

Abstract
Fused-ring and bridged-ring tetrahydrofuran scaffolds are found in a number of natural products and biologically active compounds. A new copper-catalyzed intramolecular carboetherification of alkenes for the synthesis of bicyclic tetrahydrofurans is reported herein. The reaction involves Cu-catalyzed intramolecular addition of alcohols to unactivated alkenes and subsequent aryl C-H functionalization provides the C-C bond. Mechanistic studies indicate a primary carbon radical intermediate is involved and radical addition to the aryl ring is the likely C-C bond-forming mechanism. Preliminary catalytic enantioselective reactions are promising (up to 75% ee) and provide evidence that copper is involved in the alkene addition step, likely through a cis-oxycupration mechanism. Catalytic enantioselective alkene carboetherification reactions are rare and future development of this new method into a highly enantioselective process is promising. During the course of the mechanistic studies a protocol for alkene hydroetherification was also developed.
Keywords: copper-catalyzed, alkene, alcohol, tetrahydrofuran, carboetherification
INTRODUCTION
Tetrahydrofurans are frequently found in bioactive compounds.1 These structurally complex, oftentimes chiral compounds are isolated from natural products, usually in minute quantities, so de neuvo chemical synthesis of such tetrahydrofurans has been actively pursued.2 Methods for tetrahydrofuran synthesis include intramolecular SN2 reactions involving alcohols and leaving groups, [3+2] dipolar cycloadditions, addition of alcohols to electrophile-activated alkenes, addition of nucleophiles to Lewis acid-activated lactol derivatives and hydrogenation of furans.2,3 This article reports a new method for the formation of fused-ring and bridged-ring bicyclic tetrahydrofurans via a copper-catalyzed doubly intramolecular alkene carboetherification/C-H functionalization of aryl-substituted γ-pentenols (Scheme 1). Fused-ring and bridged-ring tetrahydrofurans analogous to the ones described in this report can be found in a number of terpenoid natural products including those that possess antibiotic and anti-HIV activity (Figure 1).4
Scheme 1.
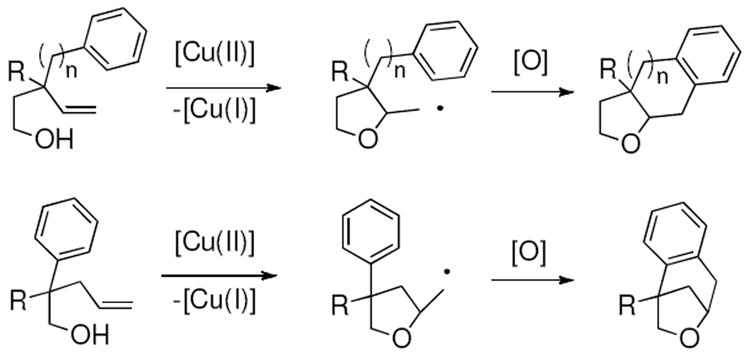
Copper-facilitated alkene carboetherifications
Figure 1.
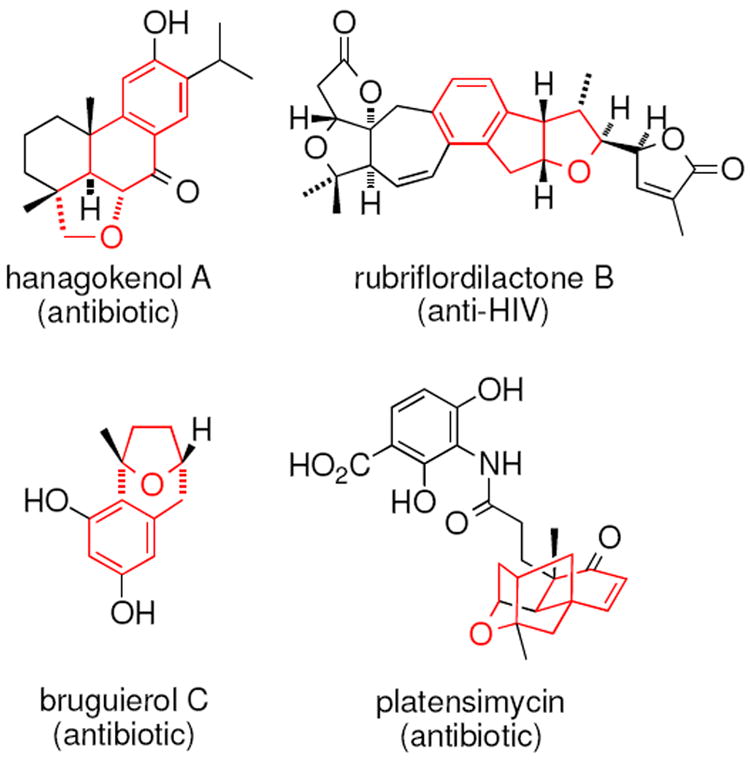
Bioactive fused-ring and bridged-ring tetrahydrofurans
Previous syntheses of fused and bridged bicyclic tetrahydrofurans include tetrahydrofuran formation followed by intramolecular electrophilic aromatic substitution,5 [4+3] annulations,6 [3+2] annulations,7 [5+2] annulations,8 ring-expanding reactions9 and palladium-catalyzed doubly intramolecular alkene carboetherification/C-H functionalization.10 While these methods are synthetic advances, most are limited in scope to either fused-ring or bridged-ring tetrahydrofuran synthesis and few are enantioselective. The method reported herein is general for both fused- and bridged-ring synthesis and the potential for development of an efficient catalytic enantioselective protocol is promising (vide infra).
The synthesis of tetrahydrofurans via transition metal catalyzed carboetherification, addition of alcohols and carbon units to alkenes, represents an efficient transformation of two very common functional groups.3a,d Transition metal catalyzed alkene carboetherification has been developed most extensively with palladium catalysts10,11 and to a lesser extent with gold catalysts.12 Similar transformations have also been reported to occur under cobalt catalysis where carbon radical intermediates are thought to be involved.13
Few catalytic asymmetric examples of carboetherification reactions of unactivated alkenes have been reported and all of them occur under palladium catalysis.3a,14 A common limitation of these methods is the potential for β-hydride elimination which is either built into the method14c,d or avoided by use of 1,1-disubstituted alkene substrates.14a The racemic Pd-catalyzed reactions most analogous to the reactions reported herein also appear to require use of 1,1-disubstituted alkene substrates.10 Herein is reported the first copper-catalyzed intramolecular alkene carboetherification. Notably, β-hydride elimination does not occur in this reaction. This reaction likely occurs via a novel cis-oxycupration and prognosis for its development into a catalytic enantioselective protocol is promising (vide infra).
There are few reports of Cu-catalyzed additions of alcohols to electronically unactivated alkenes. To date, such reactions are limited to alkene hydration and the role of Cu in these reactions is primarily as a controlled source of triflic acid [e.g. from Cu(OTf)2].15 In the presence of PhI(OAc)2, Cu(OTf)2 has also been shown to catalyze the diacetoxylation of alkenes.16 A Cu-catalyzed intermolecular addition of alkenes and anhydrides that forms γ-lactones (a net carboetherification) has recently been reported, but is quite different with respect to substrates and likely mechanism than the one reported herein.17 Likewise, Cu-promoted cyclization-acetoxylation of alkenes18 and Cu-catalyzed oxyalkylation of vinyl arenes19 have been reported but these reaction involve initial addition of carbon radicals to alkenes.
