

Background: MATE1 (multidrug and toxin extruder 1) mediates renal organic cation secretion.
Results: Mammalian MATE1 has 13, not 12, transmembrane helices, but the 13th TMH is not necessary for transport function.
Conclusion: A homology model of MATE1 can use the crystal structure of the prokaryotic MATE protein NorM (12 TMHs).
Significance: A MATE1 model is the basis for future structure/activity studies.
Keywords: Kidney, Liver, Membrane Proteins, Membrane Transport, Multidrug Transporters, Transport, Transport Drugs, Transporters
Abstract
The x-ray structure of the prototypic MATE family member, NorM from Vibrio cholerae, reveals a protein fold composed of 12 transmembrane helices (TMHs), confirming hydropathy analyses of the majority of (prokaryotic and plant) MATE transporters. However, the mammalian MATEs are generally predicted to have a 13th TMH and an extracellular C terminus. Here we affirm this prediction, showing that the C termini of epitope-tagged, full-length human, rabbit, and mouse MATE1 were accessible to antibodies from the extracellular face of the membrane. Truncation of these proteins at or near the predicted junction between the 13th TMH and the long cytoplasmic loop that precedes it resulted in proteins that (i) trafficked to the membrane and (ii) interacted with antibodies only after permeabilization of the plasma membrane. CHO cells expressing rbMate1 truncated at residue Gly-545 supported levels of pH-sensitive transport similar to that of cells expressing the full-length protein. Although the high transport rate of the Gly-545 truncation mutant was associated with higher levels of membrane expression (than full-length MATE1), suggesting the 13th TMH may influence substrate translocation, the selectivity profile of the mutant indicated that TMH13 has little impact on ligand binding. We conclude that the functional core of MATE1 consists of 12 (not 13) TMHs. Therefore, we used the x-ray structure of NorM to develop a homology model of the first 12 TMHs of MATE1. The model proved to be stable in molecular dynamic simulations and agreed with topology evident from preliminary cysteine scanning of intracellular versus extracellular loops.
Introduction
The renal proximal tubule is the primary site of secretion of a structurally diverse array of endogenous and xenobiotic compounds that share in common a net positive charge at physiological pH (organic cations (OCs)).2 Renal secretion of most Type I3 OCs requires two sequential steps. The first step involves OC uptake from the blood into renal proximal tubule cells across the basolateral membrane via electrogenic facilitated diffusion (1), a process mediated by one or more of the organic cation transporters (OCTs; in human, this process is dominated by OCT2; i.e., SLC22A2) (1–3). The second step in renal OC secretion; i.e., luminal exit into the tubular filtrate, is the active and rate-limiting element in transepithelial OC transport (4–6) and involves mediated electroneutral exchange of cellular OC for luminal H+. The molecular identity of the luminal OC/H+ exchanger remained elusive until the cloning in 2005 of the first mammalian members of the MATE (multidrug and toxin extrusion) family (i.e., SLC47) (7). Since then, substantial evidence has accumulated (e.g., Refs. 8–11), implicating MATE1, MATE2, and its kidney-specific homolog, MATE2-K, as critical players in the luminal export of OCs from renal proximal tubule cells in human kidney.
As a key element in the renal (and hepatic) secretion of cationic drugs, the human MATEs are likely targets for unwanted drug-drug interactions, as well as principal arbiters of the pharmacodynamics and pharmacokinetics of many clinically important agents. Thus, it is somewhat surprising that comparatively little attention has been given to determining the structural basis of substrate/inhibitor interaction with MATE transporters. Indeed, until recently, little has been known about the structure of any MATE family member. The structure of NorM, a Na+/OC exchanger from Vibrio and a prototypic MATE family transporter (12), was recently resolved to 3.65 Å (13). The structure revealed a protein fold consisting of 12 TMHs and supported the view, based on hydropathy analyses of several hundred prokaryotic and plant members of the MATE family, that 12 TMHs make up the functional core of these proteins (12, 14). However, hydropathy analysis of the mammalian MATEs consistently identifies 13 TMHs, arising from the presence in these proteins of a very hydrophobic series of residues at the C-terminal end of what, in most other MATE family members, is a long, hydrophilic, C-terminal strand (15). Interestingly, mouse kidney expresses two functional forms of MATE1: “full-length” MATE1 (mMate1b) and an insertion variant (mMate1) that is missing the last 35 C-terminal amino acids (7, 15, 16). We previously showed that the rabbit ortholog of MATE1 has a cytoplasmic N terminus and an extracellular C terminus (17), consistent with an odd number (e.g., 13) of TMHs. Elimination of the last 29 C-terminal amino acid residues of rbMate1 results in a protein with a cytoplasmic C terminus that retains modest transport function (17). It remains unclear (2, 18), however, whether the typical topology of mammalian MATEs, particularly the human orthologs of these proteins, includes a 13th TMH or if, instead, the hydrophobic C-terminal sequence of mammalian MATEs is cytoplasmic (10, 15), consistent with the core structure of 12 TMHs that appears to typify MATE family members (19, 20). It is also not clear whether the selectivity characteristics of MATE1 are retained in a protein lacking the 13th TMH. In the present paper, we show that the human ortholog of MATE1, like its rabbit and (full-length) mouse congeners, has an extracellular C terminus, consistent with the presence of 13 TMHs. We compare the functional characteristics of “short” and “long” forms of rabbit MATE1 and demonstrate that the 13th TMH is not necessary to retain function and that a 12 TMH structure is sufficient to support multiselective OC transport. These data indicate that the functional “core structure” of mammalian MATE1 consists of the first 12 TMHs. Based on these observations, we introduce here a homology model of hMATE1 based on the x-ray structure of NorM that can serve as the basis of future structure/function studies of this important class of human drug transporters.
EXPERIMENTAL PROCEDURES
Chemicals
[3H]1-Methyl-4-phenyl-pyridinium ([3H]MPP+;(80 Ci/mmol) was synthesized by the Synthetic Chemistry Core of the Southwest Environmental Health Science Center. Sulfosuccinimidyl 2-(biotinamido)-ethyl-1,3-dithiopropionate (NHS-biotin) was obtained from Pierce. Platinum® high fidelity DNA polymerase, Zeocin, hygromycin, Flp recombinase expression plasmid (pOG44) and the mammalian expression vector pcDNA5/FRT/V5-His TOPO were obtained from Invitrogen. Other chemicals were typically of the highest grade available and, unless otherwise indicated, were purchased from Sigma.
Generation of MATE1 Sequences
Cloning of the full-length and truncated rbMate1 sequences used in this study used methods described previously (21). The mMate1b and mMate1 sequences were cloned by RT-PCR with sequence-specific primers and mouse RNA. Briefly, mouse kidney poly(A)+ RNA was reverse transcribed using Moloney murine leukemia virus RT H− using a standard protocol (21). The RT reactions were then amplified with sequence-specific primers for mMate1b and mMate1 and subcloned into the mammalian expression vector pcDNA5/FRT/V5-His (Invitrogen). The full-length human MATE1 sequence used in this study was generously provided by Dr. Kathleen Giacomini (University of California, San Francisco) (22). The required sequences (contained in pcDNA3.1) were amplified using Platinum high fidelity DNA polymerase and sequence-specific primers with the following PCR conditions: 35 cycles of 94 °C for 1 min, 54 °C for 1 min, and 72 °C for 3.5 min. A final elongation step of 7 min was included after the last cycle. The PCR product was gel-purified and cloned into the pcDNA5/FRT/V5-His TOPO mammalian expression vector, resulting in addition of a C-terminal [V5]/[poly-His] epitope tag (amino acid sequence, [GKPIPNPLLGLDST]RTG[HHHHHH]; nucleotide sequence GGT AAG CCT ATC CCT AAC CCT CTC CTC GGT CTC GAT TCT ACG CGT ACC GGT CAT CAT CAC CAT CAC CAT). Human MATE1 truncated at residues Leu-544 or Gly-547 were created by PCR using sequence-specific primers based on the site of truncation of mMate1, resulting in a deletion of the last 27 or 24 amino acids of full-length hMATE1, respectively. Rabbit Mate1 truncated at residue Gly-545 was created using the same method resulting in a deletion of the last 23 amino acids of full-length rbMate1. Plasmid DNA was purified using standard methods (Genesee Scientific, San Diego, CA), and the sequences were confirmed with an Applied Biosystems 3730xl DNA analyzer at the University of Arizona sequencing facility.
