

Abstract
Pseudomonas exotoxin A (PE) is cytotoxic for eukaryotic cells because it enters cells by receptor-mediated endocytosis, translocates to the cell cytosol and ADP-ribosylates elongation factor 2 (EF2). However, the interaction of this toxin with eukaryotic cells and the mechanism of PE mediated cell death have not been extensively characterized. The feasibility of carrying out a genome-wide RNAi screen, makes Drosophila melanogaster S2 cells as a good model system to identify essential genes in PE-mediated cytotoxicity, provided a suitable multi-well assay is developed. Here, using the alamarBlue® viability assay, we show that Drosophila S2 cells are sensitive to PE at picomolar concentrations and that toxin treatments provoke an increase in caspase activity. This prompted us to use RNAi to characterize the mechanism of cell death. Results indicated that PE-mediated death of S2 cells was dependent on the presence of diphthamide, the post translational modification of EF2, and on the presence of Drice, the terminal caspase of insect cells. RNAi to drice or chemical inhibition of caspase action by z-VAD-fmk protected cells from PE-mediated death. Protection from death by RNAi or z-VAD-fmk did not interfere with toxin delivery to the cytosol leading to inhibition of protein synthesis. Using a convenient alamarBlue® assay, our data confirms the cytotoxicity of PE for S2 cells and establishes apoptosis as the mode of PE-mediated death. This confirms the suitability of Drosophila cells as a convenient and simple model to elucidate the role of specific genes and proteins required for PE action.
Keywords: Pseudomonas exotoxin A, apoptosis, diphthamide, RNAi, Drice
1. Introduction
The pathogenesis of P. aeruginosa, a Gram-negative soil organism and a leading cause of infections in cystic fibrosis patients, burn victims and immunocompromised individuals, has been studied in a variety of phylogenetically diverse model hosts including: vertebrates, insects, nematodes, mycetozoans and plants. (Mahajan-Miklos et al., 2000). Pseudomonas exotoxin A (PE) is a soluble exoprotein and one of the most toxic virulence factors amongst several secreted proteins important for the pathogenesis of Pseudomonas aeruginosa (Pollack, 1983). Mutations in the toxA gene, encoding PE, resulted in attenuated virulence in plants (Rahme et al., 1995), nematodes (Tan et al., 1999) and mice (Nicas and Iglewski, 1985) suggesting extensive conservation in the mechanism by which PE mediates injury, in these evolutionarily diverse hosts.
Bacterial toxins are studied to gain insights into pathogenic mechanisms. They are also studied as probes of cellular function and as components of novel therapies. In the case of the latter, it is usual to modify the toxin to allow for targeting to specific cell types. Because toxins are targeted to kill cancer cells, investigating the pathways and mechanisms of cell death becomes important. Antibody-toxin fusion proteins, also called immunotoxins, have become a valuable therapy for the targeted treatment of cancer (Pastan et al., 2006). Immunotoxins exploit the precision of antibodies and the lethality of protein toxins to target and kill cancer cells expressing specific cell surface proteins. PE-derived immunotoxins are being evaluated as anti-cancer agents in human trials (Hassan et al., 2007, Kreitman et al., 2009, Kreitman et al., 2001, Kreitman et al., 1999, Rand et al., 2000). Results of these trials reflect a divergent pattern whereby hematological malignancies respond positively, including some complete responses, while epithelial tumors exhibit less sensitivity. Whether these differences reflect problems gaining access to individual cells in a tumor mass or differences in susceptibility to death have not been established. This has prompted a detailed investigation into the mechanism of cell death caused by PE and PE immunotoxins. Moreover, for mammalian cells, investigating the mechanism of cell death has highlighted the possible involvement of several distinct pathways including apoptosis, autophagy and necrosis.
While, there is some evidence for the involvement of apoptosis in PE mediated killing in mammalian cells (Hafkemeyer et al., 1999, Jenkins et al., 2004, Keppler-Hafkemeyer et al., 2000, Komatsu et al., 1998), mechanisms of PE-mediated cell death in other hosts have not been widely studied. The genes involved in the control and execution of apoptosis are conserved throughout evolution (Cashio et al., 2005, Twomey and McCarthy, 2005). However, the actual molecular mechanisms used by these genes vary from species to species. There is a high degree of genetic redundancy in mammalian systems that has made it difficult to identify specific gene functions and to trace an exact apoptotic pathway. The fruit fly, D. melanogaster, provides a system of intermediate complexity, sharing many mammalian components and pathways, but with less redundancy. Thus the genetic conservation, low redundancy, and the feasibility for high-throughput genetic screens have made Drosophila cells a valuable and convenient model for studying biochemical pathways. Here we have chosen Drosophila S2 cells as a model tissue culture system for the investigation of the interaction of PE with cells of invertebrates eventually facilitating a better understanding of PE and PE immunotoxin-mediated cell death.
PE is a 66KDa three domain bacterial toxin. Domain I at the N-terminus binds LRP1 and/or LRP1b as the plasma membrane receptor for cell entry (Kounnas et al., 1992, Pastrana et al., 2005). Domain II harbors a site for furin processing and a poorly defined activity for translocation to the cell cytosol (Chiron et al., 1994, Jinno et al., 1989, Moehring et al., 1993). Domain III has ADP-ribosylating activity and a C-terminal KDEL-like sequence aids in retrotranslocation from the ER to the cytosol (Chaudhary et al., 1990, Jackson et al., 1999). PE mediates eukaryotic cell death by blocking protein synthesis via the ADP-ribosylation of cytosolic elongation factor 2 (EF2) (Morimoto and Bonavida, 1992). However, protein synthesis inhibition by toxins is not sufficient to mediate target cell lysis (Morimoto and Bonavida, 1992). Also, decreased ADP ribosylation activity does not always result in decreased PE-induced cytotoxicity, suggesting a possible dissociation between protein synthesis inhibition and cell death (Gallant et al., 2000). Thus, cell death mediated by PE may involve additional mechanisms.
The biochemical target of PE is the diphthamide residue of mammalian EF2. H715 is modified in a five step enzymatic process resulting in a posttranslational modification that renders EF2 susceptible to ADP-ribosylation by such bacterial toxins as PE, diphtheria toxin and cholix toxin (Liu et al., 2004, Zhang et al., 2008). There is also an endogenous enzyme that modifies diphthamide but this is poorly characterized. Toxin-mediated killing depends on the presence of diphthamide because mutants in the diphthamide pathway exhibit resistant phenotypes (Foley et al., 1992). While there is universal agreement that the ADP-ribosylation of EF2 by PE inhibits protein synthesis which may lead to cell death, the mechanism and universality of death have not been established.
