

Abstract
Precisely characterizing the breakpoints of copy number variants (CNVs) is crucial for assessing their functional impact. However, fewer than 0% of known germline CNVs have been mapped to the single-nucleotide level. We characterized the sequence breakpoints from a dataset of all CNVs detected in three unrelated individuals in previous array-based CNV discovery experiments. We used targeted hybridization-based DNA capture and 454 sequencing to sequence 324 CNV breakpoints, including 315 deletions. We observed two major breakpoint signatures: 70% of the deletion breakpoints have 1–30 bp of microhomology, whereas 33% of deletion breakpoints contain 1–367 bp of inserted sequence. The co-occurrence of microhomology and inserted sequence is low (10%), suggesting that there are at least two different mutational mechanisms. Approximately 5% of the breakpoints represent more complex rearrangements, including local microinversions, suggesting a replication-based strand switching mechanism. Despite a rich literature on DNA repair processes, reconstruction of the molecular events generating each of these mutations is not yet possible.
Structural variation in the genome, in the form of deletions, duplications, inversions, insertions and translocations, accounts for much of the difference between human genomes. Assessing the functional impact of this class of variation requires genome-wide maps of variants and reference sets of genotypes in diverse populations. Over the past 5 years, successive studies have reported increasingly large datasets of CNVs. However, only a small minority (<10%) of these has been characterized to base-pair resolution. This is despite the broad utility of this information: base-pair-resolution CNV breakpoints are required to determine the precise functional impact of a CNV, enable the development of new genotyping assays and improve our understanding of the underlying mutational mechanisms. The major barrier to high-resolution characterization of CNV breakpoints has been the lack of a high-throughout technology for breakpoint sequencing. Most known CNV breakpoints derive from genome-wide shotgun sequencing1,2. PCR-based sequencing has been used in some recent studies, but is laborious and requires assumptions about the structure of the underlying variant to enable primer design (for example, that an additional copy is tandemly duplicated in head-to-tail configuration). A recent study took a PCR-based sequencing approach to characterize breakpoints for 270 CNVs in the human genome identified by mapping paired-end 454 sequence reads3.
Our current understanding of CNV mutation processes in eukaryotes is largely based on DNA repair studies conducted on bacteria, yeast, and avian and mammalian somatic cell lines4,5. These have led to the reconstruction of two families of double-strand break (DSB) repair pathways: (i) nonhomologous end joining (NHEJ) and (ii) homology-directed repair (HDR), which includes nonallelic homologous recombination (NAHR) and single-strand annealing (SSA). Each pathway is known to recruit a distinct set of proteins, to have differing repair efficiencies and thus to have different capacities for mutation6,7. The relative contribution of each pathway to both pathogenic and nonpathogenic germline CNVs in humans has not been well characterized.
The mutational mechanism leading to the formation a CNV is typically characterized by examining the sequence context of its break-points. NAHR is thought to require 200 bp of homology8, whereas NHEJ is often associated with small stretches (1–4 bp) of microhomology and can entail the addition of short stretches of nontemplated nucleotides at the site of repair. Other end-joining processes have been suggested more recently, including microhomology-mediated end joining (MMEJ or ‘alternative NHEJ’, which requires microhomology), but their relationships as subclasses or alternatives to NHEJ have not been fully established7,9. The breakpoints of retroposed DNA often contain poly(A) sequence and flanking target site duplications, and variable number of tandem repeat (VNTR) polymorphisms are readily identified from the repetitive structure in the reference sequence10. More recently, analysis of large, complex pathogenic rearrangements has identified a replication-dependent repair of DSBs called microhomology-mediated break-induced replication11 (MMBIR), which is capable of generating complex structures through multiple rounds of template switching. Although these events are associated with microhomologies, the occurrence of templated inverted and/or inserted sequence at the breakpoints cannot be readily explained by NHEJ or MMEJ.
Recently, several studies have used array-based oligonucleotide hybridization and next-generation sequencing technologies to capture and sequence thousands of targeted genomic regions in a single experiment12,13. We hypothesized that DNA fragments containing CNV breakpoints could be captured using arrays targeted to the breakpoint region, allowing us to isolate and sequence many CNV breakpoints without PCR primer design and without requiring assumptions about the underlying structure.
