar to that found in PI-specific phospholipase C-d1I) and C-terminal UBA (ubiquitin-associated) domains. It was initially discovered as an IL-1R1-interacting protein which linked IRAK to the IL-1 receptor pathway (44), and later was implicated in suppression of the TLR2 (45) and TLR4 pathways (46). However, in vivo murine knockout models demonstrated that TOLLIP induced proinflammatory pathways, in contrast to in vitro experiments (47). To our knowledge, there are no studies examining the effects of TOLLIP on TLR signaling pathways in humans.
Several lines of evidence suggest that host genetics regulates susceptibility to TB including twin-based, linkage, candidate gene association, and genome-wide association studies. We, along with other investigators, have identified associations between common polymorphisms in innate immunity genes and susceptibility to TB and clinical phenotypes (23, 48, 49). Much of this work has focused on pattern recognition receptors (PRRs) and their associated adaptor proteins, including TLR1/2/4/6 and 9. Several candidate gene association studies have shown associations between single nucleotide polymorphisms (SNPs) in TLR2 and tuberculosis, including 597CC within a Vietnamese population (8, 50). A polymorphism in TLR1 (T1805G, I602S) regulates lipopeptide-induced cytokine secretion and TLR1 surface expression, and is associated with susceptibility to tuberculosis as well as leprosy (51–54). Genetic studies have also found associations of TLR4 polymorphisms with TB susceptibility (10, 25, 26). These studies suggest that genetic control of TLR1/2/4/6 signaling by negative regulators may alter susceptibility to TB. In contrast to TLRs, the functional and clinical significance of genetic variation of TOLLIP and other negative regulators of TLR signaling is poorly studied and only partially understood. In this study we examined TOLLIP’s function in human monocytes with knockdown studies and examination of common TOLLIP variants. We also examined whether these polymorphisms were associated with susceptibility to tuberculosis in Vietnam. Together, these studies suggest that TOLLIP is a critical regulator of the TLR pathway in humans, and that this immune regulation plays a critical role in the susceptibility to tuberculosis.
Materials and Methods
Materials
RPMI 1640 medium, L-glutamine, penicillin, and streptomycin were obtained from Life Technologies (Carlsbad, CA, USA). Ultrapure LPS (TLR4 ligand) isolated from Salmonella minnesota R595 was obtained from List Biological Labs Inc. (Campbell, CA, USA). The synthetic lipopeptides PAM2 (PAM2CSKKKK, S-[2,3-bis(palmitoyloxy)-propyl]-(R)-cysteinyl-(lysyl)3-lysine, TLR2/6 ligand) and PAM3 (Pam3CSKKKK, N-palmitoyl-S-[2,3-bis-(palmitoyloxy)-propyl]-(R)-cysteinyl-(lysyl)3-lysine, TLR2/1 ligand) were obtained from EMC Microcollections (Tuebingen, Germany). Whole cell lysate from MTb strain H37Rv was obtained as part of NIH, NIAID Contract No. HHSN266200400091C, entitled "Tuberculosis Vaccine Testing and Research Materials” (Colorado State University, Fort Collins, CO, USA).
Cell Culture and ELISA
Primary monocytes were isolated from human subjects as described previously (55). Briefly, human monocytes were isolated from peripheral blood with Ficoll gradient separation followed by positive selection using human anti-CD14 antibody associated with magnetic beads (Miltenyi Biotec, Inc, Auburn, CA). Monocytes were isolated with >95% purity, based on CD14 positivity in flow cytometric analysis and were maintained in RPMI 1640 supplemented with 10% fetal calf serum. Cells were incubated with TLR ligands PAM2, PAM3, and LPS, or with Mycobacterium tuberculosis whole cell lysates for 24 hours. Subsequently, supernatants were evaluated for levels of cytokine (IL-6, IL-10, TNF) via ELISA Duoset (R&D Systems, Minneapolis, MN). Each sample was assayed in duplicate or triplicate, and experiments shown were performed at least twice to ensure reproducibility.
Genotyping and linkage disequilibrium
Genotyping was performed with Sequenom’s MassARRAY™ technique as previously described (56). This technique uses allele-specific primer extension reactions to discriminate genotypes. We also confirmed the SNPs of interest using Taqman genotyping technology (Applied Biosystems, Inc., Carlsbad, CA). We identified haplotype-tagging SNPs from the CHB (Han Chinese in Beijing) and CEU (Utah residents with European ancestry) populations from the International HapMap Project (http://www.hapmap.org) and other public databases with the Genome Variation Server (http://www.ncbi.nlm.nih.gov/SNP/ and www.innateimmunity.net). We searched a region on chromosome 11p15.5, 10 kilobases upstream and downstream of TOLLIP for tagged SNPs using an R2 cutoff of 0.8 for linkage disequilibrium and a minor allele frequency cut-off of 5%. Stata/Intercooled v11.0 software program PWLD (StataCorp LP, College Station, TX) was used to calculate R2 and D’ as measurements of linkage disequilibrium between the polymorphisms.
Molecular Biology
TOLLIP mRNA levels were measured in monocytes from peripheral blood of healthy donors in the Seattle area. RNA was obtained after cell lysis and RNA purification (Qiagen, Valencia, CA). After synthesis of cDNA, real-time PCR was performed. TOLLIP mRNA levels were normalized with a GAPDH control using the following primer-probe sets (5’ J56-FAM/CTCCGGGAT/ZEN/GGT3’, 5’CTCAGGCTGTACCACTTG3’ 5’GGCGTGGACTCTTTCTAT3’, Integrated DNA Technologies, Coralville, IA). Quantitative PCR was performed on a Taqman machine.
Gene Silencing
Knockdown studies were performed using plasmids encoding 3 TOLLIP-specific shRNA (Santa Cruz Biotechnology, Santa Cruz, CA) that were then packaged into a non-replicating lentiviral particle to improve transfection efficiency using a standard protocol (UCSF Viracore Facility, San Francisco, CA). These lentiviral particles were then incubated with monocytes at an MOI of 2 for 24 hours before cells were stimulated and harvested for cytokine analysis. Knockdown was confirmed using quantitative PCR for TOLLIP mRNA transcript.
Human Subjects
Approval for human study protocols was obtained from the human subjects review boards at the University of Washington School of Medicine (Seattle, WA), the Hospital for Tropical Diseases (Ho Chi Minh City, Vietnam), Pham Ngoc Thach Hospital for Tuberculosis and Lung Diseases (Vietnam), Health Services of Ho Chi Minh City, Hung Vuong Hospital (Vietnam), and the Oxford Tropical Research Ethics Committee (Oxford, United Kingdom).
Genomic DNA was purified from saliva and blood samples. The Seattle study group included 56 healthy volunteers who donated blood and saliva samples. The ethnic composition of these subjects was 70% Caucasian (39/56), 27% Asian (15/56), and 4% African American (2/56). Study subjects with tuberculous meningitis (TBM) were recruited from two centers in Ho Chi Minh City, Vietnam: Pham Ngoc Thach (PNT) Hospital for Tuberculosis and the Hospital for Tropical Diseases (HTD). These 500-bed hospitals serve the local community and act as tertiary referral centers for TB (PNT) and infectious diseases (HTD) for southern Vietnam. Individuals at least 15 years of age, admitted to these centers with clinical meningitis (defined as nuchal rigidity and abnormal cerebrospinal fluid parameters), a negative HIV test, and a positive Ziehl-Neelsen stain for acid-fast bacilli and/or M. tuberculosis culture from cerebrospinal fluid (“definite TBM”) were recruited for genetics studies during 2001–2008. In addition to definite TBM, the cohort included subjects with “probable TBM”, defined as clinical meningitis plus at least one of the following: chest radiograph consistent with active TB, acid-fast bacilli found in any specimen other than cerebrospinal fluid, and clinical evidence of other extrapulmonary TB (57).