Cell death, mediated by caspase-dependent apoptosis, is driven by a set of core proteins that are evolutionarily conserved between mammals and insect cells (Hay and Guo, 2006, Twomey and McCarthy, 2005). Caspases are cysteine proteases that function as both initiators and executioners of apoptotic cell death. In Drosophila, Dronc, is a caspase-9-like enzyme that functions as an initiator caspase while DCP-1 and Drice, caspase-3-like effector caspases, are critical for the executionery response. In the absence of apoptotic signals, the inhibitor of apoptosis proteins (IAPs) such as Drosophila IAP1 (Diap1) bind to caspases and inhibit their activity. In the presence of apoptotic stimuli, the pro-apoptotic proteins, Reaper, Hid and Grim (RHG proteins) bind to Diap1 and release the caspases. Released Dronc binds to the adaptor protein Ark to form the apoptosome which subsequently cleaves and activates the effector caspases DCP-1 and Drice. These caspases then trigger a series of downstream cleavage reactions resulting in physiological changes in the cell characterized by cell shrinkage, nuclear fragmentation, chromatin condensation and DNA fragmentation.
Here we investigate PE-mediated death in S2 Drosophila cells and conclude that apoptosis is the mechanism of death.
2. Materials and Methods
2.1. Cell culture and induction of apoptosis
S2 cells (S2-DRSC, stock #181) were obtained from the Drosophila Genomics Resource Center, Indiana University, Bloomington, Indiana and propagated in M3 (Shields and Sang medium) + BPYE media (Sigma-Aldrich, St. Louis, MO) supplemented with heat inactivated 10% Fetal bovine serum (HyClone) and 100 U/ml penicillin and 100 μg/ml streptomycin (Sigma-Aldrich at 25°C. To induce apoptosis, S2 cells were treated with 150 ng/ml actinomycin D (Sigma-Aldrich), 200 μg/ml cycloheximide (Sigma-Aldrich) or 30, 50 or 100 ng/ml PE, as indicated. Caspase inhibition was achieved by including 100 μM z-VAD-fmk (Calbiochem) in the medium along with PE and cycloheximide, where indicated. To probe for the specificity of PE action, an enzymatically inactive version of PE, termed ntPE (for non-toxic PE), was used as a competitor for receptor uptake (FitzGerald et al., 1998).
2.2. Cytotoxicity assays
S2 cells were seeded at a concentration of 2 × 106 cells/ml per well in 96 well plates with or without the indicated concentrations of PE, ntPE, cycloheximide or actinomycin D. 100 μM z-VAD-fmk was added for caspase inhibition, where indicated. S2 cells were incubated for 48 h at 25°C and cell viability was measured by adding 10% alamarBlue® (Invitrogen, Carlsbad, CA) followed by incubation of 1-5 h at 37°C and measurement of fluorescence at an excitation wavelength of 540nm and emission wavelength of 590nm using the Cary eclipse fluorescence spectrophotometer (Varian). Fluorescence obtained for untreated S2 cells was set to a value of 100, indicating 100% viability, and that for variously treated S2 cells was calculated accordingly.
2.3. dsRNA synthesis
gfp dsRNA was synthesized using the plasmid, pBC-S65T-GFP (kindly provided by Dr. Bruce Patterson). Linear fragments of pBC-S65T-GFP obtained upon digestion with restriction enzymes, EagI or KpnI were used as templates for dsRNA synthesis using the MEGASCRIPT T7 transcription kit (Applied Biosystems, Foster City, CA) or MEGASCRIPT T3 transcription kit (Applied Biosystems), respectively. The T7 RNA polymerase and T3 RNA polymerase transcribed RNAs were mixed together and heated at 75°C for 5min followed by cooling at room temperature. The dsRNA was subjected to DNase and RNase (specific for single stranded RNA) treatments for 1 h at 37°C followed by purification of the sample using RNeasy MinElute columns (Ambion). Drosophila dph1 and drice were amplified by PCR using a Drosophila S2 cDNA library as a template. To generate the Drosophila S2 cDNA library, total RNA was isolated using Trizol reagent (Invitrogen) followed by reverse transcription using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Drosophila dph1 was amplified using the primers, Dph1-For1 CTGTGATAATGGGCGATGTG and Dph1-Rev1 GCAACGGGTTTGCTATCATT. In order to get a T7 promoter site on both 5’ and 3’ end of dph1, the dph1 PCR product was cloned into Topo-pcr2.1 vector in both orientations resulting in formation of Topo-Dph1-For and Topo-Dph1-Rev constructs. Similarly drice was amplified using Drice-For primer, TAAACACGGACTTTCCCCAG and Drice-Rev primer, CTTACGGTAGCTGGACGAGG and cloned into Topo-pcr2.1 in both orientations resulting in Topo-Drice-For and Topo-Drice-Rev constructs. PCR products obtained by amplifying using M13-For and Dph1-For1 primers as well as with M13-For and Dph1-Rev1 primers or using M13-For and Drice-For primers as well as with M13-For and Drice-Rev primers were used as templates for synthesizing dph1 and drice dsRNA using the MEGASCRIPT RNAi kit (Applied Biosystems), respectively. 5ng of dsRNA were analyzed by 2% agarose gel electrophoresis to ensure that the majority of the dsRNA migrated as a single band. 350bp and 444bp fragment of dsRNAs were produced corresponding to the coding transcript of drice and dph1, respectively. The dsRNA was stored at -20°C.
2.4. RNAi treatment of Drosophila S2 cells
For cytotoxicity and protein synthesis inhibition assays, S2 cells were plated in 96 well plates, at a concentration of 2×106 cells/ml in serum free M3+BPYE media. For caspase 3 activity assays, S2 cells were plated in 24 well plates, at the indicated concentration of 6×106 cells/ml or 3×106cells/ml in serum free M3+BPYE media. 20μg/ml dsRNA was mixed with 2μl of cellfectin® transfection reagent (Invitrogen) in 50 μl of serum-free M3+BPYE media and added to the S2 cells. The S2 cells were mixed vigorously with the dsRNA+cellfectin mixture. After incubating the cells for 4 hours at 25°C, equal volume of M3+BPYE media supplemented with 20% FBS was added to get a final concentration of 10% FBS supplemented M3+BPYE media. The cells were incubated for additional 48 h at 25°C to allow for turnover of protein. For cytotoxicity assays, indicated concentration of PE, cycloheximide, actinomycin D and z-VAD-fmk were added and incubated for 48 h at 25°C followed by the addition of 10% alamarBlue® (Invitrogen).