The Genome Structural Variation Consortium recently used genome-wide tiling oligo–comparative genomic hybridization (CGH) experiments to report a genome-wide map of 8,599 validated CNVs in 40 unrelated individuals, as well as reference genotypes for 4,978 of these in 450 individuals from three populations14. Here we report an attempt to sequence breakpoints for all CNVs detected in three of these unrelated individuals. Some CNV breakpoints in these genomes have been sequenced in several previous studies15–17, allowing calibration and validation of our method. We used the CGH intensity data to construct target regions for the pulldown array; DNA fragments were captured and sequenced with ~300-bp reads, which were subsequently mined for CNV breakpoints (Fig. 1), and the sequence context of the breakpoints was analyzed to provide an expanded view of the spectrum of CNV mutation processes.
Figure 1.

Experimental overview. This diagram depicts the three stages of the experiment. First, test (green) and reference (red) DNAs are cohybridized to a CGH array. Second, the intensity data generated from the CGH experiment is summarized at each probe and the distribution of probe intensities is used to identify CNVs using the GADA segmentation algorithm18. The intensity data are then used to construct confidence intervals around each putative CNV breakpoint. A hybridization-based capture array is designed to these confidence intervals. Third, test and reference samples are cohybridized to the capture array. Fragments with at least partial homology to the target regions are preferentially retained and sequenced. Sequence reads are mapped to the genome; reads without CNV breakpoints show contiguous homology to the reference across all bases, whereas reads containing breakpoints appear to be split, with partial homology to either side of the CNV.
RESULTS
Measurement of uncertainty in breakpoint placement
We previously constructed a high-resolution CNV map of 8,599 deletions and duplications in 41 individuals (20 from the CEU HapMap population, 20 from the YRI HapMap population and 1 Polymorphism Discovery Resource individual, NA15510) by applying the GADA segmentation algorithm to intensity data generated from array-CGH experiments with 42 million oligonucleotides tiling the genome14. The GADA algorithm provides a point estimate for the location of each breakpoint of a CNV, but does not provide a confidence interval18. To maximize the efficiency of our array design we developed two distinct but related methods for using the CGH intensity data to estimate a 95% confidence interval on the location of each CNV breakpoint (we refer to these methods as m1 and m2; see Online Methods, Supplementary Fig. 1 and Supplementary Note). We estimated confidence intervals for breakpoints of all 1,174 CNVs detected in the CGH experiment comparing two CEU individuals (NA12878 and NA10851; 2,348 confidence intervals in total) and for breakpoints of all 1,304 CNVs detected in the CGH experiment comparing NA15510 and NA10851 (2,608 confidence intervals), using both m1 and m2. To assess the accuracy of these confidence intervals, we compiled published sequenced breakpoints for 300 of our targeted CNVs present in either NA12878, NA15510 or both (Online Methods). We measured the accuracy of each method using these sequenced CNVs by counting the proportion of confidence intervals containing the true breakpoint location.
The two methods for estimating confidence intervals perform similarly well, with m1 perhaps slightly more efficient in terms of break-points per base pair (Supplementary Note). Each method produced confidence intervals at the combined set of 700 breakpoints that were correlated with the precision of the GADA breakpoint estimates (Fig. 2a,b) and covered the true location of the breakpoint 70% of the time. We evaluated m1, the most efficient confidence interval algorithm, using permutations in which the confidence intervals are randomly assigned to CNVs. The number of true breakpoints covered with the correctly assigned confidence intervals was 13 s.d. greater than the mean from the randomly assigned permutations (Fig. 2c). We found the magnitude of copy number difference between target and reference to be a good predictor of our ability to estimate the true location of a breakpoint (Fig. 2d), whereas the size of the event and the extent of sequence homology at the breakpoints were not good predictors (data not shown).
Figure 2.

Confidence intervals. (a) We used our array CGH data to construct confidence intervals for both the 5′ and 3′ breakpoints of 350 CNVs with published breakpoint sequences. m2 confidence intervals (shown here as 700 horizontal gray lines) are drawn in base pairs 5′ or 3′ (<0 or >0, respectively) from the GADA-estimated breakpoint location. The true location for each sequenced breakpoint is represented as a red dot. There appears to be a strong positive correlation between confidence interval size and the accuracy of the GADA breakpoint estimates, indicating the CGH data contains useful information on the uncertainty in breakpoint location. (b) We confirmed this by modeling the relationship between confidence interval size and the accuracy of our breakpoint estimates. The best-fit line from least-squares regression is shown in red (test of slope = 0, P < 10−15). (c) A permutation test of the hypothesis that our confidence intervals cover more breakpoint locations than expected by chance. As our test statistic, we used the number of true breakpoints covered by a set of confidence intervals. A null distribution for this statistic was generated using 1,000 permutations of m1 confidence intervals across CNVs (shown here as a black curve). The number of true breakpoints covered with the correctly assigned confidence intervals (indicated by a vertical red line) was 13 s.d. greater than the mean from the randomly assigned permutations. (d) The relationship between CNV log2 ratio between test and reference in the discovery CGH experiment and the breakpoint estimation error indicate that GADA breakpoint estimation accuracy decreases as the CNV signal is closer to the background.