From 2003 through 2004, subjects with pulmonary TB were recruited from a network of district TB control units within Ho Chi Minh City that provide directly observed therapy to TB patients. These subjects were outpatients who were at least 15 years of age, had no previous history of treatment for TB, no evidence of miliary or extrapulmonary TB, chest x-ray results consistent with non-miliary pulmonary TB, negative HIV test results, and sputum smear positive for acid-fast bacilli or M. tuberculosis cultured from sputum. Additionally pulmonary TB subjects enrolled were recruited from PNT hospital from 2006 through 2008.
Control subjects were enrolled at Hung Vuong Hospital in Ho Chi Minh City from blood collected from the umbilical cord of babies after birth. All case and control participants were unrelated and greater than 95% were of the Vietnamese Kinh ethnicity. Written, informed consent was obtained from patients or their relatives if the patient could not provide consent (i.e. was unconscious). Parents provided consent for cord-blood controls.
Out of the study subjects that self-identified their ancestry as Vietnamese Kinh, we formally tested for levels of background genetic differences between cases and controls by genotyping the cohort for a panel of 24 independent SNP markers (58). The mean χ2 statistic for the 24 SNPs comparing allelic frequencies between cases and control was 1.6 (p=0.20), suggesting that no significant population stratification was present in our study population (59).
Statistical Methods
All analyzed SNPs were tested for Hardy-Weinberg equilibrium in control subjects using a χ2 goodness-of-fit test. In our primary analysis, we examined whether polymorphism genotype frequencies were associated with any type of TB in a case-population study design using Stata MP 11 and the user written package “genassoc” (60). For secondary analyses, SNPs were investigated for associations under additional genetic models (dominant, recessive, and additive) and for association with the clinical sub-types of TB (pulmonary or meningeal). In the recessive model, carriers of allele 1 (00 and 01 genotypes) were compared with homozygous subjects for allele 2 (11 genotype). In the dominant model, carriers of allele 2 (01 and 11 genotypes) were compared with homozygous subjects for allele 1 (00 genotype). Measures of linkage disequilibrium (LD) were assessed in controls using “pwld” in Stata.
Nine TOLLIP polymorphisms were genotyped in the Vietnam and Seattle cohorts. Two (rs4744015 and rs5744016) were monogenic and one (rs5743890) was not in Hardy-Weinberg equilibrium and thus not analyzed further. Six are shown in all subsequent analyses. If we correct for multiple comparisons with a Bonferroni test (P value/6), only p values less than 0.008 remain significant. However, as we had two single nucleotide polymorphisms of interest based upon prior functional data, we show uncorrected P values in these tables.
The Pearson-χ2 test and Student’s t-test were used to assess categorical and continuous clinical variables, respectively. For analysis of associations between TOLLIP polymorphisms and mRNA levels or cytokines, we used an ANOVA analysis with Mann-Whitney tests for significance.
Results
TOLLIP regulates the TLR signaling pathway in humans
In order to determine the functional role of TOLLIP in the human innate immune system, we performed shRNA knockdown experiments on peripheral blood monocytes from healthy volunteers (Fig. 1). Using a lentiviral delivery system in primary monocytes, we knocked down TOLLIP mRNA expression by over 50% as compared to monocytes alone or those treated with control lentiviral particles (Fig. 1a, p<0.001 Student’s t-test). We also tested and confirmed that TLR2 and TLR4 mRNA expression were not altered by TOLLIP shRNA (Fig. 1a, p=0.78 for TLR2, p=0.14 for TLR4; Student’s t test comparing control to TOLLIP shRNA viral infection). After stimulation with PAM2 (TLR2/6 ligand) and PAM3 (TLR2/1 ligand), TOLLIP-deficient monocytes produced elevated levels of IL-6 compared with either untransfected monocytes or control-lentiviral particle-transfected monocytes (p<0.01, Fig 1b–f). Transfection with control shRNA led to a mild increase in IL-6 that we attributed to the innate inflammatory response to lentivirus infection and cytosolic DNA. In contrast, LPS (TLR4 ligand)-induced IL-6 levels were similar under all conditions (Fig 1e). TOLLIP knockdown led to increased TNF production after stimulation with PAM2, PAM3, LPS, or MTb whole cell lysate (p<0.01, Fig 1g–k). In cells stimulated with PAM2, PAM3, or MTb whole cell lysate, there were no significant differences in IL-10 production between cells stimulated with nonspecific lentiviral particles or TOLLIP-specific viral particles (Fig 1m, 1n). Intriguingly, TOLLIP knockdown in LPS-stimulated cells led to significant decreases in secreted IL-10 from monocytes (p<0.01, Fig 1o). In a separate experiment with stimulation with MTb H37Rv whole cell lysate, we found that TOLLIP knockdown led to increased IL-6 and TNF secretion, but not IL-10 compared to controls (p<0.01, Fig 1f, 1k, 1p).
Figure 1. Lentiviral shRNA knockdown of TOLLIP in primary human monocytes.

Peripheral blood monocytes were isolated and incubated with media, lentiviral particles encoding scrambled shRNA, or lentiviral particles encoding shRNA for 3 areas with in the TOLLIP gene for 24 hours. These cells were then incubated with TLR ligands for 24 hours and secreted cytokine responses were measured in the supernatant. A) mRNA levels of expression after lentiviral knockdown of peripheral blood monocytes. This data shows one representative experiment (out of a total of 2). B) IL-6, C) TNF, and D) IL-10, cytokine response after media, PAM2, PAM3, or LPS stimulation in primary monocytes incubated with nothing, control shRNA, or TOLLIP shRNA-encoding lentiviral particles. * p<0.01, ANOVA with Mann-Whitney test. MTb stimulation is from an independent experiment.
These data suggest that TOLLIP regulates the innate immune response in humans via two mechanisms – by suppressing proinflammatory cytokines via TLR2 and TLR4 and by inducing IL-10 through a TLR4-specific mechanism.
Association of TOLLIP SNPs with mRNA Expression
Next, we examined whether common variants of TOLLIP regulated its function. We genotyped nine haplotype-tagging SNPs in 84 healthy volunteers and examined their association with TOLLIP mRNA expression in monocytes. (Fig. 2, polymorphisms listed in genomic order) For SNP rs3750920, the minor homozygote was significantly associated with increased mRNA expression compared to either heterozygotes or major homozygotes (p<0.01 by genotypic model; CC = 26, CT = 32, TT = 16, Fig. 2d). This association was also significant in a recessive model comparing TT homozygotes with CT/CC genotypes (p<0.01). SNP rs5743899 genotype GG was associated with decreased mRNA transcript in a recessive model. (p<0.01; AA = 49, AG = 31, GG = 4, Fig 2a). The other 4 TOLLIP SNPs were not significantly associated with mRNA expression (Fig. 2c, 2b, 2e, 2f,). Three SNPs (rs3793964, CC = 36, CT = 24, TT = 16; rs3829223, CC = 19, CC = 32, TT = 27; and rs3168046, CC = 15, CT = 22, TT = 14) showed a trend towards an association with mRNA expression (Fig. 2c, 2e, 2f, P=0.26, 0.22, 0.14, respectively). These SNPs were in linkage disequilibrium with SNP rs3750920 (Supplemental Fig. 1a R2=0.43, 0.55, 0.73 for pairwise comparisons of 3 SNPs with rs3750920). Together these results suggest that at least one TOLLIP polymorphism is associated with mRNA expression and is a genetic marker of human TOLLIP deficiency.