2.5. Protein Synthesis inhibition assay
S2 cells (2×106 cells/ml) were transfected with dsRNA in 96 well plates followed by incubation in the presence of indicated amount of PE or z-VAD-fmk for 24 h. 1μCi of {3H}-Leucine was added to each well and incubated for 4 h at 25°C. The plate was incubated overnight at -80°C to aid with cell lysis. The effect on protein synthesis was assessed by measuring the amount of {3H}-Leucine incorporated.
2.6. Caspase activity Assay
S2 cells transfected with dsRNA followed by treatment with indicated concentrations of PE and actinomycin D for 24 h were washed thoroughly with PBS. Caspase activity was determined using the caspase-3 fluorometric-kit (Invitrogen). Specific caspase activity was determined by dividing the total fluorescence units with the amount of protein present in the whole cell lysate of each sample. Protein concentration was estimated using the BCA™ Protein Assay Kit (Pierce). The specific caspase activity of the untreated sample was set to 1.0 and that of the toxin treated samples was calculated accordingly in order to get fold increase in caspase activity.
2.7. Western Blot analysis
Equal number of S2 cells transfected with gfp dsRNA, drice dsRNA or untransfected cells were dislodged with media and washed thoroughly with PBS. The cells were incubated on ice for 20min in RIPA lysis buffer (5mM Tri pH 7.5, 150mM NaCl, 5mM EDTA, 1% NP40 1%, 0.1% SDS, 1% Sodium Deoxycholate, complete Protease inhibitor). The homogenates were passed through 28 G half inch needle several times and centrifuged at 14000g for 10min and the supernatants were stored at -20°C for western blots. Lysates from untransfected and transfected samples was boiled for 5min in Laemmli buffer and electrophoresed on 8-16% Tris Glycine SDS-PAGE gels (Invitrogen). The proteins were transferred to Immobilon-P nitrocellulose membrane (Millipore) and probed using rabbit anti-Drice (kindly provided by Dr. Andreas Bergman), diluted 1:1,000 and rabbit anti-γ-tubulin (Sigma-Aldrich) was used as a protein loading control, diluted 1:3,000. The immobilized proteins were incubated with HRP labeled secondary antibodies (Jackson immunoresearch laboratories, inc.) and the signal was detected by LumiGLO Peroxidase Chemiluminescent Substrate Kit (KPL)
2.8. Reverse transcription real time PCR
S2 cells were seeded in 24 wells at a concentration of 2×106 cells/ml and transfected with 10ug/ml gfp and dph1 dsRNA supplemented with 5μl cellfectin transfection reagent. RNA was isolated from the S2 cells by TRIzol (Invitrogen) extraction according to manufacturer’s instructions. Each RNA sample was then treated with 16 units of RNase free DNase I (Qiagen) and elute the RNA using RNeasy Minelute kit (Qiagen). The RNA pellet was resuspended in DEPC-treated water, and the nucleic acid was quantified using an ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). First strand cDNA was generated from 2μg total RNA using high capacity cDNA reverse transcription kit (Applied biosystems). Each cDNA sample was diluted 1:5 in water, and 2.5 μl was used as the template for each reaction. All primers were minor groove binding (MGB), and FAM labelled probes, purchased from Applied Biosystems, Foster City, CA. All quantitative measurements were performed in triplicate and rpl32 was used as the internal control for each sample. dph1 mRNA values obtained from gfp dsRNA treated samples was set to a value of 100% and that for dph1 dsRNA treated samples was calculated accordingly. rpl32 expression levels are not altered in either the gfp or dph1 dsRNA transfected S2 cells. All reactions were run in a 7300 Real-Time PCR system (Applied Biosystems, Foster City, CA) under standard reaction conditions.
3. Results
3.1. PE is cytotoxic for S2 Drosophila cells
To characterize the cytotoxic activity of PE for Drosophila cells in a tissue culture setting, S2 cells were incubated with various concentrations of the toxin for 48 hr and death measured using the alamarBlue® cell viability assay. AlamarBlue® was selected because, unlike other viability reagents, there was no interference from the growth media. Results indicated that PE was potently toxic with an IC50 of less than 1 ng/ml (Fig. 1) and complete cell death was achieved at 10 ng/ml or greater. In order to establish the specificity of PE cytotoxicity, a competition assay was performed where S2 cells were preincubated with an enzymatically inactive version of full length PE (ntPE). The competitor was added 20 min prior to the addition of varied concentrations of active PE. Viability results showed that 10μg/ml of ntPE was able to block completely the cytotoxic effect of the highest concentration of active PE used, 10ng/ml (Fig. 1). However, a lower concentration of competitor, 1μg/ml of ntPE, completely inhibited the cytotoxic effect of 1ng/ml active PE but failed to do so for higher concentrations of PE. This result confirms receptor-mediated uptake of PE leading to death. A further confirmation of specific uptake was established when a human-specific immunotoxin targeted to the human transferrin receptor failed to kill cells at concentrations up to 100 ng/ml (data not shown).
Fig. 1.
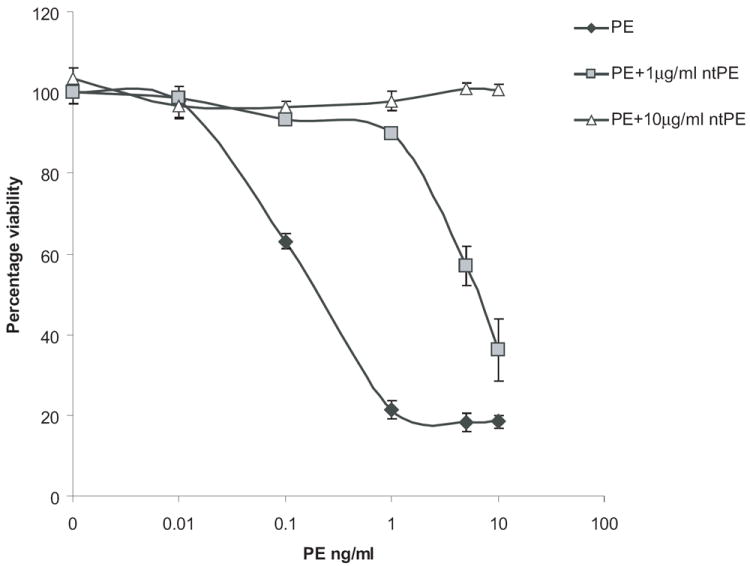
PE shows specific cytotoxicity to S2 cells in a dose-dependent manner. S2 cells were incubated with the indicated concentrations of PE for 48 h, with or without prior incubation with the indicated concentrations of ntPE for 20min, and cell viability was determined as a percentage of control, using alamarBlue®. Each point is the mean of at least three determinations. Error bars represent one standard deviation from the mean.
