

Abstract
Disturbances in corticothalamic circuitry can lead to absence epilepsy. The reticular thalamic nucleus (RTN) plays a pivotal role in that it receives excitation from cortex and thalamus and, when strongly activated, can generate excessive inhibitory output and epileptic thalamocortical oscillations that depend on postinhibitory rebound. Stargazer (stg) mice have prominent absence seizures resulting from a mutant form of the AMPAR auxiliary protein stargazin. Reduced AMPAR excitation in RTN has been demonstrated previously in stg, yet the mechanisms leading from RTN hypoexcitation to epilepsy are unknown and unexpected because thalamic epileptiform oscillatory activity requires AMPARs. We demonstrate hyperexcitability in stg thalamic slices and further characterize the various excitatory inputs to RTN using electrical stimulation and laser scanning photostimulation. Patch-clamp recordings of spontaneous and evoked EPSCs in RTN neurons demonstrate reduced amplitude and increased duration of the AMPAR component with an increased amplitude NMDAR component. Short 200 Hz stimulus trains evoked a gradual approximately threefold increase in NMDAR EPSCs compared with single stimuli in wild-type (WT), indicating progressive NMDAR recruitment, whereas in stg cells, NMDAR responses were nearly maximal with single stimuli. Array tomography revealed lower synaptic, but higher perisynaptic, AMPAR density in stg RTN. Increasing NMDAR activity via reduced [Mg2+]o in WT phenocopied the thalamic hyperexcitability observed in stg, whereas changing [Mg2+]o had no effect on stg slices. These findings suggest that, in stg, a trafficking defect in synaptic AMPARs in RTN cells leads to a compensatory increase in synaptic NMDARs and enhanced thalamic excitability.
Introduction
Absence seizures, brief losses of consciousness with bilaterally synchronous 3 Hz EEG spike-and-wave discharges (SWDs), arise from disturbances in corticothalamic circuitry (Steriade et al., 1993). Although the entire corticothalamic network is implicated in SWD, cortical output to thalamus appears critical for several aspects of seizures, including initiation, generalization, and maintenance (Steriade and Contreras, 1998; Bal et al., 2000; Blumenfeld and McCormick, 2000; Meeren et al., 2002; Polack et al., 2007; Adams et al., 2011). The reticular thalamic nucleus (RTN) plays a key role in SWD: RTN inhibition of dorsal thalamic neurons yields postinhibitory rebound bursts that promote re-entrant circuit activation of RTN (Huguenard and McCormick, 2007). Although there is debate as to whether dorsal thalamic neurons respond exclusively to RTN inhibition with postinhibitory rebound bursts during SWDs (Steriade et al., 1993; Pinault et al., 1998), periodic inhibition-mediated bursts are the characteristic response of thalamic neurons participating in seizures (Inoue et al., 1993; Pinault et al., 1998; Seidenbecher et al., 1998). A central role for RTN activation in SWD is further supported by the following: (1) enhanced inhibition (Schofield et al., 2009) or inactivation (Avanzini et al., 1992) of RTN suppresses SWDs; (2) genetic RTN disinhibition is associated with absence-like seizures (DeLorey et al., 1998; Huntsman et al., 1999); (3) RTN activity occurs early within each SWD (Slaght et al., 2002); and (4) selective attenuation of RTN bursts reduces absence seizures (Zaman et al., 2011). Because excitation-driven RTN firing is a prominent feature of SWDs (Timofeev et al., 1998; Slaght et al., 2002), the synaptic mechanisms of RTN excitation are of interest in relation to absence seizure expression and as potential treatment targets.
Cortical and/or thalamic excitation of RTN through glutamatergic AMPA receptors (AMPARs) and NMDARs is essential for synchronous thalamic oscillations (von Krosigk et al., 1993; Jacobsen et al., 2001; Huguenard and McCormick, 2007). Stargazin is an auxiliary protein that traffics/anchors AMPARs to the synapse and modulates functional properties (Hashimoto et al., 1999; Chen et al., 2000; Tomita et al., 2003, 2005, 2007; Turetsky et al., 2005; Osten and Stern-Bach, 2006; Bats et al., 2007; Sumioka et al., 2010; Jackson and Nicoll, 2011). GluA4, the main RTN AMPAR subunit (Mineff and Weinberg, 2000; Golshani et al., 2001), associates with stargazin at synaptic sites (Schnell et al., 2002; Tomita et al., 2003; Bats et al., 2007), and stargazin mutation reduces synaptic GluA4 in the RTN (Barad et al., 2012).
Mutations in stargazin (Noebels et al., 1990; Letts et al., 1998) and GluA4 (Beyer et al., 2008; Paz et al., 2011) lead to absence seizures. Reduced RTN AMPAR-excitation occurs in immature stargazer (stg) mice with mutated stargazin protein (Menuz and Nicoll, 2008), but the excitation defect does not readily explain the adult epileptic phenotype (see above and Chetkovich, 2009). We show that reduced RTN EPSCs persist in adult stg and demonstrate a shift of AMPARs from synaptic to perisynaptic zones. Furthermore, enhanced activation of NMDARs generates RTN overexcitation, promoting thalamocortical hypersynchrony that likely contributes to seizures.
Materials and Methods
All experiments were performed according to protocols approved by the Stanford Institutional Animal Care and Use Committee, and every precaution was taken to minimize stress and the number of animals used in each series of experiments. Stg mice were obtained from The Jackson Laboratory. To ensure consistency, data were collected from stg mutants and their wild-type (WT) littermates of either sex, and recordings were made on either the same day or interleaved consecutive days.
In vitro slice preparation.
Stg mutant or WT littermate controls (P19–P40) were anesthetized with pentobarbital (50 mg/kg, i.p.) and either perfused transcardially with ice-cold slicing solution (see below) before being decapitated or immediately decapitated. The brains were rapidly removed and immersed in an ice-cold (4°C) slicing solution containing the following (in mm): 234 sucrose, 2.5 KCl, 1.25 NaH2PO4, 10 MgSO4, 0.5 CaCl2, 26 NaHCO3, and 11 glucose, pH 7.4 (equilibrated with 95% O2 and 5% CO2). Horizontal thalamic slices (250–270 μm for patch-clamp recordings or 400 μm for extracellular recordings) containing the thalamic ventrobasal complex (VB) and RTN were cut as described previously (Huguenard and Prince, 1994) with a Leica VT1200 microtome. The slices were incubated, initially at 32°C for 1 h and subsequently at room temperature, in artificial CSF (ACSF) containing the following (in mm): 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl2, 26 NaHCO3, and 10 glucose, pH 7.4 (equilibrated with 95% O2 and 5% CO2).
Extracellular electrophysiology recordings.
Slices were placed in an interface chamber for recording and perfused with warmed (34°C), oxygenated normal ACSF supplemented with 0.3 mm glutamine (Bryant et al., 2009), at a rate of 2 ml/min. For some experiments, the concentration of magnesium in the ACSF was lowered to 0.5 mm (so-called “low magnesium”) to facilitate oscillations (Jacobsen et al., 2001). Extracellular multiunit field recordings were made using monopolar tungsten microelectrodes (50–100 kΩ; FHC) placed in the thalamic reticular nucleus (containing RTN neurons) and the ventrobasal somatosensory relay nucleus (VB, containing thalamocortical neurons). Signals were amplified 10,000 times and bandpass filtered between 100 Hz and 3 kHz. Electrical stimuli were delivered to the internal capsule with a pair of tungsten microelectrodes (50–100 kΩ; FHC). The stimuli were 100 μs in duration, 50 V in amplitude, and delivered once every 30 s.
Patch-clamp electrophysiology recordings.
All whole-cell patch-clamp recordings were performed at room temperature (22–25°C). Brain slices were transferred from the incubation chamber to the recording chamber and superfused with ACSF at a flow rate of 2 ml/min. Recordings were obtained from RTN neurons visually identified using differential contrast optics with a Carl Zeiss Axioskop microscope and an infrared video camera. The RTN was readily identified by proximity to the adjacent internal capsule.
Recording electrodes made of borosilicate glass had a resistance of 1.8–4 MΩ when filled with intracellular solution. For current-clamp recordings, the internal solution contained the following (in mm): 120 K-gluconate, 11 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, and 1 EGTA, pH adjusted to 7.4 with KOH (290 mOsm). The estimated ECl− was approximately −60 mV based on the Nernst equation. For EPSCs, the internal solution contained the following (in mm): 135 CsCl, 10 HEPES, 10 EGTA, 5 QX-314, and 2 MgCl2, pH adjusted to 7.3 with CsOH (290 mOsm) or, occasionally, with the same internal used for current-clamp recordings (see above). The estimated ECl− was ∼0 mV based on the Nernst equation. A liquid junction potential of approximately −15 or −4 mV, respectively, was estimated with the LJP calculator in Clampex (Molecular Devices). In all recording conditions, access resistance was monitored and cells were included for analysis only if the series resistance was <18 ΜΩ and the change of resistance was <25% over the course of the experiment. In addition, adequacy of voltage clamp was demonstrated by T-current steady-state inactivation protocols in which peak latencies remained constant after voltage-clamp steps to −60 mV from a range of hyperpolarizing conditioning potential that evoked currents. The evoked T currents, with slow activation and decay kinetics (Huguenard and Prince, 1992), were also used as a means to physiologically confirm the identity of RTN cells, especially in experiments in which current-clamp identification could not be performed (Deleuze and Huguenard, 2006; Beyer et al., 2008).
EPSCs were pharmacologically isolated by bath application of the GABAAR antagonist picrotoxin (50 μm; Tocris Bioscience). Neurons were clamped at −65 (or −80) mV or at 40 mV to study NMDAR-mediated events. Evoked EPSCs (eEPSCs) in RTN were obtained with a concentric bipolar stimulating electrode (125 μm diameter; CB-ARC75; FHC) positioned in striatum or internal capsule, activating corticothalamic and/or thalamocortical fibers. A minimal stimulation protocol (50% failures) was used to activate putative single presynaptic axons. Once threshold was determined for each cell, evoked currents were recorded with the stimulation intensity of 1.5× threshold (Huntsman et al., 1999). Single and train stimuli (five stimuli at 200 Hz) were applied. For eEPSC current–voltage (I–V) curves, neurons were held at a range of membrane potentials between −60 and +40 mV in steps of 10 mV. Individual responses were obtained with an interstimulus interval of at least 15 s. For evoked NMDAR I–V curves, from which high-resolution conductance–voltage (g–V) relationships were determined, neurons were stepped through a range of potentials between −100 and +40 mV with 5 mV steps, either with a single stimulation or trains (five stimuli at 200 Hz).
Input resistance and time constant were measured from membrane voltage responses to small (∼50 pA) current steps. Membrane capacitance was measured from the current response to voltage-clamp steps, from which the area under the curve (charge) was obtained and divided by the change in voltage to arrive at capacitance.
Data acquisition and analysis.
A Digidata 1320 digitizer and pClamp9 (Molecular Devices) were used for data acquisition and analysis. Signals were amplified with an Axopatch 200A amplifier (Molecular Devices), sampled, and filtered at 10 kHz for spontaneous recordings and filtered at 4 kHz for all other recordings. EPSCs were detected and analyzed with wDetecta, a custom-made postsynaptic current detection program (http://huguenardlab.stanford.edu/apps/wdetecta). Detection parameters were adjusted on a per cell basis to accurately detect events. For eEPSCs, the peak-to-baseline decay phase of the resulting current trace was fitted by the following double-exponential function: I = A1e−t/τ1 + A2e−t/τ2, where A1 and A2 are the slow and fast amplitude components, and τ1 and τ2 are the slow and fast decay time constants, respectively. The weighted decay time constant (τD,W) was calculated using the following equation: τD,W = (τ1A1 + τ2A2)/(A1 + A2). The same method was applied to analyzing the post-train decay current. g–V curves for NMDAR responses were best fitted with a Boltzmann function: I/Imax = (A1 − A2)/(1 + e(V−V50%)/k) + A2, where V50% and k represent the half-maximal voltage and Boltzmann slope factor, respectively, and A1 and A2 represent initial and final I/Imax values, respectively.
Focal photolysis of caged glutamate.
