

Background: HMGN1 affects the interaction of DNA repair factors with chromatin.
Results: We identified a functional interaction between HMGN1 and PARP-1 in mouse fibroblast cells.
Conclusion: HMGN1 participates in the regulation of PARP-1 catalytic activity in response to low-level genotoxic stress.
Significance: These results reveal a novel partnership between HMGN1 and PARP-1 in the response to endogenous and MMS-induced genotoxic stress.
Keywords: Base Excision Repair, DNA, DNA Repair, Fibroblast, Mouse, HMGN1, Poly(ADP-ribose), Polymerase
Abstract
In mammalian cells, the nucleosome-binding protein HMGN1 (high mobility group N1) affects the structure and function of chromatin and plays a role in repair of damaged DNA. HMGN1 affects the interaction of DNA repair factors with chromatin and their access to damaged DNA; however, not all of the repair factors affected have been identified. Here, we report that HMGN1 affects the self-poly(ADP-ribosyl)ation (i.e., PARylation) of poly(ADP-ribose) polymerase-1 (PARP-1), a multifunctional and abundant nuclear enzyme known to recognize DNA lesions and promote chromatin remodeling, DNA repair, and other nucleic acid transactions. The catalytic activity of PARP-1 is activated by DNA with a strand break, and this results in self-PARylation and PARylation of other chromatin proteins. Using cells obtained from Hmgn1−/− and Hmgn1+/+ littermate mice, we find that in untreated cells, loss of HMGN1 protein reduces PARP-1 self-PARylation. A similar result was obtained after MMS treatment of these cells. In imaging experiments after low energy laser-induced DNA damage, less PARylation at lesion sites was observed in Hmgn1−/− than in Hmgn1+/+ cells. The HMGN1 regulation of PARP-1 activity could be mediated by direct protein-protein interaction as HMGN1 and PARP-1 were found to interact in binding assays. Purified HMGN1 was able to stimulate self-PARylation of purified PARP-1, and in experiments with cell extracts, self-PARylation was greater in Hmgn1+/+ than in Hmgn1−/− extract. The results suggest a regulatory role for HMGN1 in PARP-1 activation.
Introduction
The nuclear DNA of eukaryotic cells is tightly packaged within chromatin (1–4). The fundamental structure of chromatin is the nucleosome core particle, a 147-bp segment of DNA wrapped around an octamer of two copies each of four histones: H2A, H2B, H3, and H4. In addition, the linker histone H1 binds to nucleosome core particles near the nucleosomal dyad axis and interacts with the linker DNA between two neighboring nucleosomes, thereby promoting and stabilizing chromatin compaction. The higher-order folding of DNA within chromosomal structures allows for its protection against unintended enzymatic activities, and cells maintain elaborate programs for making DNA accessible to required processes such as transcription, replication, recombination, and DNA repair. The programmed alteration of chromatin structure is regulated by a combination of events, including posttranslational modifications and by chromatin remodeling enzymes (5–11).
High-mobility group (HMG) N is one of the three HMG families of chromosomal proteins, HMGA, HMGB and HMGN. The HMGN family is found in vertebrates and consists of five members: HMGN1 (HMG14), HMGN2 (HMG17), HMGN3 (HMGN3a and HMGN3b), HMGN4, and HMGN5 (NSBP1 or NBP-45) (7, 9, 12). These proteins are small (∼10 kDa) and are highly conserved in a nucleosome binding domain and a negatively charged C-terminal domain. Among the HMGN proteins, HMGN1 and HMGN2 are expressed abundantly, although the expression levels depend on developmental stages and tissue types (13–15). HMGN1 and HMGN2 have overlapping functions in vitro but are suggested to have distinct roles in vivo (16). It has been estimated that in most vertebrate cells, the abundance of HMGN1 and HMGN2 is significantly lower than that of histones; most cells contain sufficient HMGN protein to bind ∼1–5% of the nucleosomes (12).
HMGN proteins bind to the nucleosome core particle and form homodimeric complexes. They counteract the binding of linker histone H1, thus reducing chromatin compaction. Binding of HMGN proteins to nucleosomes also alters the level of histone modification, again influencing chromatin compaction. There are conflicting reports concerning a possible role of HMGN proteins in the ATP-dependent chromatin remodeling processes. One study failed to observe an effect of HMGN proteins on SWI/SNF-dependent nucleosome remodeling (17), whereas another showed that HMGN proteins suppress chromatin remodeling by the factors ACF (ATP-utilizing chromatin assembly and remodeling factor) and BRG1 (brahma-related gene 1) (18).
The biological functions of HMGN1 have been examined using Hmgn1−/− mice and cell lines. Hmgn1−/− mice are viable and appear normal except they show minor developmental abnormalities. These mice and cell lines, however, are hypersensitive to treatment with UV and ionizing radiation (19, 20). The phenotype of the HMGN1 deficiency might be through alteration in the access of DNA lesions to the DNA repair machinery or by altering histone modifications such as acetylation of H3K14 (21, 22). There are no reports, however, of effects of HMGN1 on the base excision DNA repair pathway.
Base excision repair (BER)2 is considered the main DNA repair pathway for removal of base lesions and single-strand breaks from DNA (23–27). Base lesion BER is initiated by DNA glycosylase removal of the damaged base generating the apurinic/apyrimidinic (AP) site (28, 29). AP endonuclease cleavage of the AP site follows, and the 5′-deoxyribose phosphate group at the margin of the one-nucleotide gap is removed by DNA polymerase β (pol β). The resulting single-nucleotide gap is filled by pol β, and a DNA ligase seals this intermediate to complete the pathway. It is known that this repair pathway involves multiple accessory factors and modifying enzymes. Among them, poly(ADP-ribose) polymerase 1 (PARP-1) is an abundant nuclear enzyme that also has other activities such as chromatin structure modulation, transcriptional co-regulation, and genomic insulator function (30–32). PARP-1 has high affinity for DNA strand breaks and AP sites, including the intermediates generated during BER (33–35). Following PARP-1 binding at such BER intermediates, PARP-1 catalyzes the polymerization of ADP-ribose (PAR) using NAD+. The process results in PAR adduction of PARP-1 itself with linear and branched ADP-ribose polymers (i.e. PARylation). PARylation of PARP-1 enables recruitment of other BER proteins, such as XRCC1 (x-ray cross-complementing protein-1) (36, 37), pol β (35), and DNA ligases I and III (38), to the strand break-containing BER intermediate. PARP-1 is proposed to dissociate from the damaged site following its PARylation. In addition to a role in BER, PARylation of PARP-1 plays various roles in other cellular processes, including chromatin modification, transcription, and cell death pathways.
Here, we investigated the effect of HMGN1 deletion on BER and the possibility of a functional relationship between HMGN1 and PARP-1. We compared the self-PARylation level of PARP-1 in Hmgn1+/+ and Hmgn1−/− cells and found that it was substantially lower in the HMGN1 null cells. This decrease in self-PARylation of PARP-1 may be due to limited access to DNA lesions in the absence of HMGN1 but also could be due to a direct effect of HMGN1 on PARP-1 activity. Purified HMGN1 stimulated PARP-1 activity, and a protein-protein association between these proteins was observed. The finding of an interaction between HMGN1 and PARP1 is discussed.
EXPERIMENTAL PROCEDURES
Cell Lines and Culture
The mouse embryonic fibroblast cell lines, Hmgn1+/+ and Hmgn1−/−, used in this study were prepared and grown as described previously (20). They were maintained in Dulbecco's modified Eagle's medium (DMEM) (HyClone, Logan, UT) containing 10% fetal bovine serum (FBS) (HyClone), 4 mm GlutaMAX-1 (Invitrogen), and 25 mm HEPES buffer (Invitrogen) in a 5% CO2 incubator at 34 °C. Mycoplasma testing was performed using a MycoAlert® Mycoplasma detection kit (Lonza Group Ltd., Rockland, ME). All cells were found to be free of mycoplasma contamination.