Cell Culture and Expression of MATE1 Constructs
CHO cells containing a single integrated FRT site were acquired from Invitrogen (CHO Flp-In) and were used for stable expression of the MATE1 constructs. Prior to transfection, CHO Flp-In cells were grown in Ham's F12 Kaighn's modification medium (Sigma) supplemented with 10% fetal calf serum and Zeocin (100 μg/ml). Cultures were typically split every 3 days. The cells (2 × 106) were transfected by electroporation (BTX ECM 630, San Diego, CA; 260 volts and time constant of ∼25 ms) with 10 μg of salmon sperm, 18 μg of pOG44, and 2 μg of pcDNA5/FRT/V5-His TOPO containing the mutant constructs of MATE1. The cells were seeded in a T-75 flask following transfection and maintained under selection pressure with hygromycin (100 μg/ml). The cells were used for experiments ∼21 days after electroporation (i.e., beginning with passages 4–6).
Measurement of Transport
Transport studies were conducted on nominally confluent CHO cells in 24-well plates. The cells were incubated at room temperature (∼25 °C) in Waymouth buffer (135 mm NaCl; 13 mm HEPES-NaOH, pH 8.5; 28 mm d-glucose; 5 mm KCl; 1.2 mm MgCl2; 2.5 mm CaCl2; and 0.8 mm MgSO4), to which labeled substrate and appropriate test agents were added. Uptake was stopped by aspirating the transport buffer and rinsing the cells three times with 1 ml of ice-cold Waymouth buffer. The cells were then solubilized in 200 μl of 0.5 n NaOH with 1% (v/v) SDS, and the extract was subsequently neutralized with 100 μl of 1 n HCl. Accumulated radioactivity was determined by liquid scintillation spectrometry. Uptake is expressed as mol/cm2 of nominal cell surface of the confluent monolayer (∼0.04 mg of cell protein/cm2).
Immunocytochemistry
CHO cells were electroporated with plasmid DNA containing a C-terminal V5 epitope-tagged transporter and seeded onto coverslips. Immunocytochemistry was generally performed on a confluent monolayer 24 h after plating. The cells were fixed in ice-cold 100% methanol for 20 min (“permeabilized cells”), washed with PBS (137 mm NaCl; 2.7 mm KCl; 8.0 mm Na2HPO4; and 1.5 mm KH2PO4, pH 7; all washes were done in triplicate), and then incubated for 1 h with anti-V5 antibody (Invitrogen) diluted 1:500 in PBS. For “nonpermeabilized cells,” the cells were incubated with antibody for 45 min at room temperature first, washed with PBS, fixed in ice-cold 100% methanol for 20 min, and then washed in PBS. The permeabilized and nonpermeabilized cells were then incubated for 1 h in the dark with fluorescein isothiocyanate-conjugated goat anti-mouse antibody (Molecular Probes, Inc.) diluted 1:1000 in PBS. To visualize the nuclei, the cells were treated with propidium iodide (5 μg/ml) for 10 min. The cells were washed again, and the coverslips were mounted onto microscope slides using Dako fluorescent mounting medium (Dako Corporation, Carpenteria, CA). A confocal microscope (Nikon PCM 2000 scan head fitted to a Nikon E800 microscope) was used for detection of immunoreactivity in CHO cells.
Cell Surface Biotinylation of MATE1
The method described here is a minor modification of that described by Pelis et al. (23). All of the solutions were kept ice-cold throughout the procedure, and long incubations were conducted on ice with gentle shaking. The cells plated to confluence in a 12-well plate were initially washed three times with 2 ml of PBS solution containing calcium and magnesium (PBS/CM; 137 mm NaCl, 2.7 mm KCl, 8 mm Na2HPO4, 1.5 mm KH2PO4, 0.1 mm CaCl2, and 1 mm MgCl2, pH 7.0, with HCl). The cells were then exposed for 30 min to NHS-biotin or MTS-biotin (0.5 μg/ml), diluted in PBS/CM. After biotinylation, the cells were rinsed three times briefly with 3 ml of PBS/CM (the NHS-biotin rinse also contained 100 mm glycine). In some cases, the plasma membranes were permeabilized prior to exposure to the biotinylation reagent by treating cells for 2 min with 0.05% saponin (in PBS/CM). The cells were lysed on ice for 1 h with gentle shaking in 1 ml of lysis buffer (150 mm NaCl, 10 mm Tris-HCl, 1% Triton X-100, 1% sodium deoxycholate, and 0.1% SDS, pH 7.4) containing protease inhibitors (200 μm 4-(2-aminoethyl)-bezenesulfonyl-fluoride, 0.16 μm aprotinin, 4 μm leupeptin, 8 μm bestatin, 3 μm pepstatin A, 2.8 μm E-64; Sigma), after which 50 μl of streptavidin-agarose beads (Pierce) were added to the lysates and incubated overnight at 4 °C with constant mixing. After extensive washing with the above lysis buffer, 50 μl of Laemmli sample buffer was added, and the proteins were eluted from the beads at 100 °C for 5 min. The proteins were separated on 10% SDS-PAGE gels, transferred to PVDF membranes, and immunoreactivity corresponding to the V5-tagged MATE1 construct was visualized as previously described (17).
Homology Modeling
The homology model of hMATE1 was generated by threading the homologous regions of MATE1 onto the outward facing apo-structure of NorM from Vibrio cholerae (NorM-VC; Protein Data Bank code 3MKT) (13). The MATE1 and NorM-VC protein sequence were aligned by ClustalW (24) (supplemental Fig. S1). The coordinates of residues in NorM-VC that did not have a correspondence in MATE1 were removed. Residues in hMATE1 that did not have a correspondence in NorM-VC sequence (the first 30 and last 94) were not included in the model. The homologous sequence of hMATE1 was threaded onto NorM-VC apo-structure, using the Swiss PDB Viewer (25). The stereochemical geometry of the hMATE1 model was then energetically minimized using CNS v1.2 (26).
Molecular Dynamics Simulation
The initial structure of NorM was Protein Data Bank entry 3MKT. The protein (NorM or hMATE1) was aligned to the z axis using VMD Orient plugin (27), and protonation states for amino acid side chains were determined using the PDB2PQR/PROPKA webserver (28, 29). Based on pKa calculations, for NorM, Glu-237 (pKa = 10.88) and Glu-255 (pKa = 7.67) are protonated, and Lys-449 (pKa = 6.58) is deprotonated. For hMATE1 Asp-97 (pKa = 7.12), Glu-273 (pKa = 8.09), Glu-389 (pKa = 7.16), and His-65 (pKa = 7.09) were chosen to be protonated. An initial solvation step was carried out using the program SOLVATE. The protein (NorM or hMATE1) was then placed into a 1-palmitoyl-2-oleoyl-phosphatidylethanolamine membrane bilayer, and waters overlapping the membrane region were removed. The 1-palmitoyl-2-oleoyl-phosphatidylethanolamine bilayer was a pre-equilibrated pure lipid bilayer obtained from the CHARMM-GUI website (30) that consisted of 256 lipid molecules. Lipid molecules within 0.5 Å of the protein were removed, and additional solvation of the regions above and below the bilayer was carried out using the VMD Solvate plugin, with no water molecule placed closer than 2.4 Å to the protein. The addition of 0.1 m NaCl was done to bring the system to electrostatic neutrality using the VMD Autoionize plugin. The resulting systems consisted of 64,383 (71,260) atoms and had dimensions of ∼ 100 × 100 × 90 Å3 (100 × 100 × 100 Å3) for NorM (hMATE1).