Figure 2. TOLLIP polymorphisms and variation in mRNA expression.

Genomic DNA and mRNA were isolated from monocytes from 84 healthy individuals. TOLLIP mRNA expression was measured and normalized to GAPDH. Genotypes of six TOLLIP polymorphisms were examined for associations with TOLLIP mRNA expression. (rs5743899, AA = 49, AG = 31, GG = 4, Fig 2a; rs5743942, CC = 17, CT = 31, TT = 20, Fig 2b; ; rs3793964; CC = 36, CT = 24, TT = 16, Fig. 2c; rs3750920, CC = 26, CT = 32, TT = 16, Fig. 2d; rs3829223, CC = 19, CC = 32, TT = 27, Fig 2e; and rs3168046, CC = 15, CT = 22, TT = 14, Fig 2f). * p<0.01; # p=0.15, ANOVA analysis with Mann-Whitney test.
Association of TOLLIP SNP rs5743899 with cytokine responses to TLR ligands
We next examined whether polymorphisms rs5743899 and rs3750920 were associated with cytokine responses in monocytes. We stimulated peripheral blood mononuclear cells with TLR ligands (PAM2, PAM3, and LPS) as well as Mycobacterium tuberculosis whole cell lysate, and measured secreted IL-6 and IL-10 in the culture supernatants (Fig 3a, 3b). Minor homozygotes (GG) of polymorphism rs5743899 were associated with significantly higher levels of IL-6 compared with heterozygotes or major homozygotes (Fig 3c, 3d, 3e; AA individuals = 35, AG = 21, GG = 7, p<0.01, ANOVA with Mann-Whitney test) after stimulation with PAM2 (TLR2/TLR6) or PAM3 (TLR2/TLR1), but not LPS (TLR4). Furthermore, IL-6 levels were significantly increased after stimulation with MTb whole cell lysate (Fig 3f). In addition, IL-10 levels were significantly decreased in a recessive pattern for GG individuals after LPS stimulation (p=0.03, Fig 3g).
Figure 3. TOLLIP SNP rs5743899 and cytokine responses after TLR and MTb stimulation of peripheral blood mononuclear cells.

Peripheral blood mononuclear cells were isolated from 64 healthy volunteers in Seattle and stimulated with media or TLR ligands (LPS at 10 ng/ml, PAM2 at 250 ng/ml, PAM3 at 250 ng/ml, MTb whole cell lysate at 1 µg/ml) for 24 hours. Secreted A) IL-6 and B) IL-10 levels were measured in supernatants via ELISA. C–F) IL-6 responses after TLR stimulation, stratified by rs5743899 genotype. AA individuals = 35, AG = 21, GG = 7. G–J) IL-10 responses after TLR stimulation, stratified by rs5743899 genotype. * p<0.01; ** p=0.03 by Mann-Whitney test in a recessive model.
There were not any significant changes in IL-10 production after PAM2 or PAM3 stimulation. By way of contrast, minor homozygotes from rs3750920 were not associated with IL6 levels when compared to heterozygotes or major homozygotes (Fig 4a–d; CC = 18 individuals, CT = 24, TT 12). Furthermore, no associations were noted in IL-10 production (Fig 4e–h). These data demonstrate that the minor homozygote of rs5743899 is associated with increased levels of IL-6 after PAM2, PAM3, or MTb whole cell lysate stimulation, as well as decreased IL-10 after LPS stimulation.
Figure 4. TOLLIP polymorphism rs3750920 and cytokine responses after TLR and MTb stimulation of peripheral blood mononuclear cells.

Identical experimental details as Figure 3, except data was stratified by genotype rs3750920, CC = 18 individuals, CT = 24, TT 12. A–D) IL-6 responses and E–H) IL-10 responses after stimulation with media or TLR ligands (LPS at 10 ng/ml, PAM2 250 ng/ml, PAM3 250ng/ml, MTb whole cell lysate at 1 µg/ml) for 24 hours.
Association of TOLLIP SNPs with susceptibility to tuberculosis
To assess the role of TOLLIP deficiency in human disease, we performed a candidate gene case-population association study to examine associations between TOLLIP polymorphisms and susceptibility to TB. We examined 671 cases (394 pulmonary TB (PTB), 277 TB meningitis (TBM)) and 760 cord blood controls in our cohort. We first analyzed polymorphisms rs3750920 and rs5743899 due to their association with functional phenotypes. TOLLIP polymorphisms had similar linkage disequilibrium patterns in Seattle and Vietnam, as well as the Northern European (CEU) and Han Chinese (CHB) HapMap populations (Figure 5, Supplemental Figure 1 & 2). Polymorphisms rs3750920 and rs5743899 were associated with susceptibility to TB using a genotypic model (Table I, rs3750920 p=7.03×10−16; and rs5743899 p=6.97×10−7). Three other SNPs, rs3793964, rs3829223, and rs3168046 were associated with TB, and these SNPs were in a high degree of LD with SNP rs3750920 in the Vietnamese population (Supplemental Table I). All of these SNPs were associated with TB after a conservative Bonferroni correction for multiple comparisons. One SNP (rs5743942) was associated with TB in an unadjusted analysis, but not after a Bonferroni correction.
Figure 5.
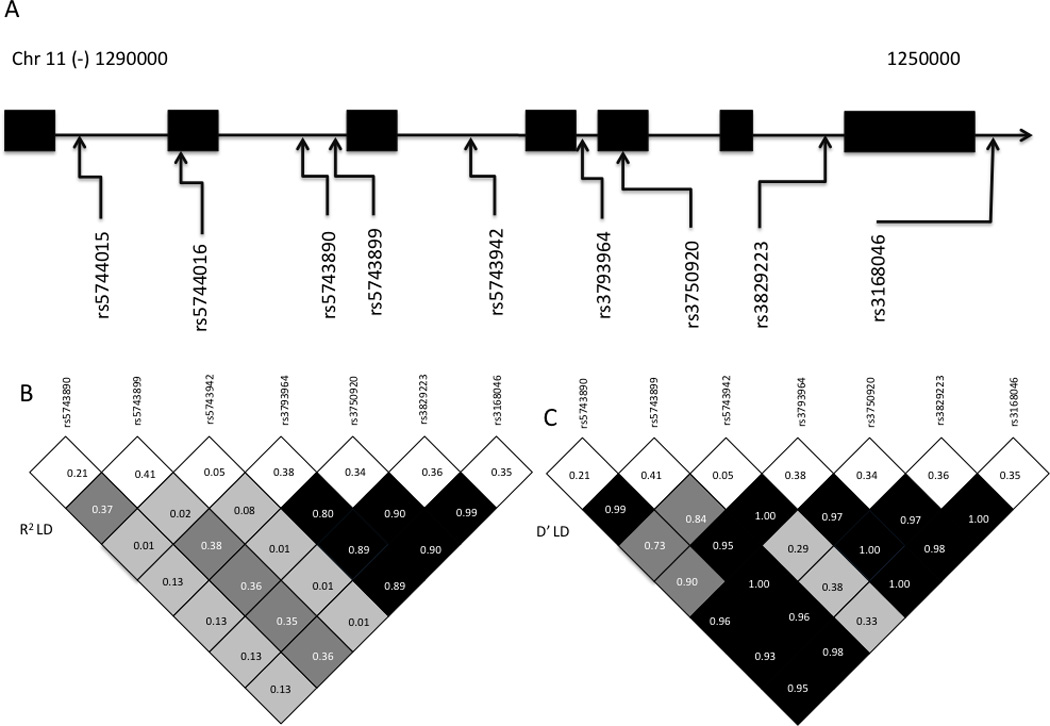
Chromosomal map and linkage disequilibrium plots of TOLLIP polymorphisms in Vietnamese cohort. (A) Chromosomal map shows genomic location of polymorphisms. Boxes show exons within gene on chromosome 11. rs3168046 was located on the 3’ UTR for the TOLLIP gene. (B, C): Linkage Disequilibrium plots with R2 and D’ values for controls in Vietnam. White boxes on top of plot show frequency of polymorphisms. Degree of shading proportionate to D’ or R2 value.