3.2. PE-mediated ADP-ribosylation of EF2 requires ‘diphthamide’
In mammalian cells, the modification of newly translated EF2 to mature EF2 involves a five enzyme (Dph1-5) post translational modification to convert the His at residue 715 to diphthamide (Liu et al., 2004). To confirm that toxin-mediated cell death is dependent on the presence of diphthamide in Drosophila cells, RNAi was used to knockdown the first enzyme, Dph1, in the pathway. In the absence of an antibody to Drosophila Dph1, the extent of knockdown was monitored via RT-PCR. An approximate 70% reduction in the endogenous dph1 mRNA expression was observed when cells were treated with a dsRNA to dph1 (Fig. 2A). To assess PE activity following dph1 knockdown, both protein synthesis and cell viability were measured. The Dph1 knockdown protected S2 cells from PE-mediated inhibition of protein synthesis (Fig. 2B); indicating that diphthamide biosynthesis is required for toxin-mediated inhibition of protein synthesis. For controls we determined that the Dph1 knockdown did not protect cells from other agents that inhibit protein synthesis via different mechanisms including actinomycin D (Fig. 2C) and cycloheximide (Fig. 2D). When cell death was measured, the addition of dsRNA to dph1, but not to gfp, protected cells from PE-mediated toxicity, thus confirming that in Drosophila S2 cells PE-mediated death was dependent on the diphthamide pathway (Fig 2 E).
Fig. 2.
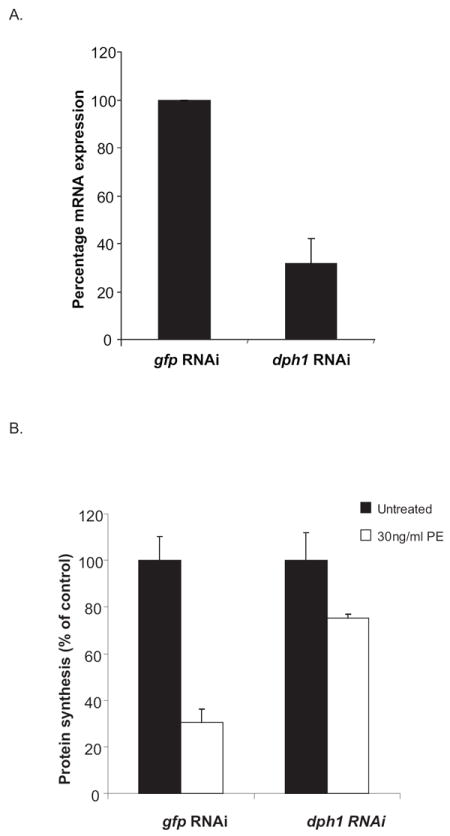
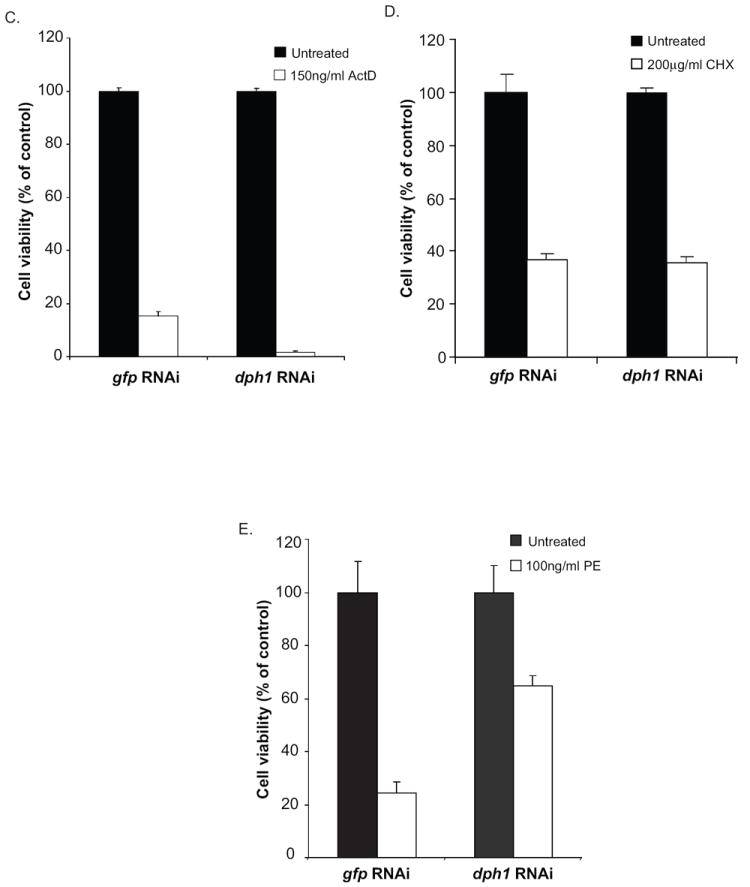
dph1 knockdown prevents both PE-mediated inhibition of protein synthesis and cell death. (A) S2 cells were incubated with dsRNA to dph1 or a non-target gene, gfp, and the expression of dph1 mRNA measured using real time PCR. Rpl32 expression was used as an internal control. The data are expressed as means of three independent experiments. (B) S2 cells were subjected to RNAi to dph1 or a control gene, followed by an incubation with 30ng/ml PE for 24 h. Protein synthesis inhibition was assessed by measuring the amount of {3H}-leucine incorporated into cellular material and is presented as the percentage value of an untreated sample. S2 cells were subjected to RNAi using dsRNA to dph1 followed by an incubation with 150ng/ml actinomycin D (C), 200μg/ml cycloheximide (D) or 100ng/ml PE (E) for 48 h. Cell viability was measured using alamarBlue® and the data is presented as percentage value of the untreated sample. Each bar is the mean of at least three determinations. Error bars represent one standard deviation from the mean.