Minimization, heating, equilibration, and ensuing 50-ns simulations of NorM and hMATE1 were all performed using NAMD version 2.7b1 (31) with the CHARMM27 force field including CMAP corrections (32). Heating and equilibration were carried out in the NPγT ensemble, with the surface tension set to 30 dyn/cm in the x-y plane and with constraint forces on the system gradually relaxed. During heating, the temperature of the system was gradually increased from 50 to 310 K. A 2-fs integration time step was used for the 50-ns simulations; nonbonded interactions were calculated every 2fs, and a full electrostatic calculation was made every 4 fs. Full electrostatics in the periodic system were calculated using the particle mesh Ewald (PME) method (33). A constant pressure of 1 atm along the z direction, and a constant temperature of 310 K was maintained using the Nose-Hoover Langevin piston method (34, 35) and Langevin dynamics in NAMD for the 50-ns simulations. Hydrogen bonds were kept rigid, which is consistent with the 2-fs integration time step. Secondary structure content was obtained using the STRIDE (36) algorithm implemented in VMD.
RESULTS
Membrane Expression Profiles of Full-length and Truncated MATE1 Orthologs
Fig. 1 shows sequence alignments for the rabbit, human, and mouse orthologs of MATE1 on which are indicated the several sites used to generate the truncated versus full-length forms of MATE1 used in the subsequent experiments. The sites selected for truncation of the rabbit and human orthologs of MATE1 were based on the length of the truncated transcript naturally expressed in the mouse (i.e., mMate1). The truncated and full-length forms of the mouse MATE1 ortholog reflect sequences described already in the literature for the naturally occurring transcripts. The initial characterization of mouse MATE1 (16) used the truncated sequence (mMate1), which was the first transcript cloned from mouse kidney (7). A subsequent study (15) reported the cloning of the full-length transcript (mMate1b) and revealed that the truncated protein was the consequence of the insertion of a single nucleotide (adenine) in exon 18 of mMate1 that results in a frameshift and introduction of an early stop codon. The overall result is a protein of 532 amino acid residues, 35 shorter than the full-length protein, in which amino acids after Lys-509 through position 532 differ from those in the full-length protein (reflecting the frameshift; Fig. 1).
FIGURE 1.

Sequence alignment (ClustalW) of the open reading frames of mMate1 (GenBankTM accession number AAH31436), mMate1b (GenBankTM accession number NP_080459), rbMate1 (GenBankTM accession number NP_001103289), and hMATE1 (GenBankTM accession number AAH10661). Red letters indicate sequence regions of identity; green letters indicate strong similarity; blue letters indicate weak similarity. Yellow highlighting indicates regions predicted to comprise a transmembrane helix (as predicted by TMHMM version 2.0). In all cases the N terminus was predicted to be intracellular. The sites of truncation of the rabbit and human orthologs are indicated and labeled at the site of truncation.
Hydropathy analysis of the full-length sequences of all three MATE1 orthologs predicts the presence of 13 TMHs (Fig. 1), including a C terminus that is exposed to the extracellular solution, whereas the elimination of the last 27–29 amino acid residues results in proteins predicted to have 12 TMHs and cytoplasmic C termini. We tested these predictions by placing a V-5 epitope tag at the C terminus of each protein and then assessing its accessibility to V-5 antibody added to the extracellular solution bathing CHO cells that stably expressed these constructs. The accessibility profiles, as determined using immunocytochemistry, of the two naturally occurring mouse MATE1 proteins were particularly revealing. Whereas the V-5 tag of the full-length mMate1b protein was fully accessible to extracellular antibody, the V-5 tag at the C terminus of mMate1 (the truncated insertion variant) was not accessible to extracellular V-5 antibody unless the plasma membrane was first rendered permeable to the antibody (Fig. 2A). A similar profile was observed for the rabbit ortholog of MATE1. Fig. 2B shows that the C terminus of full-length rbMate1 was accessible from the external face of the plasma membrane, confirming our earlier observation (17). That previous report also addressed the possibility that the addition of the hydrophilic V-5 epitope to the C-terminal end of rbMate1 was itself a causative factor in externalizing the C terminus by showing that (in the absence of the epitope) the addition of a single cysteine residue to what was otherwise an effectively cysteineless, functional full-length rbMate1 construct rendered the protein accessible to membrane-impermeant thiol reactive reagents (17). That report also showed that truncating rbMate1 at position Leu-540 results in a protein that reaches the plasma membrane but is accessible to extracellular V5 antibody only after permeabilization of the plasma membrane. However, despite being “functional,” the L540 truncation mutant supported markedly reduced levels of transport. Here we show that truncation at Gly-545 also results in a protein with an intracellular C terminus (Fig. 2B) and, as noted below, much higher levels of transport.
FIGURE 2.

Immunocytochemical identification of V5 epitope-tagged MATE1 constructs in nonpermeabilized (non-perm) and permeabilized (perm) CHO cells. The cells were stably transfected with the indicated MATE1 construct. A, mouse MATE1. B, human MATE1; C, rabbit MATE1. Nonpermeabilized cells were exposed to PBS containing the V5 antibody for 60 min prior to washing and fixation in 100% methanol. The permeabilized cells were fixed in methanol for 20 min before exposure to the V5 antibody (see text for a discussion of each truncation mutant). The red fluorescence reflects propidium iodide staining of nuclei; the green fluorescence reflects staining of the V5-tagged MATE1 constructs.
Fig. 2 also shows that, in common with mouse and rabbit MATE1, the C terminus of the full-length human ortholog of MATE1 is extracellular (Fig. 2C) and that truncation of the hydrophobic sequence (at position Leu-544) renders the C terminus accessible to the extracellular antibody only after permeabilization of the plasma membrane. A similar immunocytochemistry profile, revealing a cytoplasmic C terminus, was observed for a second truncation mutant of hMATE1 (Gly-547) as well (data not shown). Importantly, we previously showed that the N terminus of rbMate1 is within the cytoplasm (17), so the extracellular location of the C termini of the full-length sequences of the human, rabbit and mouse MATE1 orthologs argues for an odd number of TMHs and so is consistent with the presence of 13, rather than 12, TMHs. As noted earlier, hydropathy analyses of the mammalian MATE1 and MATE2-K homologs routinely predict the presence of 13 TMHs (2, 21), the consequence of the terminal hydrophobic sequence of these proteins. Therefore, the present data support the contention that the presence of 13 TMHs, including an extracellular C terminus, is a routine characteristic of the mammalian MATEs.