Table I.
TOLLIP gene region single nucleotide polymorphisms and genotype frequencies in the control and tuberculosis groups.
| Genotype | Genotype Analysis | ||||||
|---|---|---|---|---|---|---|---|
| SNP1 | Group | 00 | 01 | 11 | χ2 | P | H-W |
| rs3750920 | control | 308 (.415) | 355 (.479) | 78 (.105) | |||
| TB | 328 (.610) | 134 (.253) | 75 (.129) | 69.8 | 7.03×10−16 | 0.101 | |
| rs5743899 | control | 252 (.334) | 391 (.520) | 110 (.145) | |||
| TB | 261 (.397) | 252 (.383) | 144 (.220) | 28.4 | 6.97×10−7 | 0.163 | |
SNPs are arranged in the order that they are located on the chromosome. SNPs are listed by reference SNP ID. 0, common allele; 1, allele with minor frequency. Hardy-Weinberg equilibrium was calculated from control subjects only.
Secondary analysis of rs3750920 and rs5743899
For the 2 significant polymorphisms, we examined these associations further under different genetic models and clinical phenotypes. Testing the association with TB under different genetic models for rs3750920, we found the strongest association was consistent with a dominant model (Table II, OR 0.453, p = 6.28×10−12). In contrast, SNP rs5743899 was most strongly statistically associated with all forms of TB in a recessive model (OR 1.641, p = 0.0004). We also examined the associations with different clinical forms of tuberculosis (pulmonary tuberculosis (PTB) and meningeal TB (TBM)). Both SNPs were associated with PTB and TBM to a similar degree (Table II). Finally, we evaluated whether these two were associated with death or disability from tuberculosis, as well as with CSF and serum cytokine responses at the time of presentation (for subjects with TBM). We did not find any significant associations with these outcomes (data not shown). Together, these data suggest that 2 TOLLIP polymorphisms are associated with susceptibility to TB as well as TOLLIP expression levels or regulation of TLR-mediated cytokine secretion.
Table II.
Associations of rs3750920 and rs5743899 with TB meningitis and pulmonary tuberculosis
| Genotype | 2Dominant Analysis | Recessive Analysis | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SNP1 | Group4 | 00 | 01 | 11 | 3OR | CI (95%) | P value | OR | CI (95%) | P value |
| rs3750920 | control | 308 (.415) | 355 (.479) | 78 (.105) | ||||||
| TBM | 144 (.415) | 60 (.255) | 35 (.128) | 0.441 | .34–.64 | 5.26E-07 | 1.240 | .92 – 2.3 | 0.387 | |
| PTB | 184 (.617) | 74 (.248) | 40 (.134) | 0.445 | .33–.59 | 6.34E-09 | 1.324 | .85 – 2.0 | 0.176 | |
| TB all | 328 (.610) | 134 (.253) | 75 (.129) | 0.453 | .36–.57 | 6.28E-12 | 1.380 | .96 – 2.0 | 0.061 | |
| rs5743899 | control | 252 (.334) | 391 (.520) | 110 (.145) | ||||||
| TBM | 119 (.422) | 107 (.379) | 56 (.199) | 0.689 | .52–.920 | 0.010 | 1.448 | 1.0 – 2.09 | 0.040 | |
| PTB | 118 (.372) | 123 (.388) | 76 (.240) | 0.825 | .63–1.07 | 0.145 | 1.792 | 1.3 – 2.5 | 0.0003 | |
| TB all | 261 (.397) | 252 (.383) | 144 (.220) | 0.763 | .61–.95 | 0.015 | 1.641 | 1.23 – 2.2 | 0.0004 | |
Single nucleotide polymorphisms (SNPs) are listed by reference SNP ID.
A dominant and recessive allelic analysis was performed.
For calculations of odds ratios (OR) each group was compared with the control group. 0, common allele; 1, allele with minor frequency; CI, confidence interval;
“All TB” includes both pulmonary (PTB) and meningeal TB (TBM).
Discussion
Our study shows that TOLLIP regulates human TLR signaling pathways in monocytes by suppressing proinflammatory cytokines (IL-6 and TNF) and inducing anti-inflammatory cytokine (IL-10) secretion in peripheral blood monocytes. In addition, we found 2 common genetic variants of TOLLIP that are associated with TOLLIP mRNA expression and/or TLR-mediated cytokine release. Finally, these genetic markers of TOLLIP deficiency were associated with an increased risk for developing tuberculosis disease in a case-population study in Vietnam.
Our data from knockdown of TOLLIP in peripheral blood monocytes indicates that TOLLIP is a negative regulator of TNF and IL-6 after PAM2, PAM3, and LPS stimulation. Although these data demonstrate that TOLLIP regulates human TLR2 and TLR4 function, the findings appear to be in contrast to murine models, where Tollip−/− bone marrow-derived macrophages and dendritic cells had decreased LPS-induced IL-6 and TNF in comparison to wild type cells (47). The reasons for this discrepancy are uncertain. It is possible that the unique gene splicing profile of murine TOLLIP led to the proinflammatory profile in the knockout (61) or that cell type or assay conditions were different between the human and murine experiments. Possibly, another set of regulatory molecules may exist apart from TOLLIP that distinguish TLR2 and TLR4 signaling pathways in mice and humans. These species-specific findings underscore the well-recognized and important concept that murine data cannot always be extrapolated and assumed to be similar to humans.
In contrast to the pro-inflammatory cytokines, TLR2 and 4-mediated IL-10 secretion was decreased after TOLLIP knockdown in peripheral blood monocytes. To our knowledge, TOLLIP is the only signal transduction molecule to both downregulate IL-6 and TNF while upregulating IL-10. LPS upregulates IL-10, TNF, and IL-6, in monocytes, presumably by activation of similar transcription factors such as NF-kB (62). However, there are several examples of the IL-10 induction pathway diverting from the IL-6 or TNF induction pathway. For example, IL-10 is induced through the TLR2/TLR4 pathway (63) and modified by CARD9 as well as interferon-beta. (64) In addition, Card9−/− mice are unable to control MTb infection and do not produce significant IL-10, despite IL-6 and TNF production. (65) Furthermore, differential regulation of pro- and anti-inflammatory signaling has been observed in HIV-infected macrophages which upregulate IL-10 and downregulate IL-12. (66) Several possible mechanisms could explain how TOLLIP could differentially regulate pro- and anti-inflammatory cytokine production. For example, the C-terminal domain of TOLLIP contains a Tom-1 binding domain and likely plays a role in cellular trafficking and ubiquitination of proteins which could affect TLR signal transduction. Additionally, TOLLIP can traffic from the cytosol to the cell nucleus via sumoylation, suggesting a role for TOLLIP regulation of transcription factors (67). Although the mechanism by which TOLLIP differentially regulates cytokines is not clear, this finding may help elucidate the signaling pathway of IL-10.