A pulsed 355 nm UV laser (DPSS Lasers) was directed into the back aperture of a 5× or 63× microscope objective and was controlled with mirror galvanometers (model 6210; Cambridge Technology) using a locally developed software program. Focal photolysis of 4-methoxy-7-nitroindolinyl (MNI)-caged glutamate (100 μm; Tocris Bioscience) was triggered by UV light pulses (100–2000 μs for determination of action potential bursts in VB cells and 100–500 μs for other experiments). MNI-caged glutamate was supplied in a 30 ml recirculating bath solution. To find a location for photolysis for synaptic responses, the entire VB area visible in the 5× microscope objective was scanned until a spot was found from which bursts of EPSCs could be reliably evoked in RTN neurons. For measurement of direct glutamatergic currents in RTN neurons, the laser was directed onto the soma of the recorded RTN neuron. I–V curves were determined from synaptic responses obtained at a range of potentials between −60 and 60 mV. AMPAR conductance was calculated from the first fast EPSC in each trace, and NMDAR conductance was calculated from the peak of the slow current (if necessary after subtraction of fast currents). For NMDA/AMPA ratios of synaptic responses, the AMPA component was defined as the integrated area of all isolated fast EPSCs recorded at −60 mV, and the NMDA component was defined as the area under the curve at +60 mV 100–500 ms after the stimulus. For direct responses, NMDA/AMPA ratios were calculated by dividing the maximum current amplitude at +30 mV (NMDA component) by the maximum of the fast current at −60 mV (AMPA component). The fast current had a maximum latency of 4 ms. The AMPA current peak was readily distinguishable in all WT neurons, whereas short latency AMPA components were almost never observed in stg neurons. In these cases, the current amplitude at 4 ms after laser scanning photostimulation (LSPS) stimulus was defined as the AMPA component.
EEG recording and analysis.
Mice (P30–P45) were anesthetized using isoflurane inhalation at 1 L/min oxygen flow (3% isoflurane flow for induction, 1–2% isoflurane flow for maintenance), and carprofen (5 mg/kg) was injected intraperitoneally for postoperative analgesia. After a midline scalp incision and removal of the periosteum, small burr holes (∼0.5 mm) were drilled in the skull 5 mm lateral from the sagittal suture with a pair each in line with lambda and bregma. A fifth hole was drilled in the bone above cerebellum for ground. A plug consisting of a dual inline socket (Digi-Key Corp.) with soldered stainless steel wires (Medwire 316 SS 7/44T) each terminated with soldered stainless steel screws (2.38-mm-long #303SS; J.J. Morris). This assembly was mounted to the skull with superglue, the screws were attached to the skull through the burr holes, and dental cement was applied to secure the connection. EEG recordings were performed at 1 week of surgical recovery. Individual mice were recorded via an XLTek EEG system in which simultaneous video and multichannel EEG recordings were collected. Seizures were detected through a wavelet-based spectral analysis routine in MATLAB (Schofield et al., 2009) based on the approach of Torrence and Compo (1998).
Statistical analysis.
Numerical values are given as means ± SEM unless stated otherwise. Statistical box-and-whisker charts show the following: mean (central dot); median (large horizontal line); maximal and minimal values (short horizontal lines); 99% and 1% range (crosses); percentile 25–75 range (box); and 5–95 range (whiskers). Statistical significance was assessed by performing a Student's t test or Mann–Whitney rank-sum test. Statistical analyses were performed with SigmaStat 3.5 and Origin 7.0 (Microcal Software). Differences were considered significant when p < 0.05.
Drugs.
During recordings, AMPAR events were pharmacologically isolated by bath application of the NMDAR blocker 2-amino-5-phosphonopentanoic acid (APV; 50–100 μm; Ascent Scientific). NMDAR events were pharmacologically isolated by bath application of 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX; 20 μm). To block all ionotropic EPSCs, in some experiments, both APV and NBQX or 6,7-dinitroquinoxaline-2,3-dione (DNQX; 20 μm; Ascent Scientific) were added to the perfusate.
Array tomography.
Four WT and three stg (P35 and older) were transcardially perfused with PBS, followed by 4% paraformaldehyde (∼50 ml). The brains were removed and postfixed overnight at 4°C in 4% paraformaldehyde. Sections of thalamus (including internal capsule, reticular nucleus, and dorsal thalamus) ∼600–800 μm thick were dissected and dehydrated in a series of alcohol steps before being embedded in LR white. Ribbons of ultrathin sections (∼100 nm thick) were cut on an ultramicrotome and collected onto glycerol-coated coverslips (20–40 sections per ribbon). For more detailed methods, see Micheva and Smith (2007). Ribbons were subjected to a series of antibody “cycles.” The following proteins were targeted: parvalbumin (cell marker for RT; Calbiochem), VGLUT1, VGLUT2, NMDAR1, GluA4, pan-MAGUK, and synapsin (all purchased from Millipore). RTN borders were clearly identified by using VGLUT1 and parvalbumin, and these were then used to select RTN or VB positions for image collection. Image stacks for each cycle were collected using an Axioskop and aligned with a set of FIJI scripts (available at http://www.stanford.edu/∼bbusse/work/downloads.html). The aligned stacks were further analyzed using Volocity software to extract three-dimensional particle locations and immuno-intensities, which were used for additional radial analysis via in-laboratory-generated code (Beenhakker and Huguenard, 2010).
Results
stg thalamic networks express an epileptic phenotype
In mice harboring the spontaneous stargazin mutation, neurologic deficits, including head tossing, ataxia, and 6 Hz SWD seizures, show a developmental progression (Noebels et al., 1990). We confirm that stg at the developmental age used in our study (P35 and older), unlike their WT littermates, reliably exhibit frequent (6.2 ± 1.6/min, n = 6 mice) and prolonged (2.2 ± 0.5 s; 6.1 ± 0.5 Hz) SWDs, the EEG hallmark of absence seizures, together with behavioral absences, most evident as sudden cessations of movement interrupting normal locomotion (data not shown).
Because the thalamus contains pace-making circuitry implicated in several models of absence epilepsy (Huguenard and McCormick, 2007), we first tested whether an epileptic phenotype would be expressed in isolated thalamic networks from stg mice. Indeed, we found that robust, long-lasting synchronized oscillations can be evoked in thalamic slices prepared from homozygous young adult (P35 and older) stg and maintained under physiological conditions (n = 14; Fig. 1Aii,B). In contrast to the stg slices, such oscillations were rarely evoked in slices prepared from WT littermates under the same conditions (n = 7; Fig. 1Ai,B). In those cases in which an oscillation could be evoked in WT slices, it was significantly shorter in duration (0.40 ± 0.21 s) than in stg slices (1.67 ± 0.34 s; p < 0.05). Furthermore, the frequency of spontaneous oscillations was significantly higher in stg slices (n = 7) compared with WT (n = 5) littermates (2.134 ± 0.34 vs 0.924 ± 0.35 oscillations/min; p < 0.05; Fig. 1C). Thus, stg thalamic slices express a hyperexcitable SWD-like phenotype, indicating that the stg mutation induces pro-epileptic changes locally within thalamic circuitry.
Figure 1.

Thalamic networks are hyperexcitable in stg slices. A, Sample traces of WT (Ai) and stg (Aii) evoked thalamic (VB) multiunit oscillatory activity. Timing of stimulus applied to internal capsule is indicated by black dot. B, Evoked responses in VB and RTN show significantly longer-lasting oscillations in stg (WT, n = 7; stg, n = 14; *p < 0.01, Mann–Whitney test). C, Spontaneous, non-evoked oscillations recorded in VB occurred more frequently in stg than WT (WT, n = 5; stg, n = 7; *p < 0.01, Mann–Whitney test).
We next used whole-cell recordings from thalamic neurons to detect whether a synaptic defect might underlie thalamic hyperexcitability. All intracellular recordings were conducted in thalamic slices prepared from age-matched stg and WT littermate mice between P19 and P25 (stg mutants develop EEG correlates of absence seizures after P18; Qiao and Noebels, 1993). The RTN can be clearly identified within live horizontal thalamic slices by its position adjacent to the internal capsule (Huntsman et al., 1999; Schofield et al., 2009). RTN cells in WT (n = 15) and stg (n = 19) had indistinguishable passive and active membrane properties (Table 1).
Table 1.
Passive membrane properties of RTN cells
| RMP (mV) | Input resistance (MΩ) | Membrane time constant (ms) | Capacitance (pF) | |
|---|---|---|---|---|
| wt (24) | −72.3 ± 1.6 | 422.5 ± 36.5 | 47.1 ± 2.9 | 51.3 ± 2.8 |
| stg (29) | −70.8 ± 1.7 | 468.6 ± 32.0 | 47.0 ± 3.3 | 50.4 ± 2.1 |
RMP, Resting membrane potential.
Excitatory synaptic transmission is reduced in stg RTN
Excitation of the RTN is thought to be part of the recurrent network loop that serves to pace and synchronize large-scale thalamocortical networks during absence seizures (Huguenard and McCormick, 2007). In light of the enhanced thalamic network excitability in stg (Fig. 1) and given that these mice have a mutation in stargazin that affects the trafficking and biophysical properties of AMPAR, we first set out to identify the impact of the stargazin mutation on spontaneous EPSCs (sEPSCs) in RTN cells in vitro (Fig. 2).
Figure 2.

Frequency, amplitude, and kinetics of spontaneous EPSCs are altered in stg RTN cells. Representative traces from WT (A, D) and stg (B, E) RTN cells of continuous voltage-clamp recordings of RTN cells held at resting membrane potential (Vh = −60 mV) under conditions that isolate ionotropic sEPSCs (A–C) and AMPAR sEPSCs (D–F). Ci, Ensemble averaged sEPSCs from representative cells shown in A (black; WT) and B (gray; stg) superimposed on the same timescale. Cii, Averaged sEPSC from Ci normalized and aligned to peak to illustrate the slower kinetics of stg sEPSCs. Fi, Ensemble averaged sEPSCs from representative cells shown in D (black; WT) and E (gray; stg) superimposed on the same timescale. Fii, Averaged sEPSC from Fi normalized and aligned to peak to illustrate the slower kinetics of stg AMPAR sEPSCs. Remaining events were blocked by NBQX or DNQX at 20 μm confirming that the latter were AMPAR-mediated events (data not shown).
We found that, in contrast to what would be expected for a hyperexcitable network, isolated sEPSC frequency in stg RTN cells was significantly reduced (1.8 ± 0.4 Hz, n = 31; Fig. 2B) compared with WT (6.7 ± 0.8 Hz, n = 26; p < 0.001; Fig. 2A). The remaining stg EPSCs were significantly reduced in amplitude (WT, 29.7 ± 1.9 pA; stg, 19.8 ± 1.0 pA; p < 0.001; Fig. 2A,B). These differences in sEPSC properties were also observed under conditions in which AMPAR-mediated sEPSCs were pharmacologically isolated (50 μm APV and picrotoxin; WT, n = 15 and stg, n = 13; WT, 6.7 ± 1.5 Hz and stg, 1.4 ± 0.4 Hz, p < 0.001; and WT, 26.0 ± 2.6 pA and stg, 15.0 ± 1.0 pA, p < 0.0005; Fig. 2D,E). This is consistent with the results of a previous study (Menuz and Nicoll, 2008) that reported a congruent decrease in miniature EPSC (mEPSC) frequency and amplitude in pre-epileptic (younger than P17; cf. Qiao and Noebels, 1993) mice. Interestingly, in contrast to our results, Menuz and Nicoll (2008) reported that a statistically significant effect on mEPSC frequency did not persist at a developmental stage (P21–P23) in which seizures become apparent.
Although stg RTN sEPSCs were reduced in amplitude, their kinetics were slowed compared with WT, with both slower rise time and decay rate (rise time: WT, 0.23 ± 0.01 ms and stg, 0.28 ± 0.02 ms, p < 0.05; half-width: WT, 0.89 ± 0.05 ms and stg, 2.05 ± 0.30 ms, p < 0.005; Fig. 2C). Similarly, rise time and half-width of isolated AMPAR currents were also increased in stg (n = 13; rise time, 0.35 ± 0.02 ms; half-width, 1.72 ± 0.42 ms) compared with WT RTN cells (n = 15; rise time, 0.29 ± 0.01 ms; half-width, 0.91 ± 0.07 ms; Fig. 2F; p < 0.05). Remaining events were blocked by NBQX or DNQX (20 μm), confirming that they were AMPAR mediated (data not shown). Slowing of EPSC kinetics is not expected from a loss of stargazin function, because stargazin has been shown to slow deactivation and desensitization rates (Tomita et al., 2003, 2005, 2007; Priel et al., 2005; Turetsky et al., 2005; Kott et al., 2007). Thus, the altered EPSC kinetics in stg, which may compensate for reduced amplitude, might result not from direct modulation of synaptic AMPARs but rather from alterations in AMPAR subunit expression or synaptic trafficking.