Transport Characteristics of Truncated (12 TMH) MATE1
NorM mediates OC/Na+ exchange (37) and does so with a protein fold comprised of 12 TMHs (13). Indeed, the vast majority of MATE family members have sequences consistent with the presence of 12 TMHs (19) and what is likely a highly conserved topology. Thus, it was of interest to assess the impact of eliminating TMH 13 on OC/H+ exchange activity of a mammalian MATE. It has already been noted in the literature that the naturally occurring short (12 TMH) form of mouse MATE1 (16) retains transport function similar to that of the long (13 TMH) form (15). Fig. 3A compares the rate of 13 nm [3H]MPP transport into CHO cells stably expressing either the full-length sequence of hMATE1 or the Leu-544 truncation mutant. Although the mutant protein supported mediated transport, the level was substantially below that observed in cells expressing the full-length protein. However, that reduced level of transport was associated with substantially less protein in the plasma membrane, as shown in the Western blot of biotinylated protein below the bars in Fig. 3A. Fig. 3B compares the rates of transport supported by cells expressing either the full-length sequence of rbMate1 or one of the truncated (i.e., 12 TMH) sequences: rbMate1 (Gly-545) or the naturally occurring mMate1. In three experiments, mMate1 and the Gly-545 mutant of rbMate1 both supported substantial levels of transport, ∼50–60% of that displayed by cells expressing rbMate1. However, these rates of transport were associated with higher levels of plasma membrane expression for both truncated proteins (Western blots below the bars of Fig. 3B), suggesting that the truncated (12 TMH) sequences of rabbit and mouse MATE1 had lower turnover numbers that those of the full-length (13 TMH) proteins. Collectively, these observations show that although the 13th TMH may influence the conformational changes associated with the translocation process, the 13th TMH is not necessary for MATE1 transport function.
FIGURE 3.

Transport function and plasma membrane expression of full-length and truncated sequences of the human (A) and the rabbit and mouse orthologs (B) of MATE1. The bar graphs show 10-min uptakes of 13 nm [3H]MPP in CHO cells stably expressing the indicated sequence of MATE1. The height of each bar indicates the mean (± S.E.) of transport, measured in the presence and absence of 1 mm unlabeled MPP, in seven (A) or three (B) separate experiments. Below each bar graph, Western blots of biotinylated protein show plasma membrane expression of the indicated human, rabbit, and mouse MATE1 sequences. Each lane contained 15 μl of the protein-containing solution obtained following biotinylation reaction in a single well from a 12-well plate. The experiment was performed two (A) or three (B) times with similar results.
We used the Gly-545 truncation mutant of rbMate1 to assess the influence of eliminating TMH 13 on the qualitative and quantitative characteristics of MATE1 transport activity. As shown in Fig. 4, the kinetics of MPP transport into CHO cells that stably expressed either full-length rbMate1 (Fig. 4A) or the Gly-545 mutant protein (Fig. 4B) were described by the Michaelis-Menten equation for competitive interaction of labeled and unlabeled substrate (38),
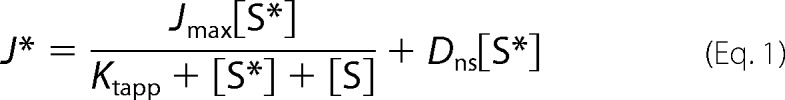 |
where J* is the rate of transport of the radiolabeled substrate (in this case, [3H]MPP) from a concentration of the labeled substrate equal to [S*]; Jmax is the maximal rate of mediated substrate transport; Ktapp is the apparent Michaelis constant of the transported substrate; [S] is the concentration of unlabeled substrate; and Dns is a rate constant that describes the nonsaturable component of labeled substrate accumulation (reflecting the combined influence of diffusion, nonspecific binding, and incomplete rinsing of [3H]MPP from the cell culture well). In three separate experiments, the Ktapp for MPP uptake in cells that expressed the Gly-545 truncation mutant was increased slightly (∼2-fold) but significantly (p < 0.005) compared with the Ktapp in cells expressing full-length rbMate1 (6.7 ± 0.42 μm versus 3.5 ± 0.34 μm). In contrast, the Jmax values for MPP uptake in the two cell lines did not differ significantly (1.78 ± 0.34 pmol cm−2 min−1 versus 2.1 ± 0.22 pmol cm−2 min−1, for full-length rbMATE1 and the Gly-545 truncation mutant, respectively). However, the robust transport capacity supported by cells expressing the Gly-545 mutant must be viewed in the context of its higher level of plasma membrane expression, compared with that of cells expressing the full-length protein.
FIGURE 4.

Kinetic characteristics of transport into CHO cells that stably expressed full-length rbMate1 (A) or rbMate1 (B) truncated at Gly-545. 2-min (rbMate1) or 5-min (Gly-545) uptakes of [3H]MPP (∼13 nm) were measured in the presence of increasing concentrations of unlabeled MPP. Each point is the mean (± S.E.) of uptakes measured in three separate experiments (each performed in triplicate).
We also assessed the influence of TMH13 on ligand binding. As noted above, truncation of rbMate1 at Gly-545 resulted in a slight (∼2-fold) increase in the Kt for MPP transport. As an OC/H+ exchanger, H+ is also a MATE1 ligand, and Fig. 5 shows the effect of extracellular pH on uptake of [3H]MPP into CHO cells that stably expressed either full-length rbMate1 or the Gly-545 truncation mutant. For both transporters, increasing extracellular pH beyond pH 7.0 resulted in an increase in the rate of [3H]MPP uptake, as expected in light of the competitive effect extracellular H+ has on activity of OC/H+ exchange (39, 40). However, the full-length transporter was substantially more sensitive to extracellular [H+], displaying an apparent pKa for the inhibition of transport of H+ of >8.0, whereas the pKa for inhibition of transport by the Gly-545 mutant was on the order of 7.7. This difference in sensitivity to H+ had the interesting result of allowing the truncated mutant to support twice the transport activity of the full-length mutant at pH 7.5 (Fig. 5).
FIGURE 5.

Effect of extracellular pH on uptake of [3H]MPP into cells expressing full-length rbMate1, the Gly-545 truncation mutant of rbMate1, or wild-type CHO cells. 10-min uptakes of 13 nm [3H]MPP were plotted as a function of extracellular pH. Each point is the mean (± S.E.) of uptakes determined in three separate experiments (n = 3), each performed in triplicate.
Another hallmark of MATE1 is its multiselectivity (41). Fig. 6 compares the effect on transport activity of full-length rbMate1 and the Gly-545 truncation mutant of a structurally diverse array of organic cations. Each of the test inhibitors (at a concentration of 1 mm, except for pyrimethamine, which was present at a concentration of 10 μm) significantly blocked uptake of [3H]MPP mediated by both the full-length and truncated forms of rbMate1. Moreover, the qualitative profile of inhibitory effectiveness was the same for both transporters; whereas MPP, atropine, and PYR reduced transport activity of both transporters by >75%, 1 mm concentrations of creatinine and midodrine blocked <50% of their transport activity.
FIGURE 6.

Effect of inhibitory ligands on uptake of [3H]MPP into CHO cells that stably expressed either full-length rbMate1 (black bars) of the Gly-545 truncation mutant of rbMate1 (gray bars). Ligands were present at a concentration of 1 mm (except for PYR, which was 10 μm). The height of each bar represents the mean (± S.E.) of 10-min uptakes determined in three separate experiments (each performed in triplicate). The uptakes were normalized to the rate for each transporter measured in the absence of inhibitor (open bar).
The kinetics of inhibitor interaction with full-length and truncated rbMate1 were assessed in more detail for “high,” “intermediate,” and “low” affinity” inhibitors (PYR, TEA, and creatinine, respectively). The inhibitors each reduced the uptake of labeled MPP in a concentration-dependent manner for both the full-length and truncated proteins (Fig. 7) that was adequately described by the following relationship,
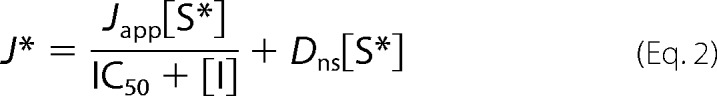 |
where Japp is defined as (IC50/Ktapp)Jmax, [I] is the concentration of the test inhibitor (e.g., PYR), and IC50 is concentration of inhibitor that blocked 50% of mediated substrate transport. Consistent with the modest (2-fold) increase in Ktapp for MPP transport observed for the Gly-545 truncation mutant (Fig. 4), the IC50 values for inhibition of [3H]MPP uptake by truncated rbMate1 produced by TEA and creatinine were modestly (but significantly; p < 0.05) increased by 2.6- and 1.6-fold, respectively, compared with those for inhibition of the full-length protein. On the other hand, the IC50 value for PYR inhibition of Gly-545-mediated transport was 50% lower than for inhibition of the full-length protein. Overall, these data suggest that elimination of the 13th TMH had comparatively little effect on ligand interaction with the binding surface of rbMATE1.