To our knowledge, this is the first description of TOLLIP polymorphisms that are associated with a loss of function as they mimic the phenotype elicited by shRNA knockdown. The mechanism that leads to its loss of function is unclear. We found that minor homozygotes of rs5743899 were associated with a trend of less TOLLIP mRNA in comparison to heterozygotes and major homozygotes, suggesting that differences in TOLLIP expression levels regulate cytokine responses. Our analysis of the case-population data supports this theory, as there is an association between rs5743899 and risk for developing TB in a recessive model. The causative polymorphisms are not known and this complicates interpretation of the data. In addition to regulation of transcription, the possible mechanisms include linkage disequilibrium of the associated SNPs with a non-synonymous coding region functional SNP that changes protein localization or conformation and leads to abnormal TOLLIP function. The C2 domain of TOLLIP contains a phosphorylation domain that is hypothesized to be important for IRAK-4 binding (68). This may affect TLR signaling transduction and lead to the cytokine differences noted in this study. A mutation in this domain is a potential location for a loss-of-function mutation, and in vitro cell culture models have shown that abolishing the phosphorylation domain reduces the regulatory capacity of TOLLIP (46). Further studies are needed to determine the molecular mechanism of action that leads to this phenotype.
In contrast to rs5743899, polymorphism rs3750920 was associated with protection from TB and increased levels of TOLLIP mRNA. The functional change that leads to the association of rs3750920 with tuberculosis is a subject of ongoing study. There are several possible mechanisms that could lead to rs3750920 affecting risk for developing tuberculosis but not IL-6 or TNF cytokine responses in monocytes. First, polymorphisms at rs3750920 may affect cytokines that were not examined during this study, such as IL-1β or IL-18, that could affect inflammasome activity. Second, rs3750920 may affect cytokines that are not expressed in monocytes, such as IL-12p70 or IL-23, that are expressed in much higher quantities in macrophages (69) and dendritic cells. Third, SNP rs3750920 may regulate protein trafficking and functions such as endocytosis and phagocytosis. TOLLIP is known to bind to ubiquitinated proteins, localize with the protein Tom1 to the early endosome, and may be important in bacterial entry (70–72). Finally, TOLLIP polymorphisms may regulate effector mechanisms that lead to bacterial killing (such as NO production). Studies are ongoing to examine these possibilities and to determine the causative SNP in linkage disequilibrium with rs3750920. The genetic and mRNA data from these 2 SNPs likely represent 2 independent effects with distinct mechanisms.
Interestingly, our genetic data suggests that a hypofunctional TOLLIP genotype (rs5743899 GG) is associated with an increased risk of tuberculosis, as well as increased levels of proinflammatory cytokines. There is a long history of observations in the TB field of the potentially deleterious effects of too much inflammation. Steroid treatment of patients with TB meningitis and pericarditis is used to dampen the immunopathologic consequences of inflammation (57, 73). In this regard, TNF has been shown to have protective and deleterious effects in different models of TB. For example, although knockout of TNF in murine models is deleterious with increased MTb replication, evidence also suggests that a hyperinflammatory state can lead to increased tuberculosis replication and worsened disease (74–76). Furthermore, studies of leukotriene A4 hydrolase (LTA4H), an enzyme which regulates eicosonoid and TNF production in zebrafish and humans, suggest that optimal control of MTb requires balanced signaling with deleterious outcomes associated with insufficient or excessive TNF secretion (75). TOLLIP’s regulation of the anti-inflammatory cytokine IL-10 provides an additional mechanism to inhibit inflammatory pathways (77). Card9-deficient mice do not produce any IL-10 and are unable to control MTb infection (65). In addition, IL-10 producing T cells promote anergy to PPD in tuberculosis patients and may alter the T-cell response and overall control of LTBI (34). Together, these data suggest that inhibition of inflammation is partially beneficial for TB clinical outcomes. However, the nuances of how and when to inhibit inflammation during clinical treatment of TB remain poorly understood.
Our study has several potential limitations. The association findings from the case-control study may not be due to polymorphisms within TOLLIP, however this seems unlikely. TOLLIP is fairly isolated on chromosome 11 in humans and is flanked by the mucin 2 precursor and BRSK2, both approximately 100kbp upstream and downstream, respectively. Neither protein has been associated with immune responses. A second limitation of any genetic study relates to multiple comparisons that require adjustment of significance thresholds for P values depending on the number of analyses performed. Even with the most conservative Bonferroni correction of our genetic association data (multiplying the P value by 6), our findings remain highly significant. Furthermore, our experimental data suggests that 2 polymorphisms are genetic markers of TOLLIP function. A third limitation of this study and case-population studies is the misclassification of controls, as some of the cord-blood subjects may develop tuberculosis in the future. Although the use of cord blood samples may lead to a modest loss of power, the misclassification of controls underestimates the genetic risk of polymorphisms. Finally, candidate gene association studies are subject to confounding due to population substructure. However, our study population was the Vietnamese Kinh, a relatively homogenous population in South East Asia. We previously found no evidence of population stratification in this population using control genomic SNPs (59).
Taken together, these data suggest that TOLLIP plays an important role in the pathogenesis of tuberculosis by regulating both pro- and anti-inflammatory pathways. Although future study is required, modulating TOLLIP activity and function may lead to important breakthroughs in tuberculosis vaccine design as well as immune drug development.
Supplementary Material
Acknowledgements
We would like to acknowledge the work of the clinical staff from the Hospital of Tropical Diseases and Pham Ngoc Thach Hospital who initially diagnosed and studied the patients with TBM and PTB. We would like to thank Dr. Nguyen Thi Hieu from Hung Vuong Obstetric Hospital Vietnam, Dr Tran Tinh Hien from the Hospital for Tropical Diseases Vietnam, Dr. Guy Thwaites from Imperial College London and all Vietnamese individuals who participated in this study. We thank Drs. Alan Aderem and Marta Janer from the Institute for Systems Biology for advice and Sarah Li for technical assistance. We thank the following individuals from the University of Washington for their assistance: Glenna Peterson, Rick Wells, Dr. William Berrington, Dr. Ann Misch, Dr. Dave Horne, and Dr. Chetan Seshadri.
Funding: This work was supported by the National Institute of Allergy and Infectious Diseases at the National Institutes of Health [1K24AI089794 to TRH; 5T32AI00704432 to JAS]; the Burroughs Wellcome Foundation [TRH]; and the Wellcome Trust of Great Britain [to JJF].