Our lab has reported the copper-catalyzed intramolecular addition of a variety of amines and carbon moieties across alkenes (carboamination) for the synthesis of pyrrolidines and to a lesser extent piperidines.20 Based on the success of these substrates, we turned our attention to γ-pentenyl alcohols to ascertain if Cu(II) could promote intramolecular addition of an alcohol to an unactivated alkene. Specifically, we designed a doubly intramolecular carboetherification wherein a projected carbon radical intermediate could add to a nearby aryl ring, thereby resulting in a fused-ring or bridged-ring tetrahydrofuran (vide supra, Scheme 1).
RESULTS AND DISCUSSION
2.1 General reaction conditions and optimization
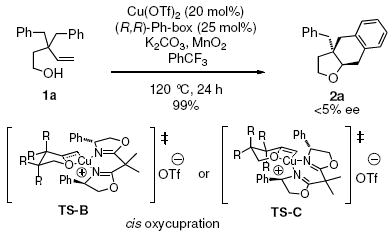
The intramolecular carboetherification of 3,3-dibenzyl-4-penten-1-ol (1a) was evaluated using a number of Cu(II) salts, ligands and reaction conditions (Table 1). The cis-fused tetrahydronaphthofuran 2a was obtained with high diastereoselectivity and variable yield depending upon the reaction conditions. We first established the feasibility of the reaction by treatment of 1a with excess of Cu(2-ethylhexanoate)2 [Cu(eh)2, 300 mol %] as the copper promoter (Table 1, entry 1). Lowering the amount of Cu(eh)2 to 150 and 100 mol % using either K2CO3 or n-BuLi as base led to roughly 50% conversion (Table 1, entries 2 and 3). While this reaction (Table 1, entry 1) was efficient in providing the cyclized adduct 2a, we sought a catalytic method. Use of MnO2 (3 equiv) as stoichiometric oxidant and Cu(eh)2 (20 mol%) as catalyst provided only 33% conversion (Table 1, entry 4). Changing to 2,2’-bipyridine-complexed Cu(OTf)2 (20 mol%) provided only a trace amount of 2a (Table 1, entry 6). [Use of Cu(OTf)2 without additional ligand also gave trace 2a (not shown).] Fortunately bis(oxazoline) 3 complexed to Cu(OTf)2 (20 mol%) provided 98% yield of 2a using MnO2 (3 equiv) as oxidant at 120 °C in CF3Ph (Table 1, entry 7).
Table 1.
Optimization of reaction conditionsa
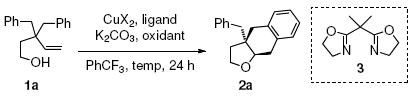
| |||||
|---|---|---|---|---|---|
| entry | CuX2 (mol%) | ligand (mol %) | oxidantb | temp (°C) | yield (%)b |
| 1 | Cu(eh)2 (300) | -- | -- | 120 | >95c |
| 2 | Cu(eh)2 (150) | -- | -- | 120 | 55c |
| 3d | Cu(eh)2 (100) | -- | -- | 120 | 46 |
| 4e | Cu(eh)2 (20) | -- | MnO2 | 120 | 33c |
| 5f | Cu(eh)2 (300) | -- | -- | 120 | 82c |
| 6e | Cu(OTf)2 (20) | Bipy (25) | MnO2 | 120 | trace |
| 7e | Cu(OTf)2 (20) | 3 (25) | MnO2 | 120 | 98 |
| 8e | Cu(OTf)2 (20) | 3 (25) | MnO2 | 100 | 98 |
| 9e | Cu(OTf)2 (20) | 3 (25) | MnO2 | 90 | 66 c |
| 10e | Cu(OTf)2 (15) | 3 (19) | MnO2 | 120 | 78 |
| 11 | Cu(OTf)2 (20) | 3 (25) | O2 (1 atm) | 100 | 20c |
| 12e,f | Cu(OTf)2 (20) | 3 (25) | MnO2 | 100 | 95 |
| 13f | Cu(OTf)2 (20) | 3 (25) | O2 (1 atm) | 100 | 20c |
| 14 | Cu(eh)2 (20) | 3 (25) | MnO2 | 100 | 0c |
| 15g | Cu(OTf)2 (20) | 3 (25) | MnO2 | 100 | 80c |
Reactions were run on 0.188 mmol scale of 1a in PhCF3 (0.1 M with respect to 1a) with K2CO3 (1 equiv), CuX2 and the designated ligand in a sealed pressure tube. For reactions using Cu(OTf)2, the Cu(OTf)2 was complexed with the ligand in CF3Ph for 1 h at 60 °C prior to substrate addition.
Yield of isolated product (>20:1 dr).
Conversion (%) based on crude 1H NMR.
Alcohol 1a was treated with n-BuLi (1.1 equiv) at 0 °C, the resulting solution was stirred at rt for 15 min, then Cu(eh)2 was added and the mixture was stirred at rt for 1 h prior to heating at 120 °C for 24 h.
300 mol% MnO2 was used.
No K2CO3 was added in this reaction.
Toluene was used instead of PhCF3. The Cu catalyst appeared less soluble in this solvent. Cu(eh)2 = Cu(2-ethylhexanoate)2, Bipy = 2,2’-bipyridine.
The reaction temperature could be lowered to 100 °C without affecting the yield (Table 1, entry 8), but lower temperature and lower catalyst loading gave lower yields (Table 1, entries 9 and 10). Oxygen (1 atm, balloon) was not an effective oxidant (Table 1, entry 11). It is also noteworthy that K2CO3 as base is not necessary for reaction to occur but provides a slightly higher yield in the reaction with Cu(eh)2 (Table 1, entries 1 and 5). K2CO3 in the Cu(OTf)2•bis(oxazoline) catalyzed reactions does not appear necessary (Table 1, compare entries 8 and 12) but was generally used in subsequent reactions for simplicity. Use of oxygen as oxidant was also attempted under neutral (no K2CO3) conditions but again, no turnover occurred (Table 1, entry 13). When Cu(eh)2 was used in conjunction with ligand 3 under the otherwise optimal reaction conditions (Table 1, entry 8) no conversion to 2a was observed, indicating the Cu(OTf)2•bis(oxazoline) catalyst is more active (Table 1, entry 14). Finally, use of the more common solvent, toluene, was investigated. The desired product was indeed obtained (Table 1, entry 15) but the conversion was lower (80%), possible due to lower solubility of the copper catalyst in this solvent.
2.2 Scope of fused-ring synthesis
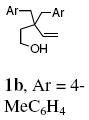
Variously substituted 3,3-dibenzyl-4-penten-1-ols were submitted to the optimized reaction conditions (Table 1, entry 8). While the electron-rich p-Me, p-OMe and p-SMe substrates provided excellent yield under these conditions (Table 2, entries 1-3), the more electron-deficient p-Br, p-Cl, p-F and p-CF3-substituted substrates required heating to 110 or 120 °C for complete conversion (Table 2, entries 4-7). All of the reactions of the 3,3-disubstituted-4-pentenols were completely selective for the cis-fused diastereomer. The cyclization reaction forms two adjacent stereocenters where one is a quaternary carbon.
Table 2.