Structural synaptic connectivity is maintained in stg thalamic circuits
Given the decreased frequency of sEPSCs in stg RTN cells at rest (Fig. 2), we were interested in determining whether this might result strictly from changes in postsynaptic receptor expression versus structural changes in the network in terms of anatomical connectivity (e.g., synapse number or presynaptic axonal branching). Changes in either synapse number or presynaptic function could explain the reduced sEPSC frequency observed in stg RTN cells, in which case we would observe a change in glutamate release. To test for this, we changed the holding potential of the voltage-clamped RTN cells to +40 mV to relieve Mg2+ block (Nowak et al., 1984) and reveal NMDAR-mediated sEPSCs. We found that, despite the lower frequency of stg sEPSCs at −60 mV (WT, 9.6 ± 1.4 Hz, n = 14; stg, 2.9 ± 0.8 Hz, n = 18; p < 0.0005; Fig. 3C), the frequency of stg sEPSCs recorded at 40 mV in the same cells was similar in stg and WT RTN cells (WT, 9.4 ± 1.4 Hz, Fig. 3A,C; stg, 6.9 ± 1.3 Hz, Fig. 3B,C; p > 0.05). The frequency of events at 40 mV was therefore significantly increased from −60 mV in stg cells (p < 0.005; Fig. 3C) but not WT cells (p > 0.05; Fig. 3C). Additionally, the mean amplitude of NMDAR-mediated events was indistinguishable between WT and stg cells (WT, 14.3 ± 1.6 pA; stg, 12.2 ± 1.1 pA; p > 0.05; Fig. 3D). Therefore, the reduction in sEPSC frequency observed at negative potentials may be attributable to a decrease in the number of available synaptic receptors resulting from inefficient trafficking of the AMPARs by the mutant stargazin.
Figure 3.

Presynaptic release of glutamate remains intact in stg RTN. Representative continuous sEPSC recordings at positive holding potential (Vh = +40 mV) in WT (A) and stg (B) RTN cells. C, Mean frequency of sEPSCs in stg (n = 14) and WT (n = 18) RTN cells is similar at positive holding potentials (p > 0.05). Furthermore, sEPSC frequency is independent of membrane potential in WT cells (p > 0.05), but in stg cells it is significantly increased by depolarization (p < 0.001). D, Population data showing mean amplitudes of sEPSCs at +40 mV are similar in WT and stg (p > 0.05). Significance assessed with Mann–Whitney test or Wilcoxon's test. **p < 0.005, ***p < 0.0005. ns, Not significant.
eEPSCs are smaller but longer in stg RTN cells
The presence of AMPAR-mediated EPSCs in stg RTN cells, albeit at reduced amplitude, suggested that synaptic AMPAR trafficking may be deficient yet not abolished in RTN cells. A reduced trafficking of AMPARs to synapses might, in part, explain the altered kinetics of sEPSCs. For example, a relative increase in extrasynaptic compared with synaptic receptors could result in greater diffusional time from synaptic release to receptor activation. According to this scenario, electrically evoked responses that are proposed to result in greater extrasynaptic receptor activation (Arnth-Jensen et al., 2002; Chen and Diamond, 2002; Zhang and Diamond, 2006) might be less affected than sEPSCs or mEPSCs, which presumably arise from single isolated release events that would primarily affect synaptic receptors. Thus, to further characterize the excitatory input to the RTN in stg, we examined eEPSCs by using near-threshold stimulation of the corticothalamic and/or thalamocortical fibers passing through the internal capsule (Fig. 4). In contrast to the very strong reduction of sEPSC amplitude in stg RTN cells, eEPSCs were reliably evoked in stg RTN cells (n = 19) with similar stimulation intensities as WT cells (n = 22; WT, 26.2 ± 4.1 V; stg, 26.4 ± 4.3 V; p > 0.05). Although eEPSC peak amplitude was indeed significantly decreased (WT, 253.8 ± 46.5 pA; stg, 117.3 ± 18.5 pA; p < 0.0005; Fig. 4A,B,Ci,D) the degree of reduction was less than expected given the strong reductions in both AMPAR-mediated sEPSC amplitude and frequency. As had been observed for sEPSCs, both the rising phase and decay of eEPSCs were significantly slowed in stg RTN cells (rise time, 0.57 ± 0.05 ms; half-width, 1.82 ± 0.18 ms; Fig. 4B,Ci,Cii,E,F) compared with WT cells (rise time, 0.46 ± 0.03 ms; half-width, 1.39 ± 0.15 ms; p < 0.005 for both; Fig. 4A,Ci,Cii,E,F). These differences persisted under conditions in which AMPAR-mediated currents were isolated (WT, n = 7 and stg, n = 5; amplitude: WT, 270.4 ± 60.9 pA and stg, 80.1 ± 14.1 pA, p < 0.005; rise time: WT, 0.41 ± 0.02 ms and stg, 0.58 ± 0.10 ms, p < 0.05; half-width: WT, 1.06 ± 0.07 ms and stg, 2.04 ± 0.02 ms, p < 0.05; along with prolonged decay of the current to baseline, 10–90 decay time: WT, 1.85 ± 0.20 ms and stg, 4.45 ± 1.25 ms, p < 0.05; Fig. 4Ciii), further supporting a change in AMPAR location, number, or subunit composition.
Figure 4.

Slow evoked excitatory currents in stg RTN cells. EPSCs evoked at threshold in the RTN by stimulating the internal capsule in WT (A) and stg (B). Black circle denotes stimulus artifact. Average of successful responses from WT (in black) and stg (in gray) aligned to response onset (Ci) or aligned and normalized to peak (Cii). Ciii, Averaged isolated AMPAR (in the presence 50 μm APV and picrotoxin) responses from WT (in black) and stg (in gray) aligned to start of event (solid gray) or aligned and normalized to peak (gray dashed). D–G, Box-and-whisker plots to show mean amplitude (D), rise time (E), half-width (F), and weighted decay time constant (G) of EPSCs (no 50 μm APV). Amplitude and kinetics of the evoked events are, respectively, smaller and slower in stg cells. H–J, Mean responses to trains of stimuli (10 trains of 5 pulses at 200 Hz) applied to internal capsule at 1.5 times threshold and the subsequent mean responses in representative WT (black) and stg (gray) RTN cells. The stimulation number and artifacts are highlighted by numbers. The dashed line illustrates baseline current. Although the initial direct EPSC responses are smaller in stg cells, the residual current 50 ms subsequent to the train is larger (see arrows; H and K) and slower to decay to baseline (arrows show time at which the current reaches baseline for each cell; I and L) in stg cells than WT cells. The residual current is NMDAR dependent because it was blocked by application of an NMDAR antagonist APV (J; stg control condition in gray and after application of APV in black). *p < 0.05, **p < 0.005, ***p < 0.0005.
The eEPSCs in stg and WT RTN cells showed a double-exponential decay, with a fast and a slow phase. The more rapidly decaying response could be blocked by AMPAR antagonists (NBQX or DNQX, 20 μm; WT, 17 of 17 cells and stg, 17 of 17 cells tested). The slower decay component was blocked by APV (50 μm), indicating that it was NMDAR mediated. The weighted decay time constant for eEPSCs were slower in stg compared with WT RTN cells (WT, 4.5 ± 0.7 ms and stg, 10.9 ± 1.4 ms, p < 0.0005; Fig. 4G). The much slower decay was predominantly attributable to a doubling of the slow time constant in stg (WT, 42.9 ± 6.1 ms and stg, 87.6 ± 10.7 ms, p < 0.0005), whereas the fast time constant was not affected (WT, 1.1 ± 0.2 ms and stg, 1.6 ± 0.2 ms, p > 0.05). This result suggests that not only is the AMPAR component of the eEPSC reduced in amplitude and increased in duration in stg cells but also that there is increased relative contribution of the NMDAR-mediated component of the eEPSC. Interestingly, although the amplitude of the eEPSC was significantly reduced in the stg RTN cells compared with WT cells, the net synaptic charge of the response was unaffected (WT, 682.0 ± 121.9 pA/ms and stg, 1259.4 ± 434.1 pA/ms; isolated AMPAR: WT, 363.1 ± 71.3 pA/ms and stg, 227.0 ± 70.4 pA/ms; both p > 0.05), suggesting that the prolonged duration of the response compensated for the decreased amplitude.
In the thalamic network, excitatory inputs to RTN cells from thalamus or cortex arise from either single action potentials or bursts. Accordingly, we determined the response of RTN cells to burst input in stg compared with WT cells (Fig. 4H–L). In these experiments, we applied trains of five stimuli at 200 Hz (producing responses similar to those produced by a burst firing thalamic cell; Bal et al., 1995) to the internal capsule (at 1.5 times threshold to eliminate failures). During the train, the first stimulation always resulted in a direct, fast EPSC (this was mostly AMPAR dependent because it was always blocked by 20 μm NBQX; data not shown), but subsequent responses within the train could be either failures or successes. During the train, most WT cells (n = 18 of 21) and stg cells (n = 25 of 25) exhibited depression of the second through fifth direct responses compared with the response of the first stimulation (3 of 21 WT cells, but none of the 25 stg cells, exhibited facilitation on the second stimulation). After the train stimulation, cells from both stg (n = 25) and WT (n = 21) exhibited a residual current that slowly decayed to baseline (Fig. 4H,I). This residual current was slower in stg cells (decay time constant: WT, 59.4 ± 4.1 ms and stg, 93.0 ± 7.9 ms; p < 0.005; Fig. 4K,L) and was blocked by an NMDAR antagonist (APV, 50 μm) in both WT cells (5 of 5 cells; data not shown) and stg cells (7 of 7 cells; Fig. 4J). It is worth noting that, although NBQX (20 μm) application could eliminate the fast direct responses during the train, it did not eliminate the slow post-train response (which was further blocked by APV application, 50–100 μm; WT, 11 of 11 cells and stg, 11 of 11 cells; data not shown; but see Fig. 6). Thus, even at membrane potentials near rest, NMDAR-mediated currents can be evoked in RTN cells.
Figure 6.

Isolated NMDAR currents are enhanced in stg RTN cells. Representative examples of isolated evoked NMDAR EPSCs obtained in the presence of 10 mm NBQX from holding potentials between −100 and 40 mV in 5 mV increments in WT (A) and stg (B) RTN cells with the respective I–V curves for WT (black squares) and stg (white squares) on the left (C). I–V curves (D) and g–V plots (E) from pooled data show an increase in isolated NMDAR-mediated responses in stg RTN cells. The voltage dependency of the current is not changed as shown by using a Boltzmann function to fit the normalized conductance (F). Quantitative data represent mean ± SEM.
The results demonstrate that, although the reduced frequency of AMPAR-mediated sEPSCs and the decreased amplitude of both AMPAR-mediated sEPSCs and eEPSCs are all paradoxical to the promotion of hyperexcitability in the circuit, the increased duration of both the fast EPSCs (mostly AMPAR mediated) and the slower component of the EPSC (mostly NMDAR mediated) can result in a longer-lasting synaptic response mediated by both AMPARs and NMDARs. We predict that this longer-lasting current could result in hyperexcitability and oscillations in the thalamic network.
Compensation of reduced AMPAR current by NMDAR current
To assess the relative contribution of AMPAR and NMDAR conductance to excitatory transmission in the stg RTN, we obtained eEPSC I–V curves from both stg (n = 35) and WT littermate (n = 33) cells (Fig. 5). Cells were held at a range of potentials between −60 and 40 mV, and a single stimulus was applied to the internal capsule at 1.5 times threshold to eliminate failures. The early, rapidly decaying EPSC component was always blocked by AMPAR antagonists (NBQX or DNQX, 20 μm; WT, 11 of 11 and stg, 8 of 8 cells tested), and the later slow component was blocked by application of NMDAR antagonist (APV at 50–100 μm; WT, 11 of 11 and stg, 11 of 11 cells tested; Fig. 4Ciii).
Figure 5.