3.3. Increase in caspase activity upon addition of PE
The data above indicate that PE treatment causes inhibition of protein synthesis in a dph1 dependent manner which results in cell death. Toxic agents kill cells via several distinct pathways. To investigate a possible role for apoptosis, post-treatment S2 cell extracts were assayed for caspase activity. The role of dph1 in altering the caspase activity in presence of PE was also monitored. Toxin treated cell extracts were tested for activated caspase by monitoring activity using DEVD-AFC as a substrate. When S2 cells were treated with a control dsRNA (targeted to gfp), there was a substantial increase in cell-associated caspase activity in the presence of PE as well as with actinomycin D (Fig. 3). This suggested that toxin-mediated killing of S2 cells occurred as a result of caspase mediated apoptosis. However, when S2 cells were treated with dsRNA to dph1, this increase in caspase activity was seen only with actinomycin D but not with PE (Fig. 3). This confirmed that PE induced caspase activation requires prior protein synthesis inhibition in a dph1-dependent manner. The actinomycin D induced caspase activation was independent of dph1.
Fig. 3.
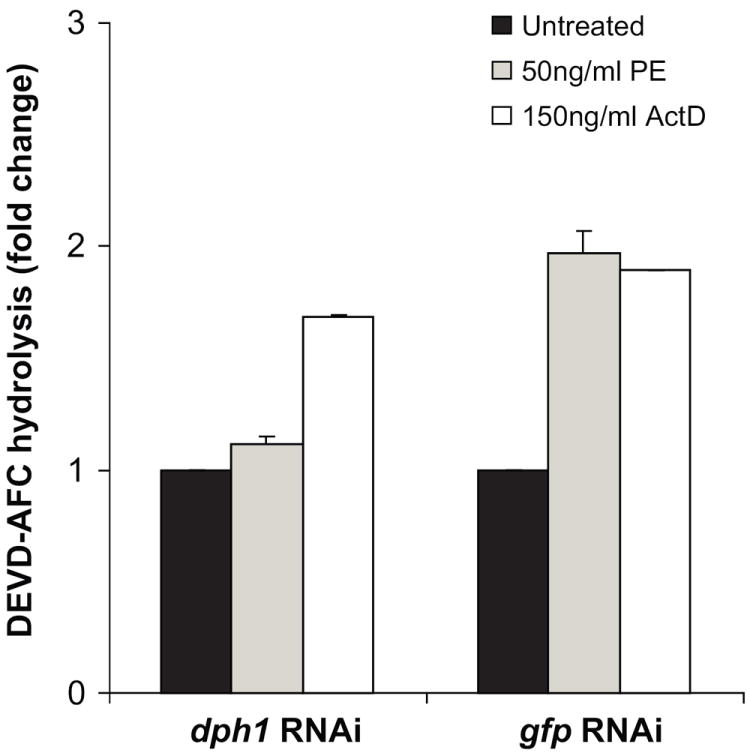
PE treatment leads to activation of caspase activity. S2 cells were subjected to RNAi using dsRNA to dph1 followed by incubation in the presence of 50ng/ml PE or 150ng/ml actinomycin D for 24 h. dsRNA to a non-specific target gene, gfp, was used as control. An increase in the caspase (presumed to be Drice, see below) activity was estimated by measuring the increase in fluorescence upon hydrolysis of DEVD-AFC. The specific caspase activity of the dph1 knockdown samples is presented as the fold-change value compared to an untreated sample. Each bar is the mean of at least three determinations. Error bars represent one standard deviation from the mean.
3.4. PE mediates S2 cell death via apoptosis
To confirm the participation of a caspase-like enzyme in toxin-mediated apoptosis, a chemical inhibitor of caspase activity, z-VAD-fmk was used. The addition of z-VAD-fmk (100 μM) protected cells from PE-mediated death (Fig. 4A) indicating that apoptosis was the probable mechanism of cell death. Inhibitors of caspase are reported to have a narrow substrate specificity and should not interfere with endocytic entry, toxin translocation to the cytosol or the ADP-ribosylation of EF2. To confirm this, the effect of z-VAD-fmk on PE-mediated inhibition of protein synthesis was determined (Fig. 4B). The addition of z-VAD-fmk did not interfere with toxin uptake or the ADP-ribosylation of EF2 leading to inhibition of protein synthesis. Thus we conclude that death occurs downstream from the ADP-ribosylation of EF2.
Fig. 4.
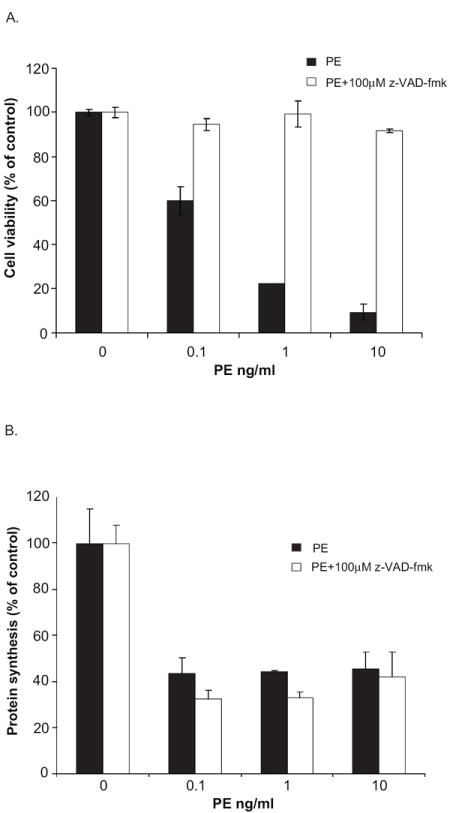
z-VAD-fmk prevents loss of viability but fails to inhibit PE-mediated inhibition of protein synthesis. S2 cells were treated with the indicated concentrations of PE in the presence or absence of 100μM z-VAD-fmk. As above, cell viability (A) was determined at 48 h using alamarBlue® and protein synthesis inhibition (B) was assessed after 24 h by measuring the amount of {3H}-Leucine incorporated into cellular material. Each point is the mean of at least three determinations. Error bars represent one standard deviation from the mean.
3.5. Drice is required for PE-mediated cell death
The terminal effector caspases in mammalian cells are caspase 3 and 7. The Drosophila homolog of these caspases is Drice. RNAi was used to downregulate drice gene expression and thereby evaluate its role in toxin-mediated death. The addition of dsRNA to drice, but not to gfp, resulted in lower levels of the Drice protein as detected by western blot analysis (Fig. 5A). In order to test the efficiency of our drice knockdown in preventing apoptotic cell death, actinomycin D and cycloheximide, compounds previously reported to kill Drosophila cells via apoptosis (Zimmermann et al., 2002) were used to test its cytotoxicity in control and drice knockdown conditions. As noted previously, dsRNA to drice prevented S2 cell death by these compounds (Fig. 5B and 5C).
Fig. 5.
