

Abstract
This review covers reservoir-based drug delivery systems that incorporate microtechnology, with an emphasis on oral, dermal, and implantable systems. Key features of each technology are highlighted such as working principles, fabrication methods, dimensional constraints, and performance criteria. Reservoir-based systems include a subset of microfabricated drug delivery systems and provide unique advantages. Reservoirs, whether external to the body or implanted, provide a well-controlled environment for a drug formulation, allowing increased drug stability and prolonged delivery times. Reservoir systems have the flexibility to accommodate various delivery schemes, including zero order, pulsatile, and on demand dosing, as opposed to a standard sustained release profile. Furthermore, the development of reservoir-based systems for targeted delivery for difficult to treat applications (e.g., ocular) has resulted in potential platforms for patient therapy.
Keywords: Controlled Release, Implant, MEMS, Microneedle, Micropump, Ocular, On Demand, Pulsatile
1. Introduction
The sales for advanced drug delivery systems were $37.9B, $54.2B, and $64.1B in 2000, 2004, and 2005, respectively [1,2]. In 2005, the largest sectors of the market consisted of sustained release implants and transdermal drug delivery systems [1]. The global market for drug delivery systems was $134.3B in 2008 and projected to increase to $196.4B in 2014. This market potential has provided the driving force behind the development of micro and nanodelivery systems [3,4].
Many therapeutically active agents, especially biomolecules, have short plasma half-lives and require frequent dosing to maintain blood levels in the therapeutic window. A more controlled therapeutic blood level can result in fewer adverse reactions because a high Cmax after injection is not required to keep the drug in the therapeutic window for the required time period. Drug delivery systems can improve therapeutic response by providing more consistent blood levels than immediate release or sustained release depot parenterals. Therefore, an objective for delivery system development is a less frequent, more efficacious dosing regimen with increased patient comfort, safety, and compliance. Such systems may also make it possible to get improved efficacy with smaller quantities of drug.
Reservoir-based delivery systems that utilize microtechnology are resulting in a decrease in the overall size/profile of delivery systems. Smaller systems can incorporate features or mechanisms that allow more precise control over the drug delivery rate, enable the patient or physician to actively start/modify/stop drug release in an interactive format, and provide ways to reach difficult to treat locations. These next generation targeted delivery systems offer the possibility that the drug can be delivered preferentially to a relatively inaccessible site, a specific tissue type, or simply to limit side effects due to systemic exposure.
Microtechnology for drug delivery can be defined as the construction or assembly of components that result in delivery aspects in the micro scale, forming either a complete device or a critical component of a larger delivery system. A more rigid definition may include only those devices where the entire unit has micro scale dimensions. For the purposes of this review, the former definition was selected and reservoir-based, microtechnology drug delivery systems including oral, dermal, and several types of implantable delivery approaches for both systemic and targeted delivery will be discussed in detail.
2. Oral Delivery
One of the first delivery platforms developed by Alza Corporation was a reservoir-based oral osmotic system (OROS®). OROS® technology can be found in many approved products including Acutrim®, Concerta®, Ditropan®, Glucotrol®, Procardia®, and Sudafed® [5,6]. An OROS® capsule is coated with a rigid semi-permeable membrane containing a precision laser-drilled orifice (0.5-1.4 mm in diameter) for drug release. After ingestion, water diffuses through the semi-permeable membrane into the tablet reservoir, and drug is released through the orifice as the osmotically active polymer excipients expand (Fig. 1). The membrane controls the rate at which water enters the tablet core, which in turn controls the rate of drug release [7]. This technology has been translated into smaller implantable systems, as described in a later section (see Section 4.1 – Passive Delivery Systems).
Fig. 1.
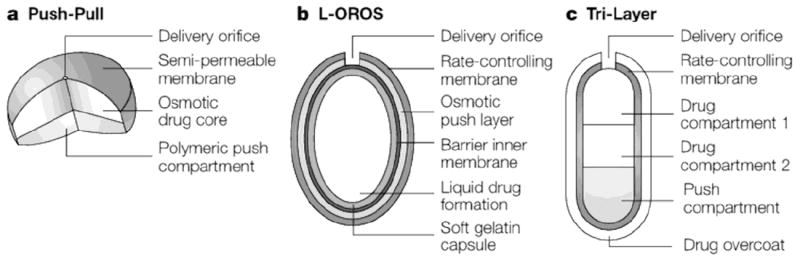
Examples of osmotic oral delivery systems, including a) push-pull, b) L-OROS®, and c) Tri-Layer designs. Reproduced with permission from Fig. 2 in [6] (©2005 Nature Publishing Group).
The application of microfabrication and MEMS (microelectromechanical systems) technologies have resulted in a new class of oral delivery systems. Silicon microparticles having dimensions of 50 μm × 50 μm × 2 μm, with 25 μm × 25 μm × 1 μm deep wells were designed for targeted oral drug delivery [8]. Polymeric based systems measuring approximately 150 μm × 150 μm × 14 μm followed (Fig. 2) [9-11]. These systems provide a protected drug reservoir, a functionalized surface for cytoadhesion, and asymmetric drug delivery to the intestinal wall instead of the lumen [11-13]. Devices have been made from polymethylmethacrylate (PMMA), poly-lactic/glycolic acid (PLGA), and poly(ethylene glycol) dimethacrylate (PEGDMA) [10-13].
Fig. 2.
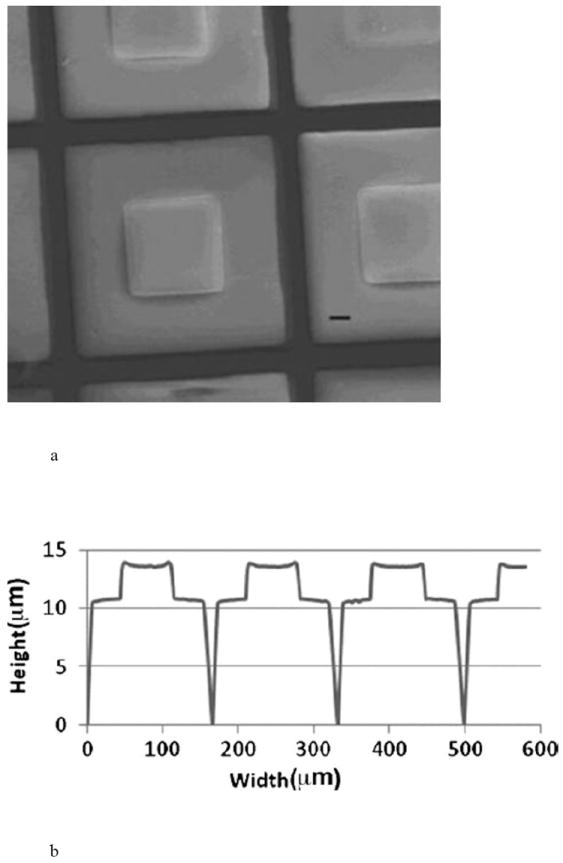
Hydrogel-loaded, polymeric Microdevices, a) SEM image where scale bar (-) is 20 μm, and b) dimensions of the microdevices measured by profilometry. Reproduced with permission from Figs. 1A and 1F in [11] (©2009 John Wiley and Sons).
3. Dermal Delivery
Microneedles were developed to increase skin permeability without causing pain. The stratum corneum makes up the top ~20 μm of the epidermal layer and provides much of the barrier to drug transport. The nerve endings in the skin are a few hundred microns below, allowing micropenetration of the skin to bypass its barrier function without causing pain.
Transdermal and intradermal delivery can be characterized into two technology categories: microneedle syringes and microneedle patches. Biodegradable needles have been formed from drug/polymer mixtures that result in a higher drug payload, but commonly result in poor drug stability [14]. Reservoir-based microneedles are hollow and allow either the delivery of drug from an external reservoir or from drug stored inside the hollow of the needle.
Jet injectors (BioJect® 2000, PharmaJet®) and thermal ablation patches (Passport™, ViaDerm™, ViaDor™) create micropores in the skin to deliver drug. However, these technologies were not included in the review because they do not utilize microtechnology to create drug reservoirs [15].
3.1 Microneedle Syringes
Many silicon-based microneedle designs are fabricated by deep reactive ion etching (DRIE) technology and include solid or hollow microneedles with tapered or beveled tips with drug coated onto the array [14,16,17]. These coated arrays are not reservoir-based, but they do allow an initial bolus dose and can be coupled with a reservoir system. Hollow microneedles provide drug reservoirs and have incorporated a wide range of geometries including barbed tips [18], microfilters at the needle base [19], bubble pumps for delivery fluids [20], blunt cylinders [21], tapered cylinders [22], volcano shapes [23,24], citadel structures [25,26] and saw tooth structures [27,28].
Saw tooth structures (Fig. 3a) are 150-350 μm in length and are ~250 μm wide at the base [27]. They have been incorporated into the NanoPass Technologies MicroPyramid MicronJet Needle and have been used in clinical trials for local anesthesia, insulin delivery, and the administration of influenza vaccine [28].
Fig. 3.

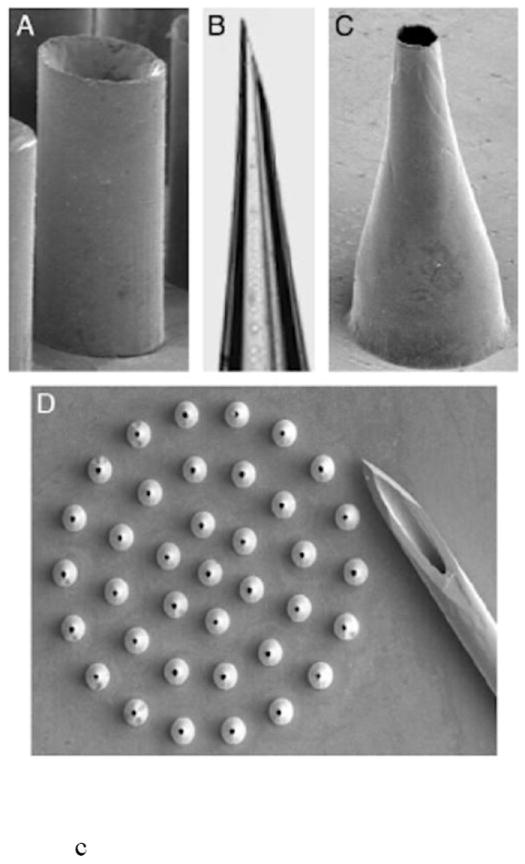
Hollow microneedle designs, including a) pyramid shaped saw tooth style, b) citadel style microneedles with side openings, and c) tapered hollow cylinders depicting (A) straight wall, (B) beveled, (C) tapered and (D) an array of tapered microneedles. Reproduced a) with permission from Fig. 2 in [28] (©2009 Elsevier), b) with permission from Debiotech SA/Switzerland, c) with permission from Fig. 2 in [22] (©2003 National Academy of Sciences, USA).