Decreased AMPA conductance but increased NMDA conductance in stg RTN eEPSCs. Representative examples of eEPSCs from several holding potentials (−60, −30, 30, and 40 mV) at different timescales in WT (Ai, Aii) and stg (Bi, Bii) RTN cells with the respective I–V curves for AMPAR (black squares) and NMDAR (white squares) components on the right (Aiii, Biii). AMPAR currents (Ci) and conductance (Cii) are reduced in stg cells (white squares) compared with WT (black squares). Ciii, Box-and-whisker plot of mean AMPAR conductance. NMDAR currents (Di) and conductance (Dii) are increased in stg cells (white squares) compared with WT (black squares). Diii, Box-and-whisker plot of maximum NMDAR conductance (mean conductance at 20–40 mV) in WT and stg RTN cells. E, Box-and-whisker plot showing that the NMDAR/AMPAR ratio at 20–40 mV is increased in stg cells. *p < 0.05, ***p < 0.0005.
In WT cells, at negative potentials, the response was composed of a large fast AMPAR component and a small NMDAR late component. At positive potentials, the fast AMPAR component remained clearly distinguishable from the later onset and larger NMDAR current component (Fig. 5A). However, in stg cells (Fig. 5B), at negative potentials, the AMPAR component was small and the NMDAR component was increased compared with the WT. This difference was more apparent at positive potentials, in which the AMPAR component was rarely discernible compared with the larger, slower NMDAR current. Indeed, the population data clearly show that the AMPAR current and conductance were significantly reduced at all potentials in the stg RTN cells compared with the WT littermates (p < 0.0005; Fig. 5C). Furthermore, in agreement with the eEPSC data above, the NMDAR conductance was significantly increased in the stg RTN cells compared with WT cells (Fig. 5D). This increase was most apparent at positive holding potentials when NMDARs were primarily relieved of magnesium block (mean NMDAR conductance, measured in the range of +20 to +40 mV: WT, 4.0 ± 0.6 nS and stg, 6.5 ± 1.0 nS; p < 0.05, Fig. 5Diii) but was also present around resting membrane potentials (measured in the range of −50 to −60 mV: WT, 0.73 ± 0.11 nS and stg, 1.11 ± 0.21 nS; p < 0.05). The decrease in AMPAR conductance, coupled with the increase in NMDAR conductance, contributes to significantly greater NMDA/AMPA ratio in stg RTN cells (ratio of NMDA/AMPA conductance averaged across responses between +20 and +40 mV; WT, 1.1 ± 0.1 and stg, 3.0 ± 0.4; p < 0.0005, Fig. 5E).
Although the above experiments suggest an increase in the NMDAR component of the composite EPSC response in RTN of stg, we wanted to examine the NMDAR EPSCs in detail without contamination of its early phase by AMPAR-mediated currents. Thus, we conducted the same experiments in the presence of the specific AMPAR antagonist, NBQX (20 μm), to isolate the NMDAR component (Fig. 6). In these experiments, we obtained eEPSCs across a wide range of potentials between −100 and +40 mV to elucidate more accurately the voltage dependency of the NMDAR current (Fig. 6A–C). Consistent with the results analyzing the presumed AMPAR and NMDAR components of the composite EPSC, we found that the peak NMDAR current and conductance was significantly increased in stg (n = 6) compared with WT (n = 7) RTN cells (mean conductance at +20 to +40 mV: WT, 4.1 ± 1.5 nS and stg, 10.6 ± 3.2 ns; p < 0.05; see Fig. 6D,E; and mean conductance −50 to −60 mV: WT, 0.43 ± 0.10 nS and stg, 1.22 ± 0.36 nS; p < 0.05). However, as shown by the Boltzmann fit to the normalized g–V curve (WT, n = 7 and stg, n = 6 cells; R2 = 0.99 for both fits; Fig. 6F), neither the slope nor half-maximal activation voltage of the NMDA conductance were different (slope: WT, 12.6 mV and stg, 11.8 mV; V50%: WT, −30.1 mV and stg, −28.7 mV), suggesting that there were no compensatory changes in the composition of the NMDARs but rather an increase in the number of receptors that could be activated by synaptically released glutamate.
These data suggest that, although the synaptic AMPAR response is decreased in stg RTN cells, the increased duration of the AMPAR response and the increased amplitude of the NMDAR component could together lead to a more hyperexcitable circuit and oscillatory activity.
Synaptic NMDAR recruitment is maximal with minimal stimulation
The data thus far provide compelling evidence that increased NMDAR activation in stg RTN cells could predispose the circuit to become hyperexcitable. After either a single or burst of presynaptic spikes that trigger glutamate release, stg RTN cells respond with an enhanced long-lasting NMDAR-mediated current (Fig. 4). Thus, we were interested in determining how NMDARs might be recruited by repetitive glutamate release during presynaptic spike trains (Fig. 7). In general, we found that NMDARs could be progressively activated during a synaptic stimulus train in WT RTN neurons such that there was a more than threefold increase in NMDA conductance (Fig. 7C; black bar, WT; white bar, stg) evoked during a five stimulus train (white squares) compared with the response to a single (black squares) stimulus (ratio of train response/single simulation response, 3.20 ± 0.72; Fig. 7Ai,Aii,C). However, in stg cells, very little if any additional NMDAR recruitment was observed during stimulus trains (white squares), in which the ratio of train response/single (black squares) response was 1.4 ± 0.13 (p > 0.5; Fig. 7Bi,Bii,C). These data suggest that, in stg, a single presynaptic spike achieves close to maximal NMDAR activation, whereas in WT cells, to achieve similar NMDAR current, a greater input, such as a burst of presynaptic activity, is necessary to achieve similar NMDAR currents. In support of this theory, the maximal current achieved during trains in WT cells (298.31 ± 109.62 pA) is similar to that achieved in stg cells after a single input (359.43 ± 109.94 pA; p > 0.5). This enhanced activation of NMDARs in stg RTN cells may make the network more hyperexcitable and lead to a condition in which epileptiform responses could be readily initiated by cortical and/or thalamic activity that would normally not strongly activate RTN.
Figure 7.

NMDAR current is maximally activated by single evoked synaptic release events in stg but not WT RTN cells. Evoked NMDAR EPSCs (holding at +40 mV) after single stimulation (black) or train of five stimuli at 200 Hz (gray) in representative WT (Ai) and stg (Bi) RTN cells. Aii, Bii, NMDAR g–V plots for single (black squares) and train (white squares). The maximum conductance is increased greatly in WT cells from single to train responses (Aii) but not in stg (Bii) cells. C, Populations data showing ratio of conductance in train/single response of WT (black bar) and stg (white bar) pooled data. Dashed line indicates unity. *p < 0.05.
Specific reductions in AMPAR responses at intrathalamic synapses
Electrical stimulation in the internal capsule activates both corticothalamic and thalamocortical axons (via orthodromic and antidromic activation, respectively) and thus does not allow to discriminate between these two sources of excitatory input onto RTN neurons. To address the possibility that AMPAR- and NMDAR-mediated EPSCs in RTN cells differ after corticothalamic versus thalamocortical axon activation synapses onto RTN cells, we probed the latter to investigate intrathalamic synapses (i.e., those arising wholly within the thalamic subcircuit) using LSPS of caged glutamate (Deleuze and Huguenard, 2006). We selectively activated thalamic relay neurons via MNI-glutamate photolysis in the VB. LSPS has some advantages over electrical stimulation because the former is relatively specific for somatodendritic activation (Katz and Dalva, 1994), whereas the latter primarily activates axons of passage (Nowak and Bullier, 1998a,b). To validate LSPS for the purposes of examining intrathalamic excitation, we characterized the direct excitation of relay neurons using cell-attached recordings and LSPS directly onto the recorded cells. In relay neurons, LSPS regularly triggered burst firing rather than single action potentials (Fig. 8Aii,Aiii) and therefore approximated train stimulation described above. Burst characteristics were similar in WT and stg relay cells (Fig. 8Aii,Aiii) with no significant differences (p > 0.05) in interspike intervals (ISIs) during bursts between WT (ISI1, 6.4 ± 0.8; ISI2, 7.8 ± 1.6; ISI3, 13.3 ± 4.0) and stg (ISI1, 7.1 ± 1.0; ISI2, 9.3 ± 1.5; ISI3, 27.4 ± 12.1), suggesting that the stg mutation did not affect relay neuron excitability. We then recorded RTN cells in slices from WT and stg and scanned VB using LSPS until we found a location from which short latency, fast EPSCs were reliably evoked. We repeatedly stimulated these spots to record I–V curves (with 15 mV steps from −75 to 60 mV). Figure 8Bi shows examples of synaptic responses obtained in representative WT and stg RTN cells at −60 and +45 mV. The responses at −60 mV were dominated by fast, presumably AMPAR-mediated, EPSCs, which were much larger in WT compared with stg cells. Responses at +45 mV displayed large, slow currents typical of NMDAR activation and were of similar magnitude in both genotypes. Analysis of the full g–V curves revealed significantly smaller conductance of AMPAR-mediated currents (Fig. 8Dii, left, first discernible fast EPSC in each burst) but no significant difference in NMDAR-mediated currents (Fig. 8Bii, right) in stg cells. The identity of AMPAR- and NMDAR-mediated components of synaptic responses was confirmed by sensitivity to APV and NBQX, respectively, in selected cells (data not shown). The lack of enhanced NMDA response with LSPS compared with evoked responses (Figs. 5, 6) presumably reflects the fact that the former activates presynaptic burst spikes in relay neurons (Fig. 8C), conditions under which the NMDAR response differences are minimized (Fig. 7). We calculated NMDA/AMPA charge ratios for EPSCs in RTN cells evoked by LSPS in VB and defined the AMPA component as the area of all isolated fast EPSCs recorded at −60 mV and the NMDA component as the area of the response at +60 mV from 100 to 500 ms after the LSPS stimulus. NMDAR/AMPAR charge ratios were significantly larger in stg (WT, 82.6 ± 16.7, n = 6; stg, 618.3 ± 154.6, n = 7; p < 0.01). These results are consistent with those obtained using electrical stimulation: individual AMPAR-mediated EPSCs had smaller conductances in stg RTN neurons, but the NMDAR-mediated currents after a burst stimulus did not differ. Therefore, characteristics of intrathalamic EPSCs are representative of electrically eEPSCs in RTN cells.
Figure 8.