Scope of fused-ring tetrahydrofuran synthesis via alkene carboetherificationa
| entry | substrate | product(s) (ratio)b | yield (%)c |
|---|---|---|---|
| 1 |
|
|
94 |
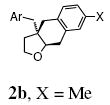
| 2 | 1c, Ar = 4-MeOC6H4 | 2c, X = OMe | 94 |
| 3 | 1d, Ar = 4-MeSC6H4 | 2d, X = SMe | 88 |
| 4d | 1e, Ar = 4-BrC6H4 | 2e, X = Br | 88 |
| 5e | 1f, Ar = 4-ClC6H4 | 2f, X = Cl | 85 |
| 6e | 1g, Ar = 4-FC6H4 | 2g, X = F | 72 |
| 7d | 1h, Ar = 4-CF3C6H4 | 2h, X = CF3 | 90 |
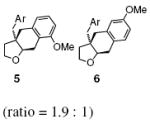
| 8 | 4, Ar = 3-MeOC6H4 |
|
98 |
| 9 | 7a, Ar = 2-MeOC6H4 |
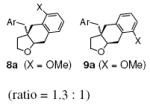
|
96 |
| 10 | 7b, Ar = 2-MeC6H4 | 8b, 9b, X = Me (ratio = 4.5 : 1) | 94 |
| 11e |
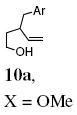
|
|
78 |
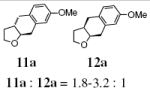
| 12f | 10a | 11a : 12a = 5-6.3 : 1 | 80 |
| 13f | 10b, X = SMe | 11b : 12b = 4-5 : 1 | 67 |
| 14 f | 10c, X = CH3 | 11c : 12c = 4 : 1 | 56 |
| 15e |
|
|
66 |
Reactions were run at 100 °C in CF3Ph with Cu(OTf)2 (20 mol%) complexed with ligand 3 (25 mol%) in the presence of K2CO3 (1 equiv), MnO2 (300 mol%) for 24 h.
Ratio determined by analysis of the 1H NMR spectra of the crude and flash chromatographed product mixtures.
Isolated yield after flash chromatography on SiO2.
Reaction was run at 110 °C.
Reaction was run at 120 °C.
Reaction was catalyzed by Cu(eh)2 instead of Cu(OTf)2•3 and was run at 140 °C. All reactions were performed in duplicate.
Regioisomers were formed in cases where substrates had ortho and meta-aryl substituents. The m-MeO substrate 4 produced a 1.9 : 1 mixture of ortho : para substituted regioisomers 5 and 6, respectively (Table 2, entry 8). This regioselectivity is consistent with addition of a carbon radical to an aryl ring (vide infra).20a,b,f,21
The o-Me and o-MeO-substituted substrates 7a and 7b provided regioisomeric mixtures as well, where in products 9a and 9b, the substituent had apparently moved over to the meta position (Table 2, entries 9 and 10). This is consistent with ipso addition of a carbon radical to the aryl ring followed by alkyl migration/rearrangement to provide either product 8 or 9, depending on which group migrates (Scheme 2).20f
Scheme 2.
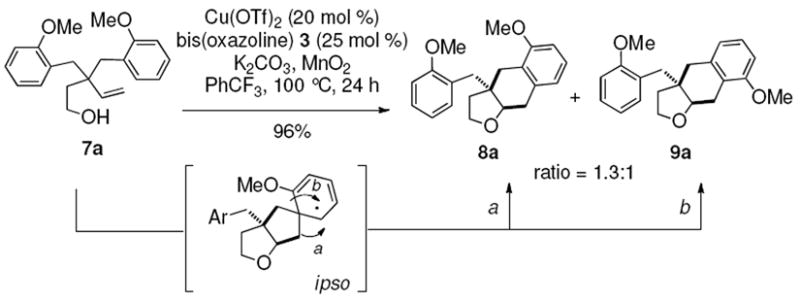
Rationale for regioisomer formation
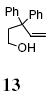
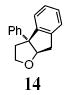
Reaction of the mono-γ-substituted pentenol 10a required heating to 120 °C and favored formation of the trans-fused diastereomer 11a with modest selectivity under the Cu(OTf)2•3 catalyzed conditions (Table 2, entry 11). The diastereoselectivity of the reaction could be improved with use of Cu(eh)2 (20 mol%) as catalyst even though higher temperature (140 °C) was required for good conversion (Table 2, entry 12). Two other mono-benzyl substrates provided analogous results (Table 2, entries 13 and 14). 3,3-Diphenyl-4-penten-1-ol 13 provided the indanofuran 14 in 66% yield upon Cu-catalyzed carboetherification (Table 2, entry 15). The mono-phenyl analog of 13, 3-phenyl-4-penten-1-ol, however, gave modest conversion (64%) of a 2:1 mixture of carboetherification and hydroetherification products even with higher Cu(OTf)2•3 loading (30 mol%) and longer reaction time (48 h) (see Supporting Information for details).
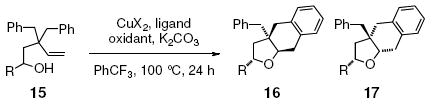
(±)-1-Phenyl and 1-allyl-3,3-dibenzyl-4-penten-1-ols 15 underwent the Cu(OTf)2•bis(oxazoline) 3 catalyzed reactions, providing moderate diastereoselectivity for the 2,5-trans-tetrahydrofuran diastereomers 16 (Table 3, entries 1 and 5). The diastereoselectivity was improved when Cu(2-ethylhexanoate)2 was used to promote the reactions (Table 3, entries 2 and 6). The diastereoselectivity further improved when the catalytic conditions with Cu(eh)2 as catalyst were used (Table 3, entries 3, 4 and 7-9). Although we have previously observed that choice of Cu(II) ligands can affect diastereoselectivity of related alkene aminooxygenation reactions,22 the oxidant contribution to diastereoselectivity was unexpected. Addition of ligand 3 to the Cu(eh)2 catalyzed reaction had little effect on the reaction (compare entries 3 and 4, Table 3).
Table 3.
Reactions of 1-substituted-3,3-dibenzyl-3-pentenolsa
| |||||
|---|---|---|---|---|---|
| entry | substrate | CuX2•ligand | oxidant | yield (%)b | drc |
| 1 | 15a, R = Ph | Cu(OTf)2•3 | MnO2 | 98 | 2.6:1 |
| 2d | 15a | Cu(eh)2 | -- | 86 | 12:1 |
| 3 | 15a | Cu(eh)2•3 | MnO2 | 81 | >20:1 |
| 4 | 15a | Cu(eh)2 | MnO2 | 79 | >20:1 |
| 5 | 15b, R = Allyl | Cu(OTf)2•3 | MnO2 | 98 | 2.5:1 |
| 6d | 15b | Cu(eh)2 | -- | 87 | 5.5:1 |
| 7 | 15b | Cu(eh)2•3 | MnO2 | 44 | >20:1 |
| 8 | 15b | Cu(eh)2 | MnO2 | 50 | >20:1 |
| 9e | 15b | Cu(eh)2 | MnO2 | 95 | >20:1 |
Same general reaction conditions as Table 2 unless otherwise noted.
Isolated yield after flash chromatography on SiO2.
Ratio determined by analysis of the 1H NMR spectra of the crude reaction mixture.
Reaction run with Cu(eh)2 (300 mol%).
Reaction run at 120 °C for 48 h.