Assay for Self-PARylation of PARP-1 and Detection of Other Proteins by Immunoblotting
Cells (2 × 105) were seeded in each well of six-well plate 24 h before treatment. The cells were then washed with Hanks' balanced salt solution (HyClone), and the medium was replaced with or without the PARP-1 inhibitor 10 μm 4-amino-1,8-naphthalimide (4-AN) (Sigma-Aldrich). After 30 min of incubation at 34 °C, cells were washed with ice-cold PBS and lysed in 100 μl of radio immunoprecipitation assay buffer (Thermo Scientific, Rockford, IL) containing protease inhibitor mixture (Roche Molecular Biochemicals, Indianapolis, IN) on ice for 15 min. Extracts were centrifuged at 14,000 rpm for 15 min at 4 °C, and the supernatant fraction was collected. For the MMS treatment, cells were incubated with medium containing 1 mm MMS (Sigma-Aldrich) with or without 4-AN (10 μm) for 30 min at 34 °C. After the incubation, cells were washed, and medium was replaced with or without 4-AN and incubated for 0 or 30 min at the same temperature. Cell extract was prepared by washing cells with PBS, followed by cell lysis. Immunoblotting analysis was performed as described previously (33). SDS-PAGE sample buffer was added to the cell extract, and it was heated for 5 min at 95 °C. Cell extract (5 μl) was loaded onto a NuPAGE 4–12% Bis-Tris gel (Invitrogen), separated by electrophoresis, and transferred to a nitrocellulose membrane. The membrane was blocked with 5% nonfat dry milk in Tris-buffered saline containing 0.5% (v/v) Tween 20. The membrane was probed either with anti-PAR (Trevigen, Gaithersburg, MD), anti-PARP-1 (BD Pharmingen, San Diego, CA), anti-HMGN1 (Abcam, Cambridge, MA), anti-pol β (18 S) (39), anti-XRCC1 (Thermo Scientific), anti-DNA ligase I (GeneTex, Irvine, CA), or anti-α-tubulin (Sigma-Aldrich) antibodies. Goat anti-mouse IgG or goat anti-rabbit IgG (H+L)-horseradish peroxidase (HRP) conjugate (Bio-Rad Laboratories) was used as a secondary antibody, and the HRP activity was detected by enhanced chemiluminescence using SuperSignal West Pico Chemiluminescent substrate (Pierce Biotechnology, Inc., Rockford, IL). Antibodies were stripped by incubation with Restore Western blot stripping buffer (Pierce Biotechnology, Inc.) and before reprobing with another antibody.
Fluorometric Analysis of Strand Breaks by DNA Unwinding (FADU)
DNA strand breaks in the Hmgn1+/+ and Hmgn1−/− cells were measured by the FADU method (40, 41). Cells were suspended in calcium- and magnesium-free PBS at a concentration of 2 × 105 cells/ml. 25 μl of this cell suspension was transferred to a well in a black 96-well plate (Thermo Scientific). An equal volume of lysis buffer (9 m urea, 0.1% SDS, 0.2 m EDTA, adjusted to pH 10 with NaOH) was added to the sample without mixing, and the sample was incubated for 20 min in the dark at room temperature. After the incubation, 25 μl of alkaline buffer (45% lysis buffer in 0.2 m NaOH) was added slowly on the top of the cell lysate so as to form a layer while avoiding any mixing. After 30 min of incubation at room temperature, samples were neutralized by addition of 50 μl of neutralization solution (14 mm β-mercaptoethanol, 1 m glucose). Next, 75 μl of 1:200 diluted PicoGreen (Invitrogen) fluorescent dye solution was added to the sample, and the solution was mixed.
The fluorescence value of the sample reflected the double-stranded DNA remaining after alkali treatment, and this value was designated as P. For a positive control, total double-stranded DNA (T) was prepared with the same treatment as the P sample, but the neutralization buffer was added before the alkaline buffer. The background value (B) was obtained from the cell lysate sample that was sonicated under alkaline conditions. The fluorescence of samples, P, T, and B, was measured by Microplate Reader SynergyTM 4 (BioTek, Winooski, VT) at excitation485 nm and emission528 nm. DNA strand breaks (F) were calculated using the expression F = (P − B)/(T − B).
Assays for Self-PARylation of PARP-1 in Vitro
The in vitro PARylation described in Fig. 2 was performed as described previously (42). Briefly, Hmgn1+/+ and Hmgn1−/− cell extracts (2 mg of protein) were prepared in a lysis buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 25 mm NaF, 0.1 mm sodium orthovanadate, 0.2% Triton X-100, 0.3% Nonidet P-40) containing protease inhibitors. The extracts were immunoprecipitated either with preimmune IgG or with anti-PARP-1 antibody as described below. The immunoprecipitated pellets were incubated in a PARylation reaction mixture (final volume 50 μl) that contained 50 mm Tris-HCl, pH 7.8, 25 mm MgCl2, 1 mm DTT, 80 μg/ml nicked calf thymus DNA, and 100 μm NAD+ along with protease inhibitor mixture. The PARylation incubation was at 37 °C for 30 min and was terminated by addition of SDS-PAGE sample buffer. The sample was separated by electrophoresis and transferred onto a nitrocellulose membrane. The membrane was probed with anti-PAR antibody and then reprobed with anti-PARP-1 antibody. Note that before reprobing with another antibody, antibodies from the membrane were stripped with Restore Western blot stripping buffer.
FIGURE 2.

Self-PARylation of PARP-1 in vitro, after its immunoprecipitation. Experiments were conducted as described under “Experimental Procedures.” HMGN1 cell extracts as indicated were prepared, PARP-1 was immunoprecipitated (IP) and then incubated in PARylation reaction mixtures. After the in vitro incubation, as outlined in the figure, reaction mixtures were subjected to SDS-PAGE and immunoblotting (IB) with anti-PAR or anti-PARP1 antibody. Lane 1, IP was with preimmune IgG; lane 10, the Hmgn1+/+ cell extract applied directly to the gel as a marker for PARP-1. Lanes 4 and 5 and 8 and 9, respectively, represent duplicate samples. The PARylation substrate NAD+ and the DNA activator were included in the reaction mixture incubation as shown.
The in vitro PARylation reactions described in Figs. 3 and 4 were performed essentially as described previously (43). Briefly, the reaction mixture (15 μl) containing 50 mm HEPES-KOH, pH 7.5, 0.5 mm EDTA, 20 mm KCl, 2 mm DTT, 5 mm MgCl2, 100 nm double-hairpin DNA, and 100 μm [32P]NAD+ was assembled on ice. The PARylation reaction was then initiated by addition of 7.5 μg of extract prepared from either Hmgn1+/+ or Hmgn1−/− cells. The reaction mixtures were incubated at 37 °C, and aliquots (4.5-μl each) were withdrawn at the indicated times. Reactions were terminated by the addition of 10 μl SDS-PAGE sample buffer and heating for 5 min at 95 °C. The reaction mixtures were analyzed by 4–12% SDS-PAGE with subsequent phosphorimaging. Note that in some cases, the reaction mixtures were supplemented with purified HMGN1. PARylation reactions under similar conditions were also performed with purified PARP-1 and HMGN1 (Fig. 4B).
FIGURE 3.

Alternative assay for PARP-1 self-PARylation capacity of extracts from the HMGN1 cell lines. Experiments were conducted as described under “Experimental Procedures.” A, self-PARylation of PARP-1 was measured after incubation (2, 5, or 10 min) with cell extracts (7.5-μg each) in the presence of 32P-labeled substrate, [32P]NAD+. Reaction mixtures were analyzed by SDS-PAGE, and an autoradiogram of the gel is shown. B, quantification of 32P-labeled PAR formed in the experiment on the left at 5 min. Relative amount of extract-based PARP-1 PARylation. The data shown are averages from three experiments ± S.E., including the experiment shown in A. Student's t test (paired data) indicated p = 0.0041 for the difference shown.
FIGURE 4.