The citadel side opened needles were licensed to Debiotech for the Microject/Nanoject platform (Fig. 3b). Hollow microneedles with side openings provide two advantages: an ultra-sharp tip for efficient skin penetration and alleviation of tissue coring/channel blockage during insertion [25,26]. The microneedles are ~210 μm long, can inject 100 μl in 2 seconds, and were fabricated in a 3 × 3 mm2 chip array [25]. Debiotech provides needles from 300-1000 μm in length in 25 microneedle arrays [29]. The drug reservoir can be sealed with a gold film that is ruptured upon application to the skin [30]. The microneedles have also been incorporated into a MEMS micropump for delivery of insulin [31].
Volcano style needles have been used to successfully deliver methyl nicotinate [23,24]. A MEMS syringe has been configured to include an array of silicon needles with a PDMS reservoir attached to the back and is called a “chiclet” [23,32]. The entire reservoir assembly is ~10 mm, holding an array of 8 needles that are 200 μm long and has been tested on mice. The dose is delivered by simply pressing the device against the skin for a few seconds.
The tapered cylinder hollow microneedles were made from silicon, metal, and polymer [22]. The silicon needles had a constant bore diameter of 60 μm and an increased wall thickness at the base (Fig. 3c). The metal needles have a constant wall thickness of 10 μm and a bore that widens at the base. The needles have been made with widths of 35-350 μm and lengths of 150-1000 μm. Insulin delivery to rats was demonstrated through a syringe [22]. An array of 16 microneedles made from polyethylene terephthalate and measuring 50 μm in length with a 75 μm tip diameter was tested in rats [33]. This passive diffusion driven system delivered a peak value of 0.43 ng/ml and decreased blood glucose levels over a 4 hour period.
Finally, hollow microneedles have been used to deliver therapeutics to the eye [34]. Solution and suspension formulations of sulforhodamine were delivered to the suprachoroidal space, suggesting that the placement of sustained release microparticles in this location is achievable. Drug volumes of 15-35 μl were delivered consistently through needles of 800-1000 μl. Small particles (20-100 nm) could easily flow through the scleral tissue, and larger particles (500-1000 nm) were successfully delivered with longer microneedles.
3.2 Microneedle Patches
Microneedle arrays have also been integrated into transdermal patches. The V-Go™ disposable insulin delivery device from Valeritas (a BioValve subsidiary) utilizes tapered-cylinder, hollow microneedles and was licensed from Georgia Tech [22,33]. V-Go™ received 510k clearance for the continuous subcutaneous delivery of insulin at preset basal rates and with as-needed bolus dosing [35]. V-Go™ utilizes the hPatch™ platform designed to deliver drugs into the subcutaneous space and the Micro-Trans™ microneedle array.
Dissolving microneedle patches for influenza vaccination have been tested in mice and pigs [36,37]. The polyvinylpyrrolidone (PVP) microneedles measured 650 μm high and encapsulated 3 μg of inactivated influenza virus per patch [36]. The patch is applied with the thumb, penetrates ~ 200 μm and deposits the drug into the epidermis. This new technology may facilitate increased vaccination in remote areas of the world.
4. Implantable Delivery
Implantable drug delivery systems can be placed into three categories [16,38-41]:
passive – a system where drug release is pre-determined by the materials, fabrication methods, or drug formulation and cannot be controlled after implantation
active – a system where drug release is controlled after implantation using mechanical, electrical, magnetic, laser or other means
Passive systems utilize diffusion, osmotic potential, or concentration gradients as their driving forces, while active systems include mechanical pumping, electrolysis, and other actuation methods. Implantable drug delivery systems that do not contain a well-defined reservoir are beyond the scope of this review and are covered elsewhere [42-44].
Reservoir-based implants can be used for both systemic and targeted drug delivery applications. These micro and nano implants offer a great deal of promise in addressing current unmet medical needs and have resulted in some of the most innovative and elegant drug delivery concepts.
4.1 Passive Delivery Systems
4.1.1 Membrane Controlled Diffusion Systems
Diffusion controlled systems, in their simplest forms, rely on diffusion of drug out of or through a polymer layer that may be nonporous or microporous. The rate determining step may be diffusion through the membrane structure or transport of drug through the static aqueous diffusion layer. For membrane controlled diffusion, the diffusion coefficient is dictated by the size of the drug molecule and the pore size or space between the polymer chains. This technology has been widely used for delivery from reservoir-based oral and transdermal systems and has recently been adapted for implants.
Ophthalmology is one field where reservoir-based, diffusion controlled implants have made an impact. The Medidur™ platform has been successfully integrated into two approved products and three more in development, suggesting that the delivery system is adaptable to a variety of drugs, acceptable to patients and physicians, and can be manufactured economically. Currently marketed, reservoir-based ocular implants utilizing drug diffusion through polyvinyl alcohol include Vitrasert® and Retisert® [7]. Both of these products were developed by pSivida based on their Medidur™ platform and are distributed by Bausch & Lomb (Fig. 4a) [42,45]. The Vitrasert® implant (approved in 1996) contains 4.5 mg ganciclovir for the treatment of CMV retinitis in patients with AIDS. The ganciclovir tablet reservoir (3.5 mm) is coated with polyvinyl alcohol (PVA) and ethylene vinyl acetate (EVA) polymers and attached to a tab, which is sutured to the inside wall of the eye. The PVA controls the drug release and the EVA controls the surface area of the device through which ganciclovir can permeate. The device releases the drug into the vitreous of the eye for a period of approximately 5-8 months [7,42,46,47].
Fig. 4.

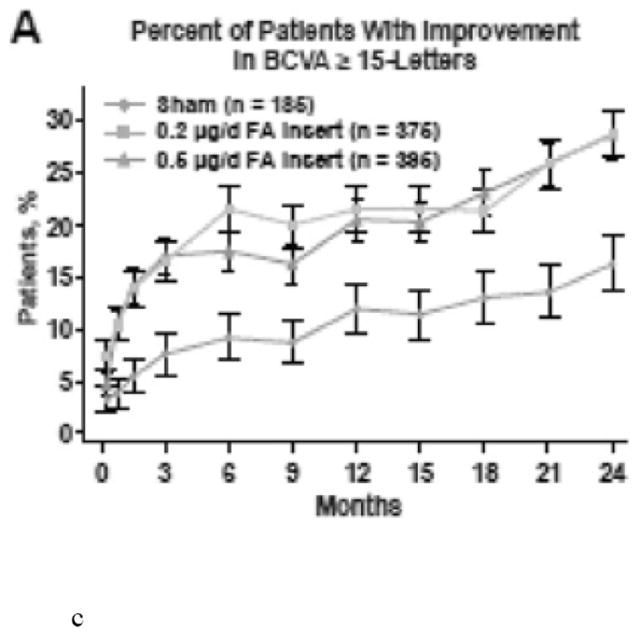
Images showing a) multiple generations of the Medidur™ delivery platform, b) Iluvien™ device next to a grain of rice for scale, and c) 24 month visual acuity clinical results for Iluvien™ in DME patients. Reproduced a) with permission from pSivida, Ltd., b) with permission from Alimera Sciences, Inc., c) with permission from Fig. 1A in [52] (©2011 Elsevier).
The Retisert® implant (approved in 2005) measures 3 mm × 2 mm × 5 mm and is the second generation Medidur™ technology. The implant consists of 0.59 mg fluocinolone acetonide in a 1.5 mm tablet reservoir. The tablet is encased in a silicone elastomer cup containing a release orifice and a PVA membrane positioned between the tablet and the orifice [7]. The silicone elastomer cup assembly is attached to a PVA suture tab with silicone adhesive that is surgically implanted thru 3-4 mm incision. Retisert® releases fluocinolone acetonide into the vitreous of the eye at a nominal initial rate of 0.6 μg/day, decreasing over the first month to a steady state between 0.3-0.4 μg/day over ~30 months, where the rate is controlled by the PVA membrane [42,48,49].
The Iluvien™ implant utilizes the third generation of Medidur™ technology and demonstrates the technological capability of fabricating smaller devices. Iluvien™ is inserted into the vitreous of the eye with a 25G needle during an outpatient procedure and is not sutured to the eye, in contrast to the surgical procedure required for the implantation of Vitrasert® and Retisert®. The implant is a cylinder 3.5 mm long and 0.37 mm in diameter and consists of a polyimide tube reservoir containing 190 μg fluocinolone in a PVA matrix (Fig. 4b) [42,43,50,51]. The tube is capped with rate controlling membranes. Implants with two release rates were considered during development: a higher dose implant with an initial in vitro release rate of 0.45 μg/day and a lower dose implant with an initial in vitro release rate 0.23 μg/day [50]. The release rates of both implants steadily decreased in vitro and were nearly the same at ~0.15 μg/day by 18 months [51]. Alimera has completed Phase 3 clinical trials with Iluvien™ for the treatment of diabetic macular edema (DME). Published 24 month clinical results showed a significant increase over sham in the number of patients that achieved an improvement in best corrected visual acuity (BVCA) of 15 letters or more (Fig. 4c) [52]. Recently reported 36 month results confirm a significant improvement for those patients having DME for three years or more [53]. Alimera’s 36 month results for Iluvien™ are currently under FDA review for the treatment of DME for 24-36 months.
pSivida followed Iluvien™ with Durasert™, a bioerodible reservoir implant for the delivery of latanoprost for high intraocular pressure, that is in Phase 2 clinical trials [45]. In general, these ocular implant platforms have been successfully applied to small molecule therapeutics. pSivida is currently developing Tethadur™ for biomolecules and delivery of more than one therapeutic agent [45]. Tethadur™ utilizes nanostructured pores to control the delivery of varying size molecules, is targeted to last 6 months, and reported to be fully bioerodible. The addition of nanopores to diffusion-based platforms provides another mechanism for controlling release rate and is discussed below (see Section 4.1.3 – MicroChannel Implants).
Iveena is developing the capsule drug ring (CDR™), reminiscent of the larger and less sophisticated pilocarpine Ocusert™ system. Intraocular lenses inserted during cataract surgery leave unfilled space in the lens capsule. The CDR™ (13 mm outside diameter, 9.2 mm inside diameter) was designed to be implanted in the peripheral lens capsule during or after cataract surgery (Fig. 5) [54]. The implant is comprised of a polymethyl methacrylate (PMMA) shell, a semipermeable membrane to control the delivery rate, and silicon valves for refilling the drug reservoir. The refillable drug reservoir holds 50 μl, and a linear delivery rate of 0.06-0.08 mg/day Avastin® (bevacizumab) was achieved [54].
Fig 5.