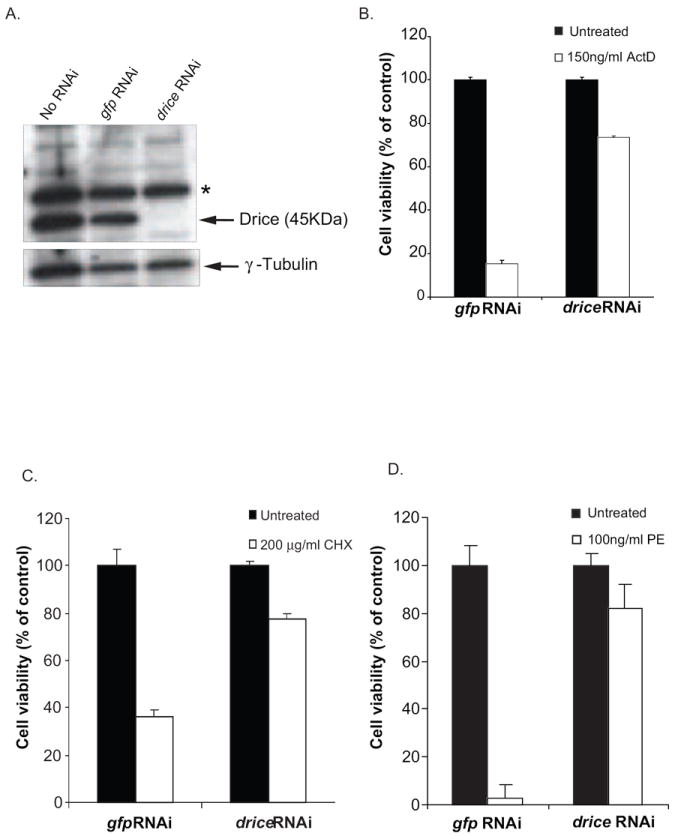
drice knockdown inhibits PE-mediated cell death. (A) S2 cells were treated with dsRNA to drice followed by western blot analysis to detect endogenous Drice. dsRNA to a non-specific target gene, gfp, was used as control. Anti-tubulin was used as a loading control. The asterisk denotes a non-specific background band. S2 cells were subjected to RNAi using dsRNA to drice and treated with 150ng/ml actinomycin D (B), 200μg/ml cycloheximide (C) or 100ng/ml PE (D) for 48 h. Cell viability was measured using alamarBlue® ® and the data is presented as percentage value of the untreated sample. Each bar is the mean of at least three determinations. Error bars represent one standard deviation from the mean.
To determine the role of Drice in PE-mediated death, cells were pretreated with dsRNA to drice or gfp and then challenged with PE. Cells treated with the gfp dsRNA were killed by greater than 90% while cells treated with dsRNA to drice showed only a 20% decrease in viability, compared to the cells that were not exposed to the toxin, further supporting a role for apoptosis in PE-mediated cell death (Fig. 5D).
Above we have shown that the downregulation or inhibition of essential genes in the ADP-ribosylation and apoptosis pathway protects cells from PE-mediated cell death. To support these ‘inhibitory approaches’ with positive evidence of PE-mediated apoptosis, we assayed treated cells for activated Drice using DEVD-AFC as a substrate. When S2 cells were treated with dsRNA to drice, there was no increase in Drice enzymatic activity when challenged with either actinomycin D or PE (Fig. 6). This confirmed that toxin-mediated activation of the drosophila caspase pathway of S2 cells occurred as a result of activation of Drice.
Fig. 6.
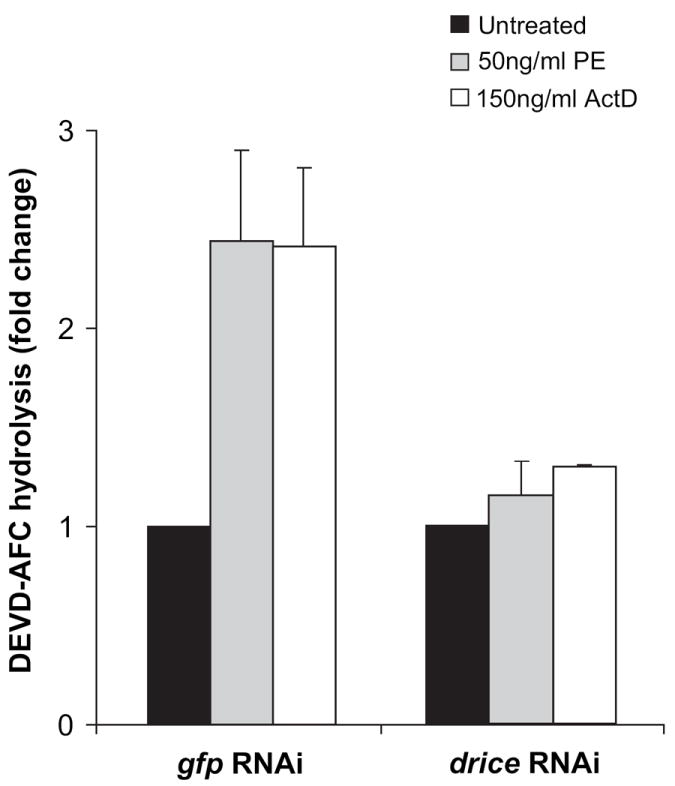
drice knockdown prevents PE-mediated increase in caspase activity. S2 cells were subjected to RNAi using dsRNA to drice followed by incubation in the presence of 50ng/ml PE or 150ng/ml actinomycin D for 24 h. An increase in the Drice activity was estimated by measuring the increase in fluorescence upon hydrolysis of DEVD-AFC. The specific caspase activity of the dsRNA to drice-treated samples is presented as the fold-change value compared to the untreated sample. Each bar is the mean of at least three determinations. Error bars represent standard deviation of the mean.
3.6. Drice is not required for PE-mediated protein synthesis inhibition
Since PE treatment causes protein synthesis inhibition via the ADP-ribosylation of diphthamide residue on EF2 and cell death via Drice dependent apoptotic pathway, the effect of downregulation of drice on protein synthesis upon PE treatment was examined. Downregulation of drice did not protect cells from PE-mediated inhibition of protein synthesis (Fig. 7) confirming that PE-mediated modification of EF2 occurred in a Drice-independent manner and PE-mediated cell death occurred in a Drice-dependent manner, subsequent to the inhibition of protein synthesis.
Fig. 7.