Stimulation of the PARP-1 self-PARylation by purified HMGN1. A, self-PARylation of PARP-1 measured after incubation (2 or 5 min) with cell extracts (7.5-μg each) in the presence of [32P]NAD+ substrate. Lanes 1 and 2, Hmgn1+/+ cell extract. Lanes 3 and 4, Hmgn1−/− cell extract. Lanes 5 and 6, Hmgn1−/− cell extract with the reaction mixture supplemented with purified HMGN1 (2 μm). 32P-Labeled PAR was quantified, and the relative amount of PARP-1 PARylation at 5 min is shown on the right. B, self-PARylation of purified PARP-1 in the presence and absence of with purified HMGN1. Lanes 1–3, PARP-1 incubated with 0, 0.25 and 0.5 μm HMGN1, respectively. 32P-Labeled PAR was quantified, and the relative amount of PARP-1 PARylation at 0 and 0.25 μm HMGN1 is shown on the right.
Cytotoxicity Assay
Cytotoxicity was determined by growth inhibition assays as described previously (44). Hmgn1+/+ and Hmgn1−/− cells were seeded at a density of 20,000 cells/well in six-well plates 24 h before treatment. The following day, they were exposed for 1 h to a range of concentration of MMS in growth medium. Cells were washed with Hanks' balanced salt solution, and fresh medium was replaced. They were incubated for 5 days in a 5% CO2 incubator at 34 °C until untreated control cells were ∼80% confluent. Cells (triplicate wells for each drug concentration) were counted by a cell lysis procedure (45), and results were expressed as the number of cells in drug-treated wells relative to untreated control cells (% control growth).
Analysis of PARP-1 in Chromatin Fraction of HMGN1 Cells
The chromatin-associated nuclear proteins were isolated as described previously (46). Briefly, 1 × 107 cells were treated or not with MMS (1 mm) for 30 min at 34 °C. The cells were then harvested, washed with cold PBS, and resuspended in 250 μl hypotonic buffer A (10 mm HEPES, pH 7.9, 10 mm KCl, 1.5 mm MgCl2, 0.34 m sucrose, 10% glycerol, 1 mm DTT) with protease inhibitor mixture. Triton X-100 was added to 0.1% final concentration, and the mixture was incubated for 5 min on ice. The nuclei were separated by centrifugation at 1300 × g for 4 min at 4 °C, and the nuclear pellet fraction was lysed by suspension in “low-stringency solution” (3 mm EDTA, 0.2 mm EGTA, 1 mm DTT) for 10 min at 4 °C. After centrifugation at 1700 × g for 4 min, the pellet fraction was suspended in 250 μl of radioimmune precipitation assay buffer. Chromatin-associated proteins were obtained by incubation for 30 min on ice. After centrifugation at 16,000 × g for 10 min at 4 °C, equal amounts of the supernatant fraction were loaded and subjected to SDS-PAGE. Proteins were transferred to a membrane, and chromatin-associated PARP-1 was analyzed using anti-PARP-1 antibody as described above.
Immunofluorescence
Hmgn1+/+ or Hmgn1−/− cells (1 × 105) were seeded on to 35-mm glass-bottomed Petri dishes (MatTek, Ashland, MA) and incubated in cell culture medium containing 10 μm BrdU (Sigma-Aldrich) for 24 h. After 24 h, medium was exchanged to complete medium, and BrdU was removed. Samples were then imaged using a 40× C-Apochromat (numerical aperture, 1.2) water immersion objective coupled to a Zeiss LSM510 META confocal microscope (Carl Zeiss MicroImaging). Strand breaks were introduced by UV laser micro-irradiation at 364 nm (Coherent Enterprise II) with intensities equivalent to 0.176 μJ. After microirradiation, cells were either immediately fixed in 4% paraformaldehyde or allowed to recover in a 37 °C incubator for the times noted. After fixation, cells were permeabilized with 0.25% Triton X-100 in PBS for 10 min, washed three times in PBS, and then further permeabilized and blocked with PBS + 1% BSA for 30 min. Cells were then incubated with a 1:50 dilution of anti-XRCC1 antibody (AB1838, Abcam) and 1:100 dilution of anti-PADPR antibody (AB14460, Abcam) for 1 h. Cells were washed three times with PBS and then incubated in 1:2000 dilution of Alexa Fluor 488-conjugated anti-mouse and Alexa Fluor 647 conjugated anti-chicken (Invitrogen) for 1 h. Finally, cells were 4′,6-diamidino-2-phenylindole (DAPI)-stained, and fluorescence images were acquired with the 40× water immersion objective on the LSM510. Recruitment of XRCC1 or PAR to sites of DNA damage was measured using ImageJ software. The mean intensity of the irradiation line was determined after subtraction of the background intensity in the irradiated cells. Each experiment was repeated on at least three cells, and data presented here represent mean values.
Co-immunoprecipitation of PARP1 and HMGN1
Hmgn1+/+ or Hmgn1−/− cells were cultured in 150-mm dishes until near confluent, washed two times with PBS, and harvested by scraping. Approximately 5 × 107 cells were suspended in 1× radio immunoprecipitation assay buffer with protease inhibitor mixture and incubated for 30 min on ice. The lysates were centrifuged at 14,000 rpm for 30 min at 4 °C, and the supernatant fraction was collected. The protein concentrations of the cell lysates were determined using a Bio-Rad protein assay (Bio-Rad) with bovine serum albumin as standard. For co-immunoprecipitations, anti-HMGN1 poly- clonal antibody (Abcam) was added to the cell lysate (1 mg of protein), and the mixture was incubated with rotation for 4 h at 4 °C. The immunocomplex was adsorbed onto protein A-Sepharose (GE Healthcare) and protein G-agarose (Roche Molecular Biochemicals) beads by incubating the mixture overnight at 4 °C. The beads were washed four times with lysis buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 25 mm NaF, 0.2% Triton X-100, 0.3% Nonidet P-40) containing protease inhibitors. Beads were resuspended in SDS-PAGE sample buffer, heated for 5 min at 95 °C, and then the soluble proteins were separated by NuPAGE in a 4–12% Bis-Tris gel (Invitrogen). Proteins were then transferred onto a nitrocellulose membrane and were analyzed as described above. Co-immunoprecipitation of purified PARP-1 protein (Enzo Life Sciences, Inc.) and purified HMGN1 protein (47) was performed in the binding buffer (25 mm Tris-HCl, pH 8.0, 10% glycerol, 100 mm NaCl, 0.1% Nonidet P-40) containing protease inhibitors. Anti-HMGN1 antibody (Abcam) was added to the mixture of purified proteins (1 μm each), in a final volume of 50 μl, and the mixture was incubated with rotation for 4 h at 4 °C. The protein complexes were adsorbed onto protein A-Sepharose and protein G-agarose beads by incubating the mixture overnight at 4 °C in a final volume of 500 μl of binding buffer. The beads were collected by centrifugation, washed, and analyzed as described for the cell lysate above. In control experiments, the antibody for immunoprecipitation was substituted with rabbit preimmune IgG (Sigma-Aldrich). Cell lysates (10 μg or 40 μg) and purified proteins (50 ng), without immunoprecipitation, were used as markers (labeled as “input”).