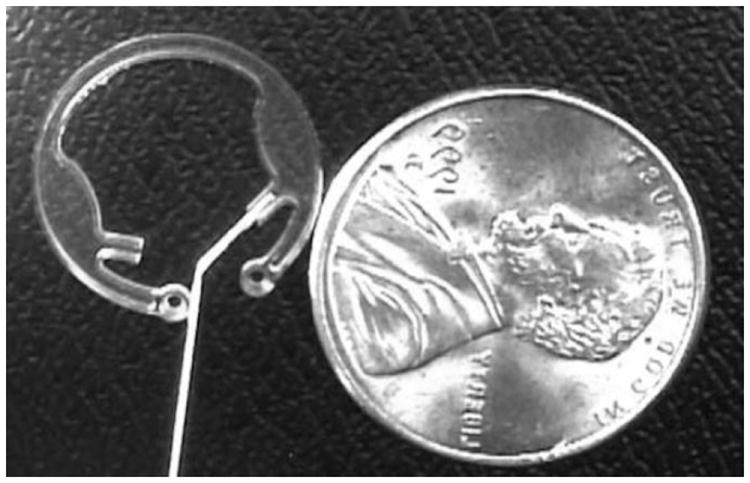
Reservoir-based, capsule drug ring with a 27 G cannula in the valve access port. Reproduced with permission from Fig. 3 in [54] (©2010 Elsevier).
4.1.2 Matrix Controlled Diffusion Systems
A subclass of diffusion controlled systems takes advantage of a combination of drug release from a polymer matrix via porosity (tortuous path) and polymer erosion.
Oncology is a therapeutic area that has taken advantage of passive, diffusion controlled implantable systems. Resorbable multi-reservoir arrays made from poly (l-lactic acid) and/or PLGA were fabricated, where each reservoir was covered with a bioerodible membrane cap comprised of varying ratios and molecular weights of PLGA [55-57]. The devices measured ~11.9 mm in diameter, were 480-560 μm thick, and contained thirty six 120-130 nl reservoirs (Fig. 6a) [55]. The incorporation of multiple reservoirs effectively provides pulsatile release kinetics by releasing individual reservoirs on a schedule determined by the properties of the bioerodible cap. A compression molded polymer microchip array (10 mm in diameter × 1 mm thick) was loaded with carmustine (BCNU) for targeted delivery to brain tumors [56]. Each microchip could hold a maximum of 1.24 mg carmustine, and this study demonstrated similar efficacy to the Gliadel® wafer in a rat model. An advantage of this approach is that initiation of drug release from each reservoir can be programmed into the device by controlling the composition and thickness of each membrane cap. Such bioerodible implants benefit from not having to be removed after use, but like other polymer systems, achieving prolonged drug stability for anything other than small molecules can be a challenge.
Fig. 6.
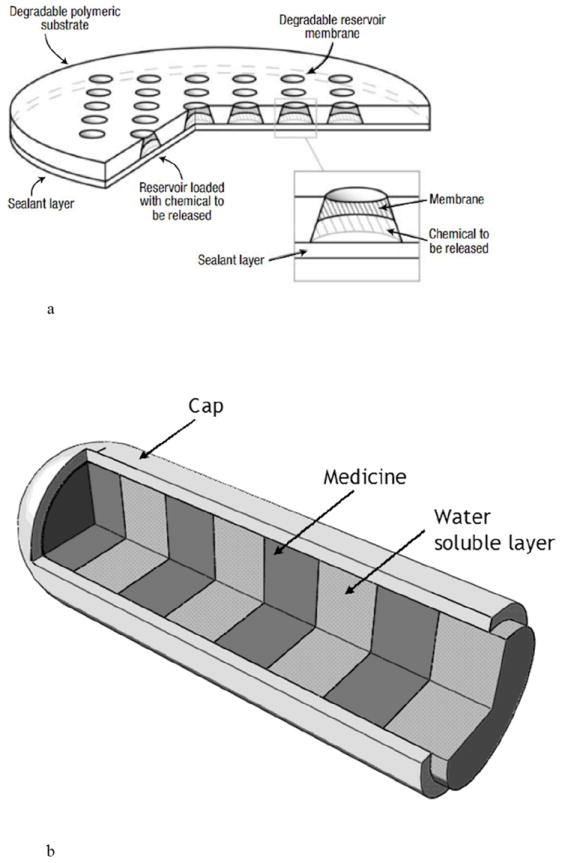
Passive, matrix controlled drug delivery systems including (a) a bioerodible PLGA implant and b) a multi-layered implant for ophthalmic use. Reproduced a) with permission from Fig. 1 in [55] (©2003 Nature Publishing Group) and b) with permission from [58].
Another approach for achieving pulsatile release from a passive system for ocular use was proposed by the Ocular Drug Delivery Group at UC Irvine. The micromachined device is designed with alternating drug-loaded and drug-free polymer zones, enabling drug levels to vary in a pre-programmed manner as each layer is sequentially exposed to the body (Fig. 6b) [42,58].
Interventional cardiology is another field where reservoir-based, diffusion controlled implants are utilized. Intravascular stents are used to hold open clogged or narrowed blood vessels and have traditionally been constructed as an expandable wire mesh structure made of metal. Many stents are simply coated with a drug/polymer matrix to reduce restenosis, a re-narrowing of the stented vessel by inflammation and tissue in growth. This coating technology does not deliver drug from a reservoir ‘per se’, but from the polymer matrix. However, several new stent technologies have incorporated reservoirs.
Debiotech developed a polymer-free nanocoating technology based on biocompatible nanostructured ceramic coatings with a variety of porosities and this coating can be manipulated to form drug reservoirs (Fig. 7a) [29,59]. The Debiostent loading capacity is defined by the size of the drug reservoirs in a lower coating layer, where reservoirs represent up to 30% of the total coating volume, and drug loading can reach up to 10 μg/mm2. An upper coating layer seals the reservoirs and controls the drug elution kinetics. The location and number of drug reservoirs can be controlled to focus the delivery of drug from specific areas of the stent (i.e., only the adluminal regions in contact with the vessel) for maximum efficacy [29].
Fig. 7.
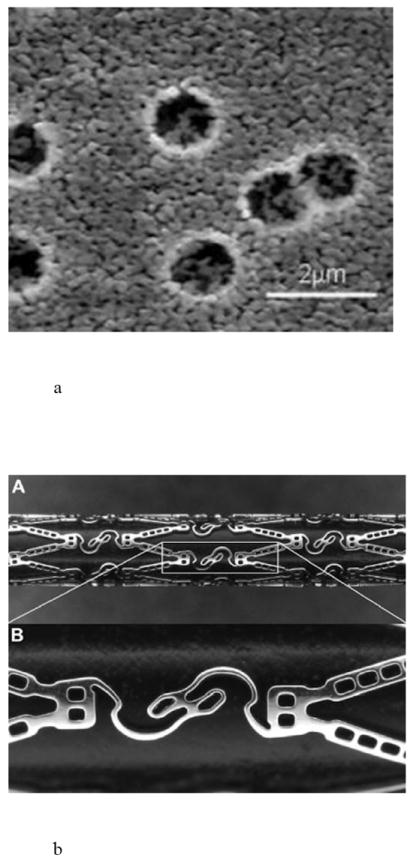
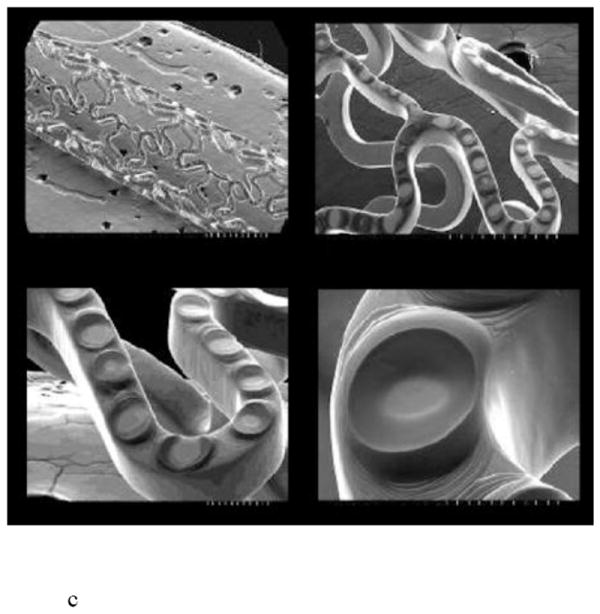
Reservoir-based coronary stents, including a) an SEM cross-section of Debiotech ceramic reservoirs, b) CoStar™ stent, and c) JACTAX™ stent with microdots. Reproduced a) with permission from Debiotech SA/Switzerland, b) with permission from Fig. 1 in [60] (©2009 Elsevier), c) with permission from Fig. 6 in [63] (©2010 Elsevier).
An alternative approach involves creating precisely defined drug reservoirs in the struts of the stent. Conor Medsystems (now part of Johnson & Johnson) developed cobalt-chromium stents with hundreds of laser cut reservoirs in the stent struts stents (Fig. 7b) [60,61]. The reservoirs are loaded with drug in a bioresorbable PLGA matrix. At the end of the biodegradation process, only the bare metal stent remains. Stents containing various drugs, Nevo™ (sirolimus), CoStar™ (paclitaxel), Corio™ (pimecrolimus), and Symbio™ (paclitaxel and pimecrolimus in adjacent reservoirs), have been tested in clinical trials [60,61].
Finally, the JACTAX™ stent by LabCoat Ltd. (now part of Boston Scientific) included its drug in localized polymer reservoirs or microdots placed on the surface of each stent strut (Fig. 7c) [62,63]. A bare metal stent is coated on the abluminal side with 2,750 discrete microdots. The microdots consist of a 50/50 mixture of polylactic acid polymer and paclitaxel.
4.1.3 Microchannel Implants
These systems utilize micro or nanochannels in a membrane-like structure, where the release rate is controlled by diffusion along a constrained channel. Specifically, the size of the pore and the size of the drug substance are correlated to ensure molecular constraint. Modifying the properties of the microfluidic devices, such as surface effects, charge interaction, concentration polarization, and streaming current phenomena, have been used to obtain a zero order release profile [64-68].
Endo Pharmaceuticals acquired the Hydron® Implant Technology from Valera Pharmaceuticals. The Hydron® technology is a cylindrical, non-bioerodible implant 3 mm in diameter by 3.5 cm in length. The implant is made of a hydrogel polymer blend (2-hydroxyethyl methacrylate, 2-hydroxypropyl methacrylate, trimethylolpropane trimethacrylate, benzoin methyl ether, Perkadox-16, Triton X-100) called MedLaunch™, and is spun cast into small tube reservoirs [7]. The implant is supplied prehydrated and contains micropores that allow drug diffusion in a zero order fashion for a year or longer. Vantas® for prostate cancer (approved 2004) and Supprelin® LA for precocious puberty (approved 2007) are both implanted with a trocar, contain 50 mg histrelin acetate, and last for one year with a release rate of 41 μg/day. The histrelin is formulated with stearic acid to form a solid drug core placed in the reservoir [7]. Endo Pharmaceuticals also has an 84 mg octreotide implant that lasts for 6 months in Phase 3 clinical trials for acromegaly [69].
iMEDD Inc. developed a small cylindrical titanium implant for continuous release of α-interferon for 3-6 months for the treatment of Hepatitis C [65]. The implant is designed to be inserted under the skin and to maintain drug plasma level above 50 pg/ml in order to maintain an antiviral effect without side effects associated with higher doses. The NanoGATE™ implant is 4-5 mm in diameter, 20-35 mm in length, and can contain a 75-300 μl reservoir (Fig. 8) [70]. The titanium implant is capped at both ends, and a 2 mm × 3 mm nanopore membrane is affixed over a small bore opening in a cylindrical methacrylate inset carrier. The carrier is fitted with two silicone O-rings and inserted into the titanium encasement. Upon aligning the membrane with the titanium grate opening, the device is filled with formulation [64]. The nanopore membrane controls diffusion of the drug from the reservoir and is made of silicon films with parallel rectangular channel arrays up to 10-100 nm in diameter [70]. The nanopore membrane provides a non-Fickian zero order release, unrelated to drug concentration. The pore size is designed to approximate the diameter of the drug substance, so that the flux of molecules occurs “single file” [65,66,68]. In vitro bovine serum albumin release rate studies with a 13 nm nominal pore size and 4 μm membrane thickness revealed a release rate of 900 μg/day. The formulations can be suspensions or solid state, while osmotic devices are solutions or suspensions [64]. Both the NanoGATE™ and Hydron® technologies are diffusion controlled instead of osmotically driven and provide more space within the implant for drug formulation.