Capture and sequencing of targeted regions
We designed a NimbleGen 385k array with oligonucleotides complementary to the 3,712 target regions constructed from the confidence intervals described above. These initial target regions corresponded to 2,049 unique CNVs (>400 bp long), but several factors lowered the number of assayable CNVs. We were able to design oligonucleotide probes for 3,263 (88%) of the target regions, corresponding to 1,785 CNVs (Online Methods). We know that approximately 15% of the targeted CNVs are false positives14, and about 9% of CNVs will not have a target region containing a breakpoint (assuming that the confidence intervals cover a breakpoint 70% of the time, (1 – 0.7)2 = 0.09). Furthermore, 25% of loci are VNTRs, at which we cannot expect to observe unique break-point sequences. Combining these figures and accounting for overlap among categories, we estimate the number of CNVs for which we could potentially sequence a breakpoint to be 1,067.
We combined genomic DNA in equimolar amounts from each of the three genomes containing the CNVs used to construct the target regions, hybridized it to the array, washed the array and eluted captured molecules. This capture eluate was then prepared for 454 sequencing (Online Methods). The data produced from 454 pyrose-quencing were generated as single reads approximately 300 bp long.
Before mapping these sequences, we devised a coding system to comprehensively categorize reads that do not align contiguously to the reference genome and thus potentially span CNV breakpoints. The system may be generally applicable in sequencing-based CNV analyses (Supplementary Note). We term these reads with discontinuous mappings ‘split reads’ if both ends of the reads are mapped to the reference; otherwise they are ‘partially aligned’. To assess the general properties of the sequence reads we generated, we created an exploratory pipeline to map sequence reads with SSAHA2 and to identify and categorize all split reads using the ontology just mentioned (Online Methods, Supplementary Fig. 2 and Supplementary Note). This pipeline can identify any class of split-read mapping to two genomic locations, irrespective of the location or orientation of the two mappings, thus allowing us to identify the full range of possible split reads. Of the 342,406 reads generated, 290,808 nonredundant reads (84.93%) mapped successfully to the genome for a total of 301,112 mappings (Fig. 3a,b). Of these mapped reads, 33% (99,826) were on target, and 91% of target regions contained mapped reads (median of 12 reads per target). A full 13.2% of mappings involved reads that were discontinuously aligned and thus potentially split; 31% of these are reads mapping to two genomic locations and thus are split reads, with the remainder annotated as partial reads. Some of these partial reads might represent breakpoint-spanning split reads in which the breakpoint falls close to the end of the read, preventing robust mapping of sequence from one side of the breakpoint.
Figure 3.

Properties of the pulldown experiment. (a) Distribution of read lengths for all sequences, mapped sequences, and mapped and targeted sequences. (b) Integration of CGH data, confidence intervals and short-read sequencing facilitates rapid identification of CNV breakpoints. Shown here is an overview of the data for a deletion observed twice in the CGH experiment and then successfully recovered by split-read analysis. (c) Power of the pulldown experiment to identify breakpoints for 1,185 validated, non-VNTR loci, plotted as a function of haploid sequence coverage. According to power simulations, the single best predictor of breakpoint sequencing success of non-VNTR loci was sequence coverage of the target region (Pearson R = 0.78). Using the BLAT pipeline, we estimated that our approach has 90% power to sequence a CNV breakpoint when both target regions of the CNV have an average of twofold haploid sequence coverage (Online Methods and Supplementary Methods).
Roughly 60% more deletion-compatible split reads (2,833) were found than duplication-compatible split reads (1,721). To permit the identification of unexpected rearrangement structures, we mined the remaining split reads for patterns indicative of a sequenced breakpoint. Notably, there were 922 split reads whose best mapping was the top strand for one end and the bottom strand (of the same chromosome) for the second end on the same chromosome, and 42 of our targeted regions contained one or more of these. These unexpected ‘between-strand’ mappings could indicate an inversion of sequence with respect to the reference genome; they are discussed in more detail below.