Detection of HMGN1 Binding to PARP-1 Immobilized Column Resin
N-terminal His-tagged human PARP-1 mutant (D214A) expression vector was a gift from Dr. Serge Desnoyers (Laval University, Quebec, Canada) (48). His-tagged PARP-1 was expressed in the Escherichia coli strain SG13009 (Qiagen, Valencia, CA) in Luria broth (LB) medium supplemented with 100 μg/ml ampicillin and 35 μg/ml kanamycin. Cells were grown until A595 nm = 0.75 at 37 °C with shaking (275 rpm), and then the temperature was dropped to 20 °C. After 1 h, isopropyl 1-thio-β-d-galactopyranoside (Affymetrix, Inc., Cleveland, OH) and ZnCl2 (Sigma-Aldrich) were added to a final concentration of 0.5 mm and 50 μm, respectively. The incubation was continued at 20 °C for 20 h with same shaking speed. E. coli cells were then centrifuged at 4000 rpm for 30 min at 4 °C, and the cell pellet was washed with 50 mm Tris-HCl, pH 8.0, and stored at −80 °C. As a control, E. coli without the His-tagged PARP-1 expression vector was also grown in LB medium as above. Both E. coli cell pellets (∼ 1-mg each) were resuspended in 10 ml of lysis buffer (50 mm Tris-HCl, pH 8.0, 500 mm NaCl, 10 mm imidizole, and 10% glycerol) with protease inhibitor mixture, and sonicated with a VirSonic (VirTis) sonifier using repeat duty cycle at 30-s pulse for 1 min in a dry ice ethanol bath. This sonication process was repeated, and the suspension was centrifuged at 14,000 rpm for 30 min at 4 °C. Supernatants were then incubated with 250 μl of Ni-NTA agarose beads (Qiagen, Valencia, CA) with rotation overnight at 4 °C. The Ni-NTA agarose beads were pre-equilibrated with the lysis buffer. After overnight incubation, Ni-NTA agarose beads were collected by centrifugation and washed sequentially with buffer 1 (25 mm Tris-HCl, pH 8.0, 500 mm NaCl, 10 mm imidazole) and buffer 2 (25 mm Tris-HCl, pH 8.0, 1 m NaCl, 20 mm imidazole) with protease inhibitor mixture three times each. Then, the beads were washed with a binding buffer (25 mm Tris-HCl, pH 8.0, 50 mm NaCl, 10% glycerol) containing protease inhibitors for five times. An equal volume (100 μl) of immobilized resin was incubated with purified HMGN1 (100 μg) with rotation at 4 °C. After 1 h, both resins were transferred to room temperature, and the incubation was continued for another 20 min. After this incubation, beads were collected by centrifugation at 1000 rpm for 1 min at 4 °C and washed with binding buffer five times. Beads were resuspended in SDS-PAGE sample buffer and heated for 5 min at 95 °C. Proteins bound to the beads were separated by electrophoresis and transferred onto a nitrocellulose membrane. Binding of HMGN1 to PARP-1 was determined by immunoblotting analysis as described above.
RESULTS
Characterization of Hmgn1+/+ and Hmgn1−/− Cells
To initially examine a possible correlation between HMGN1 deficiency and reduced BER capacity in mouse fibroblasts, we compared the BER capacity of Hmgn1+/+ and Hmgn1−/− cells using a plasmid-based in vivo assay, and we also compared extracts from the two cell lines using an oligonucleotide-based in vitro assay for repair of a site-specific uracil lesion (33, 49). The Hmgn1−/− cells showed comparable repair capacity as the Hmgn1+/+ cells in both assays (data not shown). To further confirm the BER status in the cells, expression levels of several BER factors were examined by immunoblotting analysis (Fig. 1A). The Hmgn1+/+ cells expressed HMGN1, and the protein was absent in Hmgn1−/− cells, as expected. The results confirmed expression of other BER factors including PARP-1, APE1, pol β, XRCC1, and DNA ligase I; the expression level of APE1 and XRCC1 appeared to be slightly higher in Hmgn1−/− cells, but the other proteins were expressed at similar levels in both cell lines (Fig. 1A). Next, levels of spontaneous DNA strand breaks in the two cell lines were compared. Cell suspensions from Hmgn1+/+ and Hmgn1−/− log-phase cells were subjected to an alkaline DNA unwinding assay for strand breaks. Slightly more strand breaks were observed in Hmgn1+/+ cells than Hmgn1−/− cells.
FIGURE 1.

Estimation of BER factor levels and measurement of PAR-adducted PARP-1 (self-PARylation) in HMGN1 cell lines in log-phase culture as a function of treatment of cells with the PARP-1 inhibitor 4-AN. A, immunoblot analysis of HMGN1 cell extracts. Antibodies against HMGN1, APE1, pol β, PARP-1, XRCC1, and DNA ligase I were used; an equal amount of cell extract was used in each lane. As a loading control, the blots were probed with anti-tubulin antibody. Lanes 1 and 3, mock-treated; lanes 2 and 4, 4-AN-treated. B, an example of immunoblotting of PAR-adducted PARP-1 (self-PARylation) and PARP-1 protein; an equal amount of extract was used in each case. PARP-1 activation in the presence of spontaneous or endogenous DNA damage was confirmed by PAR synthesis. Lanes 1 and 3, mock-treated cells; lanes 2 and 4, 4-AN treated cells. 4-AN treatment was for 30 min prior to extract preparation. C, summary of quantification of PAR-adducted PARP-1 in Hmgn1+/+ (filled bar) and Hmgn1−/− (open bar) cells in multiple experiments. The results represent averages from five independent experiments ± S.E., including the experiment shown in A. Student's t test (paired data) indicated p = 0.0001 for the difference shown.
We next evaluated the status of PARP-1 PARylation by immunoblotting of extracts from the two cell lines. Stronger PARylation of PARP-1 was observed in Hmgn1+/+ cells than in Hmgn1−/− cells (Fig. 1B, lane 1). Although not visible in the experiment shown (Fig. 1B), the Hmgn1−/− cells consistently exhibited a detectable, but modest level of PAR-adducted PARP-1 in multiple experiments, as summarized in Fig. 1C. As expected, PARylation was completely inhibited by treatment of cells with the PARP-1 inhibitor 4-AN (Fig. 1B, lane 2). Overall, these results indicated a reduced level PARylation of PARP-1 in Hmgn1−/− cells. PARP-1 activation in these cells was presumably secondary to endogenous genotoxic stress.
PARP-1 Activation Is Stimulated by HMGN1
To confirm the role of PARP-1 in PARylation and further explore an influence of HMGN1 on PARP-1 activation, we immunoprecipitated PARP-1 from cell extracts and then conducted in vitro PARylation reactions. Cell extracts from Hmgn1+/+ and Hmgn1−/− cells were prepared and immunoprecipitated with anti-PARP-1 antibody.
Note that the PAR adduction of PARP-1 observed in Fig. 1 was lost during the immunoprecipitation procedure (Fig. 2, lane 2). The immunoprecipitates were then incubated in PARylation reaction mixtures containing NAD+ and calf thymus DNA as co-factor. The reaction products were separated by SDS-PAGE and subjected to immunoblotting with anti-PAR antibody. The results in Fig. 2 showed that both extracts were capable of performing self-PARylation, and longer PAR polymers were observed. However, the level of PAR polymers was lower with the Hmgn1−/− extract than the Hmgn1+/+ extract (Fig. 2, compare lanes 4 and 5 with lanes 8 and 9). When the same membrane was immunoblotted for PARP-1 protein, comparable levels of PARP-1 were observed in both extracts (Fig. 2, bottom panel). Thus, the lower amount of self-PARylation with PARP-1 from the Hmgn1−/− cell extract appeared to be due to the absence of HMGN1.
Next, extract-based self-PARylation of PARP-1 was examined using 32P-labeled NAD+ as substrate and double-hairpin DNA with a uracil lesion as co-factor (Fig. 3A). After the incubation in an equal amount of the respective HMGN1 cell extract, self-PARylation of PARP-1 was greater in the Hmgn1+/+ cell extract than the Hmgn1−/− cell extract (Fig. 3A and B), whereas the extracts contained a similar level of PARP-1 protein (not shown).
To evaluate whether purified HMGN1 could complement the lower PARP-1 self-PARylation activity of the Hmgn1−/− cell extract, purified HMGN1 was added to the reaction mixture (Fig. 4, lanes 5 and 6). The reaction mixture with Hmgn1−/− extract supplemented with purified HMGN1 showed more PARP-1 self-PARylation, indicating that HMGN1 could stimulate extract-based activity of PARP-1 (Fig. 4A, lanes 3–6). The capacity for HMGN1 to stimulate the self-PARylation activity of PARP-1 was confirmed using reaction mixtures with the two purified proteins (Fig. 4B). Purified HMGN1 exerted a modest stimulation of PARP-1 self-PARylation.
Differential MMS Sensitivity of HMGN1 Cell Lines
The results described so far suggested that HMGN1 could influence DNA repair via its effect on PARP-1 activation. Therefore, we examined the HMGN1 cell lines for a widely used base lesion repair phenotype, protection against MMS-induced cytotoxicity. Cell survival was measured after treatment with MMS, and the sensitivity of the Hmgn1+/+ and Hmgn1−/− cells is shown in Fig. 5A. The Hmgn1−/− cells were moderately, but significantly, less sensitive to MMS than the Hmgn1+/+ cells. The MMS-resistance phenotype has previously been associated with a deficiency in initiation of base lesion repair (50); presumably, a deficiency in initiating repair leads to lower production of cytotoxic strand break-containing repair intermediates. Thus, the results summarized in Fig. 5A are consistent with a deficiency in access to MMS-induced lesions by the repair machinery. It is noted that another explanation of the results in Fig. 5A is that the HMGN1 deficiency is associated with a more efficient repair process but that possibility was not evaluated.