FIGURE 7.

The effect of three representative inhibitory ligands (PYR, TEA, or creatinine) on the rate of [3H]MPP uptake into CHO cells that stably expressed either full-length rbMate1 (solid circles) or the Gly-545 truncation mutant of rbMate1 (open circles). Each point represents the mean value (determined in three separate experiments) of either 2 min (rbMate1) or 5 min (Gly-545) uptake of [3H]MPP (∼13 nm) measured in the presence of increasing concentration of test inhibitor; uptakes were normalized to a 1-min uptake measured in the absence of inhibitor.
hMATE1 Homology Model
The data presented above support the following conclusions: (i) the 13th TMH is not necessary for rbMATE1 function; (ii) the 13th TMH may influence transporter turnover; and (iii) the first 12 TMHs of mammalian MATE1 support comparatively normal ligand binding. There is a widely held view that structural homology is highly conserved within a phylogenetically linked protein family, as shown by the highly conserved fold evident in the five MFS structures determined to date (42), and the similar fold evident in sarcoplasmic/endoplasmic reticulum Ca2+-ATPase and the Na,K-ATPase (43). Thus, it is likely that the first 12 TMHs in all mammalian MATEs make up the functional core structure that shares the protein-fold evident in the x-ray structure of NorM. Moreover, the binding of ligands (substrates and inhibitors) is largely controlled by the fold comprised by first 12 TMH. Consequently, we elected to construct homology models of hMATE1 and rbMate based on the 12 TMH x-ray structure of NorM (Fig. 8A) and its fold (Fig. 8B). Fig. 8 (C and D) shows the model for hMATE1. The model, which is missing the first 30 (long cytoplasmic N-terminal sequence) and last 94 (the 13th TMH and a portion of the long C-terminal cytoplasmic element) amino acids of hMATE1, shares the overall structural features of NorM-VC, including the same transmembrane helical fold, and spans ∼50 Å in the plane of the lipid bilayer. Twelve transmembrane helices are arranged as two bundles of six transmembrane helices (TMH1–TMH6 in the N-terminal half and TMH7–TMH12 in the C-terminal half) forming an internal cavity ∼4000 Å3 (Fig. 8, C and D) open to the extracellular space. The two halves are related by an intramolecular 2-fold symmetry shared by all members of the MATE family. A relatively short cytoplasmic loop (residues 238–250) between TMH6 and TMH7 connects the two halves and is consistent with the hydropathy plots and protein sequence alignment between hMATE1 and NorM-VC. The alignment between NorM and MATE1 was particularly weak in the region of TMH 12, resulting in the presence in the hMATE1 model of nine residues not present in the NorM sequence (supplemental Fig. S1). Consequently, our confidence in the positional information of residues in this TMH is lower. However, in our model, TMH 12 is peripheral (Fig. 8B) and does not appear to be a major element in the proposed translocation pathway.
FIGURE 8.

X-ray structure of NorM and hMATE1 homology model with the molecular surface of the translocation cleft. A and B, view of the cytoplasmic face of NorM (A) and a schematic representation (B) of its helical organization. C and D, stereo views of the side (C) and the extracellular face (D) of the hMATE1 homology model. The N-terminal half is shaded in light blue, and the C-terminal half is colored light green. The N terminus is noted, and the C-terminal short helix is omitted for clarity. E and F, stereo views of the electrostatic surface rendering of the internal cavity of hMATE1 homology model, showing the side composed of transmembranes 1, 5, 6, 8, 9, and 10 (E) and the other side comprising transmembranes 2, 3, 4, 7, 11, and 12 (F). Positively charged areas are shaded in deep blue, negatively charged areas are shown in red, and neutral areas are colored light gray. Stereo images and electrostatics representation were generated by PyMOL.
To provide a preliminary test of the topology predicted by the MATE1 homology model, we used the substituted cysteine accessibility method (44) with a version of rbMate1, mentioned earlier, from which 13 native cysteine residues were replaced with alanine residues (17). The resulting protein (Δ13C) is both functional and refractory to interaction with thiol-reactive reagents (17). Substituted cysteine accessibility method analysis was conducted on Δ13C constructs in which single residues in one of the first two intracellular (intracellular loops 1 and 2) and extracellular loops (extracellular loops 1 and 2) of rbMate1 were replaced by a cysteine residue and the accessibility of the resulting free thiol group to an impermeant thiol-reactive reagent (MTS-biotin) was determined before or after permeabilization (with saponin) of the plasma membrane. Fig. 9 shows that all four “single-cysteine” mutants were expressed at the plasma membrane, as shown by the accessibility of each to the impermeant lysine-reactive reagent, NHS-biotin (first lane of the Western blots in Fig. 9). Of the four single-Cys proteins, only the two containing cysteine residues in the predicted extracellular loops (Leu-70 in extracellular loop 1 and Leu-142 in extracellular loop 2) showed substantial interaction with MTS-biotin prior to permeabilization of the plasma membrane (second lane of the blots in Fig. 9). Interaction of the other two residues (Ser-105 in intracellular loop 1 and Leu-178 in intracellular loop 2) with MTS-biotin required that the membrane first be permeabilized by exposure to 0.05% saponin for 2 min (third lane of Fig. 9). This profile of residue accessibility is consistent with the location of each residue within the MATE1 homology model (Fig. 9).
FIGURE 9.

Substituted cysteine accessibility method profiles for selected rbMate1 residues (single-Cys replacement in the Δ13C mutant). A, Western blots of the accessibility profiles for selected MATE1 residues. Each three-lane “triplet” displays immunoreactive rbMate1 pulled down through reaction with (from left to right) NHS-biotin (NB), MTS-biotin (MB), or MTS-biotin following permeabilization with saponin (MB/Perm; see text for discussion). Accessibility profiles mapped on the homology model for rbMate1. Blue residues were accessible to MTS-biotin from the extracellular solution; orange residues were accessible to MTS-biotin only after permeabilization of the plasma membrane.
Computational Assessment of the Stability of the MATE1 Homology Model Using Molecular Dynamics (MD) Simulation
Combining homology modeling with MD simulation can be used to derive (plausible) homology models (35). Root mean square deviation (RMSD) has been shown to be an indicator of protein stability (45). The RMSDs were calculated for the transmembrane portions of the 12 helices in each protein, excluding the more flexible loop regions from the calculation. Protein structures were relatively stable over the course of the simulation with ∼3.5 Å RMSD (supplemental Fig. S2). Conformational stability, measured in terms of Cα RMSDs and conservation of helical content of the core fold, can be used to discriminate models (35). In our simulations, those values are very similar between the hMATE1 homology model and the NorM x-ray structure, indication of a stable structure and feasible model for hMATE1 (46).
Secondary structure was well conserved in the hMATE1 homology model (Fig. 10) because the α-helical content remained 78% over the course of the 50-ns simulation. In comparison, the α-helical content in the NorM x-ray structure started at 70% and increased to 78% at the end of the simulation (data not shown).
FIGURE 10.

Snapshots from the MD simulation (50 ns after the start of the simulation) of hMATE1 (panels b, d, and f) and views of the original homology model (panels a, c, and e). Coloring is from the N terminus (blue) to the C terminus (red). Panels a and b show views from the cytoplasmic side of the membrane, panels c and d show views from the extracellular side, and panels e and f show a side view.