Across all split reads, we identified 194 nonredundant deletions and 3 nonredundant duplications, 142 of these supported by two or more split reads. We also identified 6 potential interchromosomal duplications, one of which has been described previously19 and represents our sole positive control for this class of variant. The repetitive sequence context of these interchromosomal breakpoints makes PCR-based validation difficult.
Power simulations motivate a second mapping approach
Of the total 1,067 assayable CNVs targeted on the capture array, we succeeded in detecting a breakpoint for 205 (19%) during the initial analysis. To explore the reasons for this low yield, we conducted simulations modeling the details of our experiment to assess the variables affecting its power to detect CNV breakpoints (Online Methods, Supplementary Fig. 3 and Supplementary Note). Specifically, we simulated hundreds of breakpoints at random locations within each validated, non-VNTR target region, and we estimated the probability that each CNV breakpoint was, first, captured by a read and, second, correctly identified by SSAHA2, given the number, mapping locations and lengths of reads in the real data and the frequency of the CNV at that locus (estimated by the call frequency in the two CGH experiments used for CNV discovery). We were surprised to see that conditional on simulating a single split read, the SSAHA2 pipeline correctly identified the breakpoint only 14% of the time, with equal power for deletion- or duplication-compatible split reads, implying that many more CNV breakpoints could possibly be recovered with a different mapping approach. We hypothesized that the low power of our approach resulted from the flexibility of the model we were fitting with SSAHA2: allowing the two ends of a read to map in different orientations, on different strands and indeed on different chromosomes requires stringent filtering to avoid spurious hits.
To increase sensitivity to breakpoint structures, we constructed a second mapping pipeline based on the BLAT alignment algorithm (Online Methods). Whereas our implementation of SSAHA2 is able to identify split reads separated by a single gap and having <16 bp of breakpoint microhomology, BLAT can make alignments with multiple gaps and more extensive microhomology; however, the BLAT alignment approach detects only deletion-compatible splits. Using the same simulation framework as before, we found that a BLAT-based pipeline had an average mapping power for deletion-compatible split reads of 57%, nearly four times higher than the SSAHA2 pipeline, but, as expected, no mapping power for duplication-compatible split reads.
With two complementary mapping approaches in hand, we revisited the simulations, this time with the aim of assessing the total power of the experiment and estimating the total number of CNV breakpoint sequences we might expect to obtain with each mapping pipeline (Fig. 3c and Supplementary Fig. 4). These simulations considered a range of mutation models, including deletions and tandem duplications. In total we expected to find 300–320 breakpoints with SSAHA2. The number of breakpoints found by BLAT is a function of the (unknown) proportion of CNVs that are deletions; assuming that all interrogated CNVs were deletions, we would expect to find 590–610 breakpoints with BLAT. In summary, making realistic and conservative modeling assumptions, and given the sequencing coverage we obtained, we found we should expect a minority of CNVs targeted by the experiment to yield sequenced breakpoints.
Satisfied with this exploration of power using simulated data, we turned to the analysis of real data with the BLAT pipeline. We identified 302 deletions with this second pipeline; SSAHA2 detected 177 of these and 22 additional breakpoints, bringing the total number of breakpoints sequenced to 324. In the Discussion we consider various factors that might explain why the yield of breakpoints from the empirical data was lower than the yield from the simulated data.
Characterization of breakpoints
We sequenced breakpoints for a wide distribution of CNV sizes, including CNVs as small as 420 bp and as large as 184 kb. However, our capture was biased toward smaller events: whereas 30% of the CNVs targeted with the array were larger than 10 kb, only 8% of our sequenced breakpoints came from CNVs of that size range. This may reflect the fact that the breakpoints of larger CNVs are more often embedded in complex sequence contexts that lower the sensitivity of this approach. Notably, reads containing duplication breakpoints were far more likely to have multiple high-quality mappings than reads containing deletion breaks, underscoring the role of repetitive sequence in mediating duplications14.