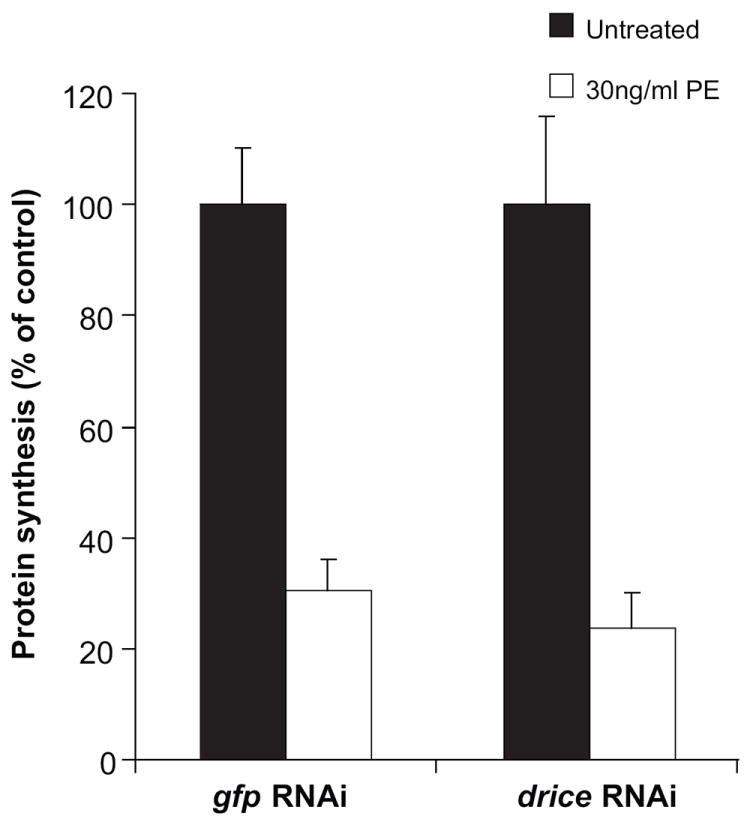
drice knockdown fails to prevent PE-mediated protein synthesis inhibition. S2 cells were subjected to dsRNA to drice followed by an incubation with 30ng/ml PE for 24 h. Protein synthesis inhibition was assessed by measuring the amount of {3H}-leucine incorporated and is presented as the percentage value of an untreated sample. Each bar is the mean of at least three determinations. Error bars represent one standard deviation from the mean.
4. Discussion
Here we show that PE-mediated cell death is via apoptosis in Drosophila S2 cells. Treatment with the pan caspase inhibitor, z-VAD-fmk preserved cell viability in the presence of PE. However, z-VAD-fmk did not prevent toxin-mediated inhibition of protein synthesis, confirming that inhibition of protein synthesis and cell death are distinct events and that apoptosis follows the inhibition of protein synthesis. In Drosophila, Drice is the effector caspase that, upon cleavage and activation by Dronc, mediates apoptosis. Depletion of Drice from S2 cells has been shown to inhibit apoptotic events in response to a variety of stimuli (Muro et al., 2006). Likewise, our study shows that PE-mediated cell death can be prevented using dsRNA targeted to drice, indicating a pivotal role for Drice in PE-mediated cell death. And like the inhibitor studies with z-VAD-fmk, the knockdown of drice did not prevent PE-mediated inhibition of protein synthesis confirming that Drice functions downstream of the ADP-ribosylation of EF2.
PE-mediated cell death is dependent on the presence of diphthamide-modified EF2 (Liu and Leppla, 2003, Pavlovskis et al., 1978). Diphthamide is well conserved amongst all eukaryotes and archaebacteria. The absence of the appropriate ‘diphthamide modification’ in mammalian cells renders EF2 refractory to modification by PE and hence resistant to toxin-mediated cell death (Foley et al., 1995). Our study with S2 cells shows that Dph1 knockdown prevents both toxin-mediated inhibition of protein synthesis and apoptosis, indicating that the ADP-ribosylation of EF2 is both necessary and sufficient for PE-mediated cell death. However, the Dph1 knockdown did not prevent cycloheximide-mediated inhibition of protein synthesis (data not shown). Cycloheximide acts via inhibition of 80S ribosome function (Schneider-Poetsch et al., 2010) and does not use the diphthamide pathway. These kinds of observations further confirm the diphthamide residue of EF2 as the single terminal target of exotoxin A.
The enzyme specificity of Drice so closely resembles that of mammalian caspase 3, we were able to use a caspase 3 substrate to monitor cell death. In order to provide positive evidence of Drice-dependent cell death, we measured caspase-3 activity in S2 cells treated with either PE or actinomycin D. While non-specific dsRNA-treated cells showed a significant increase in caspase-3 activity upon PE or actinomycin D treatment, a drice knockdown prevented this increase. However, when Dph1 levels were knocked-down the increase in caspase 3 activity was seen only with actinomycin D, confirming the requirement of Dph1 specifically in the PE pathway. Additionally, Dph1 mediated protein synthesis inhibition by PE is necessary to induce apoptosis that results in Drosophila cell death.
The potency of PE (IC50 of less than 100 pM) and the lack of cytotoxicity in the presence of an inactive version of PE, suggests that receptor mediated binding rather than nonspecific uptake is responsible for the internalization of toxin into cells. High potency also suggests that components of the intoxication pathway are well conserved between Drosophila cells and sensitive mammalian cells. Having a potent agent that kills S2 cells by a well defined pathway may be useful as a probe in future studies of apoptosis. In mammalian cells apoptosis can be blocked not only by the chemical inhibition of caspase activity but also by the overexpression of prosurvival Bcl2 proteins. The identification of the equivalent factors is currently under investigation in Drosophila and this toxin system should provide a convenient model to test these via overexpression of candidate prosurvival proteins. In fact, toxin mediated death of S2 cells lends itself to global searches for genes that are essential for binding, endocytic uptake, retrograde transport to the ER and retrotranslocation to the cytosol. Gene knockdowns of the toxin pathway, using RNAi, should produce survivors, which would be monitored easily using our alamarBlue® assay. Additionally, since the toxin interacts with highly conserved eukaryotic proteins, the resulting survivors may help identify genes whose role cannot be discovered using gene knockout strategies (because of lethality). For example, here we show that the knockdown of Dph1 results in EF2 that cannot be ADP-ribosylated. However, Dph1 -/- and Dph3-/- mice are embryonic lethal (Liang et al., 2008, Liu et al., 2006) and effects related to the absence of Dph1 or Dph3 would be difficult to discover using a gene knockout strategy.
Bacterial toxins serve as valuable probes for elucidating eukaryotic cell functions. Our findings facilitate the use of this potent protein toxin in combination with RNAi as a way to monitor a complex pathway beginning with binding and endocytosis and ending with the modification of EF2 leading to apoptotic cell death.