Fig. 8.
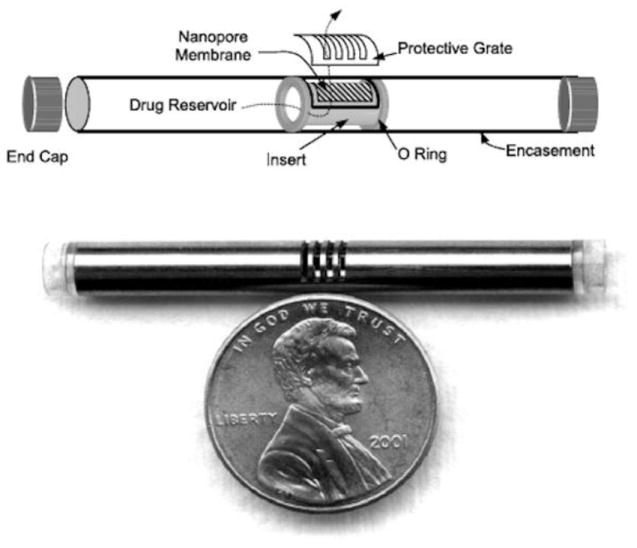
An example microchannel delivery systems called the NanoGATE™ implant. Reproduced with permission from Fig. 4 in [64] (©2005 Elsevier).
An interesting application of molecular constraint has been used deliver α-INF from a nanochannel delivery system (nDS). The silicon-based nanodevice can be implanted in close proximity to unresectable metastatic melanoma lesions. Local, targeted delivery of α-INF to the tumor site may alleviate the need for multiple injections and side effects observed with systemic administration [65].
Drug delivery studies with somatostatin have demonstrated delivery rate control by altering pore diameter and surface properties. Debiotech offers the DebioStar™ technology, a silicon nanoporous membrane used in an implant over a reservoir to deliver drug from weeks to months [29]. The pores can be controlled between 1-250 nm, where the number of pores per membrane can be as high as one billion pores/cm2 and the total thickness of the membrane can range 50 nm to ~200 μm.
4.1.4 Osmotic Pumps
Osmotic pumps are comprised of a drug reservoir, a piston, a semipermeable membrane, and an osmotic engine. In operation, water is drawn through the semipermeable membrane in response to an osmotic gradient between an osmotic engine and moisture in the surrounding interstitial fluid. Expansion of the osmotic engine drives a piston forward, expelling the drug formulation through an orifice.
The Viadur® leuprolide acetate implant was developed by Alza (now part of Johnson & Johnson) and marketed by Bayer until it was discontinued in 2007 due to generic competition. Viadur® gained FDA approval in 2000 for the treatment of prostate cancer and utilized the DUROS® platform. The DUROS® titanium implant is 4 mm in diameter by 45 mm in length and provides osmotically driven, zero-order drug delivery (Fig. 9) [71,72]. The implant is placed under the skin on the inside of the upper arm with the aid of a trocar. At the end of the one year delivery duration, the empty system is explanted. Viadur® delivered leuprolide (370 mg/ml leuprolide in dimethyl sulfoxide) continuously over 1 year at ~120 μg/day (0.4 μl/day) from a 150 μl drug reservoir [73]. In vitro and in vivo (rats and beagles) release rate data demonstrated zero-order delivery for 1 year [72,74]. Serum testosterone and leuprolide levels were also monitored and showed steady release rates for 12 months.
Fig. 9.
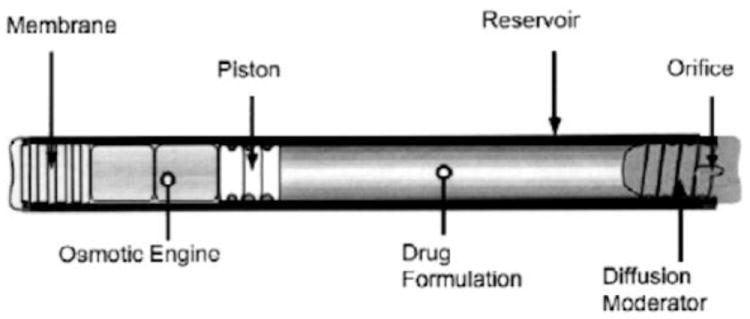
Diagram of a DUROS® osmotic implant. Reproduced with permission from Fig. 1 in [72] (©2001 Elsevier).
Durect Corp. licensed the DUROS® platform from Alza for pain indications. The CHRONOGESIC® reservoir contained ~155 μl sufentanil in benzyl alcohol and provided zero-order release at 5 μg/hr [75]. Durect initiated Phase 3 trials with sufentanil for moderate to severe chronic pain. However, the trial was suspended in 2003 due to premature shutdown of devices [76].
Intarcia Therapeutics uses the DUROS® implant for the delivery of exenatide (type 2 diabetes) and ω-interferon (Hepatitis C) [77]. Exenatide was formulated for a 6 month (45 μg/day), 9 month (30 μg/day), and 12 month (10 μg/day) duration and showed good stability at 25°C and 37°C for 12 months [78]. Furthermore, exenatide was dosed at 20-80 μg/day for 6 months in Phase 2b trials [79]. Intarcia also has ω-interferon in Phase 1b (25 μg/day and 50 μg/day) for Hepatitis C [80,81]. ω-interferon was formulated as a suspension and showed good stability for 2 years at 30°C. The protein was delivered from the device at 9 μg/day and 22 μg/day for 6 months at 37°C.
Finally, a bioerodible, micro-osmotic implant has also been constructed for the local delivery of basic fibroblast growth factor (bFGF) for tissue regeneration. The implants were micromolded and thermally assembled from PLGA sheets. The bottom layer contained the drug reservoir and microchannel array for drug release. An osmotic potential drives water into the reservoir through a semipermeable membrane. The inflated reservoir exerts pressure delivery drug from the microarrays at a rate of 40 ng/day for 4 weeks [82].
4.2 Active Delivery Systems
4.2.1 Single Reservoir Active Systems
Pumps generally include a drug reservoir, an actuator, and one or more valves in order to accurately control the delivery of small volumes of a drug in solution. They may operate by manual actuation, electrolysis, piezoelectric actuation, resistive heating, magnetic actuation, or by incorporation of reversible polymeric valves.
The simplest example of an active drug delivery pump is one that is actuated manually by pressing on it with an instrument or a finger. Such a system was designed for the treatment of glaucoma, age-related macular degeneration, and diabetic retinopathy [44,83]. The pump is fabricated from three layers of polymethylsiloxane (PDMS) using soft lithography. The device was sutured to the outside of the eye and contained a drug reservoir of approximately 200 μl, a check valve, and a cannula (10 mm × 1 mm × 1 mm) placed through the wall of the eye (Fig. 10). When the pump was manually actuated by pressing on the drug reservoir, the increase in pressure in the reservoir caused the check valve in the cannula to open, dispensing the drug.
Fig. 10.
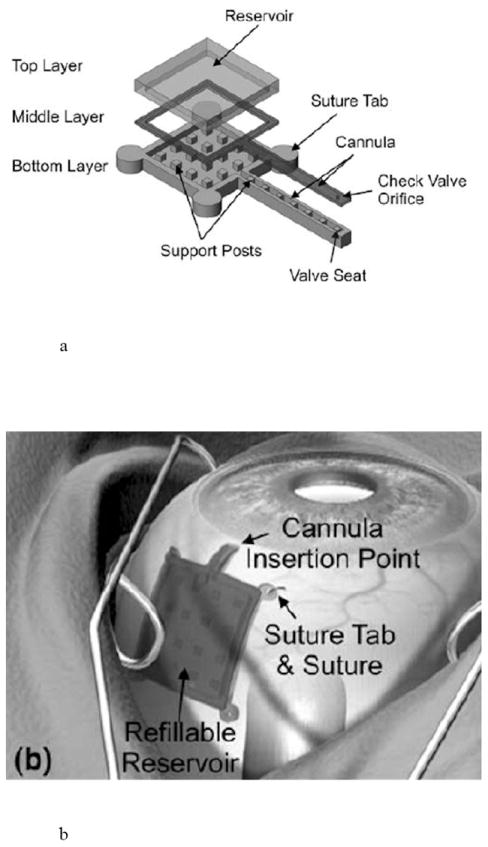
Manually actuated drug delivery pump for ophthalmic use, including a) a diagram showing the components of the pump and b) an illustration of device placement on the eye. Reproduced with permission from Figs. 1b and 2 in [83] (©2009 Springer).
Replenish has developed an electrochemically driven drug delivery pump based on silicon MEMS technology that is designed to allow variable drug delivery rates. It contains a refillable reservoir for a drug solution and a one way check valve made of parylene. The device is surgically implanted beneath the conjunctiva with a flexible parylene cannula inserted through the eye wall. An electric current is passed between two electrodes located on the silicon in contact with the drug solution when a dose is required. The gas generated by electrolysis of the water increases the pressure on the flexible membrane of the drug reservoir, pushing drug solution out through the cannula and into the eye (Fig. 11) [84].
Fig. 11.