DISCUSSION
The core structure of the prokaryotic and plant members of the MATE family of transport proteins is generally considered to involve 12 transmembrane helices (19, 20), a view supported by the recent solution of the x-ray structure of NorM, the prototypic prokaryotic MATE transporter, which revealed a protein fold composed of 12 TMHs (13). The results presented here support the contention that the structure of mammalian members of the MATE family includes a C-terminal 13th TMH. The reasoning is as follows. The C termini of the human, rabbit, and (full-length) mouse orthologs of MATE1 are extracellular (Fig. 2). That result, when combined with the previous observation that the N terminus (of rbMate1) (17) is intracellular, argues that mammalian MATEs have an odd (i.e., 13 TMHs) rather than an even (12 TMHs) number of TMHs. Furthermore, elimination of the terminal hydrophobic sequence of MATE1, either through truncation as in the present case with the human and rabbit transporters (Fig. 2, B and C) or as in the naturally occurring, short mouse MATE1 variant (Fig. 2A), results in proteins with cytoplasmic C termini, effectively mimicking the prototypic MATE family structure. Taken together, these observations support the view that, whereas proteins of the MATE family generally have 12 TMHs, the mammalian MATEs deviate from this norm.
The core structure of 12 TMHs, which clearly supports transport function in NorM (37) (and presumably in other prokaryotic MATE family members), was also sufficient to support transport in truncated mammalian MATEs. The naturally occurring truncated mouse sequence (mMate1) supports rates of TEA transport similar to that supported by the full-length sequence (mMate1b) (15, 16). Here, we show that truncation mutants of both the rabbit and human orthologs of MATE1 also support transport (Fig. 3). Indeed, the Gly-545 truncation mutant of rbMate1 supported robust pH-sensitive transport (Figs. 4 and 5) and a profile of selectivity that closely parallels that of the full-length protein (Figs. 6 and 7).
Although these observations show that the 13th TMH is not necessary to support transport activity markedly similar to that of full-length MATE1, the results also suggest that elimination of the terminal helix can influence quantitative characteristics of transport. The robust rates of transport into cells expressing truncated rabbit and mouse MATE1 sequences appear to reflect higher levels of plasma membrane expression than those of full-length MATE1 (Fig. 3B). This observation is consistent with a previous result (17) that the modest transport (90% reduction of Jmax for MPP uptake) supported by a different truncation mutant of rbMate1 (at Leu-540, rather than Gly-545) was nevertheless associated with a much higher level of plasma membrane expression, compared with that of full-length rbMate1. The most likely explanation for reduced rates of transport in the presence of more transport protein (assuming that protein in the plasma membrane reflects functional transporters) is a reduction in catalytic efficiency (kcat) of proteins lacking the 13th TMH.
rbMate1(G545) also displayed a modest (but significant) difference in apparent affinity for MPP (Fig. 4) and for several inhibitory ligands (Fig. 5). However, these differences did not reflect a consistent pattern; i.e., whereas the truncated transporter displayed 2–3-fold decreases in apparent affinity for TEA and creatinine, the apparent affinity for PYR was 50% greater. Of perhaps greater relevance was the observation that the overall profile of ligand interaction was quite consistent between the truncated and full-length sequences, suggesting that the presence of the 13th TMH has little impact on the structure of the substrate-binding pocket of MATE1.
With this latter idea in mind, we used the x-ray structure of NorM as a template to develop homology models of hMATE1 and rbMate1. There is ∼21% identity and 60% sequence similarity between NorM and mammalian MATE1 (supplemental Fig. S1), which is substantially greater than the degree of “identity/similarity” generally observed between MFS members that have been shown to be structurally homologous (42). The human MATE1 homology model inherited the overall fold of NorM and represents an outward-facing conformation arranged as two bundles of six transmembrane helices (TMH1–TMH6 and TMH7–TMH12) forming a large internal cavity open to the extracellular space (Fig. 8).
Analysis of the MD simulations (RMSD, secondary structure content, helical tilt angle) of hMATE1 and NorM provided evidence that the hMATE1 homology model is at least as stable as the NorM x-ray structure from which it is derived. In particular, it is interesting to note that helical tilt angles for several helices (for example, H6 and H9) changed in opposite directions for NorM and hMATE1 (supplemental Fig. S3), demonstrating that the x-ray structure and homology model can possess different dynamics from one another over 50 ns of simulation time. These changes in the helix orientations of hMATE1 led to partial closure of the internal cavity, which was originally open to the extracellular space, as demonstrated by calculations of the channel profile using the program HOLE (47) (supplemental Fig. S4).
Previous studies identified several amino acid residues in MATE1 that influence transport activity, and the current structural model of MATE1 provides insight into the possible bases of these effects. The glutamate residues that reside at positions 273, 278, 300, and 389 in hMATE1 are conserved as acidic amino acid residues at homologous loci in all mammalian MATEs. Replacement of these residues in hMATE1 with aspartate residues exerts large effects on substrate affinity, leading to the suggestion that they “form a part of the substrate binding site” (48). In the current model of MATE1 structure, each of these residues is, in fact, exposed to the hydrophilic cleft that forms the presumptive translocation pathway (Fig. 11). Interestingly, two of these residues (Glu-273 and Glu-389) are broadly conserved in prokaryotic MATE family members, and the homologous sites in NorM-VC (Glu-255 and Asp-371) form part of the cavity that stabilizes the interaction of this Na+/OC exchanger with monovalent metal cations (13). The surface of the cleft exposed to the external solution is characterized by large regions dominated by the presence of anionic or cationic residues (red and blue surfaces, respectively, in Fig. 8, C and D). Interestingly, the cleft also contains relatively large regions dominated by hydrophobic residues (gray surfaces in Fig. 8, C and D), which is consistent with the long standing view that hydrophobicity is a significant determinant of substrate/inhibitor interaction with renal OC/H+ exchange (49–52).
FIGURE 11.

Key residues identified in previous studies (57), mapped in the proposed three-dimensional structure of hMATE1. Purple, His; yellow, Cys; dark red, Glu. The side view is canted slightly to emphasize the position of these residues. N and C indicate the position of the N and C termini of the model, respectively. Note that the cytoplasmic location of the C terminus reflects the absence of the 13th TMH in this homology model.
Substantial interest has also been directed toward the influence of H+-titratable histidine residues on the activity of MATE1. In fact, the histidine-specific reagent, diethyl pyrocarbonate (DEPC) inhibited activity of the OC/H+ exchanger of rat renal brush border membrane vesicles (53), although the presence of TEA in excess did not protect the transporter from DEPC inhibition. Asaka et al. (54) individually replaced each of the seven conserved histidine residues in rMate1 with glutamine residues and found that only the H385Q mutation resulted in a change (50% decrease) in TEA transport. They also found that DEPC inhibition of rMate1-mediated TEA transport is not eliminated by coexposure of DEPC with excess TEA, leading to the suggestion that His-385 is not part of the TEA binding site but, instead, may be part of the H+ interaction site (54). Mutations of Cys-62 and Cys-126 in rMate1 also resulted in substantial decreases in TEA transport, resulting in the suggestion that these residues have a role in substrate recognition (54). Whereas Cys-63 (the human homolog of Cys-62 in rat MATE1) lies within TMH1, which forms a portion of the translocation pathway, Cys-127 is in TMH3 and its position, well away from the hydrophilic cleft (Fig. 11), makes it an unlikely candidate for direct substrate interaction. Nevertheless, the clear influence of this residue on transport activity (54) emphasizes the “long range influence” that amino acid residues can have on the selectivity of membrane transporters (for example, Ref. 55).