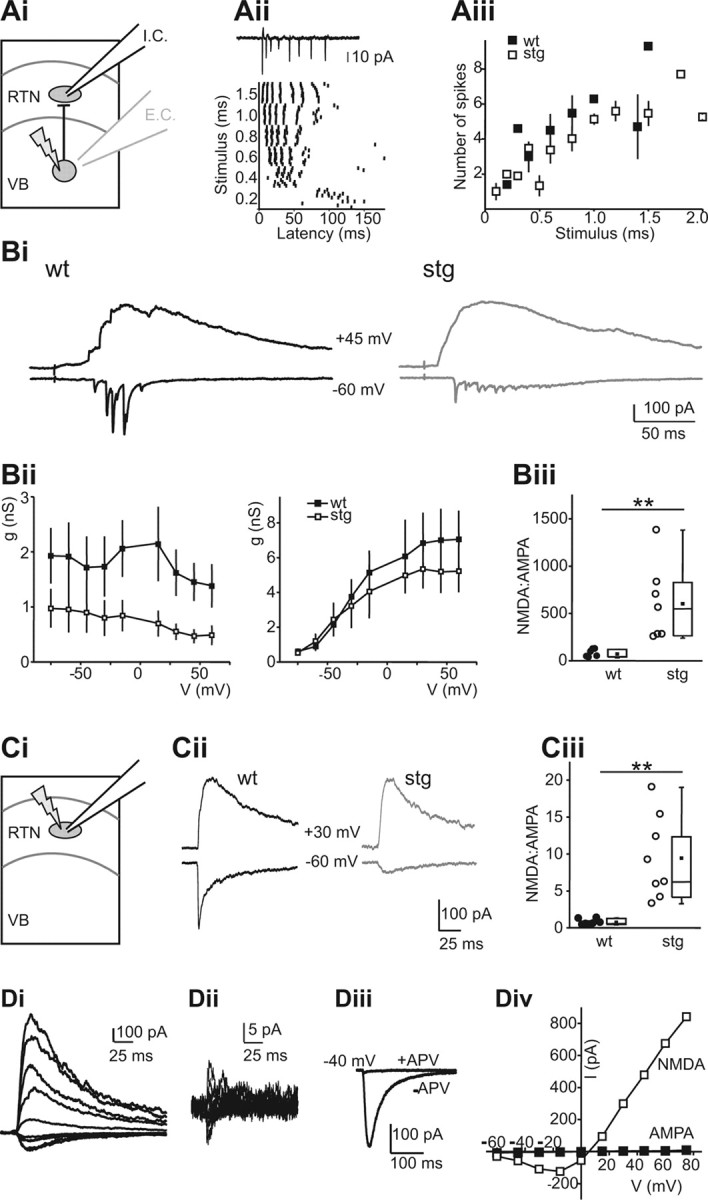
Specific reductions in AMPAR responses at intrathalamic synapses. Ai, Activation of synaptic currents in RTN neurons resulting from LSPS induced burst firing in VB. E.C. indicates extracellular recording from VB neurons (gray electrode in VB, Aii, Aiii), and I.C. indicates intracellular recording from RTN neurons (black electrode in RTN, Bi–Biii). Aii, Action potentials in a WT VB neuron in response to LSPS. Action potentials were recorded via an extracellular recording in cell-attached mode (see trace on top). The graph (bottom) shows spike times at different duration stimuli in that neuron. VB neurons typically fired action potential bursts in response to LSPS. Aiii, Comparison of action potential bursts generated in VB neurons of WT (n = 4) and stg (n = 6) reveals no significant differences. Bi, LSPS eEPSCs in WT (left) and stg (right) RTN neurons. EPSCs were evoked at −60 mV (bottom traces, mainly AMPA) and +45 mV (top traces, mainly NMDA). Bii, AMPAR (left) and NMDAR (right) conductance at different holding potentials in WT (n = 6) and stg (n = 5) RTN cells. AMPA, but not NMDAR, conductance is reduced in stg. Biii, NMDA/AMPA ratio EPSCs in WT and stg RTN neurons. Higher ratio in stg is attributable to a smaller AMPA component (WT, 82.6 ± 16.7, n = 6; stg, 618.3 ± 154.6, n = 7; p < 0.01). Ci, Direct activation of RTN neurons by LSPS. Cii, Direct uncaging responses evoked at holding potentials of −60 and +30 mV in representative WT and stg (stg−/−) neurons. Note absence of fast, presumably AMPAR-mediated, component in stg. Ciii, NMDA/AMPA ratios were significantly greater in stg, reflecting the decreased AMPA component (WT, 0.76 ± 0.15, n = 7; stg, 9.44 ± 2.0, n = 8; p < 0.01). Di, Direct currents in an stg RTN neuron at holding potentials between −60 and +60 mV. Dii, Direct current in the same neuron, after application of 50 mm d-APV, evoked as in Bi. Diii, Comparison of glutamatergic currents evoked at −40 mV before and after NMDAR block. Div, I–V curve for NMDAR- and AMPAR-mediated currents (peak currents in Di and Dii, respectively). **p < 0.01.
We also examined the properties of AMPAR- and NMDAR-mediated direct responses in WT and stg RTN cells (Fig. 8C,D), by directly releasing glutamate onto RTN cells that were recorded in voltage-clamp mode. This type of stimulation eliminates the presynaptic component, but it primarily activates nonsynaptic neurotransmitter receptors because synapses are apparently shielded from LSPS-released glutamate, perhaps by surrounding glia (Brill and Huguenard, 2008). In contrast to electrically eEPSCs, we could not easily detect a fast AMPAR-mediated component at negative membrane potentials in stg cells, whereas the AMPA component was readily identifiable in WT cells (Fig. 8Cii). Consequently, the NMDA/AMPA ratio in stg cells was greatly increased (Fig. 8Ciii). NMDAR blockade revealed an extremely small residual AMPAR-mediated current in stg cells (Fig. 8, Di before and Dii after application of 50 μm APV; Fig. 8Diii shows comparison of currents recorded at −40 mV), resulting in an almost negligible AMPAR-mediated component of the directly evoked nonsynaptic response in stg cells at all membrane potentials tested (Fig. 8Div). This further underscores that the stg mutation leads to a severe trafficking defect of AMPARs in RTN but leaves NMDAR responses intact.
Increasing NMDAR activity in WT slices mimics stg slice hyperexcitability
The above experiments with single RTN cells suggest that compensation of reduced AMPAR activity by increased NMDAR activation in stg could, in part, create a hyperexcitable thalamic network. Because of these results and the fact that MK-801 (a noncompetitive NMDAR antagonist) can reduce seizures in stg but not lethargic mice or in the γ-hydroxybutyric acid pharmacological model of absence seizures (Aizawa et al., 1997), we tested whether blocking NMDAR activity could eliminate the hyperoscillatory activity seen in the stg slice network preparation (Fig. 9). Lowering the concentration of magnesium in the bath solution promotes NMDAR activation and thalamic oscillations (Jacobsen et al., 2001). We found that, in conditions of low extracellular magnesium, we could reliably evoke oscillations in WT (n = 14) and stg (n = 10) slices. Under these pro-oscillatory conditions, the duration of the evoked oscillations in WT was increased to the point (1.82 ± 0.25 s) that it was indistinguishable from the responses in stg slices (1.42 ± 0.12 s; p > 0.05). Thus, a manipulation that would be expected to increase NMDAR recruitment was able to phenocopy in WT slices the stg epileptic phenotype. Interestingly, although the duration of the oscillations was significantly increased from normal to low magnesium concentrations in WT (p < 0.005), the change of concentration had no effect on stg oscillation duration (p > 0.05; Fig. 9B), again consistent with the idea that NMDAR activation is near maximal under baseline conditions in stg RTN cells (Fig. 7). We found, as reported previously (see rat and ferret: von Krosigk et al., 1993), that application of APV (50 μm) reversibly reduced oscillation duration similarly in the thalamic network in both WT slices (n = 9; control, 1.38 ± 0.14 s; APV, 0.62 ± 0.06 s; wash, 0.85 ± 0.10 s; p < 0.0001; Fig. 9Ai) and stg slices (n = 6; control, 1.98 ± 0.38 s; APV, 0.40 ± 0.17 s; wash, 0.69 ± 0.27 s; p ≤ 0.005; Fig. 9Aii). Under normal magnesium conditions, APV strongly reduced the duration of the oscillation in stg slices (n = 6; control, 1.73 ± 0.61 s; APV, 0.37 ± 0.11 s; wash, 0.75 ± 0.21 s; p ≤ 0.0005; data not shown) and abolished spontaneous oscillatory activity (data not shown), but the affect of APV on normal magnesium oscillations was not tested in WT because oscillations were rarely obtained under these conditions. These results suggest that, in thalamic slices from stg, elevated NMDAR activity is a critical factor in the generation of epileptiform oscillations.
Figure 9.
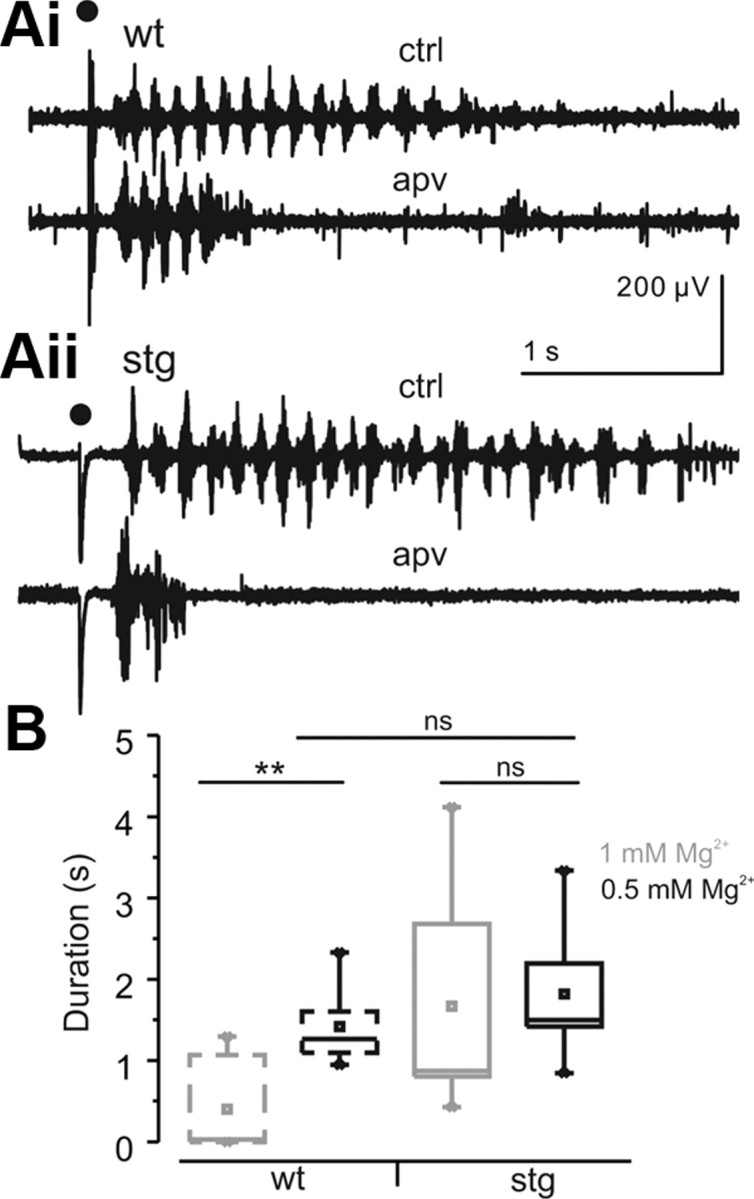
Hyperexcitability in stg thalamic slices can be phenocopied in WT thalamic slices by activating NMDAR. A, Sample traces of thalamic (RTN and VB) WT (Ai) and stg (Aii) control oscillations and oscillations evoked after application of APV (50 μm). ACSF contains low concentrations of magnesium (0.5 mm). Stimulus is indicated by black dot. B, Averaged population responses show low concentrations of magnesium (0.5 mm) selectively prolonged WT oscillation (WT, 1 mm Mg2+, n = 7; WT, 0.5 mm Mg2+, n = 14; stg, 1 mm Mg2+, n = 14; stg, 0.5 mm Mg2+, n = 10). **p < 0.001. ns, Not significant.
Redistribution of AMPARs away from synapses in stg RTN
Given the physiological results that suggest a change in number and/or location of synaptic AMPAR and NMDAR in the RTN of stg, we took advantage of the powerful, recently developed, anatomical technique of array tomography (AT; Micheva and Smith, 2007; Micheva et al., 2010) to directly address this issue. We made ultrathin horizontal sections from both WT and stg thalamus that included dorsal thalamus, RTN, and internal capsule. The ultrathin sections made it impossible to determine the location of the borders of the RTN within the section with simple visual inspection. Therefore, we took advantage of the known differences in protein density of VGLUT1 to clearly delineate RTN from both the internal capsule (very low to no VGLUT1 immunolabeling) and dorsal thalamus (VB; high VGLUT1 immunolabeling) (Fig. 10Ai,Aii). To further verify localization, we stained with parvalbumin and confirmed the presence of densely packed parvalbumin-positive cells in the RTN (Fig. 10Aiii).
Figure 10.

AT reveals reductions in synaptic but increases in perisynaptic AMPARs in stg RTN. Low-magnification (10×) AT image of VGLUT1 immunolabeling of one 100 nm section of a ribbon from WT (Ai) and stg (Aii) thalamus. In each case, RTN can be clearly distinguished (white dotted lines) from the dorsal thalamus (VB; right) and internal capsule (ic; left) by the different intensities of VGLUT1 immunofluorescence. Extent of RTN was further verified at 63× by parvalbumin (PARV) labeling. Scale bars: Ai, Aii, 200 μm; Aiii, 10 μm). AT image from stack of ∼40 sections (100 nm) from WT (Bi) and stg (Bii) RTN immunostained for synaptic markers pan-MAGUK (pseudocolor red) and synapsin (psuedocolor blue) and AMPAR subunit GluA4 (pseudocolor green). Colocalization of the red and blue channels is displayed as magenta (“synapses”), and colocalization of all three proteins is displayed as white (white arrows depict some colocalized GluA4 containing “synapses”). VGLUT1 and VGLUT2 are not shown for clarity. Scale bars, 1 μm. C, Radial analysis of GluA4 particle density in relation to VGLUT1 (Ci) and VGLUT2 (Cii) positive “synapses,” defined as those containing pan-MAGUK, synapsin, and either VGLUT1 or VGLUT2 (black squares, WT; white squares, stg). D, Average density of “perisynaptic” (0.3–0.5 μm) GluA4 particles associated with VGLUT1 or VGLUT2 synapses in WT (black) and stg (gray). E, Ratio of perisynaptic/synaptic GluA4 puncta in WT (black) and stg (gray). *p < 0.05, **p < 0.005. ns, Not significant.