FIGURE 5.

Sensitivity of HMGN1 cell lines to treatment with MMS and assessment of chromatin. Experiments were conducted as described under “Experimental Procedures.” A, survival curves in the presence of MMS for Hmgn1+/+ (closed circles) and Hmgn1−/− (open circles) cells. S.D. is shown corresponding to multiple experiments. B, chromatin-associated PARP-1 in HMGN1 cells with or without MMS treatment. After the treatment, chromatin-associated protein was subjected to immunoblotting with anti-PARP-1 or anti-HMGN1 antibody. Lane 1, Hmgn1+/+ cells without MMS treatment. Lane 2, Hmgn1+/+ cells with MMS treatment. Lane 3, Hmgn1−/− cells without MMS treatment. Lane 4, Hmgn1−/− cells with MMS treatment.
We wished to know whether there was a lower level of chromatin-associated PARP-1 in the Hmgn1−/− cells, as compared with wild-type cells. We evaluated the amount of PARP-1 associated with chromatin in the two HMGN1 cell lines. Nuclei were isolated from the respective cells both mock-treated and MMS-treated, and the chromatin fraction was separated from the soluble nuclear protein fraction. The amount of PARP-1 in the chromatin fraction was then quantified by immunoblotting with anti-PARP-1 antibody (Fig. 5B). The amount of chromatin-associated PARP-1 was similar in the Hmgn1−/− cells and Hmgn1+/+ cells; significant differences were not observed (Fig. 5B, lanes 1 and 3). The MMS treatment of the cells failed to alter this picture (Fig. 5B, lanes 2 and 4). Immunoblotting of the chromatin fraction with anti-HMGN1 antibody yielded similar results with the Hmgn1+/+ cells. Thus, we conclude from these experiments that the amount of chromatin-associated PARP-1 was not affected strongly as a function of either HMGN1 expression or the MMS treatment used.
Self-PARylation after MMS Treatment in HMGN1 Cell Lines
PARP-1 self-PARylation was measured in cell extracts by immunoblotting after MMS treatment of cells (Fig. 6). As described above, self-PARylation in mock-treated cells was observed in Hmgn1+/+ cells, and this also was considerably less in Hmgn1−/− cells (Fig. 6A, lanes 1 and 4). Following 1 mm MMS treatment of Hmgn1+/+ cells, the level of self-PARylation was higher (1.7-fold) compared with mock treated cells. In these Hmgn1+/+ cells, self-PARylation increased as a function of MMS concentration (data not shown). In experiments with Hmgn1−/− cells (Fig. 6A, lanes 4–6), self-PARylation was below the detection limit. However, when results from this and multiple other experiments (not shown) were considered, the Hmgn1−/− cells exhibited a low level of self-PARylation, and this increased with MMS treatment (Fig. 6B). The presence of 4-AN in the culture medium blocked self-PARylation, confirming the identity of the PAR signal (Fig. 6A, lanes 3 and 6). The increase in PARP-1 self-PARylation with MMS treatment in Hmgn1+/+ cells is consistent with more repair and recruitment of PARP-1 to repair sites, as compared with mock treated cells.
FIGURE 6.

Measurement of PAR-adducted PARP-1 in HMGN1 cell lines in log-phase culture after MMS treatment. Experiments were conducted as described under “Experimental Procedures.” After 1 mm MMS treatment for 1 h, cells were harvested immediately. A, the PAR-adducted PARP-1 level was examined as a function of treatment of cells with the PARP-1 inhibitor 4-AN. The same amount of cell extract was added to each lane as shown, and representative immunoblots are shown. In lanes 3 and 6, 4-AN was present during the MMS treatment. As a loading control, the blots were probed with anti-tubulin antibody. B, quantification of PAR-adducted PARP-1 in Hmgn1+/+ (filled bar) and Hmgn1−/− (open bar) cells. The results represent averages from three experiments ± S.D., including the experiment shown in A. Student's t test (paired data) was calculated by comparison with Hmgn1+/+ cell without MMS treatment. *, p = 0.0290; **, p = 0.0010; ***, p = 0.0115.
Immunofluorescence Visualization of PAR in HMGN1 Cells after Laser-induced DNA Damage
We wished to apply an alternative approach for studying PARylation as a function of genotoxic stress-induced DNA damage in live cells. Laser microirradiation producing focal regions of base damage in the HMGN1 cell lines was used. By making use of anti-γH2AX antibody, we confirmed that double-strand breaks were negligible under the conditions used (data not shown). DNA lesions, corresponding to intermediates of BER, were visualized by immunofluorescence imaging using anti-XRCC1 and anti-PAR antibodies (Fig. 7). In the Hmgn1+/+ cells, XRCC1 was recruited maximally 60 s after irradiation, and the signal then decreased over 240 s (Fig. 7, A and D). The signal with anti-PAR antibody was also highest 60 s after irradiation and decreased over 240 s (Fig. 7, A and C). In Hmgn1−/− cells, the PAR signal was lower (open circles) compared with Hmgn1+/+ cells (closed circles), even though the same irradiation was applied. Similarly, XRCC1 recruitment was lower in the Hmgn1−/− cells. These results with the laser irradiation and imaging approach indicated that DNA damage-induced PARP-1 activation was lower in Hmgn1−/− cells than in Hmgn1+/+ cells and are consistent with the hypothesis that there is less initiation of repair as a function of HMGN1 deficiency.
FIGURE 7.

Fluorescence microscope images of HMGN1 cells irradiated with a scanning laser microirradiation system combined with a confocal microscope. Experiments were conducted as described under “Experimental Procedures.” Immunofluorecsence with anti-PAR and anti-XRCC1 antibodies is shown for measurement of endogenous protein and PAR. Time-dependent (15, 30, 60, 120, and 240 s) PAR signals at laser stripes, along with XRCC1 recruitment and a merger of the images (Merge). A, Hmgn1+/+ cells. B, Hmgn1−/− cells. C and D, quantification of PAR and XRCC1 signals, respectively, in Hmgn1+/+ cell (closed circles) and Hmgn1−/− (open circles); S.D. is shown corresponding to measurements of four cells at each point.
Protein-Protein Interaction between HMGN1 and PARP-1
To examine the possibility of an interaction between HMGN1 and PARP-1, we first conducted co-immunoprecipitation experiments with extracts from untreated Hmgn1−/− and Hmgn1+/+ cells. Cell extracts were prepared and subjected to a co-immunoprecipitation protocol using anti-HMGN1 antibody. PARP-1 co-immunoprecipitated with HMGN1 from the Hmgn1+/+ extract, but this was not observed with Hmgn1−/− extract (Fig. 8A, compare lanes 1 and 2). A control immunoprecipitation of the Hmgn1+/+ extract using preimmune IgG was negative, as expected (Fig. 8A, lane 3), and immunoprecipitation of HMGN1 was confirmed.
FIGURE 8.

Immunoprecipitation of PARP-1 and HMGN1. Experiments were conducted as described under “Experimental Procedures.” A, equal amounts of cell extracts were immunoprecipitated with anti-HMGN1 antibody, and the immunoprecipitates were subjected to immunoblotting (IB) with anti-PARP-1 or anti-HMGN1 antibody. Lane 1, Hmgn1+/+ cell extract; lane 2, Hmgn1−/− cell extract; lane 3, Immunoprecipitation of the Hmgn1+/+ cell extract was with preimmune IgG; lane 4, the Hmgn1+/+ cell extract applied directly to the gel as a marker for PARP-1 and HMGN1. B, purified HMGN1 and PARP-1 were used in the immunoprecipitation protocol. Lane 1, protein mixture of HMGN1 and PARP-1; lane 2, the protein mixture was immunoprecipitated with preimmune IgG; lane 3, the immunoprecipitation protocol with PARP-1 only; lane 4, the purified proteins were applied directly to the gel as a marker for PARP-1 and HMGN1.