In summary, the present study confirmed that the full-length sequence of mammalian MATE1 consists of 13 TMHs, i.e., one more than the typical structure of MATE protein family members. However, transport activity of human, rabbit, and mouse MATE1 does not require the presence of the terminal, 13th TMH, supporting the contention that the functional core structure of MATE proteins, including the mammalian MATEs, is comprised of 12 TMHs (see also Ref. 19). Whereas the 13th TMH may influence turnover activity of the transporter, ligand binding of truncated (12 TMH) proteins is essentially normal. A homology model of hMATE1, based upon the 12 TMH structure of NorM, displayed a structural stability in MD simulations comparable with that of NorM, consistent with the conclusion that the MATE1 model can serve as a useful basis for the design and interpretation of future studies on ligand (substrate/inhibitor) interactions with this key element in renal and hepatic drug secretion.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants 1R01DK080801, 1U54GM094610, and 5P30ES006694. This work was also supported by the Skaggs Institute for Chemical Biology.

This article contains supplemental Figs. S1–S4.
Given the structural diversity of organic cations, it is useful to refer to the “Type I” and “Type II” classifications for different structural classes of organic cations developed to describe OC secretion in the liver (56). In general, Type I OCs are comparatively small (generally <400 Da) monovalent compounds, such as TEA, tributylmethylammonium, and procainamide ethobromide. Importantly, the majority of cationic drugs from a wide array of clinical classes, including antihistamines, skeletal muscle relaxants, antiarrhythmics, and β-adrenoceptor blocking agents, are adequately described as being Type I OCs. Type II OCs are usually bulkier (generally >500 Da) and frequently polyvalent, including d-tubocurarine, vercuronium, and hexafluorenium.
- OC
- organic cation
- TMH
- transmembrane helix
- MPP
- 1-methyl-4-phenyl-pyridinium
- NHS-biotin
- sulfosuccinimidyl 2-(biotinamido)-ethyl-1,3-dithiopropionate (sulfo-NHS-SS-biotin
- FRT
- Flp recombination target
- MD
- molecular dynamics
- TEA
- tetraethylammonium
- RMSD
- root mean square deviation
- PYR
- pyrimethamine.
REFERENCES
- 1. Wright S. H., Dantzler W. H. (2004) Molecular and cellular physiology of renal organic cation and anion transport. Physiol. Rev. 84, 987–1049 [DOI] [PubMed] [Google Scholar]
- 2. Nies A. T., Koepsell H., Damme K., Schwab M. (2011) Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. Handb. Exp. Pharmacol. 201, 105–167 [DOI] [PubMed] [Google Scholar]
- 3. Koepsell H., Lips K., Volk C. (2007) Polyspecific organic cation transporters. Structure, function, physiological roles, and biopharmaceutical implications. Pharm. Res. 24, 1227–1251 [DOI] [PubMed] [Google Scholar]
- 4. Dantzler W. H., Wright S. H., Chatsudthipong V., Brokl O. (1991) Basolateral tetraethylammonium transport in intact tubules. Specificity and trans-stimulation. Am. J. Physiol. 261, F386–F392 [DOI] [PubMed] [Google Scholar]
- 5. Montrose-Rafizadeh C., Roch-Ramel F., Schäli C. (1987) Axial heterogeneity of organic cation transport along the rabbit renal proximal tubule. Studies with brush-border membrane vesicles. Biochim. Biophys. Acta 904, 175–177 [DOI] [PubMed] [Google Scholar]
- 6. Schäli C., Schild L., Overney J., Roch-Ramel F. (1983) Secretion of tetraethylammonium by proximal tubules of rabbit kidneys. Am. J. Physiol. 245, F238–F246 [DOI] [PubMed] [Google Scholar]
- 7. Otsuka M., Matsumoto T., Morimoto R., Arioka S., Omote H., Moriyama Y. (2005) A human transporter protein that mediates the final excretion step for toxic organic cations. Proc. Natl. Acad. Sci. U.S.A. 102, 17923–17928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tsuda M., Terada T., Mizuno T., Katsura T., Shimakura J., Inui K. (2009) Targeted disruption of the multidrug and toxin extrusion 1 (mate1) gene in mice reduces renal secretion of metformin. Mol. Pharmacol. 75, 1280–1286 [DOI] [PubMed] [Google Scholar]
- 9. Masuda S., Terada T., Yonezawa A., Tanihara Y., Kishimoto K., Katsura T., Ogawa O., Inui K. (2006) Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J. Am. Soc. Nephrol. 17, 2127–2135 [DOI] [PubMed] [Google Scholar]
- 10. Komatsu T., Hiasa M., Miyaji T., Kanamoto T., Matsumoto T., Otsuka M., Moriyama Y., Omote H. (2011) Characterization of the human MATE2 proton-coupled polyspecific organic cation exporter. Int. J. Biochem. Cell Biol. 43, 913–918 [DOI] [PubMed] [Google Scholar]
- 11. Watanabe S., Tsuda M., Terada T., Katsura T., Inui K. (2010) Reduced renal clearance of a zwitterionic substrate cephalexin in MATE1-deficient mice. J. Pharmacol. Exp. Ther. 334, 651–656 [DOI] [PubMed] [Google Scholar]
- 12. Kuroda T., Tsuchiya T. (2009) Multidrug efflux transporters in the MATE family. Biochim. Biophys. Acta 1794, 763–768 [DOI] [PubMed] [Google Scholar]
- 13. He X., Szewczyk P., Karyakin A., Evin M., Hong W. X., Zhang Q., Chang G. (2010) Structure of a cation-bound multidrug and toxic compound extrusion transporter. Nature 467, 991–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Omote H., Hiasa M., Matsumoto T., Otsuka M., Moriyama Y. (2006) The MATE proteins as fundamental transporters of metabolic and xenobiotic organic cations. Trends Pharmacol. Sci. 27, 587–593 [DOI] [PubMed] [Google Scholar]
- 15. Kobara A., Hiasa M., Matsumoto T., Otsuka M., Omote H., Moriyama Y. (2008) A novel variant of mouse MATE-1 H+/organic cation antiporter with a long hydrophobic tail. Arch. Biochem. Biophys. 469, 195–199 [DOI] [PubMed] [Google Scholar]
- 16. Hiasa M., Matsumoto T., Komatsu T., Moriyama Y. (2006) Wide variety of locations for rodent MATE1, a transporter protein that mediates the final excretion step for toxic organic cations. Am. J. Physiol. Cell Physiol 291, C678–C686 [DOI] [PubMed] [Google Scholar]
- 17. Zhang X., Wright S. H. (2009) MATE1 has an external COOH terminus, consistent with a 13-helix topology. Am. J. Physiol. Renal Physiol 297, F263–F271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koepsell H. (2011) Substrate recognition and translocation by polyspecific organic cation transporters. Biol. Chem. 392, 95–101 [DOI] [PubMed] [Google Scholar]
- 19. Hvorup R. N., Winnen B., Chang A. B., Jiang Y., Zhou X. F., Saier M. H., Jr. (2003) The multidrug/oligosaccharidyl-lipid/polysaccharide (MOP) exporter superfamily. Eur. J. Biochem. 270, 799–813 [DOI] [PubMed] [Google Scholar]
- 20. Saier M. H., Jr., Paulsen I. T. (2001) Phylogeny of multidrug transporters. Semin. Cell Dev. Biol. 12, 205–213 [DOI] [PubMed] [Google Scholar]
- 21. Zhang X., Cherrington N. J., Wright S. H. (2007) Molecular identification and functional characterization of rabbit MATE1 and MATE2-K. Am. J. Physiol. Renal Physiol 293, F360–F370 [DOI] [PubMed] [Google Scholar]
- 22. Chen Y., Zhang S., Sorani M., Giacomini K. M. (2007) Transport of paraquat by human organic cation transporters and multidrug and toxic compound extrusion family. J Pharmacol. Exp. Ther. 322, 695–700 [DOI] [PubMed] [Google Scholar]