References
- 1.WHO. Geneva: World Health Organization; 2011. Global tuberculosis control - surveillance, planning, financing. [Google Scholar]
- 2.Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC. Consensus statement. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and Monitoring Project. JAMA. 1999;282:677–686. doi: 10.1001/jama.282.7.677. [DOI] [PubMed] [Google Scholar]
- 3.Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 4.Thuong NTT, Dunstan SJ, Chau TTH, Thorsson V, Simmons CP, Quyen NTH, Thwaites GE, Thi Ngoc Lan N, Hibberd M, Teo YY, Seielstad M, Aderem A, Farrar JJ, Hawn TR. Identification of tuberculosis susceptibility genes with human macrophage gene expression profiles. PLoS Pathog. 2008;4 doi: 10.1371/journal.ppat.1000229. e1000229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thoma-Uszynski S, Stenger S, Takeuchi O, Ochoa MT, Engele M, Sieling PA, Barnes PF, Rollinghoff M, Bolcskei PL, Wagner M, Akira S, Norgard MV, Belisle JT, Godowski PJ, Bloom BR, Modlin RL. Induction of direct antimicrobial activity through mammalian toll-like receptors. Science. 2001;291:1544–1547. doi: 10.1126/science.291.5508.1544. [DOI] [PubMed] [Google Scholar]
- 6.Means TK, Lien E, Yoshimura A, Wang S, Golenbock DT, Fenton MJ. The CD14 ligands lipoarabinomannan and lipopolysaccharide differ in their requirement for Toll-like receptors. J Immunol. 1999;163:6748–6755. [PubMed] [Google Scholar]
- 7.Lien E, Sellati TJ, Yoshimura A, Flo TH, Rawadi G, Finberg RW, Carroll JD, Espevik T, Ingalls RR, Radolf JD, Golenbock DT. Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J Biol Chem. 1999;274:33419–33425. doi: 10.1074/jbc.274.47.33419. [DOI] [PubMed] [Google Scholar]
- 8.Thuong NT, Hawn TR, Thwaites GE, Chau TT, Lan NT, Quy HT, Hieu NT, Aderem A, Hien TT, Farrar JJ, Dunstan SJ. A polymorphism in human TLR2 is associated with increased susceptibility to tuberculous meningitis. Genes Immun. 2007;8:422–428. doi: 10.1038/sj.gene.6364405. [DOI] [PubMed] [Google Scholar]
- 9.Shey MS, Randhawa AK, Bowmaker M, Smith E, Scriba TJ, de Kock M, Mahomed H, Hussey G, Hawn TR, Hanekom WA. Single nucleotide polymorphisms in toll-like receptor 6 are associated with altered lipopeptide- and mycobacteria-induced interleukin-6 secretion. Genes Immun. 2010;11:561–572. doi: 10.1038/gene.2010.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pulido I, Leal M, Genebat M, Pacheco YM, Saez ME, Soriano-Sarabia N. The TLR4 ASP299GLY polymorphism is a risk factor for active tuberculosis in Caucasian HIV-infected patients. Curr HIV Res. 2010;8:253–258. doi: 10.2174/157016210791111052. [DOI] [PubMed] [Google Scholar]
- 11.Davila S, Hibberd ML, Hari Dass R, Wong HE, Sahiratmadja E, Bonnard C, Alisjahbana B, Szeszko JS, Balabanova Y, Drobniewski F, van Crevel R, van de Vosse E, Nejentsev S, Ottenhoff TH, Seielstad M. Genetic association and expression studies indicate a role of toll-like receptor 8 in pulmonary tuberculosis. PLoS Genet. 2008;4 doi: 10.1371/journal.pgen.1000218. e1000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brightbill HD, Libraty DH, Krutzik SR, Yang RB, Belisle JT, Bleharski JR, Maitland M, Norgard MV, Plevy SE, Smale ST, Brennan PJ, Bloom BR, Godowski PJ, Modlin RL. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science. 1999;285:732–736. doi: 10.1126/science.285.5428.732. [DOI] [PubMed] [Google Scholar]
- 13.Means TK, Wang S, Lien E, Yoshimura A, Golenbock DT, Fenton MJ. Human toll-like receptors mediate cellular activation by Mycobacterium tuberculosis. J Immunol. 1999;163:3920–3927. [PubMed] [Google Scholar]
- 14.Underhill DM, Ozinsky A, Smith KD, Aderem A. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc Natl Acad Sci U S A. 1999;96:14459–14463. doi: 10.1073/pnas.96.25.14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scanga CA, Bafica A, Feng CG, Cheever AW, Hieny S, Sher A. MyD88-deficient mice display a profound loss in resistance to Mycobacterium tuberculosis associated with partially impaired Th1 cytokine and nitric oxide synthase 2 expression. Infect Immun. 2004;72:2400–2404. doi: 10.1128/IAI.72.4.2400-2404.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng CG, Scanga CA, Collazo-Custodio CM, Cheever AW, Hieny S, Caspar P, Sher A. Mice lacking myeloid differentiation factor 88 display profound defects in host resistance and immune responses to Mycobacterium avium infection not exhibited by Toll-like receptor 2 (TLR2)- and TLR4-deficient animals. J Immunol. 2003;171:4758–4764. doi: 10.4049/jimmunol.171.9.4758. [DOI] [PubMed] [Google Scholar]
- 17.Fremond CM, Yeremeev V, Nicolle DM, Jacobs M, Quesniaux VF, Ryffel B. Fatal Mycobacterium tuberculosis infection despite adaptive immune response in the absence of MyD88. J Clin Invest. 2004;114:1790–1799. doi: 10.1172/JCI21027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bafica A, Scanga CA, Feng CG, Leifer C, Cheever A, Sher A. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med. 2005;202:1715–1724. doi: 10.1084/jem.20051782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bulut Y, Michelsen KS, Hayrapetian L, Naiki Y, Spallek R, Singh M, Arditi M. Mycobacterium tuberculosis heat shock proteins use diverse Toll-like receptor pathways to activate pro-inflammatory signals. J Biol Chem. 2005;280:20961–20967. doi: 10.1074/jbc.M411379200. [DOI] [PubMed] [Google Scholar]
- 20.Abel B, Thieblemont N, Quesniaux VJ, Brown N, Mpagi J, Miyake K, Bihl F, Ryffel B. Toll-like receptor 4 expression is required to control chronic Mycobacterium tuberculosis infection in mice. J Immunol. 2002;169:3155–3162. doi: 10.4049/jimmunol.169.6.3155. [DOI] [PubMed] [Google Scholar]
- 21.Reiling N, Holscher C, Fehrenbach A, Kroger S, Kirschning CJ, Goyert S, Ehlers S. Cutting edge: Toll-like receptor (TLR)2- and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. J Immunol. 2002;169:3480–3484. doi: 10.4049/jimmunol.169.7.3480. [DOI] [PubMed] [Google Scholar]
- 22.Underhill DM, Ozinsky A, Smith KD, Aderem A. Toll-like receptor-2 mediates mycobacteria-induced pro-inflammatory signaling in macrophages. Proc Nat Acad Sci. 1999;96:14459–14463. doi: 10.1073/pnas.96.25.14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berrington WR, Hawn TR. Mycobacterium tuberculosis, macrophages, and the innate immune response: does common variation matter? In Immunol Rev. 2007:167–186. doi: 10.1111/j.1600-065X.2007.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamath AB, Alt J, Debbabi H, Behar SM. Toll-like receptor 4-defective C3H/HeJ mice are not more susceptible than other C3H substrains to infection with Mycobacterium tuberculosis. Infect Immun. 2003;71:4112–4118. doi: 10.1128/IAI.71.7.4112-4118.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferwerda B, Kibiki GS, Netea MG, Dolmans WM, van der Ven AJ. The toll-like receptor 4 Asp299Gly variant and tuberculosis susceptibility in HIV-infected patients in Tanzania. AIDS. 2007;21:1375–1377. doi: 10.1097/QAD.0b013e32814e6b2d. [DOI] [PubMed] [Google Scholar]