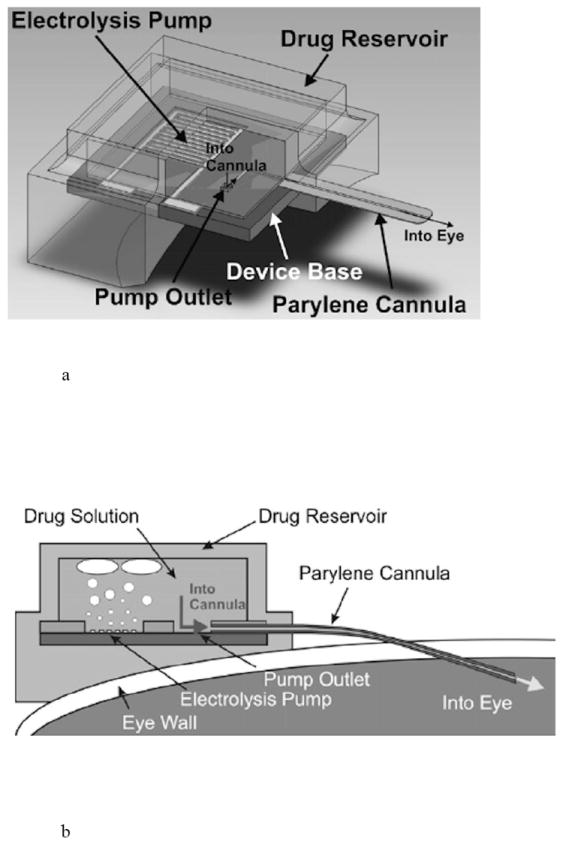
Active delivery pump for ophthalmic use, including a) a diagram showing the components of the pump and b) a cross-section of the pump showing electrolysis enabled pumping of drug into the eye. Reproduced with permission from Figs. 1 and 2 in [84] (©2008 Elsevier).
A magnetically actuated ocular implant has been reported by the University of British Columbia for the treatment of diabetic retinopathy [85]. The device is designed to be surgically implanted behind the eye. A prototype implant consisted of a reservoir (6 mm × 550 μm) containing docetaxel, sealed with an elastic magnetic polydimethylsiloxane (PDMS) membrane (6 mm × 40 μm), and a laser drilled orifice (100 × 100 μm2) (Fig. 12). Upon application of a magnetic field (255 mT), the membrane deforms, causing expulsion of drug solution (~171 ng/actuation) from the implant.
Fig. 12.
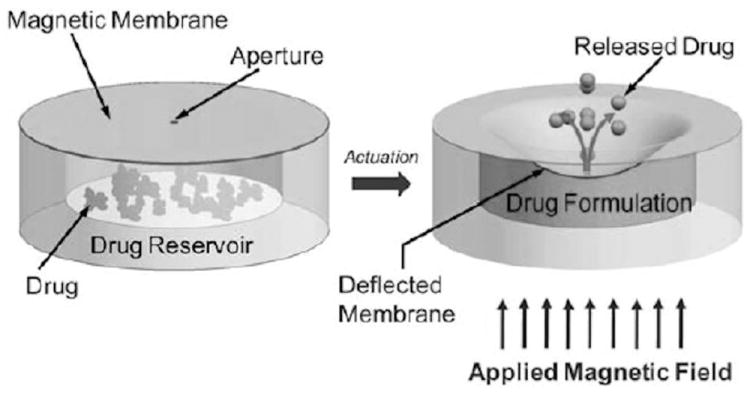
Ocular drug delivery implant actuated by a magnetic field. Reproduced with permission from Fig. 1 in [85] (©2011 Royal Society of Chemistry).
The MIP implantable pump from Debiotech is a piezo-actuated silicon micropump for drug delivery. The pump consists of pair of check valves and a reciprocating pumping membrane to guide liquid flow in the proper direction from a drug reservoir to the target location. The pump is fabricated from multiple bonded layers of silicon and glass, with a piezoelectric ceramic disk and titanium fluid connectors and has a typical flow rate of 1 ml/min (Fig. 13) [29].
Fig. 13.

An implantable active drug delivery pump, including a) a view of the piezoelectric membrane pumping mechanism and check valves and b) a view of the fluidic connections to the pump. Reproduced with permission from Debiotech SA/Switzerland.
Burst release devices have also been proposed for emergency care [39,86,87]. As an example, the IRD3 (implantable rapid drug delivery device) developed at the Massachusetts Institute of Technology (MIT) is comprised of three layers: a drug reservoir layer, a membrane layer that seals the drug reservoir, and an actuation layer where bubbles are formed. Micro-resistors in the actuation layer heat the drug solution, generating bubbles that ruptured the membrane over the reservoir and drove the drug out of the reservoir. The device delivered approximately 20 μl of a vasopressin solution in 45 seconds (Fig. 14). Another group developed a similar microfluidic implant (4.4 mm × 2.3 mm × 22 mm) to deliver vasopressin from a 15 μl reservoir [87].
Fig. 14.

An implantable, active drug delivery pump, including a) a diagram of the implant cross-section showing membrane (A), reservoir (B), and actuation (C) layers and b) the packaged pump next to a coin for scale. Reproduced with permission from Figs. 1 and 4 in [86] (©2008 Springer).
Valves made from polymer actuators, sometimes referred to as “artificial muscles,” have been proposed to modulate release of a drug solution from a single-reservoir “smart pill” implant [88]. The implant would contain micrometer sized polymer rings that expand and contract in response to an electrical signal transmitted through a conducting polymer, thereby allowing the modulation of flow of drug solution out of the reservoir.
4.2.2 Multi-Reservoir Active Systems
The advantage of one active delivery mechanism versus another, and the need for more than one drug reservoir, is highly dependent on the treatment modality of the specific disease state and the preferred delivery profile (sustained release or pulsatile). Active delivery from multi-reservoir array implants can be actuated by electrochemical, electrothermal, or laser means [41,89-94]. Drug is sealed in each reservoir of the array to isolate the drug from the body until it is needed. The initiation of drug release is actively controlled by the application of the electrical or laser stimulus to create an opening in the sealing material, thereby exposing the drug to the body. The rate of release is then passively controlled by the dissolution and diffusion characteristics of the drug formulation in the reservoir. Thus, it is possible to deliver nanoliter drug payloads in their most stable form, including solid formulations, using active systems.
The first demonstration of electrochemically activated microchip implants occurred at MIT and involved arrays of gold membrane capped drug reservoirs in silicon. When an electric potential was applied to the gold cap in a solution containing physiological levels of saline, the gold membrane was converted to a soluble gold salt and dissolved away, thereby exposing the drug to the surrounding environment and releasing the drug (Fig. 15) [16,89,90,92,93]. These devices were the first to allow the precisely-timed release of solid materials from an implant. Although the reproducibility of an electrochemical dissolution process in the in vivo environment can be a challenge, these devices established the feasibility of pulsatile delivery of macromolecule drugs and the ability to achieve complex release profiles with multiple therapeutics compounds [38,95].
Fig. 15.
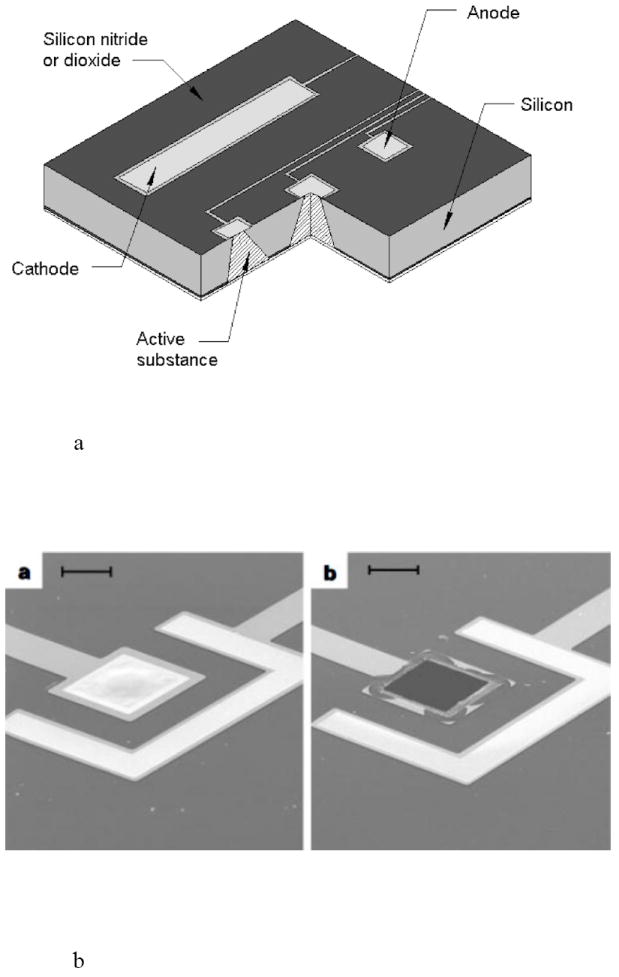
An electrochemically actuated drug delivery implant, including a) a cut away view showing several reservoirs of the implant and b) reservoir caps of the microchip (a) before and (b) after electrochemical actuation. Reproduced a) and b) with permission from Figs. 1A and 2 in [89] (©1999 Nature Publishing Group).
MicroCHIPS developed reservoir arrays where the metal membranes coating the reservoirs were opened electrothermally instead of electrochemically [96,97]. Metal membranes composed of either gold or a platinum and titanium laminate were removed by resistive heating from an applied current [96]. This electrothermal activation method was much faster than the electrochemical method, and it provided a method independent of the drug formulation inside the reservoir or the environment surrounding the device. Microchip arrays measuring 15 × 15 × 1 mm3 containing 100 reservoirs capable of holding 300 nl drug formulation per reservoir have been fabricated [96]. The fully assembled devices included a drug filled array, microprocessor, implantable battery, and wireless antenna in a titanium housing that measured 4.5 × 5.5 × 1 cm3 with a volume of ~ 30 ml. Initial studies with leuprolide formulated at 100 mg/ml and filled into 300 nl reservoirs showed good stability and an excellent in vitro-in vivo release rate correlation [98]. Similarly, an osteoporosis implant delivering pulsatile parathyroid hormone (PTH) was formulated at 500 mg/ml (~ 20 μg PTH/reservoir) and demonstrated good stability after 6 months at 37°C (Fig. 16) [99].
Fig. 16.
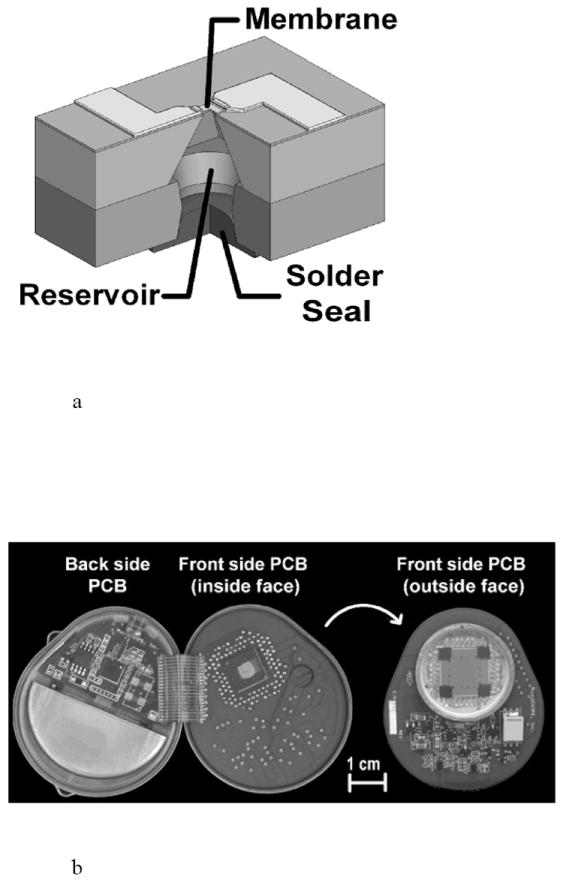

An electrothermally actuated drug delivery implant, including a) a cut away view of a single reservoir from a multi-reservoir implant, b) a view of the implant housing, drug containing microchip, electronic and wireless communication components, and battery, and c) a fully assembled implant. Reproduced a), b), c) with permission from Figs. 1B, 1C, 1D in [96] (©2006 Nature Publishing Group).