Researchers’ understanding of mutational processes changes over time, and the processes have primarily been studied in model organisms and somatic tissues. Given this, it is important to first describe the phenomenology of germline breakpoint sequences in humans that we directly observed through experimentation, before attempting to ascribe mechanisms to different classes of breakpoints. We assembled and manually curated contigs containing the newly sequenced breakpoints, then characterized the sequence context at each break by ascertaining the extent of homology at the ancestral edges of the break and the number and nature of base pairs inserted in the break (Online Methods, Fig. 4a). These properties clearly delineate four different classes of CNV breakpoint in our data (Supplementary Table 1). Of the 315 sequenced deletion breakpoints, 103 (33%) showed 1–367 bp of inserted sequence at the breakpoints in addition to the deletion. Two hundred and nineteen (70%) of the sequenced deletion breakpoints showed 1–30 bases of homology at the ends, consistent with a microhomology-mediated process such as MMBIR or NHEJ. Only 32 of the deletions with sequence insertion at the breakpoints are flanked by microhomologies; this is significantly less than would be expected if the two signatures arose independently of one another (P < 10−15; χ2 test; Fig. 4b). Moreover, 21 (66%) of these microhomologies are of only a single base and may have occurred by chance. Twenty-five breaks were simple blunt-end joins, with no homology or inserted sequence, which is also less than would be expected if CNV ends were placed at random on the genome (Fig. 4c). We did not observe a correlation between size of deletion and the frequency of the four different breakpoint signatures, nor was there a correlation between deletion size and the number of inserted bases or the length of microhomology (Fig. 4d).
Figure 4.

Summary of sequence content at deletion breaks. (a) Histogram summarizing the number of breakpoints showing blunt ends (red), microhomology (blue) or inserted sequence (red). For each class of breakpoint, events are binned by the number of bases in each feature; in the case of blunt ends, all events are in the same bin of 0 bases. (b) Nonrandom distribution of microhomology observed at deletion breakpoints. We derived an expected distribution of microhomology length by simulating random breakpoints while conditioning on the base content of CNV breakpoint regions. Here we have plotted the difference between the observed and expected amount of microhomology for our deletion breakpoints, which reveals two notable features of our data: (i) there are more deletion breakpoints showing microhomology than expected by chance; (ii) conditional on the presence of microhomology, there is an enrichment of breakpoints with 2–9 bases of microhomology. (c) The presence of inserted sequence within deletion breakpoints is more common in the absence of microhomology (P < 10−15, χ2 test). (d) Each deletion sequenced in the pulldown experiment is represented with a horizontal line. The deletions are parsed by sequence features into three groups: the top group shows no microhomology or inserted sequence, the second group shows at least 1 bp of inserted sequence, represented by a blue line, and the third groups shows at least 1 bp of microhomology at the breakpoints, represented by green lines. CNVs and sequence features are plotted on a log scale, and CNVs are sorted by size within groups.
We aligned (using BLAT) the 22 insert sequences larger than 20 bp against the reference genome to identify their likely origins, which serve as clues to the mutation process that generated them. Five sequences had no clear matches, but the other 17 inserts strongly matched local genomic sequence and appear to result from more complex mutational events. In 13 instances (4.5% of all sequenced deletions), the inserted sequence (39–377 bp) is actually from the other strand nearby in the same chromosome, representing an inversion of local sequence (Fig. 5). We validated the structures of these more complex apparent rearrangements by PCR and capillary sequencing (Supplementary Table 2) to confirm that they were not sequencing artifacts. These findings explain, in part, the between-strand mappings described above and have important implications for understanding structural mutation. In some cases, it is likely that the inverted sequence was formed at the same time as the deletion, an event compatible with a replication-based mechanism involving local template switching, such as MMBIR, as has been described recently20,21,22. Another possibility is that the inversion was present before the deletion, which raises the possibility that the inversion may have had a mutagenic effect. This phenomenon has previously been observed only in the formation of rare, pathogenic rearrangements caused by NAHR23,24, but it could occur more widely. Similarly, we identified small indels within 300 bp of at least seven breakpoints, consistent with emerging evidence that indels may increase local genome instability14,25.
Figure 5.