- 26.Newport MJ, Allen A, Awomoyi AA, Dunstan SJ, McKinney E, Marchant A, Sirugo G. The toll-like receptor 4 Asp299Gly variant: no influence on LPS responsiveness or susceptibility to pulmonary tuberculosis in The Gambia. Tuberculosis (Edinb) 2004;84:347–352. doi: 10.1016/j.tube.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 27.Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, Schreiber R, Mak TW, Bloom BR. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 1995;2:561–572. doi: 10.1016/1074-7613(95)90001-2. [DOI] [PubMed] [Google Scholar]
- 28.Redford PS, Murray PJ, O'Garra A. The role of IL-10 in immune regulation during M. tuberculosis infection. Mucosal Immunol. 2011:261–270. doi: 10.1038/mi.2011.7. [DOI] [PubMed] [Google Scholar]
- 29.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 30.Redford PS, Boonstra A, Read S, Pitt J, Graham C, Stavropoulos E, Bancroft GJ, O'Garra A. Enhanced protection to Mycobacterium tuberculosis infection in IL-10-deficient mice is accompanied by early and enhanced Th1 responses in the lung. Eur J Immunol. 2010:2200–2210. doi: 10.1002/eji.201040433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Higgins DM, Sanchez-Campillo J, Rosas-Taraco AG, Lee EJ, Orme IM, Gonzalez-Juarrero M. Lack of IL-10 alters inflammatory and immune responses during pulmonary Mycobacterium tuberculosis infection. Tuberculosis (Edinb) 2009;89:149–157. doi: 10.1016/j.tube.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 32.North RJ. Mice incapable of making IL-4 or IL-10 display normal resistance to infection with Mycobacterium tuberculosis. Clin Exp Immunol. 1998;113:55–58. doi: 10.1046/j.1365-2249.1998.00636.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verbon A, Juffermans N, Van Deventer SJ, Speelman P, Van Deutekom H, Van Der Poll T. Serum concentrations of cytokines in patients with active tuberculosis (TB) and after treatment. Clin Exp Immunol. 1999;115:110–113. doi: 10.1046/j.1365-2249.1999.00783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boussiotis VA, Tsai EY, Yunis EJ, Thim S, Delgado JC, Dascher CC, Berezovskaya A, Rousset D, Reynes JM, Goldfeld AE. IL-10-producing T cells suppress immune responses in anergic tuberculosis patients. J Clin Invest. 2000;105:1317–1325. doi: 10.1172/JCI9918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bellamy R, Ruwende C, Corrah T, McAdam KP, Whittle HC, Hill AV. Assessment of the interleukin 1 gene cluster and other candidate gene polymorphisms in host susceptibility to tuberculosis. Tuber Lung Dis. 1998;79:83–89. doi: 10.1054/tuld.1998.0009. [DOI] [PubMed] [Google Scholar]
- 36.Delgado JC, Baena A, Thim S, Goldfeld AE. Ethnic-specific genetic associations with pulmonary tuberculosis. J Infect Dis. 2002;186:1463–1468. doi: 10.1086/344891. [DOI] [PubMed] [Google Scholar]
- 37.Ates O, Musellim B, Ongen G, Topal-Sarikaya A. Interleukin-10 and tumor necrosis factor-alpha gene polymorphisms in tuberculosis. J Clin Immunol. 2008;28:232–236. doi: 10.1007/s10875-007-9155-2. [DOI] [PubMed] [Google Scholar]
- 38.Shibolet O, Podolsky DK. TLRs in the Gut. IV. Negative regulation of Toll-like receptors and intestinal homeostasis: addition by subtraction. Am J Physiol Gastrointest Liver Physiol. 2007:G1469–G1473. doi: 10.1152/ajpgi.00531.2006. [DOI] [PubMed] [Google Scholar]
- 39.Ostuni R, Zanoni I, Granucci F. Deciphering the complexity of Toll-like receptor signaling. Cell Mol Life Sci. 2010:4109–4134. doi: 10.1007/s00018-010-0464-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liew FY, Xu D, Brint EK, O'Neill LAJ. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 41.Oshima N, Ishihara S, Rumi MAK, Aziz MM, Mishima Y, Kadota C, Moriyama I, Ishimura N, Amano Y, Kinoshita Y. A20 is an early responding negative regulator of Toll-like receptor 5 signalling in intestinal epithelial cells during inflammation. Clin Exp Immunol. 2010:185–198. doi: 10.1111/j.1365-2249.2009.04048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wald D, Qin J, Zhao Z, Qian Y, Naramura M, Tian L, Towne J, Sims JE, Stark GR, Li X. SIGIRR, a negative regulator of Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. 2003;4:920–927. doi: 10.1038/ni968. [DOI] [PubMed] [Google Scholar]
- 43.Kobayashi K, Hernandez LD, Galan JE, Janeway CA, Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 44.Burns K, Clatworthy J, Martin L, Martinon F, Plumpton C, Maschera B, Lewis A, Ray K, Tschopp J, Volpe F. Tollip, a new component of the IL-1RI pathway, links IRAK to the IL-1 receptor. Nat Cell Biol. 2000;2:346–351. doi: 10.1038/35014038. [DOI] [PubMed] [Google Scholar]
- 45.Bulut Y, Faure E, Thomas L, Equils O, Arditi M. Cooperation of Toll-like receptor 2 and 6 for cellular activation by soluble tuberculosis factor and Borrelia burgdorferi outer surface protein A lipoprotein: role of Toll-interacting protein and IL-1 receptor signaling molecules in Toll-like receptor 2 signaling. J Immunol. 2001:987–994. doi: 10.4049/jimmunol.167.2.987. [DOI] [PubMed] [Google Scholar]
- 46.Li T, Hu J, Li L. Characterization of Tollip protein upon Lipopolysaccharide challenge. Mol Immunol. 2004;41:85–92. doi: 10.1016/j.molimm.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 47.Didierlaurent A, Brissoni B, Velin D, Aebi N, Tardivel A, Kaslin E, Sirard JC, Angelov G, Tschopp J, Burns K. Tollip regulates proinflammatory responses to interleukin-1 and lipopolysaccharide. Molecular and Cellular Biology. 2006;26:735–742. doi: 10.1128/MCB.26.3.735-742.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Casanova J-L, Abel L. Genetic Dissection of Immunity to Tuberculosis: The Human Model. Annu. Rev. Immunol. 2002:581–620. doi: 10.1146/annurev.immunol.20.081501.125851. [DOI] [PubMed] [Google Scholar]
- 49.Misch EA, Hawn TR. Toll-like receptor polymorphisms and susceptibility to human disease. Clinical Science. 2008;114:347–360. doi: 10.1042/CS20070214. [DOI] [PubMed] [Google Scholar]
- 50.Caws M, Thwaites G, Dunstan S, Hawn TR, Lan NTN, Thuong NTT, Stepniewska K, Huyen MNT, Bang ND, Loc TH, Gagneux S, van Soolingen D, Kremer K, van der Sande M, Small P, Anh PTH, Chinh NT, Quy HT, Duyen NTH, Tho DQ, Hieu NT, Torok E, Hien TT, Dung NH, Nhu NTQ, Duy PM, Chau NV, Farrar J. The influence of host and bacterial genotype on the development of disseminated disease with Mycobacterium tuberculosis. Plos Pathogens. 2008;4 doi: 10.1371/journal.ppat.1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Misch EA, Hawn TR. Toll-like receptor polymorphisms and susceptibility to human disease. Clin Sci. 2008:347–360. doi: 10.1042/CS20070214. [DOI] [PubMed] [Google Scholar]