Our first aim was to determine the location of AMPARs in relationship to excitatory synapses in stg and WT, and we targeted the GluA4 subunit because this is the most abundant AMPAR subunit in the thalamus. The density and location of GluA4 subunits in relation to excitatory synapses was calculated by means of a radial analysis. Synapses were defined as triple colocalizations of immunopositive particles of synapsin, pan-MAGUK (a broad-spectrum antisera for membrane-associated guanylate kinase proteins, such as PSD95, SAP97, etc.; Gardoni, 2008) and either VGLUT1 (corticothalamic) or VGLUT2 (subcortical origin; Fremeau et al., 2004).
Consistent with our physiological results, the density of GluA4 subunits in RTN synapses (defined as within 0.05 μm of a synaptic colocalization) was less in stg (n = 5; white squares) compared with WT (n = 6; black squares) for both VGLUT1-positive (WT, 68.66 ± 7.88 μm3; stg, 34.87 ± 5.27 μm3; Fig. 10Ci) and VGLUT2-positive (WT, 63.31 ± 7.66 μm3; stg, 32.82 ± 5.35 μm3; Fig. 10Cii) synapses. Interestingly, for both VGLUT1- and VGLUT2-containing synapses, the radial profiles for WT and stg intersect: GluA4 consistently clustered at lower densities in stg synapses (all radial distances <0.2 μm) yet concentrated at higher densities in stg perisynaptic zones (between 0.3 and 0.5 μm from the center of the synapse). Accordingly, perisynaptic GluA4 was higher in stg (average density, 0.3–0.5 mm from synapses; mean VGLUT1 synapses, 1.86 ± 0.12 μm3; mean VGLUT2 synapses, 1.84 ± 0.12 μm3; Fig. 10D) compared with WT (mean VGLUT1 synapses, 1.37 ± 0.11 μm3; mean VGLUT2 synapses, 1.48 ± 0.10 μm3; both p < 0.05; Fig. 10D). In addition, the ratio of perisynaptic (all GluA4 particles >0.5 μm from any excitatory synapse, as defined as a VGLUT/synapsin/pan-MAGUK colocalization) to “synaptic” GluA4 receptors was twofold to threefold higher in stg (gray) than WT (black) for both VGLUT1 (WT, 0.03 ± 0.00; stg, 0.07 ± 0.02; p < 0.05; Fig. 10E) and VGLUT2 (WT, 0.02 ± 0.00; stg, 0.06 ± 0.01; p < 0.05; Fig. 10E) synapses. These results are consistent with an AMPAR trafficking defect in stg such that a reduction in centri-synaptic receptors is associated with greater perisynaptic receptors distal to the synapse, likely contributing to delayed and prolonged postsynaptic response kinetics. Importantly, using immunofluorescent confocal microscopy, we did not find a change in overall number of VGLUT boutons, synapsin puncta, or GluA4 puncta numbers in stg RTN (data not shown).
Next we wanted to identify whether there were any changes in NMDAR location and/or targeting. We found no differences overall in the location of the subunit NMDAR1 to excitatory synapses or amount of perisynaptic receptors, which is consistent with the finding that stargazin has not been shown to directly traffic NMDARs (Chen et al., 2000) and the fact that the maximally activated synaptic NMDAR response was unaffected in stg (Fig. 7). Cortical inputs to the RTN (as defined by VGLUT1-positive terminals and synapses; WT, 18.71 ± 3.46 μm3; stg, 23.56 ± 3.67 μm3; p > 0.05; data not shown) and thalamic inputs to the RTN (as defined by VGLUT2-positive terminals and synapses; WT, 21.53 ± 3.47 μm3; stg, 23.61 ± 3.8 μm3; p > 0.05; data not shown) were similar in terms of distribution and density of NMDAR1 to the synapse.
Discussion
stg display frequent SWDs, the physiological correlate of absence seizures. Stargazin can act as either a voltage-gated calcium channel subunit or an AMPAR auxiliary subunit (Letts, 2005; Osten and Stern-Bach, 2006; Tselnicker and Dascal, 2010), and its mutation in stg leads to suppression of AMPAR-mediated synaptic excitation in cerebellum (Chen et al., 2000) and RTN (Menuz and Nicoll, 2008), along with increased VB tonic inhibition (Cope et al., 2009). Absence seizures require RTN (Avanzini et al., 1992), and epileptiform network activity depends on RTN excitation to either sustain intrathalamic (Bal and McCormick, 1993; von Krosigk et al., 1993; Huguenard and Prince, 1994; Warren and Jones, 1994) or enhance thalamocortical (Bal et al., 2000; Blumenfeld and McCormick, 2000) seizure-related activity. Given the necessity for RTN excitation, how does the loss of stargazin and resultant decrease in RTN AMPAR excitation paradoxically lead to seizures? We demonstrate enhanced excitatory drive onto RTN neurons through augmented NMDAR excitation and increased activation of perisynaptic AMPARs. We suggest that such increased excitation of RTN (from cortex or thalamus) leads to increased phasic inhibitory output to VB, which in combination with the enhanced tonic inhibitory currents that are a common feature in absence models (Cope et al., 2009), results in enhanced synchronous oscillations that are critical for absence seizures.
Hypersynchronous oscillatory activity in stg slices: a thalamic locus
We show that adult stg thalamus displays aberrant hypersynchronous spontaneous and evoked oscillations in vitro, indicating that a thalamic excitability defect likely contributes to seizures in these animals. These enhanced oscillations result from excessive RTN excitation, from corticothalamic and/or thalamocortical projections, consistent with prominent and early RTN neuronal involvement during SWD (Slaght et al., 2002). Indeed, in vitro intrathalamic rhythmic oscillations are dependent on reciprocal synaptic interactions between glutamatergic dorsal thalamus and GABAergic RTN (von Krosigk et al., 1993; Huguenard and Prince, 1994; Warren and Jones, 1994; Jacobsen et al., 2001), and excitation of RTN is mediated by AMPARs and NMDARs (Liu, 1997; Mineff and Weinberg, 2000; Golshani et al., 2001).
Reduced AMPAR excitation of stg RTN is paradoxical to seizures
We demonstrate that AMPAR sEPSCs onto adult stg neurons are reduced in both frequency and amplitude (cf. Menuz and Nicoll, 2008). This reduction could indicate a general reduction in RTN AMPAR excitation, which would in turn reduce dorsal thalamic inhibition and the resulting postinhibitory rebound bursts. However, given that absence networks appear to be strictly dependent on inhibition (Huguenard, 2001) and that enhancing inhibition can actually worsen absence seizures (Hosford et al., 1997; Danober et al., 1998; Perucca et al., 1998; Ettinger et al., 1999; Cope et al., 2009), such a reduction seems unlikely to be solely responsible for stg hyperexcitability. Indeed, we found that, despite RTN AMPAR amplitude reductions (spontaneous and evoked), total AMPAR eEPSC charge transfer at resting membrane potential onto stg RTN cells remained unchanged as a result of increased duration. The loss of stargazin does not directly account for the prolonged AMPAR kinetics because stargazin itself modulates AMPAR biophysical properties to increase channel open time (also reducing desensitization and increasing glutamate affinity; Tomita et al., 2003, 2005, 2007; Turetsky et al., 2005; Kott et al., 2007), so that its loss would be associated with briefer synaptic responses. We show that the maintained eEPSC charge transfer is accompanied by a shift in AMPAR synaptic localization and augmented NMDAR activation.
Loss of stargazin disrupts AMPAR synaptic localization
Stargazin forms a complex with AMPARs and interacts with PSD-95 and other MAGUKs to maintain AMPARs at the synapse (Schnell et al., 2002; Deng et al., 2006; Bats et al., 2007). Thus, in stg RTN, loss of functional stargazin is expected to reduce the synaptic stabilization of membrane-bound AMPARs (Opazo et al., 2011), and indeed a recent study demonstrated a reduction of synaptic GluA4 immunogold particles in stg RTN (Barad et al., 2012).
Our AT data supports this hypothesis, revealing a shift of GluA4 away from synaptic regions, accounting for both the frequency reduction and slower kinetics of stg RTN AMPAR sEPSCs. A predicted consequence of this shift is that responses to evoked glutamate release will be less affected than spontaneous single vesicle release events. This distinction arises because evoked synaptic activation promotes pooling of neurotransmitter in intersynaptic regions, allowing more reliable activation of nonsynaptic receptors (Huang et al., 1998; Kullmann, 2000). Our electrophysiological results support this hypothesis; evoked AMPAR EPSCs were reliably recorded in stg RTN neurons, and observed reductions in amplitude were less than expected given the large changes in AMPAR sEPSCs.
However, the stargazin mutation causes AMPAR localization defects that are not entirely accounted for by reduced synaptic stabilization. Notably, preferential activation of extrasynaptic glutamate receptors, achieved via LSPS (Brill and Huguenard, 2010), failed to produce a significant AMPAR-mediated response in stg RTN cells, indicating an additional loss of AMPARs from extrasynaptic regions, consistent with the role of transmembrane AMPA receptor regulatory proteins (TARPs) in trafficking AMPARs to the membrane (Chen et al., 2000; Tomita et al., 2003, 2005; Turetsky et al., 2005; Bats et al., 2007; Ziff, 2007; Jackson and Nicoll, 2011). Immunofluorescent confocal microscopy revealed no overall reduction in the number of GluA4 puncta in RTN (cf. Barad et al., 2012) suggesting that, without stargazin, AMPARs still accumulate at or near the membrane, consistent with AMPARs associating with non-stargazin TARPs (Tomita et al., 2003; Cho et al., 2007; Kott et al., 2007; Kato et al., 2010), cornichons (Schwenk et al., 2009; Kato et al., 2010), or SAP97 (Howard et al., 2010), or being “TARPless” (Bats et al., 2012). Non-stargazin interactions may cause trafficking of AMPARs into “perisynaptic” regions, in which glutamate metabolism is well regulated by astrocytes (Schousboe et al., 2004), and AMPARs would be unresponsive to both spontaneous glutamate release events and to extrasynaptic glutamate release via LSPS.
NMDAR current is the critical source of excitatory drive in stg RTN
We demonstrate prolonged and increased NMDAR eEPSCs in stg RTN cells at rest, and we propose that this enhancement in NMDAR-mediated drive acts as a major contributing factor to hyperexcitability in stg, consistent with stg seizures being particularly susceptible to NMDAR blockade (Aizawa et al., 1997). In WT RTN cells, the slow NMDAR eEPSCs summate during presynaptic trains as a result of cumulative glutamate release, such that the composite response to train stimulation is threefold larger than a single evoked response. In contrast, nearly the entire pool of NMDARs is activated by single eEPSC in stg RTN cells. Consistent with a role of greater NMDAR activation in enhanced thalamic excitability, we found that lowering [Mg2+]o resulted in fourfold longer oscillations in WT slices (cf. Jacobsen et al., 2001), shifting WT toward the same NMDAR dependence as stg slices (stg thalamic oscillations were not enhanced by reducing [Mg2+]o). Indeed, an in vivo study demonstrated that a noncompetitive selective antagonist of NMDAR (MK-801) greatly reduced stg seizures but not other mouse models (Aizawa et al., 1997) and is further supported in rat absence models (Koerner et al., 1996). In combination, our data lead us to propose that enhanced RTN NMDAR excitation is a main contributor to the generation of seizures in stg.
Although there has been little evidence to suggest that stargazin directly affects NMDARs (Chen et al., 2000; Menuz and Nicoll, 2008), synaptic targeting of NMDARs may be dependent on synaptic AMPAR/stargazin interactions, especially in inhibitory cells (Mi et al., 2004), and NMDAR-related increases in calcium influx suppresses stargazin-mediated AMPAR lateral diffusion (Opazo et al., 2010). NMDARs have a high affinity for glutamate (∼500-fold higher than AMPARs; Patneau and Mayer, 1990) and may respond more effectively than AMPARs to glutamate in extrasynaptic locations. Larger glutamate “spillover” (Atasoy et al., 2008) may occur in stg RTN that lack synaptic AMPARs, resulting in more activation of NMDARs, and this high NMDAR activity may result in additional immobilization of synaptic AMPARs (cf. Opazo et al., 2011). Alternatively, lack of stargazin anchoring of AMPARs to the synapse might result in reduced competition with NMDARs for postsynaptic scaffolding proteins, resulting in more centri-synaptic NMDARs and larger recruitment in stg (cf. Bard et al., 2010). Our AT results suggest comparable NMDAR number/location in stg and WT RTN, but the resolution may be insufficient to detect small changes. The NMDAR-current enhancements may be attributable to a larger pool of activated NMDARs, perhaps as a secondary compensatory mechanism during development resulting from reduced AMPAR activity (Ben-Ari et al., 1997).