Next, purified HMGN1 and PARP-1 proteins were used in co-immnoprecipitation analysis. The proteins were mixed and subjected to the co-immunoprecipitation protocol with anti-HMGN1 antibody (Fig. 8B). PARP-1 was co-immunoprecipitated by the anti-HMGN1 antibody (Fig. 8B, lane 1). No signal was detected in an immunoprecipitate with preimmune IgG or with PARP-1 alone (Fig. 8B, lanes 2 and 3); immunoprecipitation of HMGN1 was confirmed.
The interaction of HMGN1 and PARP-1 was confirmed using a column-based binding assay (Fig. 9). His-tagged PARP-1 was prepared and loaded onto a nickel-charged column; an identical column was prepared without PARP-1, for use as a negative control. Purified HMGN1 was loaded onto the columns, and the columns were extensively washed. HMGN1 remaining bound to the columns after washing was analyzed by SDS-PAGE. Much more HMGN1 was bound to the PARP-1 column (closed bar) than the column without PARP-1 (open bar). These results suggested a direct interaction between purified HMGN1 and PARP-1.
FIGURE 9.
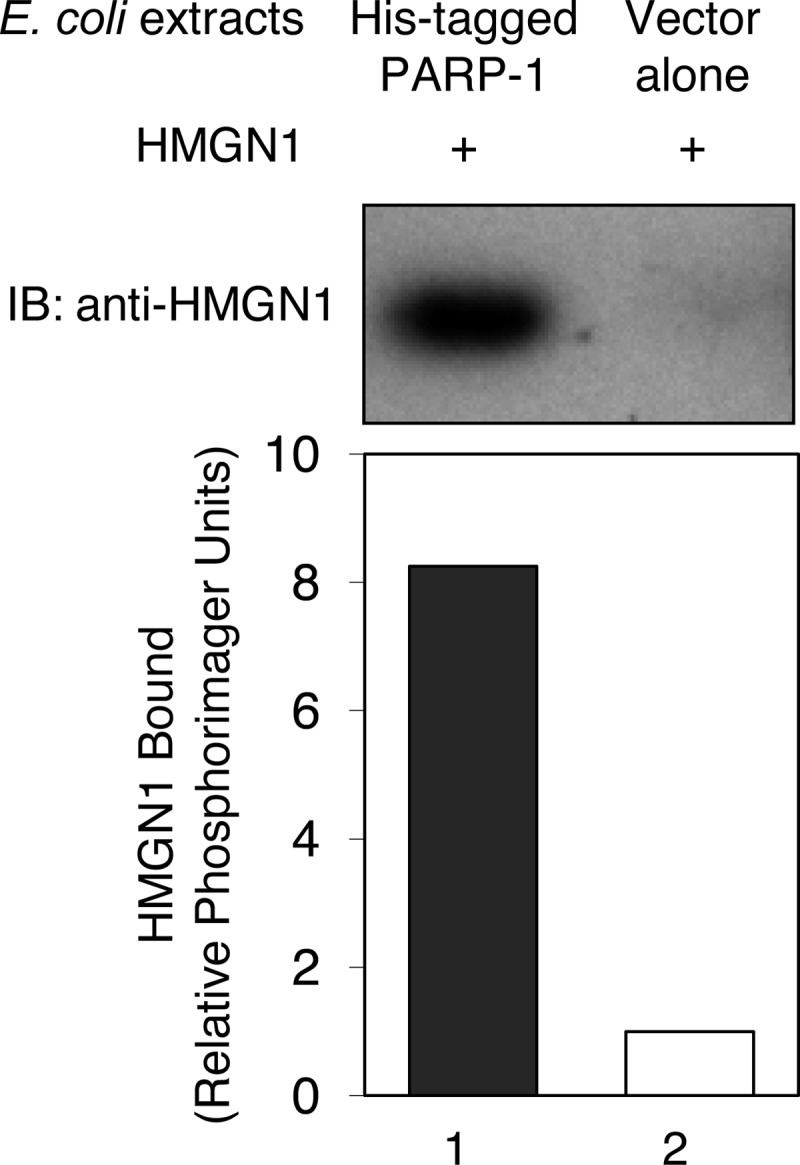
Binding of HMGN1 to PARP-1. Experiments were conducted as described under “Experimental Procedures.” Columns immobilized with E. coli crude extracts with or without expressed His-tagged PARP-1 were prepared and then incubated with purified HMGN1. Bound HMGN1 was detected by SDS-PAGE and immunoblotting (IB) using anti-HMGN1 antibody.
DISCUSSION
It is known that HMGN1 can regulate chromatin structure and various chromatin functions through its nucleosome binding. Similarly, the chromatin-associated enzyme PARP-1 regulates chromatin functions through its nucleosome binding and poly(ADP-ribose) synthesis activity here termed PARylation. Yet, functional and protein-protein interactions between these chromatin-associated factors is not well understood, although HMGN1 is known as one of the PARylation targets (51). In this study, we examined self-PARylation of PARP-1 as a function of HMGN1 expression in mouse fibroblasts in culture. Self-PARylation is considered to be a component in the catalytic activation of PARP-1, along with DNA co-factor binding and the presence of the substrate NAD+. We found PARP-1 self-PARylation to be relatively low in HMGN1-deficient cells, both for untreated log-phase cells and after treatment of cells with a dose of MMS resulting in low level toxicity. Similarly, PARylation was lower at sites of low-energy laser-induced DNA damage in Hmgn1−/− cells than in Hmgn1+/+ cells. These results suggested to us that HMGN1 functions as an activator of the self-PARylation activity of PARP-1. We obtained further evidence of HMGN1 activation of PARP-1 self-PARylation activity in experiments with cell extracts and also with purified PARP-1 and HMGN1, and these two proteins were found to interact in protein-protein interaction assays. Taken together, the results point to a functional partnership between HMGN1 and PARP-1. Self-PARylation of PARP-1, however, was not completely dependent on HMGN1 expression in cells or its presence in in vitro assays for self-PARylation activity. Instead, HMGN1 stimulated self-PARylation in both types of experiments.
The differences in self-PARylation between the Hmgn1+/+ and Hmgn1−/− cells were observed under conditions of limited DNA damage, such as MMS treatment at 4 °C (Fig. 6) or low-energy laser irradiation (Fig. 7) or conditions of endogenous damage (Fig. 1). In the case of MMS treatment, the self-PARylation level increased depending on MMS concentration in Hmgn1−/− cells (data not shown), although the level was consistently much lower than the level in Hmgn1+/+ cells. It is known that PARylation is stimulated by MMS-induced strand break-containing DNA (30). In the laser irradiation experiments with Hmgn1+/+ cells, PAR was synthesized rapidly and degraded within minutes, probably as a function of DNA repair. This cycle was different in Hmgn1−/− cells (Fig. 7C). Nevertheless, the results of these laser irradiation experiments illustrated a correlation between generation of DNA damage, initiation of repair, as revealed by XRCC1 recruitment, and coincident PAR synthesis at the irradiated site. The results support the idea of a less efficient DNA repair process in Hmgn1−/− cells than Hmgn1+/+ cells. That is, the difference in PARylation in the cell lines may reflect a difference in the speed of formation of strand break-containing repair intermediates. Overall, this is consistent with the idea that HMGN1 may regulate base lesion repair via decompaction of chromatin to expose a damaged site enabling initiation of repair.
HMGN1 promotes chromatin decompaction by binding to nucleosomes and reducing the binding of histone H1 to chromatin. Chromatin decompaction may enable PARP-1 to gain more efficient access to a damaged site, resulting in PARylation that further facilitates relaxation of chromatin structure. PARylation of histone H1 and core histones is known to induce de-compaction of chromatin, and the recruitment of repair enzymes is also regulated by PARylation (52).
It was reported that HMGN1 affects ataxia telangiectasia mutated (ATM) activation via histone modification (22). Our experiments suggest the possibility of an additional mechanism of ATM activation via PARP-1 activation by HMGN1. PARP-1 is known to interact with ATM, and PARylation of ATM affects the phosphorylation cascade (53). Less activity of PARP-1 by virtue of loss of HMGN1 could introduce a delay in phosphorylation of p53 that is primarily achieved by upstream ATM. This could result in a change in the p53 pathway, including its transcriptional regulation (54–57). Furthermore, activation of PARP-1 modulates the activity of transcription factors such as NF-kB, AP-1, and YY1 (30, 58, 59). Thus, the role of HMGN1 appears to be linked to several cellular processes in addition to the regulation of chromatin structure.