2.3 Bridged-ring tetrahydrofuran synthesis
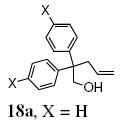
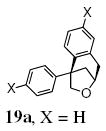
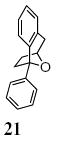
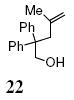
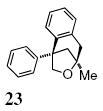
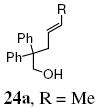
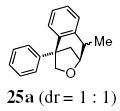
A transannular carboetherification for the formation of oxabicyclo[3.2.1]octanes was next explored (Table 4). These are rare examples of transannular additions of carbon radicals to arenes (vide infra). These reactions occurred best at 120 °C using the Cu(OTf)2•bis(oxazoline) 3 catalyzed reaction conditions. The oxabicyclo[3.2.1]octane 21 formed in entry 6 resembles the natural product bruguierol (Figure 1). We also examined the reactivity of substrates with higher alkene substitution in this carboetherification reaction (Table 4, entries 7-9). 2,2-Diphenyl-4-methyl-4-penten-1-ol 22 underwent the oxidative cyclization to afford the 2-methyl-oxabicyclo[3.2.1]octane 23 in 87% yield. (E)-2,2-diphenyl-4-hexen-1-ol 24a also underwent the cyclization efficiently, producing adducts 25a as a 1:1 diastereomeric mixture. This result was surprising as we have previously observed poor reactivity with internal alkene substrates in copper-promoted carboamination reactions.20b Substrate 24b, with phenyl substitution on the terminal carbon of the alkene, reacted to give a complex mixture that included aldehyde peaks in the crude 1H NMR (Table 4, entry 9).
Table 4.
Synthesis of bridged bicyclic tetrahydrofuransa
| entry | substrate | product(s) | yield (%)b |
|---|---|---|---|
| 1 |
|
|
93 |
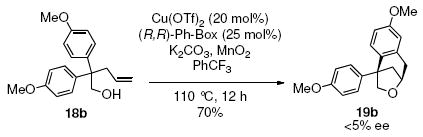
| 2 | 18b, X = OMe | 19b, X = OMe | 91 |
| 3 | 18c, X = SMe | 19c, X = SMe | 87 |
| 4 | 18d, X = Cl | 19d, X = Cl | 93 |
| 5 | 18e, X = F | 19e, X = F | 92 |
| 6c |
|
|
67 |
| 7 |
|
|
87 |
| 8 |
|
|
81 |
| 9 | 24b, R = Ph | complex mixture | -- |
Reactions were run at 120 °C in CF3Ph (0.1 M with respect to substrate) with Cu(OTf)2 (20 mol%) complexed with ligand 3 (25 mol%) in the presence of K2CO3 (1 equiv), MnO2 (300 mol%) for 12 h unless otherwise noted.
Isolated yield after flash chromatography on SiO2.
18% of starting alcohol 20 was recovered.
2.4 Catalytic enantioselective carboetherification
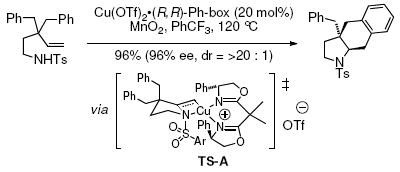
Development of an enantioselective carboetherification protocol was next explored. The [Cu(R,R)-Ph-box](OTf)2 catalyst was explored due to our previous success in rendering the copper-catalyzed alkene carboamination reaction enantioselective (Eq 1).20f Unfortunately γ-alkenyl alcohols 1a and 18b provided racemic carboetherification products using this catalyst (Eqs 2 and 3).
We believe the enantioselectivity in the carboamination reaction (Eq. 1) is due in large part to the steric interaction between the substrate’s N-tosyl substituent and the phenyl group on the ligand’s oxazoline ring (e.g., TS-A). Assuming an analogous cyclic cis-oxycupration transition state (e.g. TS-B) in the carboetherification reaction, this steric interaction is not present, which could contribute to loss of enantioselectivity (the enantiomeric TS-C may not be energetically very different from TS-B).
 |
(1) |
 |
(2) |
 |
(3) |
We reasoned that increasing the steric demands of the substrate near the alcohol region could render a cyclic oxycupration step more enantioselective by increasing steric interactions between the substrate and ligand. Thus, substrates with mono and disubstitution at the hydroxyl-bearing carbon were examined (Table 5). While 1-phenyl-3,3-dibenzyl-4-pentenol 15a gave high diastereoselectivity for 16 with Cu(eh)2•(R,R)-Ph-box as catalyst, the enantioselectivity was <5%. Changing to the [Cu(R,R)-Ph-box](OTf)2 catalyst, we obtained a 2:1 diastereomeric ratio of 16a to 17a where 16a was formed in 30% ee and 17a was formed in 67% ee. This reaction is likely a kinetic resolution of the racemic alcohol substrate (absolute configurations tentatively assigned as shown, vide infra). The analogous 1-phenyl-3,3-di-(p-chlorobenzyl)-4-pentenol 15c reacted with similar selectivity (Table 5, entry 3). The 1,1-diphenyl-3,3-dibenzyl-4-pentenol 15d provided one carboetherification diastereomer in ca. 75% ee (conservative estimate based on incompletely resolved enantiomers) and its 1,1-diphenyl-3,3-di-(p-chlorobenzyl)-4-pentenol derivative (15e) gave a product with a comparable optical rotation (see supporting information) but the enantiomers could not be separated for %ee determination. The data in Table 5 clearly indicate that development of a catalytic enantioselective copper-catalyzed alkene carboetherification is feasible. Mechanistically, the enantioselective reactions provide strong support for the presence of Cu in the alkene addition step.
Table 5.
Catalytic asymmetric carboetherificationsa
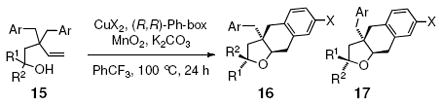
| |||||
|---|---|---|---|---|---|
| entry | substrate | CuX2 | yield (%) | dr (16:17)b | ee (%)c |
| 1 | 15a, R1 = Ph, R2 = H, Ar = Ph | Cu(eh)2 | 62 | >20:1 | <5 |
| 2 | 15a | Cu(OTf)2 | 89 | 2:1 | 30, 67 |
| 3 | 15c, R1 = Ph, R2 = H, Ar = 4-Cl-C6H4 | Cu(OTf)2 | 77 | 2:1 | 32, 60 |
| 4 | 15d, R1 =Ph, R2 = Ph, Ar = Ph | Cu(OTf)2 | 89 | -- | ca 75% |
| 5 | 15e, R1 = Ph, R2 = Ph, Ar = 4-Cl-C6H4 | Cu(OTf)2 | 85 | -- | nd |
Reaction conditions: CuX2 (0.376 mmol) and (R,R)-Ph-box (0.0470 mmol) were combined in PhCF3 (0.6 mL) and heated in a sealed tube for 2 h. Alcohol 15 (0.188 mmol) in 0.9 mL PhCF3, K2CO3, (0.188 mmol) and MnO2 (0.564 mmol) were added and the reaction was stirred at 100 °C in a sealed tube for 24 h.
Substrates are racemic or achiral.
Isolated yield after flash chromatography on SiO2.
Ratio determined by analysis of the 1H NMR spectra of the crude reaction mixture. c Enantiomeric excess determined by chiral HPLC. Nd = not determined. Enantiomers could not be separated on chiral HPLC or gc.