Supplementary Material
Acknowledgments
We would like to thank Dr. Bruce Patterson and Dr. Concetta Lipardi for pBC-S65T-GFP plasmid and their valuable suggestions on RNAi technique with S2 cells. We thank Robert Sarnovsky for providing ntPE and to Dr. Antonella Antignani for reviewing the manuscript.
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Abbreviations
- LRP1
low density lipoprotein-related protein 1
- ADP
Adenosine diphosphate
- PE
Pseudomonas exotoxin A
- EF2
elongation factor-2
- Act D
actinomycin D
- CHX
cycloheximide
- RNAi
RNA interference
- dsRNA
double stranded RNA
- GFP
green fluorescence protein
- Dph1
Diphthamide biosynthesis-like 1
- Drice
Drosophila Interlukin-1B-converting enzyme
- DCP-1
death caspase-1
- Dronc
Drosophila Nedd2-like caspase
- Hid
head involution defective
- Bcl-2
B cell lymphoma-2
- z-VAD-fmk
carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]- fluoromethylketone
- DEVD-AFC
Asp-Glu-Val-Asp-7-amino-4-trifluoromethyl coumarin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Cashio P, Lee TV, Bergmann A. Genetic control of programmed cell death in Drosophila melanogaster. Semin Cell Dev Biol. 2005;16:225–235. doi: 10.1016/j.semcdb.2005.01.002. [DOI] [PubMed] [Google Scholar]
- FitzGerald DJ, Fryling CM, McKee ML, Vennari JC, Wrin T, Cromwell ME, Daugherty AL, Mrsny RJ. Characterization of V3 loop-Pseudomonas exotoxin chimeras. Candidate vaccines for human immunodeficiency virus-1. J Biol Chem. 1998;273:9951–9958. doi: 10.1074/jbc.273.16.9951. [DOI] [PubMed] [Google Scholar]
- Hafkemeyer P, Brinkmann U, Gottesman MM, Pastan I. Apoptosis induced by Pseudomonas exotoxin: a sensitive and rapid marker for gene delivery in vivo. Hum Gene Ther. 1999;10:923–934. doi: 10.1089/10430349950018328. [DOI] [PubMed] [Google Scholar]
- Hassan R, Bullock S, Premkumar A, Kreitman RJ, Kindler H, Willingham MC, Pastan I. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin Cancer Res. 2007;13:5144–5149. doi: 10.1158/1078-0432.CCR-07-0869. [DOI] [PubMed] [Google Scholar]
- Jenkins CE, Swiatoniowski A, Issekutz AC, Lin TJ. Pseudomonas aeruginosa exotoxin A induces human mast cell apoptosis by a caspase-8 and -3-dependent mechanism. J Biol Chem. 2004;279:37201–37207. doi: 10.1074/jbc.M405594200. [DOI] [PubMed] [Google Scholar]
- Keppler-Hafkemeyer A, Kreitman RJ, Pastan I. Apoptosis induced by immunotoxins used in the treatment of hematologic malignancies. Int J Cancer. 2000;87:86–94. [PubMed] [Google Scholar]
- Komatsu N, Oda T, Muramatsu T. Involvement of both caspase-like proteases and serine proteases in apoptotic cell death induced by ricin, modeccin, diphtheria toxin, and pseudomonas toxin. J Biochem. 1998;124:1038–1044. doi: 10.1093/oxfordjournals.jbchem.a022197. [DOI] [PubMed] [Google Scholar]
- Kreitman RJ, Wilson WH, Robbins D, Margulies I, Stetler-Stevenson M, Waldmann TA, Pastan I. Responses in refractory hairy cell leukemia to a recombinant immunotoxin. Blood. 1999;94:3340–3348. [PubMed] [Google Scholar]
- Kreitman RJ, Wilson WH, Bergeron K, Raggio M, Stetler-Stevenson M, FitzGerald DJ, Pastan I. Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N Engl J Med. 2001;345:241–247. doi: 10.1056/NEJM200107263450402. [DOI] [PubMed] [Google Scholar]
- Kreitman RJ, Stetler-Stevenson M, Margulies I, Noel P, Fitzgerald DJ, Wilson WH, Pastan I. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J Clin Oncol. 2009;27:2983–2990. doi: 10.1200/JCO.2008.20.2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan-Miklos S, Rahme LG, Ausubel FM. Elucidating the molecular mechanisms of bacterial virulence using non-mammalian hosts. Mol Microbiol. 2000;37:981–988. doi: 10.1046/j.1365-2958.2000.02056.x. [DOI] [PubMed] [Google Scholar]
- Nicas TI, Iglewski BH. The contribution of exoproducts to virulence of Pseudomonas aeruginosa. Can J Microbiol. 1985;31:387–392. doi: 10.1139/m85-074. [DOI] [PubMed] [Google Scholar]
- Pastan I, Hassan R, Fitzgerald DJ, Kreitman RJ. Immunotoxin therapy of cancer. Nat Rev Cancer. 2006;6:559–565. doi: 10.1038/nrc1891. [DOI] [PubMed] [Google Scholar]
- Pollack M. The role of exotoxin A in pseudomonas disease and immunity. Rev Infect Dis. 1983;5(Suppl 5):S979–984. doi: 10.1093/clinids/5.supplement_5.s979. [DOI] [PubMed] [Google Scholar]
- Rahme LG, Stevens EJ, Wolfort SF, Shao J, Tompkins RG, Ausubel FM. Common virulence factors for bacterial pathogenicity in plants and animals. Science. 1995;268:1899–1902. doi: 10.1126/science.7604262. [DOI] [PubMed] [Google Scholar]
- Rand RW, Kreitman RJ, Patronas N, Varricchio F, Pastan I, Puri RK. Intratumoral administration of recombinant circularly permuted interleukin-4-Pseudomonas exotoxin in patients with high-grade glioma. Clin Cancer Res. 2000;6:2157–2165. [PubMed] [Google Scholar]
- Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B, Liu JO. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol. 2010;6:209–217. doi: 10.1038/nchembio.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MW, Rahme LG, Sternberg JA, Tompkins RG, Ausubel FM. Pseudomonas aeruginosa killing of Caenorhabditis elegans used to identify P. aeruginosa virulence factors. Proc Natl Acad Sci U S A. 1999;96:2408–2413. doi: 10.1073/pnas.96.5.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey C, McCarthy JV. Pathways of apoptosis and importance in development. J Cell Mol Med. 2005;9:345–359. doi: 10.1111/j.1582-4934.2005.tb00360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.