Significance
We have established a novel and powerful mechanism by which thalamic excitability is generated in stg. Despite reductions in amplitude of AMPAR-mediated responses in stg, we report increased NMDAR activation of RTN neurons and suggest that this is a critical source of hyperexcitability in the thalamocortical network. These changes are accompanied by shifts in AMPARs to less centri-synaptic locations in stg RTN neurons in which they can still be activated, although somewhat less effectively, by synaptically released glutamate. Our data strongly suggest that the enhanced NMDAR response in RTN to excitatory inputs (be it cortical or thalamic) can lead to hypersynchronous oscillatory activity in the corticothalamic circuit and, potentially, seizures. Our findings suggest that regulation of TARP-mediated AMPAR function is likely to be important in understanding of epilepsy and potential treatments.
Footnotes
This work was supported by National Institutes of Health Grant NS34774. C.J.L. is the recipient of an Epilepsy Foundation Eric Lotham Postdoctoral Research Fellowship. We thank Isabel Parada for expert histological assistance and advice, Tim Lew and Kathy Peng for genotyping and breeding assistance, and Koji Takahashi and Nick Weiler for invaluable discussion and advice on array tomography data analysis.
References
- Adams BE, Kyi M, Reid CA, Myers DE, Xu S, Williams DA, O'Brien TJ. Seizure-like thalamocortical rhythms initiate in the deep layers of the cortex in a co-culture model. Exp Neurol. 2011;227:203–209. doi: 10.1016/j.expneurol.2010.11.001. [DOI] [PubMed] [Google Scholar]
- Aizawa M, Ito Y, Fukuda H. Pharmacological profiles of generalized absence seizures in lethargic, stargazer and gamma-hydroxybutyrate-treated model mice. Neurosci Res. 1997;29:17–25. doi: 10.1016/s0168-0102(97)00066-7. [DOI] [PubMed] [Google Scholar]
- Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci. 2002;5:325–331. doi: 10.1038/nn825. [DOI] [PubMed] [Google Scholar]
- Atasoy D, Ertunc M, Moulder KL, Blackwell J, Chung C, Su J, Kavalali ET. Spontaneous and evoked glutamate release activates two populations of NMDA receptors with limited overlap. J Neurosci. 2008;28:10151–10166. doi: 10.1523/JNEUROSCI.2432-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avanzini G, de Curtis M, Marescaux C, Panzica F, Spreafico R, Vergnes M. Role of the thalamic reticular nucleus in the generation of rhythmic thalamo-cortical activities subserving spike and waves. J Neural Transm Suppl. 1992;35:85–95. doi: 10.1007/978-3-7091-9206-1_6. [DOI] [PubMed] [Google Scholar]
- Bal T, McCormick DA. Mechanisms of oscillatory activity in guinea-pig nucleus reticularis thalami in vitro: a mammalian pacemaker. J Physiol. 1993;468:669–691. doi: 10.1113/jphysiol.1993.sp019794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal T, von Krosigk M, McCormick DA. Role of the ferret perigeniculate nucleus in the generation of synchronized oscillations in vitro. J Physiol. 1995;483:665–685. doi: 10.1113/jphysiol.1995.sp020613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal T, Debay D, Destexhe A. Cortical feedback controls the frequency and synchrony of oscillations in the visual thalamus. J Neurosci. 2000;20:7478–7488. doi: 10.1523/JNEUROSCI.20-19-07478.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barad Z, Shevtsova O, Arbuthnott GW, Leitch B. Selective loss of AMPA receptors at corticothalamic synapses in the epileptic stargazer mouse. Neuroscience. 2012;217:19–31. doi: 10.1016/j.neuroscience.2012.05.011. [DOI] [PubMed] [Google Scholar]
- Bard L, Sainlos M, Bouchet D, Cousins S, Mikasova L, Breillat C, Stephenson FA, Imperiali B, Choquet D, Groc L. Dynamic and specific interaction between synaptic NR2-NMDA receptor and PDZ proteins. Proc Natl Acad Sci U S A. 2010;107:19561–19566. doi: 10.1073/pnas.1002690107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bats C, Groc L, Choquet D. The interaction between Stargazin and PSD-95 regulates AMPA receptor surface trafficking. Neuron. 2007;53:719–734. doi: 10.1016/j.neuron.2007.01.030. [DOI] [PubMed] [Google Scholar]
- Bats C, Soto D, Studniarczyk D, Farrant M, Cull-Candy SG. Channel properties reveal differential expression of TARPed and TARPless AMPARs in stargazer neurons. Nat Neurosci. 2012 doi: 10.1038/nn.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beenhakker MP, Huguenard JR. Astrocytes as gatekeepers of GABAB receptor function. J Neurosci. 2010;30:15262–15276. doi: 10.1523/JNEUROSCI.3243-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Khazipov R, Leinekugel X, Caillard O, Gaiarsa JL. GABAA, NMDA and AMPA receptors: a developmentally regulated “menage a trois.”. Trends Neurosci. 1997;20:523–529. doi: 10.1016/s0166-2236(97)01147-8. [DOI] [PubMed] [Google Scholar]
- Beyer B, Deleuze C, Letts VA, Mahaffey CL, Boumil RM, Lew TA, Huguenard JR, Frankel WN. Absence seizures in C3H/HeJ and knockout mice caused by mutation of the AMPA receptor subunit Gria4. Hum Mol Genet. 2008;17:1738–1749. doi: 10.1093/hmg/ddn064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenfeld H, McCormick DA. Corticothalamic inputs control the pattern of activity generated in thalamocortical networks. J Neurosci. 2000;20:5153–5162. doi: 10.1523/JNEUROSCI.20-13-05153.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brill J, Huguenard JR. Sequential changes in AMPA receptor targeting in the developing neocortical excitatory circuit. J Neurosci. 2008;28:13918–13928. doi: 10.1523/JNEUROSCI.3229-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brill J, Huguenard JR. Enhanced infragranular and supragranular synaptic input onto layer 5 pyramidal neurons in a rat model of cortical dysplasia. Cereb Cortex. 2010;20:2926–2938. doi: 10.1093/cercor/bhq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant AS, Li B, Beenhakker MP, Huguenard JR. Maintenance of thalamic epileptiform activity depends on the astrocytic glutamate-glutamine cycle. J Neurophysiol. 2009;102:2880–2888. doi: 10.1152/jn.00476.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- Chen S, Diamond JS. Synaptically released glutamate activates extrasynaptic NMDA receptors on cells in the ganglion cell layer of rat retina. J Neurosci. 2002;22:2165–2173. doi: 10.1523/JNEUROSCI.22-06-02165.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetkovich D. Thalamic reticular neurons are unexcited by new stargazer seizure mechanism. Epilepsy Curr. 2009;9:59–61. doi: 10.1111/j.1535-7511.2008.01293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho CH, St-Gelais F, Zhang W, Tomita S, Howe JR. Two families of TARP isoforms that have distinct effects on the kinetic properties of AMPA receptors and synaptic currents. Neuron. 2007;55:890–904. doi: 10.1016/j.neuron.2007.08.024. [DOI] [PubMed] [Google Scholar]
- Cope DW, Di Giovanni G, Fyson SJ, Orbán G, Errington AC, Lorincz ML, Gould TM, Carter DA, Crunelli V. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med. 2009;15:1392–1398. doi: 10.1038/nm.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danober L, Deransart C, Depaulis A, Vergnes M, Marescaux C. Pathophysiological mechanisms of genetic absence epilepsy in the rat. Prog Neurobiol. 1998;55:27–57. doi: 10.1016/s0301-0082(97)00091-9. [DOI] [PubMed] [Google Scholar]
- Deleuze C, Huguenard JR. Distinct electrical and chemical connectivity maps in the thalamic reticular nucleus: potential roles in synchronization and sensation. J Neurosci. 2006;26:8633–8645. doi: 10.1523/JNEUROSCI.2333-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLorey TM, Handforth A, Anagnostaras SG, Homanics GE, Minassian BA, Asatourian A, Fanselow MS, Delgado-Escueta A, Ellison GD, Olsen RW. Mice lacking the beta3 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J Neurosci. 1998;18:8505–8514. doi: 10.1523/JNEUROSCI.18-20-08505.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng F, Price MG, Davis CF, Mori M, Burgess DL. Stargazin and other transmembrane AMPA receptor regulating proteins interact with synaptic scaffolding protein MAGI-2 in brain. J Neurosci. 2006;26:7875–7884. doi: 10.1523/JNEUROSCI.1851-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettinger AB, Bernal OG, Andriola MR, Bagchi S, Flores P, Just C, Pitocco C, Rooney T, Tuominen J, Devinsky O. Two cases of nonconvulsive status epilepticus in association with tiagabine therapy. Epilepsia. 1999;40:1159–1162. doi: 10.1111/j.1528-1157.1999.tb00835.x. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, Chaudhry FA, Nicoll RA, Edwards RH. Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science. 2004;304:1815–1819. doi: 10.1126/science.1097468. [DOI] [PubMed] [Google Scholar]
- Gardoni F. MAGUK proteins: new targets for pharmacological intervention in the glutamatergic synapse. Eur J Pharmacol. 2008;585:147–152. doi: 10.1016/j.ejphar.2008.01.048. [DOI] [PubMed] [Google Scholar]
- Golshani P, Liu XB, Jones EG. Differences in quantal amplitude reflect GluR4-subunit number at corticothalamic synapses on two populations of thalamic neurons. Proc Natl Acad Sci U S A. 2001;98:4172–4177. doi: 10.1073/pnas.061013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Fukaya M, Qiao X, Sakimura K, Watanabe M, Kano M. Impairment of AMPA receptor function in cerebellar granule cells of ataxic mutant mouse stargazer. J Neurosci. 1999;19:6027–6036. doi: 10.1523/JNEUROSCI.19-14-06027.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosford DA, Wang Y, Cao Z. Differential effects mediated by GABAA receptors in thalamic nuclei in lh/lh model of absence seizures. Epilepsy Res. 1997;27:55–65. doi: 10.1016/s0920-1211(97)01023-1. [DOI] [PubMed] [Google Scholar]
- Howard MA, Elias GM, Elias LA, Swat W, Nicoll RA. The role of SAP97 in synaptic glutamate receptor dynamics. Proc Natl Acad Sci U S A. 2010;107:3805–3810. doi: 10.1073/pnas.0914422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CL, Feng S, Hilgemann DW. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβgamma. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- Huguenard J. Thalamocortical circuits and excitability. Epilepsy Curr. 2001;1:13. doi: 10.1046/j.1535-7597.2001.00008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR, McCormick DA. Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends Neurosci. 2007;30:350–356. doi: 10.1016/j.tins.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Huguenard JR, Prince DA. A novel T-type current underlies prolonged Ca2+-dependent burst firing in GABAergic neurons of rat thalamic reticular nucleus. J Neurosci. 1992;12:3804–3817. doi: 10.1523/JNEUROSCI.12-10-03804.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR, Prince DA. Intrathalamic rhythmicity studied in vitro: nominal T current modulation causes robust anti-oscillatory effects. J Neurosci. 1994;14:5485–5502. doi: 10.1523/JNEUROSCI.14-09-05485.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntsman MM, Porcello DM, Homanics GE, DeLorey TM, Huguenard JR. Reciprocal inhibitory connections and network synchrony in the mammalian thalamus. Science. 1999;283:541–543. doi: 10.1126/science.283.5401.541. [DOI] [PubMed] [Google Scholar]
- Inoue M, Duysens J, Vossen JM, Coenen AM. Thalamic multiple-unit activity underlying spike-wave discharges in anesthetized rats. Brain Res. 1993;612:35–40. doi: 10.1016/0006-8993(93)91641-5. [DOI] [PubMed] [Google Scholar]