2.5 Absolute stereochemistry assignment
X-ray quality crystals of enantiomerically enriched heavy atom (Cl)-containing carboetherification products could not be obtained, unfortunately. We have tentatively assigned the absolute stereochemistry of the products as shown in Table 5 and equations 4 and 5 (vide infra) by analogy to the enantioselective carboamination products (e.g. Eq. 1 and see optical rotation correlation discussion in the Supporting Information for 21, vide infra).20f
2.6 Mechanistic probes
We further explored the enantioselective variant of the transannular carboetherification of γ-pentenyl alcohol 20. Although the reaction did not go to completion at 100 °C, the bicyclic furan 21 was obtained in 50% ee (Scheme 3). We next introduced 1,4-cyclohexadiene to the reaction and obtained tetrahydrofuran 26 in 85% yield (along with 10% unreacted alcohol 20, Scheme 3). Tetrahydrofuran 26 was obtained in 76% enantiomeric excess. Taken together these reactions indicate (1) a carbon radical, one that can perform H-atom abstraction, is a likely intermediate in the reaction, (2) the H-atom abstraction is faster than transannular C-C bond formation and (3) the carboetherification reaction may suffer some ring-opening/unselective ring-closing prior to C-C bond formation which could account for its diminished enantioselectivity relative to the apparently less reversible alkene hydroetherification involving H-atom abstraction. One potential reversibility scenario is depicted in Scheme 3. This reversibility may be less prominent when addition to a more proximal aryl ring, e.g. as in Table 5, occurs. Formation of the favored cis-fused products, e.g. 16 and 17, may be faster than furan ring opening. In the case of α-substituted substrates 15a and 15c-e, selectivity can be as high as 75% ee, which is comparable to the hydroetherification reaction (Scheme 3).
Scheme 3.
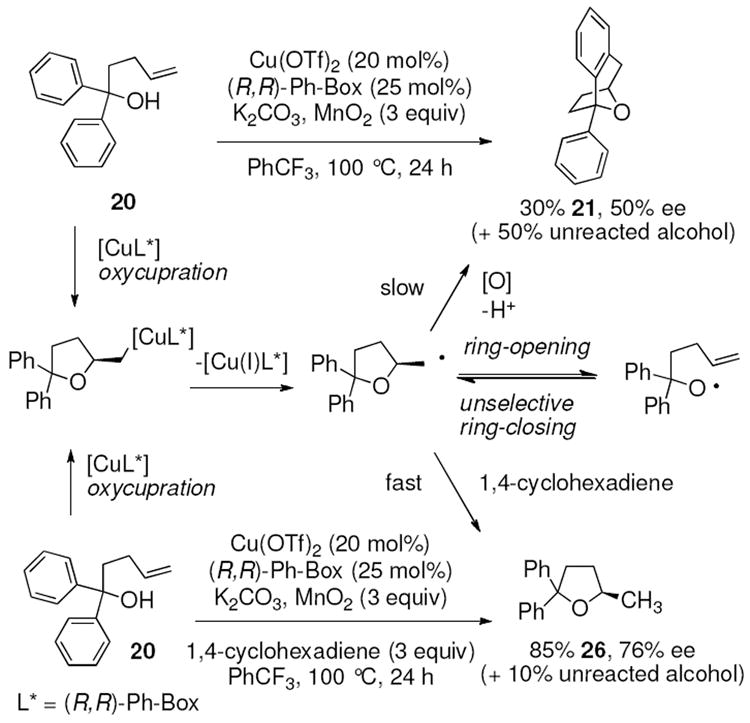
Enantioselective transannular carboetherification and hydroetherification
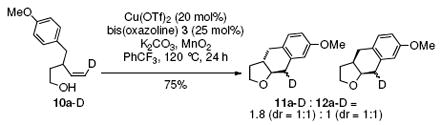
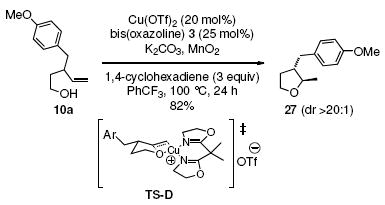
The intermediacy of an sp2-hybridized carbon intermediate (e.g. a carbon radical, see Schemes 1-3, was further probed by the reaction of the isotopically labeled 3-p-methoxybenzyl-4-penten-1-ol (10a-D). Carboetherification provided a 1.8:1 ratio of trans-fused 11a-D and cis-fused 12a-D, both of which were composed of a 1:1 diastereomeric mixture at the deuterated carbon (Eq. 4). The presence of the carbon radical was further confirmed by running the reaction in the presence of 1,4-cyclohexadiene (Eq. 5). This hydroetherification reaction was highly diastereoselective and produced the 2,3-trans-tetrahydrofuran 27 in 82% yield. No carboetherification products were formed, indicating the H-atom transfer process is faster. This relative rate data, in conjunction with the much higher trans-diastereoselectivity for the hydroetherification, seems to indicate again that addition to the alkene is less reversible in the hydroetherification reaction. The 2,3-trans kinetic selectivity can be rationalized by TS-D, which places the allylic substituent in a pseudo-equatorial position in the cyclization transition state. The 2,3-trans diastereomer should experience less sterics in the H-atom abstraction step than its corresponding 2,3-cis diastereomer, which can contribute to a highly trans-selective hydroetherification (Eq. 5). Conversely, the C-C bond-forming step of the corresponding carboetherification reaction (radical addition to arene after O-C bond formation) in Eq. 4 should be more facile for the minor 2,3-cis carbon radical intermediate that leads to formation of the cis-fused diastereomer 12a-D, than for the major 2,3-trans carbon radical to form the trans-fused diastereomer 11a-D. The preference for cis-fused cyclization is clear in the highly selective conversion of 1 to 2 (vide supra). Thus, the 2,3-trans carbon radical or organocopper intermediate could suffer ring opening and closing prior to C-C bond formation, resulting in a lower overall reaction diastereoselectivity.
Based on the labeling and H-atom abstraction reactions, the intermediacy of a primary carbon radical is reasonable. The C-C bond forming process in the aryl addition step of the carboetherification could, however, still feasibly take place either by a radical addition or an electrophilic aromatic substitution mechanism. Aryl addition to an organocopper(III) intermediate, either directly at carbon or via addition to copper(III) followed by reductive elimination, could feasibly lead to the carboetherification product. Analogous mechanisms have been proposed for Pd- and Au-catalyzed reactions.23 (Carbon-carbon bond formation through a Friedel-Crafts type addition to a primary carbocation intermediate is less likely as such an intermediate is expected to be very high energy and unlikely to be formed under the reaction conditions.)
 |
(4) |
 |
(5) |
The stereochemistry of the deutero-alkene cannot be used in our analysis since stereochemical information is lost due to the presence of the carbon radical intermediate. Regiochemical trends in electrophilic and radical aryl substitution reactions are known, however, and can be used to infer the likely reaction mechanism. It is known that radicals favor addition ortho to substituents on aromatic rings, possibly due to electronic stabilization of aryl radical intermediates (e.g. Eq. 6).21 Conversely, electrophilic addition of metals, electrophilic organometallics or activated carbonyls (Friedel-Crafts acylation) to aromatic rings generally occurs at the less sterically hindered para-position (e.g. Eq 7).23
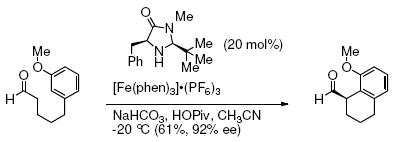 |
(6) |
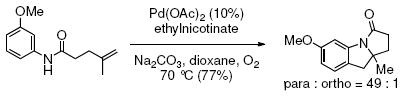 |
(7) |
Our reaction provides a regioisomeric mixture where the ortho-substituted product is formed as the major product (Table 2, entry 8, ortho : para = 1.9:1). This regioselectivity is most consistent with a carbon radical addition to the aromatic ring.