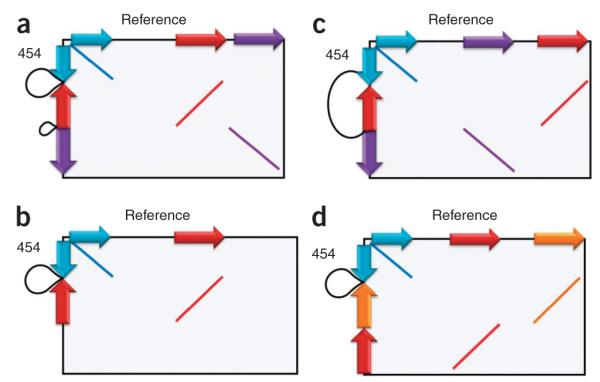
Inverted sequence at complex CNV breakpoints. These schematic homology plots summarize into four classes the 12 cases of deletions with inverted sequence we observed. The plots represent the regions of similarity and orientation of these sequences within the CNV region as if we had plotted a dot plot of the reference (x axis) against the new allelic structure from assembly of the 454 reads (y axis). Sequences inverted within the new allele relative to the reference are colored red and orange; those in the same orientation are blue and purple. The black loops represent the deleted sequence. (a) A deletion plus an inverted sequence originating from within the larger deleted region; n = 8. (b) Deletion plus inverted sequence originating from the local vicinity; n = 2. (c) Deletion plus inverted sequence originating from the local vicinity, but owing to an incomplete assembly it is not clear whether it comes from within or outside the deletion region; n = 2. (d) In a single case, a deletion plus two separate inversions with sequence originating from the local vicinity of the breakpoint.
Several mutation processes are associated with a genomic signature that can be identified without base-pair resolution. The CNVs identified in these three individuals were generated from a CGH platform with 50-bp resolution, which is sufficient to confidently assign CNVs as VNTRs or the products of NAHR between large blocks of homologous sequence (>200 bp). We can therefore combine the results of the sequencing and the array data to provide a more comprehensive overview of the spectrum of mutations forming these array-detectable CNVs in normal individuals (Table 1).
Table 1.
CNV breakpoint signatures
| Signature | Possible mechanism(s) |
Data required | Estimated proportion |
|---|---|---|---|
| >100 bp sequence homology at breakpoints |
NAHR | ~100-bp breakpoint resolution |
10–15% |
| Tandem repeat array in reference sequence |
VNTR | ~100-bp breakpoint resolution |
10–15% |
| Blunt ends | NHEJ, others | Precise sequence | ~5% |
| Insertion of <20 bp non- templated sequence but no microhomology |
NHEJ | Precise sequence | 20–25% |
| Insertion of >20 bp local sequence |
MMBIR | Precise sequence | 5–10% |
| Microhomology (<10 bp) |
NHEJ, MMEJ, MMBIR |
Precise sequence | 40–50% |
| Dispersed duplication | Retrotransposition | Evidence of dispersion |
~1% |
This table combines the analysis of array data described elsewhere14 with analysis of the sequencing data presented here. The information is relevant for CNVs larger than 500 bp outside of the highly repetitive (more than ten matches) regions of the genome.
DISCUSSION
Although we have focused here on the insights into mutational mechanisms that can be gained when CNV breakpoints are mapped to base-pair resolution, there are two other important applications of this knowledge. Mapping CNVs to base-pair resolution allows precise annotation of function, including whether each CNV overlaps functional sequences and the likely the impact on those sequences. In addition, base-pair resolution enables the development of breakpoint-specific genotyping assays, which, by virtue of their qualitative nature, are likely to be more robust than quantitative assays for the same variants and thus more useful in locus-specific population surveys, such as association studies.
Genome-wide resequencing has recently become possible, but the cost still prohibits the ascertainment of CNV breakpoints from a large number of samples. Many fundamental research questions require approaches to sampling that differ from those of large international genome-resequencing projects (such as the 1,000 Genomes Project), including sampling a variety of tissues, individuals or organisms. As the technology matures, targeted resequencing could be the gold standard for validation in CNV studies. Moreover, we have shown that not predicating breakpoint sequencing on prior assumptions of the underlying allelic structure allows complex events to be discerned that may have been missed by PCR-based approaches.
Although we were able to increase the number of sequenced breakpoints by using two mapping pipelines, we did not exhaustively explore all possible mapping strategies. There are likely to be additional breakpoint sequences to be mined from these data, perhaps corresponding to complex rearrangements. The vast majority of events we have identified here are deletions, despite our expectation that at least 20% of targeted events are duplications14. A modified strategy for capturing duplications—by targeting additional sequence within the breakpoints and using de novo assembly of all targeted reads—seems particularly appropriate, considering the enrichment of repetitive contexts at duplication breakpoints14.
Our experimental approach may not have ascertained all classes of CNV. We discovered the target CNVs by array CGH, a platform that is not well suited for identifying polymorphism of extremely high–copy number repeats or heterochromatin. Moreover, breakpoints embedded in repeats much larger than 300 bp cannot be sequenced with the approach used here. In the short term, the most complete picture of mutation processes will come from integrating information from multiple experiments.