On Demand Therapeutics utilizes laser activation to achieve ophthalmic drug delivery for treatment of retinal diseases, such as wet age-related macular degeneration or diabetic retinopathy. The preclinical product is a small, injectable rod containing multiple reservoirs that are hermetically sealed to protect the stability of anti-VEGF or other drugs in the reservoirs. The drug-filled device is implanted into the periphery of the vitreous in the region of the pars plana using a standard intravitreal injection technique. When the ophthalmologist decides to release drug from the implant, a slit lamp and a Goldmann mirrored lens are used to locate the implant and focus the laser beam on the selected reservoir. Upon laser activation, an opening is created that allows the drug formulation to elute into the vitreous and diffuse to the retina. The drug doses in the unopened reservoirs remain intact, such that additional drug doses can be released during subsequent visits. This approach provides a less invasive dosing regimen than monthly injections (Fig. 17) [94].
Fig. 17.

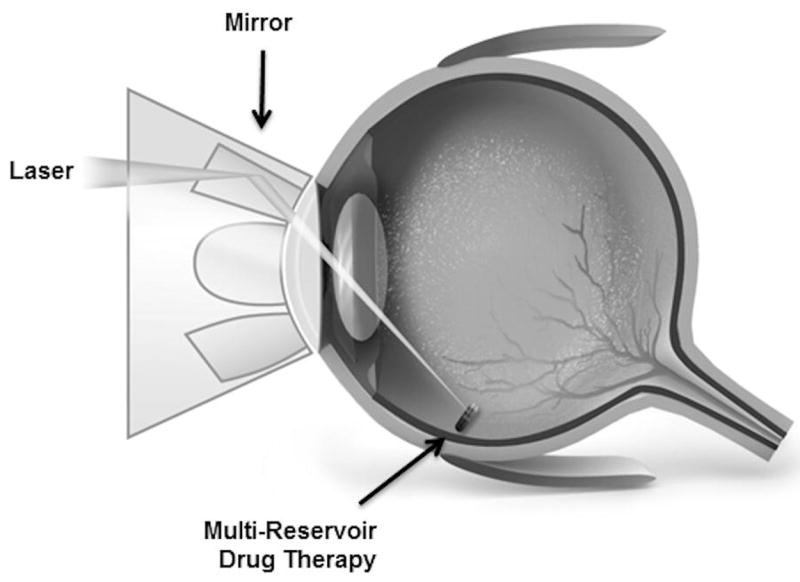
A laser activated system for on demand ophthalmic drug delivery showing a) a multi-reservoir, intravitreal implant and b) initiation of drug release from the implant using an ophthalmic laser [94]. Reproduced with permission from On Demand Therapeutics, Inc.
5. Outlook
As these reservoir delivery systems utilizing microtechnologies gain maturity, acceptance by the FDA, and find their niche in the marketplace, several factors will impact their success. First, the “marrying” of a drug substance in need of a more controlled delivery profile and the appropriate delivery system is critical. The ultimate success of the product is not dictated by a good technical match between the drug and the delivery method, but by the product’s ability to meet true patient and physician unmet needs.
Second, in the current economic environment, the combination product must also provide a distinct advantage over current therapies, with only a small premium in price. The impact of pricing can now be felt early in the R&D process, as these budgets are increasingly curtailed. The ultimate outcome is that engineers and scientists must provide innovative products and cost effective processes at every stage of development.
Third, development of successful drug delivery products requires forethought into the regulatory hurdles in order to receive timely market approval. These include adequate shelf life stability and in vivo use life stability to assure that the final dosing period was as efficacious as the initial dosing period. The development of drug release tests used to demonstrate the lack of dose dumping, percentage of total formulation released from the delivery system, in vivo stability, and an in vivo-in vitro correlation are all critical.
Fourth, extra care should be given to supply chain issues early in development. Drug excipients should be Generally Regarded As Safe (GRAS). Device materials that are drug or patient contacting should be Medical Grade with acceptable physicochemical characteristics, biocompatibility and leachables/extractables profile. These tests will apply to incoming raw materials and the final product. Although beyond the scope of this review, the selection of biocompatible device materials is critical [10,100-103]. This is especially important for long term or permanent implants, where tissue adhesion or polymeric degradation products can cause inflammation. Furthermore, supplier selection should be done with care to assure that the vendor has or will be able to provide a Drug Master File (DMF) and that they can demonstrate control of their suppliers. For example, if an upstream supplier changes a “trade-secret” slip agent, antioxidant or dye package in a commodity material, the downstream drug product producer must understand the impact on the product before it is sold to a patient (and prevent the need for a product recall).
6. Conclusions
The knowledge and tools enabling the development of reservoir-based drug delivery systems utilizing microtechnology have come from number of diverse fields of study including chemistry, materials science, mechanics, information technology, and microelectronics. The innovations at the core of such novel oral, dermal, and implantable delivery systems come from the intersection of these disparate fields. Devices combining reservoirs and microtechnology driven by passive or active mechanisms are enabling the delivery of both small and macromolecule drugs with increased specificity and control.
While tremendous progress continues on the development of micro and nanotechnology for reservoir-based drug delivery devices, much additional work will be required to translate the promising technology into safe and effective, well-controlled, patient acceptable products.
Acknowledgments
Robert Langer would like to acknowledge grant support for reservoir-based, microchip drug delivery systems from the U.S. National Institutes of Health, grant #EB006365.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mandal DC, Mandal M. Current status and future prospects of new drug delivery system. Pharma Times. 2010 Apr;42 [Google Scholar]
- 2.Kaparissides C, Alexandridou S, Kotti K, Chaididou S. Advanced drug delivery systems: New developments, new technologies. AzoM com Pvt L Ltd. 2006 Oct; [Google Scholar]
- 3.Langer R, Peppas NA. Advances in biomaterials, drug delivery and bionanotechnology. AIChE J. 2003;49:2990–3006. [Google Scholar]
- 4.Hilt JZ, Peppas NA. Microfabricated drug delivery devices. Int J Pharm. 2005;306:15–23. doi: 10.1016/j.ijpharm.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 5.Verma RK, Garg S. Drug delivery technologies and future directions. Pharmaceutical Technology. 2001;25:1–14. [Google Scholar]
- 6.Rosen H, Abribat T. The rise and rise of drug delivery. Nat Rev Drug Discov. 2005;4:381–385. doi: 10.1038/nrd1721. [DOI] [PubMed] [Google Scholar]
- 7.Thomson Physicians’ Desk Reference. 63. Thomson Publishing; Montvale, N.J: 2009. [Google Scholar]
- 8.Ahmed A, Bonner C, Desai T. Bioadhesive microdevices with multiple reservoirs: a new platform for oral drug delivery. J Control Release. 2002;81:291–306. doi: 10.1016/s0168-3659(02)00074-3. [DOI] [PubMed] [Google Scholar]
- 9.Tao SL, Desai TA. Micromachined devices: the impact of controlled geometry for cell-targeting to bioavailability. J Control Release. 2005;109:127–138. doi: 10.1016/j.jconrel.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 10.Ainslie KM, Desai TA. Microfabricated implant for applications in therapeutic delivery, tissue engineering and biosensing. Lab Chip. 2008;8:1864–1878. doi: 10.1039/b806446f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ainslie KM, Lowe RD, Beaudette TT, Petty L, Bachelder EM, Desai TA. Microfabricated devices for enhanced bioadhesive drug delivery: attachment to and small-molecule release through a cell monolayer under flow. Small. 2009;5:2857–2863. doi: 10.1002/smll.200901254. [DOI] [PubMed] [Google Scholar]
- 12.Tao SL, Desai TA. Microfabrication of multilayer, assymetric, polymeric devices for drug delivery. Adv Mater. 2005;17:1625–1630. [Google Scholar]
- 13.Tao SL, Desai TA. Gastrointestinal patch systems for oral drug delivery. Drug Discov Today. 2005;10:909–915. doi: 10.1016/S1359-6446(05)03489-6. [DOI] [PubMed] [Google Scholar]
- 14.Nuxoll EE, Siegel RA. BioMEMS devices for drug delivery. IEEE Eng Med Biol. 2009 Jan-Feb;:31–39. doi: 10.1109/MEMB.2008.931014. [DOI] [PubMed] [Google Scholar]
- 15.Aroro A, Prausnitz M, Mitragotri S. Micro-scale devices for transdermal drug delivery. Int J Pharm. 2008;8:227–236. doi: 10.1016/j.ijpharm.2008.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Staples M, Daniel K, Cima MJ, Langer R. Application of micro- and nano-electromechanical devices to drug delivery. Pharm Res. 2006;23:847–863. doi: 10.1007/s11095-006-9906-4. [DOI] [PubMed] [Google Scholar]
- 17.Matriano J, Cormier M, Johnson J, Young W, Buttery M, Nyam K, Daddona P. Macroflux microprojection array patch technology: A new and efficient approach for intracutaneous immunization. Pharm Res. 2002;19:1653–1664. doi: 10.1023/a:1013607400040. [DOI] [PubMed] [Google Scholar]
- 18.Chandrasekara S, Brazzle J, Frazier A. Surface machined metallic microneedles. J Microelectromech S. 2003;12:281–288. [Google Scholar]
- 19.Zahn J, Talbot N, Liepmann D, Pisano A. Microfabricated polysilicon microneedles for minimally invasive biomedical devices. Biomed Microdevices. 2000;2:295–303. [Google Scholar]
- 20.Zahn J, Deshmekh A, Pisano A, Liepmann D. Continuous on-chip micropumping from microneedled enhance drug delivery. Biomed Microdevices. 2004;6:183–190. doi: 10.1023/B:BMMD.0000042047.83433.96. [DOI] [PubMed] [Google Scholar]
- 21.Teo MAL, Shearwood C, Ng KC, Lu J, Moochhala S. In vitro and in vivo characterization of MEMS microneedles. Biomed Microdevices. 2005;7:47–52. doi: 10.1007/s10544-005-6171-y. [DOI] [PubMed] [Google Scholar]