2.7 Proposed mechanism
A proposed catalytic cycle is illustrated in Scheme 4. A cis-oxycupration transition state (TS-E) is proposed in analogy to related aminocuprations.20 For α-substituted pentenols 15, the R group is placed in an equatorial position in the cyclic transition state (TS-E) resulting in formation of the major 2,5-trans-tetrahydrofuran diastereomer 16. The electronic effect observed in the substrate reactivity in Table 1 seems to indicate that more electron-rich alkenes and/or alcohols are more reactive. Homolysis of the unstable C-Cu(II) bond of intermediate 28 results in the primary carbon radical intermediate 29. Addition of the resulting carbon radical to the nearest (cis) aryl ring produces the new C-C bond in aryl radical 30, and aryl oxidation provides the product 16. Oxidation of Cu(I) with MnO2 regenerates the Cu(II) catalyst.
Scheme 4.
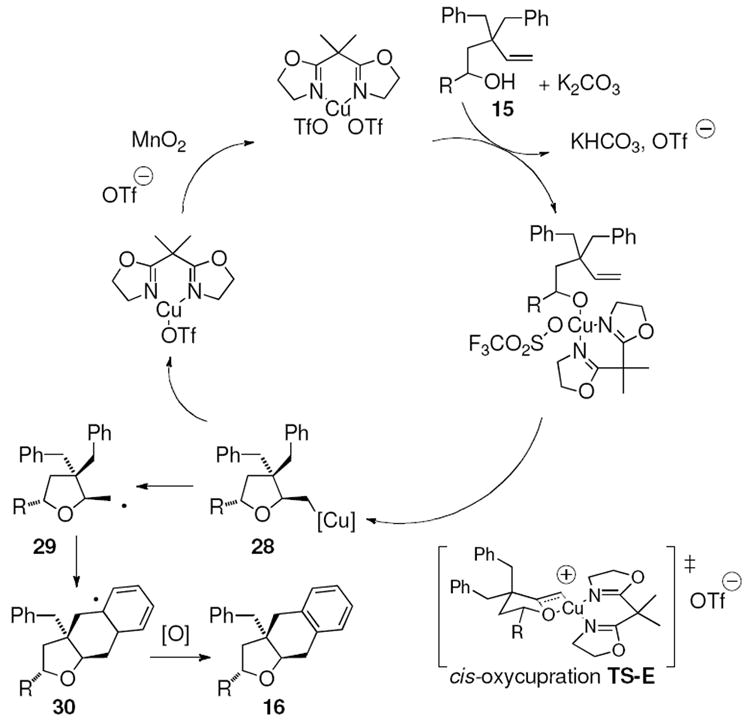
Proposed carboetherification catalytic cycle
We have observed that the reaction diastereoselectivity can vary depending upon the nature of the copper(II) ligands, the presence or absence of MnO2 and the presence or absence of 1,4-cyclohexadiene (Table 2, Table 3 and Eqs. 4 and 5). Reaction reversibility due to a ring-opening and unselective ring-closing process should be less prevalent if the subsequent step, reaction termination by H atom abstraction or addition of the carbon radical to the aromatic ring, is rapid. The Cu(eh)2 catalyst and the presence of MnO2 increase reaction diastereoselectivity which may indicate these reactions are also less reversible. This is possibly due to an increase in stability of the primary carbon radical intermediate by its reversible conversion to an organocopper(III) species. This species can be formed by direct oxidation of the organocopper(II) species (e.g. 28) or addition of the primary carbon radical (e.g. 29) to Cu(II). Both processes should be increased by greater concentration of oxidizing species or greater propensity of the Cu(I) or Cu(II) species to undergo oxidation. A ligand affect on copper oxidation potential is feasible.24
The extent of the reversibility of the tetrahydrofuran ring formation is also highly dependent upon the rate of the subsequent C-C or C-H bond forming step. As we observed in our enantioselective reactions, 1-substituted-4-pentenol substrates that are able to form cis-fused tetrahydronapthofuran products, e.g. 16, and reactions involving C-H bond formation via H-atom abstraction (e.g. formation of 26 in Scheme 3) give the highest enantioselectivities (up to 76%). It can be inferred with these substrates and reactions that the oxycupration step is enantioselective and suffers little if any ring-opening/unselective ring-closing.
Carbon-oxygen bond formation via trans-oxycupration is a mechanistic alternative to the proposed cis-oxycupration shown in Scheme 4. The product stereochemistry cannot be used to differentiate the two mechanisms since stereochemical information from the C-Cu bond formation is lost (vide supra, Eq. 4). The cis-oxycupration mechanism requires coordination of the alcohol to the copper center prior to alkene addition. This seems likely due to the high affinity the hard alcohol nucleophile should have for the hard Cu(II) catalyst. The olefin, in comparison, is a softer nucleophile.
An alternative, oxygen radical alkene addition mechanism (from homolysis of an O-Cu bond) can be ruled out since we have observed that the ligand on Cu(II) can influence the enantioselectivity of the reaction. A mechanism involving internal charge transfer of the RO-Cu(II) intermediate to give an RO• Cu(I) complex where both the metal and alkoxyradical participate in the olefin addition step, in analogy to that proposed for related Co-catalyzed reactions,13 cannot be ruled out, but seems unlikely since such N to Cu charge transfer does not appear to occur in the analogous aminocupration reaction.25
CONCLUSIONS
In conclusion, the first copper-catalyzed intramolecular carboetherification of γ-unsaturated alcohols has been described. This reaction efficiently provides fused and bridged-ring tetrahydrofurans from common and unactivated alkenols. Evidence for oxycupration as the alkene addition mechanism is presented, notably in the form of ligand-based asymmetric catalysis. The C-C bond-forming step in the carboetherification reaction is thought to occur via carbon radical arene addition and this step constitutes an efficient C-H functionalization. A novel, efficient, diastereoselective and enantioselective hydroetherification reaction, the result of trapping the carbon radical intermediate with 1,4-cyclohexadiene in an intermolecular H-atom abstraction step, was also presented. This hydroetherification reaction could prove useful in generating 5-methyl-2,2-disubstituted tetrahydrofurans currently of interest to the perfume industry.26 The novel reactivity reported herein will serve as the basis for the development of other alkene oxidative difunctionalization reactions. We anticipate we can broaden the scope of tetrahydrofurans formed through carboetherification by introducing external alkene radical acceptors20g to enable intermolecular C-C bond formation. Application of this method in target-oriented synthesis and optimization of the enantioselective protocol are underway and will be reported in due course.
Supplementary Material
Acknowledgments
The National Institute of General Medical Science (GM078383) is gratefully acknowledged for support of this research. We thank William W. Brennessel and the X-ray Crystallographic Facility at the University of Rochester for the X-ray structures of 11a and 16e. We thank Barbara Casavant, Shulendu Kariakarte and Timothy Liwosz for kind assistance in the synthesis of five substrates.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. / All authors have given approval to the final version of the manuscript.
Supporting Information. Experimental procedures, spectroscopic data, and crystallographic data for 11a and 16e (CIFs). The material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Reviews: Boivin TLB. Tetrahedron. 1987;43:3309–3362.; Cardillo G, Orena M. Tetrahedron. 1990;46:3321–3408.; Harmange J-C, Figadere B. Tetrahedron: Asym. 1993;4:1711–1754.; Miura K, Hosomi A. Synlett. 2003:143–155.