- 52.Misch EA, Macdonald M, Ranjit C, Sapkota BR, Wells RD, Siddiqui MR, Kaplan G, Hawn TR. Human TLR1 deficiency is associated with impaired mycobacterial signaling and protection from leprosy reversal reaction. PLoS Negl Trop Dis. 2008:e231. doi: 10.1371/journal.pntd.0000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johnson CM, Lyle EA, Omueti KO, Stepensky VA, Yegin O, Alpsoy E, Hamann L, Schumann RR, Tapping RI. Cutting edge: A common polymorphism impairs cell surface trafficking and functional responses of TLR1 but protects against leprosy. J Immunol. 2007;178:7520–7524. doi: 10.4049/jimmunol.178.12.7520. [DOI] [PubMed] [Google Scholar]
- 54.Uciechowski P, Imhoff H, Lange C, Meyer CG, Browne EN, Kirsten DK, Schroder AK, Schaaf B, Al-Lahham A, Reinert RR, Reiling N, Haase H, Hatzmann A, Fleischer D, Heussen N, Kleines M, Rink L. Susceptibility to tuberculosis is associated with TLR1 polymorphisms resulting in a lack of TLR1 cell surface expression. J Leukoc Biol. 2011;90:377–388. doi: 10.1189/jlb.0409233. [DOI] [PubMed] [Google Scholar]
- 55.Hawn TR, Verbon A, Lettinga KD, Zhao LP, Li SS, Laws RJ, Skerrett SJ, Beutler B, Schroeder L, Nachman A, Ozinsky A, Smith KD, Aderem A. A common dominant TLR5 stop codon polymorphism abolishes flagellin signaling and is associated with susceptibility to legionnaires' disease. J Exp Med. 2003:1563–1572. doi: 10.1084/jem.20031220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Storm N, Darnhofer-Patel B, van den Boom D, Rodi CP. MALDI-TOF mass spectrometry-based SNP genotyping. Methods Mol Biol. 2003;212:241–262. doi: 10.1385/1-59259-327-5:241. [DOI] [PubMed] [Google Scholar]
- 57.Thwaites GE, Nguyen DB, Nguyen HD, Hoang TQ, Do TT, Nguyen TC, Nguyen QH, Nguyen TT, Nguyen NH, Nguyen TN, Nguyen NL, Vu NT, Cao HH, Tran TH, Pham PM, Nguyen TD, Stepniewska K, White NJ, Farrar JJ. Dexamethasone for the treatment of tuberculous meningitis in adolescents and adults. N Engl J Med. 2004;351:1741–1751. doi: 10.1056/NEJMoa040573. [DOI] [PubMed] [Google Scholar]
- 58.Barreiro LB, Neyrolles O, Babb CL, Tailleux L, Quach H, McElreavey K, Helden PD, Hoal EG, Gicquel B, Quintana-Murci L. Promoter Variation in the DC-SIGN-Encoding Gene CD209 Is Associated with Tuberculosis. PLoS Med. 2006;3:e20. doi: 10.1371/journal.pmed.0030020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Horne DJ, RA, Chau TTH, Bang ND, Hieu NT, Farrar JJ, Dunstan SJ, Hawn TR. Common Polymorphisms in the PKP3-SIGIRR-TMEM16J Gene Region Are Associated with Susceptibility to Tuberculosis. Journal of Infectious Diseases. doi: 10.1093/infdis/jir785. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shephard N. GENASS: Stata module to perform Genetic Case-control Association tests. Boston College Department of Economics; 2005. [Google Scholar]
- 61.Lo YLS, Beckhouse AG, Boulus SL, Wells CA. Diversification of TOLLIP isoforms in mouse and man. Mammalian Genome. 2009;20:305–314. doi: 10.1007/s00335-009-9188-3. [DOI] [PubMed] [Google Scholar]
- 62.Higgins SC, Lavelle EC, McCann C, Keogh B, McNeela E, Byrne P, O'Gorman B, Jarnicki A, McGuirk P, Mills KH. Toll-like receptor 4-mediated innate IL-10 activates antigen-specific regulatory T cells and confers resistance to Bordetella pertussis by inhibiting inflammatory pathology. J Immunol. 2003;171:3119–3127. doi: 10.4049/jimmunol.171.6.3119. [DOI] [PubMed] [Google Scholar]
- 63.Saraiva M, O'Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10:170–181. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- 64.Kaiser F, Cook D, Papoutsopoulou S, Rajsbaum R, Wu X, Yang HT, Grant S, Ricciardi-Castagnoli P, Tsichlis PN, Ley SC, O'Garra A. TPL-2 negatively regulates interferon-beta production in macrophages and myeloid dendritic cells. J Exp Med. 2009;206:1863–1871. doi: 10.1084/jem.20091059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dorhoi A, Desel C, Yeremeev V, Pradl L, Brinkmann V, Mollenkopf HJ, Hanke K, Gross O, Ruland J, Kaufmann SH. The adaptor molecule CARD9 is essential for tuberculosis control. J Exp Med. 2010;207:777–792. doi: 10.1084/jem.20090067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yoo J, Chen H, Kraus T, Hirsch D, Polyak S, George I, Sperber K. Altered cytokine production and accessory cell function after HIV-1 infection. J Immunol. 1996;157:1313–1320. [PubMed] [Google Scholar]
- 67.Ciarrocchi A, D'Angelo R, Cordiglieri C, Rispoli A, Santi S, Riccio M, Carone S, Mancia AL, Paci S, Cipollini E, Ambrosetti D, Melli M. Tollip Is a Mediator of Protein Sumoylation. Plos One. 2009;4 doi: 10.1371/journal.pone.0004404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ankem G, Mitra S, Sun F, Moreno AC, Chutvirasakul B, Azurmendi HF, Li L, Capelluto DG. The C2 domain of Tollip, a Toll-like Receptor Signaling Regulator, Exhibits Broad Preference to Phosphoinositides. The Biochemical journal. 2011 doi: 10.1042/BJ20102160. [DOI] [PubMed] [Google Scholar]
- 69.Nau GJ, Schlesinger A, Richmond JF, Young RA. Cumulative Toll-like receptor activation in human macrophages treated with whole bacteria. J Immunol. 2003;170:5203–5209. doi: 10.4049/jimmunol.170.10.5203. [DOI] [PubMed] [Google Scholar]
- 70.Yamakami M, Yokosawa H. Tom1 (target of Myb 1) is a novel negative regulator of interleukin-1- and tumor necrosis factor-induced signaling pathways. Biol Pharm Bull. 2004:564–566. doi: 10.1248/bpb.27.564. [DOI] [PubMed] [Google Scholar]
- 71.Yamakami M, Yoshimori T, Yokosawa H. Tom1, a VHS domain-containing protein, interacts with tollip, ubiquitin, and clathrin. J Biol Chem. 2003;278:52865–52872. doi: 10.1074/jbc.M306740200. [DOI] [PubMed] [Google Scholar]
- 72.Visvikis O, Boyer L, Torrino S, Doye A, Lemonnier M, Lorès P, Rolando M, Flatau G, Mettouchi A, Bouvard D, Veiga E, Gacon G, Cossart P, Lemichez E. Escherichia coli producing CNF1 toxin hijacks Tollip to trigger Rac1-dependent cell invasion. Traffic (Copenhagen, Denmark) 2011 doi: 10.1111/j.1600-0854.2011.01174.x. [DOI] [PubMed] [Google Scholar]
- 73.Strang JI, Kakaza HH, Gibson DG, Girling DJ, Nunn AJ, Fox W. Controlled trial of prednisolone as adjuvant in treatment of tuberculous constrictive pericarditis in Transkei. Lancet. 1987;2:1418–1422. doi: 10.1016/s0140-6736(87)91127-5. [DOI] [PubMed] [Google Scholar]
- 74.Mohan VP, Scanga CA, Yu K, Scott HM, Tanaka KE, Tsang E, Tsai MM, Flynn JL, Chan J. Effects of tumor necrosis factor alpha on host immune response in chronic persistent tuberculosis: possible role for limiting pathology. Infect Immun. 2001;69:1847–1855. doi: 10.1128/IAI.69.3.1847-1855.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tobin DM, JCV, Ray JP, Walsh GS, Dunstan SJ, Bang ND, Hagge DA, Khadge S, King M-C, Hawn TR, Moens CB, Ramakrishnan L. The lta4h Locus Modulates Susceptibility to Mycobacterial Infection in Zebrafish and Humans. Cell. 2010:717–730. doi: 10.1016/j.cell.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Agarwal N, Lamichhane G, Gupta R, Nolan S, Bishai WR. Cyclic AMP intoxication of macrophages by a Mycobacterium tuberculosis adenylate cyclase. Nature. 2009:98–102. doi: 10.1038/nature08123. [DOI] [PubMed] [Google Scholar]
- 77.Saraiva M, O'Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010:170–181. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.