- 23. Pelis R. M., Suhre W. M., Wright S. H. (2006) Functional influence of N-glycosylation in OCT2-mediated tetraethylammonium transport. Am. J. Physiol. Renal Physiol 290, F1118–F1126 [DOI] [PubMed] [Google Scholar]
- 24. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics. 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 25. Guex N., Peitsch M. C. (1997) SWISS-MODEL and the Swiss-PdbViewer. An environment for comparative protein modeling. Electrophoresis 18, 2714–2723 [DOI] [PubMed] [Google Scholar]
- 26. Brunger A. T. (2007) Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2, 2728–2733 [DOI] [PubMed] [Google Scholar]
- 27. Humphrey W., Dalke A., Schulten K. (1996) VMD. Visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 28. Bas D. C., Rogers D. M., Jensen J. H. (2008) Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins 73, 765–783 [DOI] [PubMed] [Google Scholar]
- 29. Li H., Robertson A. D., Jensen J. H. (2005) Very fast empirical prediction and rationalization of protein pKa values. Proteins 61, 704–721 [DOI] [PubMed] [Google Scholar]
- 30. Jo S., Kim T., Im W. (2007) Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS. ONE. 2, e880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mackerell A. D., Jr., Feig M., Brooks C. L., 3rd (2004) Extending the treatment of backbone energetics in protein force fields. Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 25, 1400–1415 [DOI] [PubMed] [Google Scholar]
- 33. Darden T., York D., Pedersen L. (1993) An N-log(N) method for Ewald sums in large systems. J. Chem. Physics 98, 10089–10092 [Google Scholar]
- 34. Martyna G. J., Tobias D. J., Klein M. L. (1994) Constant pressure molecular dynamics algorithms. J. Chem. Physics 101, 4177–4189 [Google Scholar]
- 35. Feller S. E., Zhang Y., Pastor R. W., Brooks B. R. (1995) Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Physics 103, 4613–4621 [Google Scholar]
- 36. Frishman D., Argos P. (1995) Knowledge-based protein secondary structure assignment. Proteins 23, 566–579 [DOI] [PubMed] [Google Scholar]
- 37. Morita Y., Kataoka A., Shiota S., Mizushima T., Tsuchiya T. (2000) NorM of Vibrio parahaemolyticus is an Na+-driven multidrug efflux pump. J. Bacteriol. 182, 6694–6697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Malo C., Berteloot A. (1991) Analysis of kinetic data in transport studies. New insights from kinetic studies of Na+-d-glucose cotransport in human intestinal brush-border membrane vesicles using a fast sampling, rapid filtration apparatus. J. Membr. Biol. 122, 127–141 [DOI] [PubMed] [Google Scholar]
- 39. Dangprapai Y., Wright S. H. (2011) Interaction of H+ with the extracellular and intracellular aspects of hMATE1. Am. J. Physiol. Renal Physiol 301, F520–F528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wright S. H., Wunz T. M. (1988) Mechanism of cis- and trans-substrate interactions at the tetraethylammonium/H+ exchanger of rabbit renal brush-border membrane vesicles. J. Biol. Chem. 263, 19494–19497 [PubMed] [Google Scholar]
- 41. Tanihara Y., Masuda S., Sato T., Katsura T., Ogawa O., Inui K. (2007) Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H+-organic cation antiporters. Biochem. Pharmacol. 74, 359–371 [DOI] [PubMed] [Google Scholar]
- 42. Vardy E., Arkin I. T., Gottschalk K. E., Kaback H. R., Schuldiner S. (2004) Structural conservation in the major facilitator superfamily as revealed by comparative modeling. Protein Sci. 13, 1832–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Morth J. P., Pedersen B. P., Toustrup-Jensen M. S., Sørensen T. L., Petersen J., Andersen J. P., Vilsen B., Nissen P. (2007) Crystal structure of the sodium-potassium pump. Nature 450, 1043–1049 [DOI] [PubMed] [Google Scholar]
- 44. Zhu Q., Casey J. R. (2007) Topology of transmembrane proteins by scanning cysteine accessibility mutagenesis methodology. Methods 41, 439–450 [DOI] [PubMed] [Google Scholar]
- 45. Law R. J., Capener C., Baaden M., Bond P. J., Campbell J., Patargias G., Arinaminpathy Y., Sansom M. S. (2005) Membrane protein structure quality in molecular dynamics simulation. J. Mol. Graph. Model. 24, 157–165 [DOI] [PubMed] [Google Scholar]
- 46. Holyoake J., Caulfeild V., Baldwin S. A., Sansom M. S. (2006) Modeling, docking, and simulation of the major facilitator superfamily. Biophys. J. 91, L84-L86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Smart O. S., Goodfellow J. M., Wallace B. A. (1993) The pore dimensions of gramicidin A. Biophys. J. 65, 2455–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Matsumoto T., Kanamoto T., Otsuka M., Omote H., Moriyama Y. (2008) Role of glutamate residues in substrate recognition by human MATE1 polyspecific H+/organic cation exporter. Am. J. Physiol. Cell Physiol 294, C1074–C1078 [DOI] [PubMed] [Google Scholar]
- 49. Wright S. H., Wunz T. M. (1999) Influence of substrate structure on substrate binding to the renal organic cation/H+ exchanger. Pfluegers Arch. Eur. J. Physiol. 437, 603–610 [DOI] [PubMed] [Google Scholar]
- 50. Wright S. H., Wunz T. M., Wunz T. P. (1995) Structure and interaction of inhibitors with the TEA/H+ exchanger of rabbit renal brush border membranes. Pfluegers Arch. Eur. J. Physiol. 429, 313–324 [DOI] [PubMed] [Google Scholar]
- 51. Ullrich K. J., Rumrich G. (1996) Luminal transport system for choline+ in relation to the other organic cation transport systems in the rat proximal tubule. Kinetics, specificity. Alkyl/arylamines, alkylamines with OH, O, SH, NH2, ROCO, RSCO and H2PO4-groups, methylaminostyryl, rhodamine, acridine, phenanthrene and cyanine compounds. Pfluegers Arch. Eur. J. Physiol. 432, 471–485 [DOI] [PubMed] [Google Scholar]
- 52. David C., Rumrich G., Ullrich K. J. (1995) Luminal transport system for H+/organic cations in the rat proximal tubule. Kinetics, dependence on pH. Specificity as compared with the contraluminal organic cation-transport system. Pfluegers Arch. Eur. J. Physiol. 430, 477–492 [DOI] [PubMed] [Google Scholar]
- 53. Hori R., Maegawa H., Kato M., Katsura T., Inui K. (1989) Inhibitory effect of diethyl pyrocarbonate on the H+/organic cation antiport system in rat renal brush-border membranes. J. Biol. Chem. 264, 12232–12237 [PubMed] [Google Scholar]
- 54. Asaka J., Terada T., Tsuda M., Katsura T., Inui K. (2007) Identification of essential histidine and cysteine residues of the H+/organic cation antiporter multidrug and toxin extrusion (MATE). Mol. Pharmacol. 71, 1487–1493 [DOI] [PubMed] [Google Scholar]
- 55. Lee Y. H., Li L., Lasalde J., Rojas L., McNamee M., Ortiz-Miranda S. I., Pappone P. (1994) Mutations in the M4 domain of Torpedo californica acetylcholine receptor dramatically alter ion channel function. Biophys. J 66, 646–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Meijer D. K., Mol W. E., Müller M., Kurz G. (1990) Carrier-mediated transport in the hepatic distribution and elimination of drugs, with special reference to the category of organic cations. J. Pharmacokin. Biopharmaceut. 18, 35–70 [DOI] [PubMed] [Google Scholar]
- 57. Tandon S. K., Singh S., Jain V. K., Prasad S. (1996) Chelation in metal intoxication. XXXVIII: Effect of structurally different chelating agents in treatment of nickel intoxication in rat. Fundam. Appl. Toxicol. 31, 141–148 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.