Through power simulations, we showed that breakpoints for only a minority of targeted CNVs were likely to be found by this experiment. Nonetheless, substantially fewer breakpoints were recovered than we predicted through simulations. Several properties of real data may account for this. First, we did not simulate our reads with sequencing error, and the assumption of error-free sequencing allows a higher proportion of simulated reads to be mapped with confidence. Second, breakpoint-spanning reads have shorter contiguous matches to the reference genome than unsplit reads, and we did not attempt to model the effect that this lower sequence homology may have on capture efficiency. Third, the several mutation models we considered were only simple models of deletion and duplication; more complex models will presumably lower both the sampling and mapping power. Finally, it is possible that the locations of CNV breakpoints within target regions are biased toward sequences within the target region that have lower probe densities, and thus sampling power is not uniform across the target region.
In a single experiment, we sequenced more CNV breakpoints than have been reported in any previous study, to our knowledge, excepting genome-wide sequencing projects. Until now, the prohibitive cost and effort required to sequence CNV breakpoints has limited the number of events described at base-pair resolution. An analysis of 270 deletion breakpoints found that 40% of the breaks show microhomology and 14% contain small amounts of inserted bases3. A study looking at 227 CNVs larger than 7 kb concluded that 38% of their events were formed by NAHR, 39% by NHEJ and 17.5% by retrotransposition, and 4.5% were VNTRs26. In a screen of structural variants from individuals with lung cancer, 306 germline structural variants were sequenced27. We reanalyzed this dataset, removing 226 inversions and likely transposable element polymorphisms. We found insertion of nontemplated sequence in 22.5% of events and microhomology in 40% of events, but only 7.5% of events showed both signatures; the remainder were blunt ends. In total, these figures accord reasonably closely with what we observed in the present study: microhomology at 70% of deletion breakpoints, inserted sequence in 33%, but just 10% of breaks showing both microhomology and inserted sequence. Thus, in contrast to previous studies that have disagreed over the relative proportions of different breakpoint signatures3,26, once CNVs are detected at high (<3 kb) resolution and obvious differences in ascertainment accounted for, distinct studies agree relatively closely on the proportions of different breakpoint signatures, and thus on the relative contributions of different mutational mechanisms.
There are still hurdles between the generation of copious CNV breakpoint information and the use of that information to make rigorous inferences about germline CNV mutation processes. It cannot be taken for granted that insights derived from experiments on somatic cells (which often have mutations affecting other components of DNA repair) are comprehensive with respect to germline mutation processes. There may be additional mechanisms operating in the germline, and the relative contributions of mutational mechanisms may be different. One example of the former is the developmentally programmed homologous recombination that takes place preferentially at recombination hotspots in the germline, which drives mutation at some VNTR loci28 and can cause NAHR29,30.
The second challenge is to develop a rigorous, statistically driven framework for mapping the breakpoint signatures we observed to the mutation processes that formed them. There are multiple mutational mechanisms that can generate similar breakpoint signatures: for example, MMEJ, MMBIR and NHEJ are all capable of generating deletions with microhomology at the breakpoints. There are thought to be subtle distinctions, however, in the properties of breakpoints produced by NHEJ and MMEJ; for example, MMEJ is thought to require longer stretches of microhomology (>5 bp) than NHEJ (1–4 bp). If these preferences can be precisely characterized, we envisage being able to use statistical analysis of large collections of CNV breakpoints to estimate the relative contributions of different pathways or sub-pathways to in vivo CNV formation. This could be done, for example, by modeling the empirical distribution of microhomology lengths as a mixture of contributions from different pathways.
There is not universal agreement as to whether certain mutational mechanisms are biologically distinct. For example, some view NHEJ and MMEJ as distinct pathways9, whereas others see them as two strands of a more general and flexible NHEJ mechanism7. The phenomenology is static, but researchers’ understanding of mutational mechanisms is dynamic, so the mapping of signatures to mechanisms is subject to change over time. Large amounts of data from targeted experiments, coupled with statistical analyses, should help crystallize these issues and establish population-based studies of CNV mutation as less of a descriptive exercise and more of an inference-based one.
METHODS
Methods and any associated references are available in the online version of the paper at http://www.nature.com/naturegenetics/.
Supplementary Material
ACKNOWLEDGMENTS
This research was funded by Wellcome Trust grant 077014/Z/05/Z. We thank the Wellcome Trust Sanger Institute for informatics and sequencing support, H. Lam for sharing sequenced CNV breakpoint locations and colleagues at NimbleGen for support and discussions.
Footnotes
Note: Supplementary information is available on the Nature Genetics website.