- 2.(a) Koert U. Synthesis. 1995:115–132. [Google Scholar]; (b) Bates RH, Chen M, Roush WR. Current Opinion in Drug Discovery & Development. 2008;11:778–792. [PubMed] [Google Scholar]; (c) Kuwano R. Heterocycles. 2008;76:909–922. [Google Scholar]; (d) Wolfe JP, Hay MB. Tetrahedron. 2007;63:261–290. doi: 10.1016/j.tet.2006.08.105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Li N, Shi Z, Tang Y, Chen J, Li X. Beilstein J Org Chem. 2008;4(48) doi: 10.3762/bjoc.4.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McDonald RI, Liu G, Stahl SS. Chem Rev. 2011;111:2981–3019. doi: 10.1021/cr100371y.. Piccialli V. Synthesis. 2007:2585–2607.. Majumdar KC, Debnath P, Roy B. Heterocycles. 2009;78:2661–2728.. Wolfe JP. Synlett. 2008;19:2913–2937.. Selected electrophile-activated additions/cyclization of ROH and alkenes: Kang SH, Lee SB, Park CM. J Am Chem Soc. 2003;125:15748–15749. doi: 10.1021/ja0369921.. Zhou L, Tan CK, Jiang X, Chen F, Yeung Y–Y. J Am Chem Soc. 2010;132:15474–15476. doi: 10.1021/ja1048972.. Chen G, Ma S. Angew Chem Int Ed. 2010;49:8306–8308. doi: 10.1002/anie.201003114.. Veitch GE, Jacobsen EN. Angew Chem Int Ed. 2010;49:7332–7335. doi: 10.1002/anie.201003681.. Murai K, Matsushita KT, Nakamura A, Fukushima S, Shimura M, Fujioka H. Angew Chem Int Ed. 2010;49:9174–9177. doi: 10.1002/anie.201005409.. Whitehead DC, Yousefi R, Jaganathan A, Borhan B. J Am Chem Soc. 2010;132:3298–3299. doi: 10.1021/ja100502f.. Yousefi R, Whitehead DC, Mueller JM, Staples RJ, Borhan B. Org Lett. 2011;13:608–611. doi: 10.1021/ol102850m.. Hennecke U, Muller CH, Frohlich R. Org Lett. 2011;13:860–863. doi: 10.1021/ol1028805.. Denmark SE, Burk MT. Org Lett. 2012;14:256–259. doi: 10.1021/ol203033k.
- 4.(a) Yoshikawa K, Kokudo N, Tanaka M, Nakano T, Shibata H, Aragaki N, Higuchi T, Hashimoto T. Chem Pharm Bull. 2008;56:89–92. doi: 10.1248/cpb.56.89. [DOI] [PubMed] [Google Scholar]; (b) Yao S, Tang CP, Ke CQ, Ye Y. J Nat Prod. 2008;71:1242–1246. doi: 10.1021/np8002063. [DOI] [PubMed] [Google Scholar]; (c) Peranza-Sanchez SR, Chavez D, Chai H-B, Shin YG, Garcia R, Mejia M, Fairchild CR, Lane KE, Menendez AT, Farnsworth NR, Cordell GA, Pezzuto JM, Kinghorn AD. J Nat Prod. 2000;63:492–495. doi: 10.1021/np990528l. [DOI] [PubMed] [Google Scholar]; (d) Xiao W-L, Yang L-M, Gong N-B, Wu L, Wang R-R, Pu J-X, Li X-L, Huang S-X, Zheng Y-T, Li R-T, Lu Y, Zheng Q-T, Sun H-D. Org Lett. 2006;8:991–994. doi: 10.1021/ol060062f. [DOI] [PubMed] [Google Scholar]; (e) Han L, Huang X, Sattler I, Moellmann U, Fu H, Lin W, Grabley S. Planta Med. 2005;71:160. doi: 10.1055/s-2005-837784. [DOI] [PubMed] [Google Scholar]; (f) Wang J, Soisson SM, Young K, Shoop W, Kodali S, Galgoci A, Painter R, Parthasarathy G, Tang YS, Cummings R, Ha S, Dorso K, Motyl M, Jayasuriya H, Ondeyka J, Herath K, Zhang C, Hernandez L, Allocco J, Basilio A, Tormo JR, Genilloud O, Vincente F, Pelaez F, Colwell L, Lee SH, Michael B, Felcetto T, Gill C, Silver LL, Hermes JD, Bartizal K, Barrett J, Schmatz D, Becker JW, Cully D, Singh SB. Nature. 2006;441:358–361. doi: 10.1038/nature04784. [DOI] [PubMed] [Google Scholar]
- 5.(a) Solorio DM, Jennings MP. J Org Chem. 2007;72:6621–6623. doi: 10.1021/jo071035l. [DOI] [PubMed] [Google Scholar]; (b) Fananas FJ, Fernandez A, Cevic D, Rodriguez F. J Org Chem. 2009;74:932–934. doi: 10.1021/jo8021204. [DOI] [PubMed] [Google Scholar]
- 6.Molander GA, Cameron KO. J Am Chem Soc. 1993;115:830–846. [Google Scholar]
- 7.(a) Padwa A, Hornbuckle SF, Fryxell GE, Stull PD. J Org Chem. 1989;54:817–824. [Google Scholar]; (b) Hodgson DM, Labande AH, Pierard FYTM, Castro MAE. J Org Chem. 2003;68:6153–6159. doi: 10.1021/jo0343735. [DOI] [PubMed] [Google Scholar]; (c) Hodgson DM, Stupple PA, Pierard FYTM, Labande AH, Johnstone C. Chem Eur J. 2001;7:4465–4476. doi: 10.1002/1521-3765(20011015)7:20<4465::aid-chem4465>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]; (d) Ishida K, Kusama H, Iwasawa N. J Am Chem Soc. 2010;132:8842–8843. doi: 10.1021/ja102391t. [DOI] [PubMed] [Google Scholar]
- 8.Rodriguez JR, Rumbo A, Castedo L, Mascarenas JL. J Org Chem. 1999;64:4560–4563. [Google Scholar]
- 9.(a) McGrath NA, Bartlett ES, Sittihan S, Njardarson JT. Angew Chem Int Ed. 2009;48:8543–8546. doi: 10.1002/anie.200903347. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Loy RN, Jacobsen EN. J Am Chem Soc. 2009;131:2786–2787. doi: 10.1021/ja809176m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Matuura BS, Condie AG, Buff RC, Karahalis GJ, Stephenson CRJ. Org Lett. 2011;13:6320–6323. doi: 10.1021/ol202881q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhu R, Buchwald SL. Angew Chem Int Ed. 2012;51:1926–1929. doi: 10.1002/anie.201108129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Representative Pd-catalyzed alkene carboetherifications: Semmelhack MF, Bodurow C. J Am Chem Soc. 1984;106:1496–1498.. Wolfe JP, Rossi MA. J Am Chem Soc. 2004;126:1620–1621. doi: 10.1021/ja0394838.. Hay MB, Wolfe JP. J Am Chem Soc. 2005;127:16468–16476. doi: 10.1021/ja054754v.. Ward AF, Wolfe JP. Org Lett. 2009;11:2209–2212. doi: 10.1021/ol900594h.. Nakhla JS, Kampf JW, Wolfe JP. J Am Chem Soc. 2006;128:2893. doi: 10.1021/ja057489m.. Nicolai S, Erard S, Fernandez-Gonzales D, Waser J. Org Lett. 2010;12:384–387. doi: 10.1021/ol9027286.. Jiang D, Peng J, Chen Y. Org Lett. 2008;10:1689–1698. doi: 10.1021/ol8002173.. Hayashi S, Yorimitsu H, Oshima K. J Am Chem Soc. 2009;131:2052–2053. doi: 10.1021/ja8084065.. Han X, Lu X. Org Lett. 2009;11:2381–2384. doi: 10.1021/ol9007348.. Coy B, E D, Jovanovic L, Sefkow M. Org Lett. 2010;12:1976–1979. doi: 10.1021/ol100433z.. Satterfield AD, Kubota A, Sanford MS. Org Lett. 2011;13:1076–1079. doi: 10.1021/ol103121r.