- 22.McAllister DV, Wang PM, Davis SP, Park JH, Canatella PJ, Allen MG, Prausnitz MR. Microfabricated needles for transdermal delivery of macromolecules and nanoparticles: fabrication methods and transport studies. P Natl Acad Sci USA. 2003;100:13755–13760. doi: 10.1073/pnas.2331316100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stoeber B, Liepmann D. Arrays of hollow out-of-plane microneedles for drug delivery. J Microelectromech S. 2005;14:472–479. [Google Scholar]
- 24.Sivamani R, Stoeber B, Wu G, Zhai H, Liepmann D, Maibach H. Clinical microneedle injection of methyl nicotinate: stratum corneum penetration. Skin Res Technol. 2005;11:152–156. doi: 10.1111/j.1600-0846.2005.00107.x. [DOI] [PubMed] [Google Scholar]
- 25.Griss P, Stemme G. Side-opened out-of-plane microneedles for microfluidic transdermal liquid transfers. J Microelectromech S. 2003;12:296–301. [Google Scholar]
- 26.Roxhed N, Gasser C, Griss P, Holzapfel G, Stemme G. Penetration enhanced ultrashorp microneedles and prediction in skin interaction for efficient transdermal drug delivery. J Microelectromech S. 2007;16:1429–1440. [Google Scholar]
- 27.Gardeniers H, Luttge R, Bereschot E, de Boer M, Yeshurun S, Hefetz M, van’t Oever R, van den Berg A. Silicon micromachines hollow microneedles for transdermal liquid transport. J Microelectromech S. 2003;12:855–862. [Google Scholar]
- 28.Van Damme P, Oosterhuis-Kafeja F, Van der Wielen M, Almagor Y, Sharon O, Levin Y. Safety and efficacy of a novel microneedle device for dose sparing intradermal influenza vaccination in healthy adults. Vaccine. 2009;27:454–459. doi: 10.1016/j.vaccine.2008.10.077. [DOI] [PubMed] [Google Scholar]
- 29.http://www.debiotech.com/
- 30.Roxhed N, Griss P, Stemme G. Membrane-sealed hollow microneedles and related administration schemes for transdermal drug delivery. Biomed Microdevices. 2008;10:271–279. doi: 10.1007/s10544-007-9133-8. [DOI] [PubMed] [Google Scholar]
- 31.Nordquist L, Roxhed N, Griss P, Stemme G. Novel microneedle patches for active insulin delivery are efficient in maintaining glycaemic control: an initial comparison with subcutaneous administration. Pharm Res. 2007;24:1381–1387. doi: 10.1007/s11095-007-9256-x. [DOI] [PubMed] [Google Scholar]
- 32.Hafeli UO, Mikhtari A, Liepmann D. In vivo evaluation of a microneedle-based miniature syringe for intradermal drug delivery. Biomed Microdevices. 2009;11:943–950. doi: 10.1007/s10544-009-9311-y. [DOI] [PubMed] [Google Scholar]
- 33.Davis SP, Martanto W, Allen MG, Prausnitz MR. Hollow metal microneedles for insulin delivery to diabetic rats. IEEE T Bio-Med Eng. 2005;52:909–915. doi: 10.1109/TBME.2005.845240. [DOI] [PubMed] [Google Scholar]
- 34.Patel SR, Lin ASP, Edelhauser HF, Prausnitz MR. Suprachoroidal drug delivery to the back of the eye using hollow microneedles. Pharm Res. 2010;28:166–176. doi: 10.1007/s11095-010-0271-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.http://www.valeritas.com/
- 36.Sullivan SP, Koutsonanos DG, Martin MDP, Lee JW, Zarnitsyn V, Choi SO, Murthy N, Compans RW, Skountzou I, Prausnitz MR. Dissolving polymer microneedle patches for influenza vaccination. Nature Medicine. 2010;16:915–920. doi: 10.1038/nm.2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kommareddy S, Boudner BC, Oh S, Kwon SY, Singh M, O’Hagan DT. Dissolvable microneedle patches for the delivery of cell culture derived influenza vaccine antigens. J Pharm Sci. 2012;101:1021–1027. doi: 10.1002/jps.23019. [DOI] [PubMed] [Google Scholar]
- 38.Grayson ACR, Shawgo RS, Johnson AM, Flynn NT, Li Y, Cima MJ, Langer R. A BioMEMS review: MEMS technology for physiologically integrated devices. P IEEE. 2009;92:6–21. [Google Scholar]
- 39.Sakamoto JH, van de Ven AL, Godin B, Blanco E, Serda RE, Grattoni A, Ziemys A, Bouramrani A, Hu T, Ranganathan SI, De Rosa E, Martinez JO, Smid CA, Chchana RM, Lee SY, Srinavasan S, Landry M, Meyn A, Tasciotti E, Liu X, Decuzzi P, Ferrari M. Enabling individualized therapy through nanotechnology. Pharmacol Res. 2010;62:57–89. doi: 10.1016/j.phrs.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nisar A, Afzulpurkar N, Mahaisavariya B, Tuantranont A. MEMS-based micropumps in drug delivery and biomedical applications. Sensor Actuat B-Chem. 2008;130:917–942. [Google Scholar]
- 41.Receveur RAM, Linemans FW, de Rooij NF. Microsystem technologies for implantable applications. J Micromech Microeng. 2007;17:R50–R80. [Google Scholar]
- 42.Choonara YE, Pillay V, Danckwerts MP, Carmichael TR, Du Toit LC. A review of implantable intravitreal drug delivery technologies for the treatment of posterior segment eye diseases. J Pharm Sci. 2010;99:2219–2239. doi: 10.1002/jps.21987. [DOI] [PubMed] [Google Scholar]
- 43.Eljarrat-Binstock E, Pe’er J, Domb AJ. New techniques for drug delivery to the posterior eye segment. Pharm Res. 2010;27:530–543. doi: 10.1007/s11095-009-0042-9. [DOI] [PubMed] [Google Scholar]
- 44.Mansoor S, Kuppermann BD, Kenney MC. Intraocular sustained release delivery systems for triamcinolone acetonide. Pharm Res. 2009;26:770–784. doi: 10.1007/s11095-008-9812-z. [DOI] [PubMed] [Google Scholar]
- 45.http://www.psivida.com/products.html
- 46.Sanborn GE, Anand R, Torti RE, Nightingale SD, Cal SX, Yates B, Aston P, Smith TJ. Sustained release ganciclovir therapy for treatment of cytomegalovirus retinitis; uses of an intravitreal device. Arch Ophthamol. 1992;110:188–195. doi: 10.1001/archopht.1992.01080140044023. [DOI] [PubMed] [Google Scholar]
- 47.Smith TJ, Pearson PA, Blandford DL, Brown JD, Goins KA, Hollins JL, Schmiesser ET, Glavinos P, Baldwin LB, Ashton P. Intravitreal sustained release ganciclovir. Arch Ophthamol. 1992;110:255–258. doi: 10.1001/archopht.1992.01080140111037. [DOI] [PubMed] [Google Scholar]
- 48.Mruthyunjaya P, Khalatbari D, Yang P, Stinnett S, Tano R, Ashton P, Guo H, Nazzar M, Jaffe GJ. Efficacy of low release rate fluocinolone acetonide intravitreal implants to treat experiments uveitis. Arch Ophthalmol. 2006;124:1012–1018. doi: 10.1001/archopht.124.7.1012. [DOI] [PubMed] [Google Scholar]
- 49.Callanan DG, Jaffe GJ, Martin DF, Pearson PA, Comstock TL. Treatment of posterior uveitis with a flucinolone acetonide implant. Arch Ophthalmol. 2008;126:1191–1201. doi: 10.1001/archopht.126.9.1191. [DOI] [PubMed] [Google Scholar]
- 50.http://www.alimerasciences.com/Products/iluvien.aspx
- 51.Kane FE, Burdan J, Cutino A, Green KE. Iluvien: a new sustained delivery technology for posterior eye disease. Expert Opin Drug Del. 2008;5:1039–1046. doi: 10.1517/17425247.5.9.1039. [DOI] [PubMed] [Google Scholar]
- 52.Campochiaro PA, Brown DM, Pearson A, Ciulla T, Boyer D, Holz FG, Tolentino M, Gupta A, Duarte L, Madreperla S, Gonder J, Kapik B, Billman K, Kane F. Long-term benefit of sustained-delivery fluocinolone acetonide vitreous inserts for diabetic macular edema. Ophthalmology. 2011;118:626–635. doi: 10.1016/j.ophtha.2010.12.028. [DOI] [PubMed] [Google Scholar]
- 53.http://investor.alimerasciences.com/releasedetail.cfm?ReleaseID=574002
- 54.Molokhia SA, Sant H, Simonis J, Bishop CJ, Burr RM, Gale BK, Ambati BK. The capsule drug device: novel approach for drug delivery to the eye. Vision Res. 2010;50:680–685. doi: 10.1016/j.visres.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 55.Grayson ACR, Choi IS, Tyler BM, Wang PP, Brem H, Cima MJ, Langer R. Multi-pulse drug delivery from a resorbable polymeric microchip device. Nat Mater. 2003;2:767–772. doi: 10.1038/nmat998. [DOI] [PubMed] [Google Scholar]
- 56.Kim GY, Tyler BM, Tupper MM, Karp JM, Langer RS, Brem H, Cima MJ. Resorbable polymer microchips releasing BCNU inhibit tumor growth in the rat 9L flank model. J Control Release. 2007;123:172–178. doi: 10.1016/j.jconrel.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grayson ACR, Cima MJ, Langer R. Molecular release from a polymeric microreservoir device: influence of chemistry, polymer swelling and loading on device performance. J Biomed Mater Res A. 2004;69A:502–512. doi: 10.1002/jbm.a.30019. [DOI] [PubMed] [Google Scholar]
- 58.Kuppermann BD. Implant delivery of corticosteroids and other pharmacologic agents, Retina 2006: Emerging New Concepts. American Academy of Ophthalmology Annual Meeting; Las Vegas, NV. Nov 10-11 2006. [Google Scholar]
- 59.Piveteau LD, Hofmann H, Neftel F. Anisotropic nanoporous coatings for medical implants. 1,891,988. EP Patent. 2008
- 60.Verhey S, Adostoni P, Dawkins KD, Dens J, Rutsch W, Carrie D, Schofer J, Lotan C, Dubois CL, Cohen SA, Fitzgerald PJ, Lansky AJ. The GENESIS (randomized, multicenter study of the pimecrolimus-eluting and pimecrolimus/paclitaxel-eluting coronary stent system in patients with de novo lesions of the native coronary arties) Trial. J Am Coll Cardiol Intv. 2009;2:205–214. doi: 10.1016/j.jcin.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 61.Capodanno D, Dipasqua F, Tamburino C. Novel drug-eluting stents in the treatment of de novo coronary lesions. Vasc Health Risk Manag. 2011;7:103–118. doi: 10.2147/VHRM.S11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grube E, Schofer J, Hauptmann KE, Nickenig G, Curzen N, Allocco DJ, Dawkins KD. A novel paclitaxel-eluting stent with an ultrathin abluminal biodegradable polymer. J Am Coll Cardiol Intv. 2010;3:431–438. doi: 10.1016/j.jcin.2009.12.015. [DOI] [PubMed] [Google Scholar]