As noted above, self-PARylation of PARP-1 was not completely lacking in HMGN1 null cells. A low level was observed in untreated Hmgn1−/− cells and also with MMS treatment. This suggests that there may be HMGN1-independent pathways toward activation of PARP-1. Recently, several reports indicated PARP-1 activation by the externally regulated kinase signaling pathway, and this activation is unrelated to DNA damage or involvement of PARP-1 binding to DNA (60). Also, other HMGN family members may contribute to the self-PARylation of PARP-1. Nevertheless, there was reduced PARylation in the absence of HMGN1 in mouse fibroblasts.
Acknowledgment
We thank Bonnie Earnhardt for editorial assistance.
- BER
- base excision repair
- PARP-1
- poly(ADP-ribose) polymerase-1
- AP
- apurinic/apyrimidinic
- pol β
- polymerase β
- PAR
- polymerization of ADP-ribose
- 4-AN
- 4-amino-1,8-naphthalimide
- Ni-NTA
- nickel-nitrilotriacetic acid
- ATM
- ataxia telangiectasia mutated.
REFERENCES
- 1. Finch J. T., Lutter L. C., Rhodes D., Brown R. S., Rushton B., Levitt M., Klug A. (1977) Structure of nucleosome core particles of chromatin. Nature 269, 29–36 [DOI] [PubMed] [Google Scholar]
- 2. Kornberg R. D. (1977) Structure of chromatin. Annu. Rev. Biochem. 46, 931–954 [DOI] [PubMed] [Google Scholar]
- 3. Luger K., Mäder A. W., Richmond R. K., Sargent D. F., Richmond T. J. (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260 [DOI] [PubMed] [Google Scholar]
- 4. McGhee J. D., Felsenfeld G. (1980) Nucleosome structure. Annu. Rev. Biochem. 49, 1115–1156 [DOI] [PubMed] [Google Scholar]
- 5. Bannister A. J., Kouzarides T. (2011) Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ehrenhofer-Murray A. E. (2004) Chromatin dynamics at DNA replication, transcription, and repair. Eur. J. Biochem. 271, 2335–2349 [DOI] [PubMed] [Google Scholar]
- 7. Gerlitz G., Hock R., Ueda T., Bustin M. (2009) The dynamics of HMG protein-chromatin interactions in living cells. Biochem. Cell Biol. 87, 127–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Groth A., Rocha W., Verreault A., Almouzni G. (2007) Chromatin challenges during DNA replication and repair. Cell 128, 721–733 [DOI] [PubMed] [Google Scholar]
- 9. Reeves R. (2010) Nuclear functions of the HMG proteins. Biochim. Biophys. Acta 1799, 3–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Postnikov Y., Bustin M. (2010) Regulation of chromatin structure and function by HMGN proteins. Biochim. Biophys. Acta 1799, 62–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Luger K. (2006) Dynamic nucleosomes. Chromosome Res. 14, 5–16 [DOI] [PubMed] [Google Scholar]
- 12. Pogna E. A., Clayton A. L., Mahadevan L. C. (2010) Signaling to chromatin through post-translational modifications of HMGN. Biochim. Biophys. Acta 1799, 93–100 [DOI] [PubMed] [Google Scholar]
- 13. Crippa M. P., Nickol J. M., Bustin M. (1991) Developmental changes in the expression of high mobility group chromosomal proteins. J. Biol. Chem. 266, 2712–2714 [PubMed] [Google Scholar]
- 14. Pash J. M., Bhorjee J. S., Patterson B. M., Bustin M. (1990) Persistence of chromosomal proteins HMG-14/-17 in myotubes following differentiation-dependent reduction of HMG mRNA. J. Biol. Chem. 265, 4197–4199 [PubMed] [Google Scholar]
- 15. West K. L. (2004) HMGN proteins play roles in DNA repair and gene expression in mammalian cells. Biochem. Soc. Trans. 32, 918–919 [DOI] [PubMed] [Google Scholar]
- 16. Hock R., Furusawa T., Ueda T., Bustin M. (2007) HMG chromosomal proteins in development and disease. Trends Cell Biol. 17, 72–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hill D. A., Peterson C. L., Imbalzano A. N. (2005) Effects of HMGN1 on chromatin structure and SWI/SNF-mediated chromatin remodeling. J. Biol. Chem. 280, 41777–41783 [DOI] [PubMed] [Google Scholar]
- 18. Rattner B. P., Yusufzai T., Kadonaga J. T. (2009) HMGN proteins act in opposition to ATP-dependent chromatin remodeling factors to restrict nucleosome mobility. Mol. Cell 34, 620–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Birger Y., Catez F., Furusawa T., Lim J. H., Prymakowska-Bosak M., West K. L., Postnikov Y. V., Haines D. C., Bustin M. (2005) Increased tumorigenicity and sensitivity to ionizing radiation upon loss of chromosomal protein HMGN1. Cancer Res. 65, 6711–6718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Birger Y., West K. L., Postnikov Y. V., Lim J. H., Furusawa T., Wagner J. P., Laufer C. S., Kraemer K. H., Bustin M. (2003) Chromosomal protein HMGN1 enhances the rate of DNA repair in chromatin. EMBO J. 22, 1665–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lim J. H., Catez F., Birger Y., West K. L., Prymakowska-Bosak M., Postnikov Y. V., Bustin M. (2004) Chromosomal protein HMGN1 modulates histone H3 phosphorylation. Mol. Cell 15, 573–584 [DOI] [PubMed] [Google Scholar]
- 22. Kim Y. C., Gerlitz G., Furusawa T., Catez F., Nussenzweig A., Oh K. S., Kraemer K. H., Shiloh Y., Bustin M. (2009) Activation of ATM depends on chromatin interactions occurring before induction of DNA damage. Nat. Cell Biol. 11, 92–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Asagoshi K., Wilson S. H. (2011) Base-excision repair: Role of DNA polymerase β in late-stage base excision repair in Chemical Carcinogenesis (Penning T. M., ed.) pp. 297–319, Springer, New York [Google Scholar]
- 24. Wilson S. H., Beard W. A., Shock D. D., Batra V. K., Cavanaugh N. A., Prasad R., Hou E. W., Liu Y., Asagoshi K., Horton J. K., Stefanick D. F., Kedar P. S., Carrozza M. J., Masaoka A., Heacock M. L. (2010) Base excision repair and design of small molecule inhibitors of human DNA polymerase β. Cell Mol. Life Sci. 67, 3633–3647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lindahl T., Wood R. D. (1999) Quality control by DNA repair. Science 286, 1897–1905 [DOI] [PubMed] [Google Scholar]
- 26. Krokan H. E., Nilsen H., Skorpen F., Otterlei M., Slupphaug G. (2000) Base excision repair of DNA in mammalian cells. FEBS Lett. 476, 73–77 [DOI] [PubMed] [Google Scholar]
- 27. Izumi T., Wiederhold L. R., Roy G., Roy R., Jaiswal A., Bhakat K. K., Mitra S., Hazra T. K. (2003) Mammalian DNA base excision repair proteins: Their interactions and role in repair of oxidative DNA damage. Toxicology 193, 43–65 [DOI] [PubMed] [Google Scholar]
- 28. Masaoka A., Terato H., Kobayashi M., Honsho A., Ohyama Y., Ide H. (1999) Enzymatic repair of 5-formyluracil. I. Excision of 5-formyluracil site-specifically incorporated into oligonucleotide substrates by alka protein (Escherichia coli 3-methyladenine DNA glycosylase II). J. Biol. Chem. 274, 25136–25143 [DOI] [PubMed] [Google Scholar]
- 29. Terato H., Masaoka A., Asagoshi K., Honsho A., Ohyama Y., Suzuki T., Yamada M., Makino K., Yamamoto K., Ide H. (2002) Novel repair activities of AlkA (3-methyladenine DNA glycosylase II) and endonuclease VIII for xanthine and oxanine, guanine lesions induced by nitric oxide and nitrous acid. Nucleic Acids Res. 30, 4975–4984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. D'Amours D., Desnoyers S., D'Silva I., Poirier G. G. (1999) Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 342, 249–268 [PMC free article] [PubMed] [Google Scholar]