- 12.Au-catalyzed alkene carboetherifications: Zhang L, Kozmin SA. J Am Chem Soc. 2005;127:6962–6963. doi: 10.1021/ja051110e.. Sherry BD, Maus L, Laforteza BN, Toste FD. J Am Chem Soc. 2006;128:8132–8133. doi: 10.1021/ja061344d.. Zhang G, Cui L, Wang Y, Zhang L. J Am Chem Soc. 2010;132:1474–1475. doi: 10.1021/ja909555d.. Melhado AD, Brenzovich WE, Lackner AD, Toste FD. J Am Chem Soc. 2010;132:8885–8887. doi: 10.1021/ja1034123.. Ball LT, Green M, Lloyd-Jones GC, Russell CA. Org Lett. 2010;12:4724–4727. doi: 10.1021/ol1019162.. Brenzovich WE, Jr, Brazeau J-F, Toste FD. Org Lett. 2010;12:4728–4731. doi: 10.1021/ol102194c.
- 13.Cobalt-catalyzed 4-pentenol cyclizations: Schuch D, Fries P, Donges M, Menendez Perez B, Hartung J. J Am Chem Soc. 2009;131:12918–12920. doi: 10.1021/ja904577c.. Inoki S, Mukaiyama T. Chem Lett. 1990:67–70.. Fries P, Halter D, Kleinschek A, Hartung J. J Am Chem Soc. 2011;133:3906–3912. doi: 10.1021/ja108403s.. Palmer C, Morra NA, Stevens AC, Bajtos B, Machin BP, Pagenkopf BL. Org Lett. 2009;11:5614–5617. doi: 10.1021/ol9023375.
- 14.Catalytic enantioselective alkene carboetherification-type reactions: Tietze LF, Sommer KM, Zinngrebe J, Stecker F. Angew Chem Int Ed. 2005;44:257–259. doi: 10.1002/anie.200461629.. Pathak TP, Gligorich KM, Welm BE, Sigman MS. J Am Chem Soc. 2010;132:7870–7871. doi: 10.1021/ja103472a.. Arai MA, Kuraishi M, Arai T, Sasai H. J Am Chem Soc. 2001;123:2907. doi: 10.1021/ja005920w.. Kawamura Y, Kawano Y, Matsuda T, Ishitobi Y, Hosokawa T. J Org Chem. 2009;74:3048. doi: 10.1021/jo802807w.. Selected other catalytic enantioselective additions of alcohols to alkenes to form tetrahydrofurans: Burke SD, Jiang L. Org Lett. 2001;3:1953–1955. doi: 10.1021/ol0160304.
- 15.(a) Taylor JG, Whittall N, Hii KKM. Chem Comm. 2005:5103–5105. doi: 10.1039/b509933a. [DOI] [PubMed] [Google Scholar]; (b) Tschan MJ–L, Thomas CM, Strub H, Carpentier J-F. Adv Synth Catal. 2009;351:2496–2504. [Google Scholar]; (c) Adrio LA, Quek LS, Taylor JG, Hii KKM. Tetrahedron. 2009;65:10334–10338. [Google Scholar]
- 16.(a) Seayad J, Seayad AM, Chai CLL. Org Lett. 2010;12:1412–1415. doi: 10.1021/ol902813m. [DOI] [PubMed] [Google Scholar]; (b) Kang Y-B, Gade LH. J Am Chem Soc. 2011;133:3658–3667. doi: 10.1021/ja110805b. [DOI] [PubMed] [Google Scholar]
- 17.Huang L, Jiang H, Qi C, Liu X. J Am Chem Soc. 2010;132:17652–17654. doi: 10.1021/ja108073k. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Du H. J Org Chem. 2010;75:3503–3506. doi: 10.1021/jo100413p. [DOI] [PubMed] [Google Scholar]
- 19.Cheng K, Huang L, Zhang Y. Org Lett. 2009;11:2908–2911. doi: 10.1021/ol900947d.. An Fe-catalyzed radical carboetherification: Liu W, Li Y, Liu K, Li Z. J Am Chem Soc. 2011;133:10756–10759. doi: 10.1021/ja204226n.
- 20.Cu-promoted carboamination: Sherman ES, Chemler SR, Tan TB, Gerlits O. Org Lett. 2004;6:1573–1575. doi: 10.1021/ol049702+.. Sherman ES, Fuller PH, Kasi D, Chemler SR. J Org Chem. 2007;72:3896–3905. doi: 10.1021/jo070321u.. Fuller PH, Chemler SR. Org Lett. 2007;9:5477–5480. doi: 10.1021/ol702401w.. Cu-catalyzed carboamination: Zeng W, Chemler SR. J Am Chem Soc. 2007;129:12948–12949. doi: 10.1021/ja0762240.. Sherman ES, Chemler SR. Adv Synth Catal. 2009;351:467–471. doi: 10.1002/adsc.200800705.. Miao L, Haque I, Manzoni MR, Tham WS, Chemler SR. Org Lett. 2010;12:4739–4741. doi: 10.1021/ol102233g.. Liwosz TW, Chemler SR. J Am Chem Soc. 2012;134:2020–2023. doi: 10.1021/ja211272v.
- 21.Addition of radicals to arenes: Ito R, Migita T, Morikawa N, Simamura O. Tetrahedron. 1965;21:955.. Pryor WA, Davis WH, Jr, Gleaton JH. J Org Chem. 1975;40:2099–2102.. Conrad JC, Kong J, Laforteza BN, MacMillan DWC. J Am Chem Soc. 2009;131:11640–11641. doi: 10.1021/ja9026902.
- 22.Paderes MC, Chemler SR. Eur J Org Chem. 2011:3679–3684. doi: 10.1002/ejoc.201100444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Yip K-T, Yang D. Org Lett. 2011;13:2134–2137. doi: 10.1021/ol2006083. [DOI] [PubMed] [Google Scholar]; (b) Sibbald PA, Rosewall CF, Swartz RD, Michael FE. J Am Chem Soc. 2009;131:15945–15951. doi: 10.1021/ja906915w. [DOI] [PubMed] [Google Scholar]; (c) de Haro T, Nevado C. Angew Chem Int Ed. 2011;50:906–910. doi: 10.1002/anie.201005763. [DOI] [PubMed] [Google Scholar]; (d) Chiche B, Finiels A, Gauthier C, Geneste P. J Org Chem. 1986;51:2128–2130. [Google Scholar]
- 24.Jones GO, Liu P, Houk KN, Buchwald SL. J Am Chem Soc. 2010;132:6205–6213. doi: 10.1021/ja100739h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paderes MC, Belding L, Fanovic B, Dudding T, Keister JB, Chemler SR. Chem Eur J. 2012;18:1711–1726. doi: 10.1002/chem.201101703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kraft P, Givaudan SA. Trisubstituted furans suitable for the preparation of fragrance compositions. 7,632,790. US patent. 2009 Dec 15;
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.