- 63.Garg S, Serruys PW. Coronary stents: looking forward. J Am Coll Cardiol. 2010;56:S43–S78. doi: 10.1016/j.jacc.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 64.Martin F, Walczak R, Boiarski A, Cohen M, West T, Casentino C, Ferrari M. Tailoring width of microfabricated nanochannels to solute size can be used to control diffusion kinetics. J Control Release. 2005;102:123–133. doi: 10.1016/j.jconrel.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 65.Lesinski G, Sharma S, Varker K, Sinha P, Ferrari M, Carson W. Release of biologically functional interferon-alpha from a nanochannel delivery system. Biomed Microdevices. 2005;7:71–79. doi: 10.1007/s10544-005-6174-8. [DOI] [PubMed] [Google Scholar]
- 66.Pricl S, Ferrone M, Fermeglia M, Amato F, Cosentino C, Cheng MM, Walczak R, Ferrari M. Multiscale modeling of protein transport in silicon membrane nanochannels. Part 1. Derivation of molecular parameters from computer simulations. Biomed Microdevices. 2006;8:277–290. doi: 10.1007/s10544-006-0031-2. [DOI] [PubMed] [Google Scholar]
- 67.Eijkel JCT, van den Berg A. Nanofluidics: what is it and what can we expect from it? Microfluid Nanofluid. 2005;5:249–267. [Google Scholar]
- 68.Sinha PM, Valco G, Sharma S, Liu X, Ferarri M. Nanoengineered device for drug delivery application. Nanotechnology. 2004;15:S585–S589. [Google Scholar]
- 69.Paisley AN, Trainer PJ. Recent advances in the treatment of acromegaly. Recent Advances in Investigational Drugs. 2006;15:251–256. doi: 10.1517/13543784.15.3.251. [DOI] [PubMed] [Google Scholar]
- 70.Gardner P. Use of a nanopore membrane in novel a drug delivery device. Future Drug Delivery. 2006 Jun;:59–60. [Google Scholar]
- 71.Stevenson CL, Theeuwes F, Wright JC. Osmotic implantable delivery systems. In: Wise D, editor. Handbook of Pharmaceutical Controlled Release Technology. Marcel Dekker, Inc.; New York: 2000. pp. 225–253. [Google Scholar]
- 72.Wright JC, Leonard ST, Stevenson CL, Beck JC, Chen G, Jao RM, Johnson PA, Leonard J, Skowronski RJ. An in vivo/in vitro comparison with a leuprolide osmotic implant for the treatment of prostate cancer. J Control Release. 2001;75:1–10. doi: 10.1016/s0168-3659(01)00358-3. [DOI] [PubMed] [Google Scholar]
- 73.Stevenson CL. Formulation of Leuprolide at High Concentration for Delivery from a One Year Implant. In: McNally EJ, Hastedt JE, editors. Protein Formulation and Delivery. 2. Marcel Dekker, Inc.; NY: 2007. pp. 153–175. [Google Scholar]
- 74.Cukierski MJ, Johnson PA, Beck JC. Chronic (60 week) toxicity study of DUROS leuprolide implants in dogs. Int J Toxicol. 2001;20:369–381. doi: 10.1080/109158101753333659. [DOI] [PubMed] [Google Scholar]
- 75.Fisher DM, Kellett N, Lenhardt R. Pharmacokinetics of an implanted osmotic pump delivering sufentanil for the treatment of chronic pain. Anesthesiology. 2003;99:929–937. doi: 10.1097/00000542-200310000-00028. [DOI] [PubMed] [Google Scholar]
- 76.Durect Corp. Press Release from 16-Oct-2003 ( http://phx.corporate-ir.net/phoenix.zhtml?c=121590&p=irol-newsArticle&ID=459693&highlight)
- 77.www.intarcia.com
- 78.Yang B, Negulescu C, D’vas R, Eftimie C, Carr J, Lautenbach S, Horwege K, Mercer C, Ford D, Alessi T. Stability of ICTA 560 for continuous subcutaneous delivery of Exenatide at body temperature for 12 months. Diabetes Technology Meeting; Long Beach, CA. April 3, 2009. [Google Scholar]
- 79.Henry RR, Cuddig R, Rosenstock J, Alessi T, Luskey K. Comparing ITCA 650, continuous subcutaneous delivery of exenatide via DUROS® device, vs. twice daily exenatide injections in metformin-treated type 2 diabetes. European Association for the Study of Diabetes (EASD) 46th Annual Meeting: Abstract 78, presented; September 22, 2010. [Google Scholar]
- 80.Yang B, Alessi T, Rohloff C, Mercer R, Negulescu C, Lautenbach S, Gumucio J, Guo M, Weeks E, Carr J, Ford D. Continuous delivery of proteins and peptides at consistent rates for at least 3 months form the DUROS® device. AAPS 2008 Meeting; Atlanta Georgia. Nov 18, 2008; Poster T3150. [Google Scholar]
- 81.Rohloff CM, Alessi TR, Yang B, Dahms J, Carr JP, Lautenbach SD. DUROS® technology delivers peptides and proteins at consistent rate continuously for 3 to 12 months. Journal of Diabetes Science & Technology. 2008;2:461–467. doi: 10.1177/193229680800200316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ryu WH, Huang Z, Prinz FB, Goodman SB, Fasching R. Biodegradable microosmotic pump for long-term and controller lease of basic fibroblast growth factor. J Control Release. 2007;124:98–105. doi: 10.1016/j.jconrel.2007.08.024. [DOI] [PubMed] [Google Scholar]
- 83.Lo R, Kuwahar K, Li PY, Saati S, Agrawal RN, Humayun MS, Meng E. A passive MEMS drug delivery pump for treatment of ocular diseases. Biomed Microdevices. 2009;11:959–970. doi: 10.1007/s10544-009-9313-9. [DOI] [PubMed] [Google Scholar]
- 84.Li PY, Shih J, Lo R, Saati S, Agrawal R, Humayun MS, Tai YC, Meng E. An electrochemical intraocular drug delivery device. Sensor Actuat A-Phys. 2008;143:41–48. [Google Scholar]
- 85.Pirmoradi FN, Jackson JK, Burt HM, Chaio M. On-demand controlled release of docetaxel from a battery-less MEMS drug delivery device. Lab Chip. 2011;11:2744–2752. doi: 10.1039/c1lc20134d. [DOI] [PubMed] [Google Scholar]
- 86.Elman NM, Ho Duc HL, Cima MJ. An implantable MEMS drug delivery device for rapid delivery in ambulatory emergency care. Biomed Microdevices. 2009;11:625–631. doi: 10.1007/s10544-008-9272-6. [DOI] [PubMed] [Google Scholar]
- 87.Chung AJ, Huh YS, Erickson D. A robust, electrochemically driven microwell drug delivery system for controlled vasopressin release. Biomed Microdevices. 2009;11:861–867. doi: 10.1007/s10544-009-9303-y. [DOI] [PubMed] [Google Scholar]
- 88.Low LM, Seetharaman S, He KQ, Madou MJ. Microactuators toward microvalves for responsive controlled drug delivery. Sensor Actuat B-Chem. 2000;1:149–160. [Google Scholar]
- 89.Santini JT, Jr, Cima MJ, Langer R. A controlled-release microchip. Nature. 1999;397:335–338. doi: 10.1038/16898. [DOI] [PubMed] [Google Scholar]
- 90.Santini JT, Jr, Richards AC, Scheidt RA, Cima MJ, Langer R. Microchips as implantable drug delivery devices. Agnew Chem Int Edit. 2000;39:2396–2407. [PubMed] [Google Scholar]
- 91.Shawgo R, Grayson ACR, Li Y, Cima MJ. BioMEMS for drug delivery. Curr Opin Solid St M. 2002;6:329–334. [Google Scholar]
- 92.Li Y, Ho Duc HL, Tyler B, Williams T, Tupper M, Langer R, Brem H, Cima MJ. In vivo delivery of BCNU from a MEMS device to a tumor model. J Control Release. 2005;106:138–145. doi: 10.1016/j.jconrel.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 93.Li Y, Shawgo R, Tyler B, Henderson P, Gobel J, Rosenberg A, Storm PB, Langer R, Brem H, Cima MJ. In vivo release from a drug delivery MEMS device. J Control Release. 2004;100:211–291. doi: 10.1016/j.jconrel.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 94.www.ondemandtx.com
- 95.Grayson ACR, Shawgo RS, Li Y, Cima MJ. Electronic MEMS for triggered delivery. Adv Drug Deliver Rev. 2004;56:173–184. doi: 10.1016/j.addr.2003.07.012. [DOI] [PubMed] [Google Scholar]
- 96.Prescott JH, Lipka S, Baldwin S, Sheppard NF, Jr, Maloney JM, Coppeta J, Yomtov B, Stables MA, Santini JT., Jr Chronic, programmed polypeptide delivery from an implanted, multireservoir microchip device. Nat Biotechnol. 2006;24:437–438. doi: 10.1038/nbt1199. [DOI] [PubMed] [Google Scholar]
- 97.Maloney JM, Uhland SA, Polito BF, Sheppard NF, Jr, Pelta CM, Santini JT., Jr Electrothermally activated microchips for implantable drug delivery and biosensing. J Control Release. 2005;109:244–255. doi: 10.1016/j.jconrel.2005.09.035. [DOI] [PubMed] [Google Scholar]
- 98.Prescott JH, Krieger TJ, Lipka S, Staples MA. Dosage form development, in vitro release kinetics, and in vitro-in vivo correlation for leuprolide released from an implantable multi-reservoir array. Pharm Res. 2007;24:1252–1261. doi: 10.1007/s11095-007-9243-2. [DOI] [PubMed] [Google Scholar]
- 99.Proos ER, Prescott JH, Staples MA. Long-term stability and in vitro release of hPTH(1-34) from a multi-reservoir array. Pharm Res. 2008;25:1387–1395. doi: 10.1007/s11095-008-9544-0. [DOI] [PubMed] [Google Scholar]
- 100.Ainslie KM, Thakar RG, Bernards DA, Desai TA. Inflammatory Response to Implanted Nanostructured Materials. In: Puleo DA, Bizios R, editors. Biological Interactions on Material Surfaces. Springer; New York: 2009. pp. 355–371. [Google Scholar]
- 101.Ainslie KM, Tao SL, Popat KC, Desai TA. In vitro immunogenicity of silicon based micro and nanostructured surfaces. ACS Nano. 2008;2:1076–1084. doi: 10.1021/nn800071k. [DOI] [PubMed] [Google Scholar]
- 102.Muthusubramaniam L, Lowe R, Fissell WH, Li L, Marchant RE, Desai TA, Roy S. Hemocompatibility of silicon based substrates for biomedical implant applications. Annals of Biomedical Engineering. 2011;39:1296–1305. doi: 10.1007/s10439-011-0256-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Voskerician G, Shive MS, Shawgo RS, von Recum H, Anderson JM, Cima MJ, Langer R. Biocompatibility and biofouling of MEMS drug delivery devices. Biomaterials. 2003;24:1959–1967. doi: 10.1016/s0142-9612(02)00565-3. [DOI] [PubMed] [Google Scholar]