AUTHOR CONTRIBUTIONS D.F.C. and M.E.H. designed the study. L.M. and D.J.T. performed pulldown and sequencing experiments. D.F.C., B.B., C.B., S.L. and M.E.H. analyzed the data. C.B. performed validation experiments. D.F.C. and M.E.H. wrote the paper with contributions from C.B., B.B. and C.L.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/.
References
- 1.Mills RE, et al. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 2006;16:1182–1190. doi: 10.1101/gr.4565806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levy S, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim PM, et al. Analysis of copy number variants and segmental duplications in the human genome: evidence for a change in the process of formation in recent evolutionary history. Genome Res. 2008;18:1865–1874. doi: 10.1101/gr.081422.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wyman C, Kanaar R. DNA double-strand break repair: all’s well that ends well. Annu. Rev. Genet. 2006;40:363–383. doi: 10.1146/annurev.genet.40.110405.090451. [DOI] [PubMed] [Google Scholar]
- 5.Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of change in gene copy number. Nat. Rev. Genet. 2009;10:551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iliakis G, et al. Mechanisms of DNA double strand break repair and chromosome aberration formation. Cytogenet. Genome Res. 2004;104:14–20. doi: 10.1159/000077461. [DOI] [PubMed] [Google Scholar]
- 7.Lieber MR. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- 8.Inoue K, Lupski JR. Molecular mechanisms for genomic disorders. Annu. Rev. Genomics Hum. Genet. 2002;3:199–242. doi: 10.1146/annurev.genom.3.032802.120023. [DOI] [PubMed] [Google Scholar]
- 9.Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okou DT, et al. Microarray-based genomic selection for high-throughput resequencing. Nat. Methods. 2007;4:907–909. doi: 10.1038/nmeth1109. [DOI] [PubMed] [Google Scholar]
- 13.Albert TJ, et al. Direct selection of human genomic loci by microarray hybridization. Nat. Methods. 2007;4:903–905. doi: 10.1038/nmeth1111. [DOI] [PubMed] [Google Scholar]
- 14.Conrad D, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2009 Oct 7; doi: 10.1038/nature08516. advance online publication, doi:10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korbel JO, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Redon R, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tuzun E, et al. Fine-scale structural variation of the human genome. Nat. Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- 18.Pique-Regi R, et al. Sparse representation and Bayesian detection of genome copy number alterations from microarray data. Bioinformatics. 2008;24:309–318. doi: 10.1093/bioinformatics/btm601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong Z, Wilson V, Patel I, Povey S, Jeffreys AJ. Characterization of a panel of highly variable minisatellites cloned from human DNA. Ann. Hum. Genet. 1987;51:269–288. doi: 10.1111/j.1469-1809.1987.tb01062.x. [DOI] [PubMed] [Google Scholar]
- 20.Lee JA, Carvalho CM, Lupski JRA. DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 21.Carvalho CM, et al. Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum. Mol. Genet. 2009;18:2188–2203. doi: 10.1093/hmg/ddp151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang F, et al. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet. 2009;41:849–853. doi: 10.1038/ng.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jobling MA, et al. A selective difference between human Y-chromosomal DNA haplotypes. Curr. Biol. 1998;8:1391–1394. doi: 10.1016/s0960-9822(98)00020-7. [DOI] [PubMed] [Google Scholar]
- 24.Sharp AJ. Emerging themes and new challenges in defining the role of structural variation in human disease. Hum. Mutat. 2009;30:135–144. doi: 10.1002/humu.20843. [DOI] [PubMed] [Google Scholar]
- 25.Tian D, et al. Single-nucleotide mutation rate increases close to insertions/deletions in eukaryotes. Nature. 2008;455:105–108. doi: 10.1038/nature07175. [DOI] [PubMed] [Google Scholar]
- 26.Kidd JM, et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campbell PJ, et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat. Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bois P, Jeffreys AJ. Minisatellite instability and germline mutation. Cell. Mol. Life Sci. 1999;55:1636–1648. doi: 10.1007/s000180050402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lindsay SJ, Khajavi M, Lupski JR, Hurles ME. A chromosomal rearrangement hotspot can be identified from population genetic variation and is coincident with a hotspot for allelic recombination. Am. J. Hum. Genet. 2006;79:890–902. doi: 10.1086/508709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Myers S, Freeman C, Auton A, Donnelly P, McVean G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat. Genet. 2008;40:1124–1129. doi: 10.1038/ng.213. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.