- 31. Krishnakumar R., Kraus W. L. (2010) The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 39, 8–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Oei S. L., Keil C., Ziegler M. (2005) Poly(ADP-ribosylation) and genomic stability. Biochem. Cell Biol. 83, 263–269 [DOI] [PubMed] [Google Scholar]
- 33. Masaoka A., Horton J. K., Beard W. A., Wilson S. H. (2009) DNA polymerase β and PARP activities in base excision repair in living cells. DNA Repair 8, 1290–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sukhanova M. V., Khodyreva S. N., Lebedeva N. A., Prasad R., Wilson S. H., Lavrik O. I. (2005) Human base excision repair enzymes apurinic/apyrimidinic endonuclease1 (APE1), DNA polymerase β, and poly(ADP-ribose) polymerase 1: Interplay between strand-displacement DNA synthesis and proofreading exonuclease activity. Nucleic Acids Res. 33, 1222–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dantzer F., de La Rubia G., Ménissier-De Murcia J., Hostomsky Z., de Murcia G., Schreiber V. (2000) Base excision repair is impaired in mammalian cells lacking poly(ADP-ribose) polymerase-1. Biochemistry 39, 7559–7569 [DOI] [PubMed] [Google Scholar]
- 36. Caldecott K. W., Aoufouchi S., Johnson P., Shall S. (1996) XRCC1 polypeptide interacts with DNA polymerase β and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular “nick-sensor” in vitro. Nucleic Acids Res. 24, 4387–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Masson M., Niedergang C., Schreiber V., Muller S., Menissier-de Murcia J., de Murcia G. (1998) XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell Biol. 18, 3563–3571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nash R. A., Caldecott K. W., Barnes D. E., Lindahl T. (1997) XRCC1 protein interacts with one of two distinct forms of DNA ligase III. Biochemistry 36, 5207–5211 [DOI] [PubMed] [Google Scholar]
- 39. Srivastava D. K., Rawson T. Y., Showalter S. D., Wilson S. H. (1995) Phorbol ester abrogates up-regulation of DNA polymerase β by DNA-alkylating agents in Chinese hamster ovary cells. J. Biol. Chem. 270, 16402–16408 [DOI] [PubMed] [Google Scholar]
- 40. Schröder H. C., Batel R., Schwertner H., Boreiko O., Müller W. E. (2006) Fast micromethod DNA single-strand-break assay. Methods Mol. Biol. 314, 287–305 [DOI] [PubMed] [Google Scholar]
- 41. Moreno-Villanueva M., Pfeiffer R., Sindlinger T., Leake A., Müller M., Kirkwood T. B., Bürkle A. (2009) A modified and automated version of the “fluorimetric detection of alkaline DNA unwinding” method to quantify formation and repair of DNA strand breaks. BMC Biotechnol. 9, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kedar P. S., Stefanick D. F., Horton J. K., Wilson S. H. (2008) Interaction between PARP-1 and ATR in mouse fibroblasts is blocked by PARP inhibition. DNA Repair 7, 1787–1798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Khodyreva S. N., Prasad R., Ilina E. S., Sukhanova M. V., Kutuzov M. M., Liu Y., Hou E. W., Wilson S. H., Lavrik O. I. (2010) Apurinic/apyrimidinic (AP) site recognition by the 5′-dRP/AP lyase in poly(ADP-ribose) polymerase-1 (PARP-1). Proc. Natl. Acad. Sci. U.S.A. 107, 22090–22095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Horton J. K., Joyce-Gray D. F., Pachkowski B. F., Swenberg J. A., Wilson S. H. (2003) Hypersensitivity of DNA polymerase β null mouse fibroblasts reflects accumulation of cytotoxic repair intermediates from site-specific alkyl DNA lesions. DNA Repair 2, 27–48 [DOI] [PubMed] [Google Scholar]
- 45. Butler W. B. (1984) Preparing nuclei from cells in monolayer cultures suitable for counting and for following synchronized cells through the cell cycle. Anal. Biochem. 141, 70–73 [DOI] [PubMed] [Google Scholar]
- 46. Kedar P. S., Stefanick D. F., Horton J. K., Wilson S. H. (2012) Increased PARP-1 association with DNA in alkylation damaged, PARP-inhibited mouse fibroblasts. Mol. Cancer Res., 10, 360–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lim J. H., Catez F., Birger Y., Postnikov Y. V., Bustin M. (2004) Preparation and functional analysis of HMGN proteins. Methods Enzymol. 375, 323–342 [DOI] [PubMed] [Google Scholar]
- 48. Gagnon S. N., Desnoyers S. (2003) Single amino acid substitution enhances bacterial expression of PARP-4D214A. Mol. Cell Biochem. 243, 15–22 [DOI] [PubMed] [Google Scholar]
- 49. Asagoshi K., Tano K., Chastain P. D., 2nd, Adachi N., Sonoda E., Kikuchi K., Koyama H., Nagata K., Kaufman D. G., Takeda S., Wilson S. H., Watanabe M., Swenberg J. A., Nakamura J. (2010) FEN1 functions in long patch base excision repair under conditions of oxidative stress in vertebrate cells. Mol. Cancer Res. 8, 204–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sobol R. W., Kartalou M., Almeida K. H., Joyce D. F., Engelward B. P., Horton J. K., Prasad R., Samson L. D., Wilson S. H. (2003) Base excision repair intermediates induce p53-independent cytotoxic and genotoxic responses. J. Biol. Chem. 278, 39951–39959 [DOI] [PubMed] [Google Scholar]
- 51. Tanuma S., Johnson G. S. (1983) ADP-ribosylation of nonhistone high mobility group proteins in intact cells. J. Biol. Chem. 258, 4067–4070 [PubMed] [Google Scholar]
- 52. Kim M. Y., Zhang T., Kraus W. L. (2005) Poly(ADP-ribosyl)ation by PARP-1: “PAR-laying” NAD+ into a nuclear signal. Genes Dev. 19, 1951–1967 [DOI] [PubMed] [Google Scholar]
- 53. Haince J. F., Kozlov S., Dawson V. L., Dawson T. M., Hendzel M. J., Lavin M. F., Poirier G. G. (2007) Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J. Biol. Chem. 282, 16441–16453 [DOI] [PubMed] [Google Scholar]
- 54. Zhou J., Ahn J., Wilson S. H., Prives C. (2001) A role for p53 in base excision repair. EMBO J. 20, 914–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Achanta G., Huang P. (2004) Role of p53 in sensing oxidative DNA damage in response to reactive oxygen species-generating agents. Cancer Res. 64, 6233–6239 [DOI] [PubMed] [Google Scholar]
- 56. Offer H., Wolkowicz R., Matas D., Blumenstein S., Livneh Z., Rotter V. (1999) Direct involvement of p53 in the base excision repair pathway of the DNA repair machinery. FEBS Lett. 450, 197–204 [DOI] [PubMed] [Google Scholar]
- 57. Hamann I., Konig C., Richter C., Jahnke G., Hartwig A. (2011) Impact of cadmium on hOGG1 and APE1 as a function of the cellular P53 status. Mutat. Res. Fundam. Mol. Mech. Mutagen. doi: 10.1016/j.mrfmmm.2011.05.006 [DOI] [PubMed] [Google Scholar]
- 58. Kraus W. L. (2008) Transcriptional control by PARP-1: Chromatin modulation, enhancer-binding, coregulation, and insulation. Curr. Opin. Cell Biol. 20, 294–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Abd Elmageed Z. Y., Naura A. S., Errami Y., Zerfaoui M. (2012) The poly(ADP-ribose) polymerases (PARPs): New roles in intracellular transport. Cell. Signal 24, 1–8 [DOI] [PubMed] [Google Scholar]
- 60. Cohen-Armon M. (2007) PARP-1 activation in the ERK signaling pathway. Trends Pharmacol. Sci. 28, 556–560 [DOI] [PubMed] [Google Scholar]