- Jackson AC, Nicoll RA. The expanding social network of ionotropic glutamate receptors: TARPs and other transmembrane auxiliary subunits. Neuron. 2011;70:178–199. doi: 10.1016/j.neuron.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen RB, Ulrich D, Huguenard JR. GABAB and NMDA receptors contribute to spindle-like oscillations in rat thalamus in vitro. J Neurophysiol. 2001;86:1365–1375. doi: 10.1152/jn.2001.86.3.1365. [DOI] [PubMed] [Google Scholar]
- Kato AS, Gill MB, Yu H, Nisenbaum ES, Bredt DS. TARPs differentially decorate AMPA receptors to specify neuropharmacology. Trends Neurosci. 2010;33:241–248. doi: 10.1016/j.tins.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Katz LC, Dalva MB. Scanning laser photostimulation: a new approach for analyzing brain circuits. J Neurosci Methods. 1994;54:205–218. doi: 10.1016/0165-0270(94)90194-5. [DOI] [PubMed] [Google Scholar]
- Koerner C, Danober L, Boehrer A, Marescaux C, Vergnes M. Thalamic NMDA transmission in a genetic model of absence epilepsy in rats. Epilepsy Res. 1996;25:11–19. doi: 10.1016/0920-1211(96)00015-0. [DOI] [PubMed] [Google Scholar]
- Kott S, Werner M, Körber C, Hollmann M. Electrophysiological properties of AMPA receptors are differentially modulated depending on the associated member of the TARP family. J Neurosci. 2007;27:3780–3789. doi: 10.1523/JNEUROSCI.4185-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM. Spillover and synaptic cross talk mediated by glutamate and GABA in the mammalian brain. Prog Brain Res. 2000;125:339–351. doi: 10.1016/S0079-6123(00)25023-1. [DOI] [PubMed] [Google Scholar]
- Letts VA. Stargazer—a mouse to seize! Epilepsy Curr. 2005;5:161–165. doi: 10.1111/j.1535-7511.2005.00051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letts VA, Felix R, Biddlecome GH, Arikkath J, Mahaffey CL, Valenzuela A, Bartlett FS, 2nd, Mori Y, Campbell KP, Frankel WN. The mouse stargazer gene encodes a neuronal Ca2+-channel gamma subunit. Nat Genet. 1998;19:340–347. doi: 10.1038/1228. [DOI] [PubMed] [Google Scholar]
- Liu XB. Subcellular distribution of AMPA and NMDA receptor subunit immunoreactivity in ventral posterior and reticular nuclei of rat and cat thalamus. J Comp Neurol. 1997;388:587–602. doi: 10.1002/(sici)1096-9861(19971201)388:4<587::aid-cne7>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Meeren HK, Pijn JP, Van Luijtelaar EL, Coenen AM, Lopes da Silva FH. Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. J Neurosci. 2002;22:1480–1495. doi: 10.1523/JNEUROSCI.22-04-01480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menuz K, Nicoll RA. Loss of inhibitory neuron AMPA receptors contributes to ataxia and epilepsy in stargazer mice. J Neurosci. 2008;28:10599–10603. doi: 10.1523/JNEUROSCI.2732-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi R, Sia GM, Rosen K, Tang X, Moghekar A, Black JL, McEnery M, Huganir RL, O'Brien RJ. AMPA receptor-dependent clustering of synaptic NMDA receptors is mediated by Stargazin and NR2A/B in spinal neurons and hippocampal interneurons. Neuron. 2004;44:335–349. doi: 10.1016/j.neuron.2004.09.029. [DOI] [PubMed] [Google Scholar]
- Micheva KD, Smith SJ. Array tomography: a new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 2007;55:25–36. doi: 10.1016/j.neuron.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva KD, Busse B, Weiler NC, O'Rourke N, Smith SJ. Single-synapse analysis of a diverse synapse population: proteomic imaging methods and markers. Neuron. 2010;68:639–653. doi: 10.1016/j.neuron.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineff EM, Weinberg RJ. Differential synaptic distribution of AMPA receptor subunits in the ventral posterior and reticular thalamic nuclei of the rat. Neuroscience. 2000;101:969–982. doi: 10.1016/s0306-4522(00)00421-8. [DOI] [PubMed] [Google Scholar]
- Noebels JL, Qiao X, Bronson RT, Spencer C, Davisson MT. Stargazer: a new neurological mutant on chromosome 15 in the mouse with prolonged cortical seizures. Epilepsy Res. 1990;7:129–135. doi: 10.1016/0920-1211(90)90098-g. [DOI] [PubMed] [Google Scholar]
- Nowak LG, Bullier J. Axons, but not cell bodies, are activated by electrical stimulation in cortical gray matter. I. Evidence from chronaxie measurements. Exp Brain Res. 1998a;118:477–488. doi: 10.1007/s002210050304. [DOI] [PubMed] [Google Scholar]
- Nowak LG, Bullier J. Axons, but not cell bodies, are activated by electrical stimulation in cortical gray matter. II. Evidence from selective inactivation of cell bodies and axon initial segments. Exp Brain Res. 1998b;118:489–500. doi: 10.1007/s002210050305. [DOI] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- Opazo P, Labrecque S, Tigaret CM, Frouin A, Wiseman PW, De Koninck P, Choquet D. CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of Stargazin. Neuron. 2010;67:239–252. doi: 10.1016/j.neuron.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Opazo P, Sainlos M, Choquet D. Regulation of AMPA receptor surface diffusion by PSD-95 slots. Curr Opin Neurobiol. 2011 doi: 10.1016/j.conb.2011.10.010. [DOI] [PubMed] [Google Scholar]
- Osten P, Stern-Bach Y. Learning from stargazin: the mouse, the phenotype and the unexpected. Curr Opin Neurobiol. 2006;16:275–280. doi: 10.1016/j.conb.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Patneau DK, Mayer ML. Structure-activity relationships for amino acid transmitter candidates acting at N-methyl-d-aspartate and quisqualate receptors. J Neurosci. 1990;10:2385–2399. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz JT, Bryant AS, Peng K, Fenno L, Yizhar O, Frankel WN, Deisseroth K, Huguenard JR. A new mode of corticothalamic transmission revealed in the Gria4(−/−) model of absence epilepsy. Nat Neurosci. 2011;14:1167–1173. doi: 10.1038/nn.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perucca E, Gram L, Avanzini G, Dulac O. Antiepileptic drugs as a cause of worsening seizures. Epilepsia. 1998;39:5–17. doi: 10.1111/j.1528-1157.1998.tb01268.x. [DOI] [PubMed] [Google Scholar]
- Pinault D, Leresche N, Charpier S, Deniau JM, Marescaux C, Vergnes M, Crunelli V. Intracellular recordings in thalamic neurones during spontaneous spike and wave discharges in rats with absence epilepsy. J Physiol. 1998;509:449–456. doi: 10.1111/j.1469-7793.1998.449bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack PO, Guillemain I, Hu E, Deransart C, Depaulis A, Charpier S. Deep layer somatosensory cortical neurons initiate spike-and-wave discharges in a genetic model of absence seizures. J Neurosci. 2007;27:6590–6599. doi: 10.1523/JNEUROSCI.0753-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priel A, Kolleker A, Ayalon G, Gillor M, Osten P, Stern-Bach Y. Stargazin reduces desensitization and slows deactivation of the AMPA-type glutamate receptors. J Neurosci. 2005;25:2682–2686. doi: 10.1523/JNEUROSCI.4834-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao X, Noebels JL. Developmental analysis of hippocampal mossy fiber outgrowth in a mutant mouse with inherited spike-wave seizures. J Neurosci. 1993;13:4622–4635. doi: 10.1523/JNEUROSCI.13-11-04622.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci U S A. 2002;99:13902–13907. doi: 10.1073/pnas.172511199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield CM, Kleiman-Weiner M, Rudolph U, Huguenard JR. A gain in GABAA receptor synaptic strength in thalamus reduces oscillatory activity and absence seizures. Proc Natl Acad Sci U S A. 2009;106:7630–7635. doi: 10.1073/pnas.0811326106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schousboe A, Sarup A, Bak LK, Waagepetersen HS, Larsson OM. Role of astrocytic transport processes in glutamatergic and GABAergic neurotransmission. Neurochem Int. 2004;45:521–527. doi: 10.1016/j.neuint.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Schwenk J, Harmel N, Zolles G, Bildl W, Kulik A, Heimrich B, Chisaka O, Jonas P, Schulte U, Fakler B, Klöcker N. Functional proteomics identify cornichon proteins as auxiliary subunits of AMPA receptors. Science. 2009;323:1313–1319. doi: 10.1126/science.1167852. [DOI] [PubMed] [Google Scholar]
- Seidenbecher T, Staak R, Pape HC. Relations between cortical and thalamic cellular activities during absence seizures in rats. Eur J Neurosci. 1998;10:1103–1112. doi: 10.1046/j.1460-9568.1998.00123.x. [DOI] [PubMed] [Google Scholar]
- Slaght SJ, Leresche N, Deniau JM, Crunelli V, Charpier S. Activity of thalamic reticular neurons during spontaneous genetically determined spike and wave discharges. J Neurosci. 2002;22:2323–2334. doi: 10.1523/JNEUROSCI.22-06-02323.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Contreras D. Spike-wave complexes and fast components of cortically generated seizures. I. Role of neocortex and thalamus. J Neurophysiol. 1998;80:1439–1455. doi: 10.1152/jn.1998.80.3.1439. [DOI] [PubMed] [Google Scholar]
- Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- Sumioka A, Yan D, Tomita S. TARP phosphorylation regulates synaptic AMPA receptors through lipid bilayers. Neuron. 2010;66:755–767. doi: 10.1016/j.neuron.2010.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeev I, Grenier F, Steriade M. Spike-wave complexes and fast components of cortically generated seizures. IV. Paroxysmal fast runs in cortical and thalamic neurons. J Neurophysiol. 1998;80:1495–1513. doi: 10.1152/jn.1998.80.3.1495. [DOI] [PubMed] [Google Scholar]
- Tomita S, Chen L, Kawasaki Y, Petralia RS, Wenthold RJ, Nicoll RA, Bredt DS. Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. J Cell Biol. 2003;161:805–816. doi: 10.1083/jcb.200212116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita S, Adesnik H, Sekiguchi M, Zhang W, Wada K, Howe JR, Nicoll RA, Bredt DS. Stargazin modulates AMPA receptor gating and trafficking by distinct domains. Nature. 2005;435:1052–1058. doi: 10.1038/nature03624. [DOI] [PubMed] [Google Scholar]
- Tomita S, Byrd RK, Rouach N, Bellone C, Venegas A, O'Brien JL, Kim KS, Olsen O, Nicoll RA, Bredt DS. AMPA receptors and stargazin-like transmembrane AMPA receptor-regulatory proteins mediate hippocampal kainate neurotoxicity. Proc Natl Acad Sci U S A. 2007;104:18784–18788. doi: 10.1073/pnas.0708970104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrence C, Compo GP. A practical guide to wavelet analysis. Bull Am Met Soc. 1998;79:61–78. [Google Scholar]
- Tselnicker I, Dascal N. Further characterization of regulation of CaV2.2 by stargazin. Channels (Austin) 2010;4:351–354. doi: 10.4161/chan.4.5.13504. [DOI] [PubMed] [Google Scholar]
- Turetsky D, Garringer E, Patneau DK. Stargazin modulates native AMPA receptor functional properties by two distinct mechanisms. J Neurosci. 2005;25:7438–7448. doi: 10.1523/JNEUROSCI.1108-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Krosigk M, Bal T, McCormick DA. Cellular mechanisms of a synchronized oscillation in the thalamus. Science. 1993;261:361–364. doi: 10.1126/science.8392750. [DOI] [PubMed] [Google Scholar]
- Warren RA, Jones EG. Glutamate activation of cat thalamic reticular nucleus: effects on response properties of ventroposterior neurons. Exp Brain Res. 1994;100:215–226. doi: 10.1007/BF00227192. [DOI] [PubMed] [Google Scholar]
- Zaman T, Lee K, Park C, Paydar A, Choi JH, Cheong E, Lee CJ, Shin HS. Cav2.3 channels are critical for oscillatory burst discharges in the reticular thalamus and absence epilepsy. Neuron. 2011;70:95–108. doi: 10.1016/j.neuron.2011.02.042. [DOI] [PubMed] [Google Scholar]
- Zhang J, Diamond JS. Distinct perisynaptic and synaptic localization of NMDA and AMPA receptors on ganglion cells in rat retina. J Comp Neurol. 2006;498:810–820. doi: 10.1002/cne.21089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziff EB. TARPs and the AMPA receptor trafficking paradox. Neuron. 2007;53:627–633. doi: 10.1016/j.neuron.2007.02.006. [DOI] [PubMed] [Google Scholar]