

Abstract
A more detailed understanding of the somatic genetic events that drive gastrointestinal adenocarcinomas is necessary to improve diagnosis and therapy. Using data from high-density genomic profiling arrays, we conducted an analysis of somatic copy-number aberrations (SCNAs) in 486 gastrointestinal adenocarcinomas including 296 esophageal and gastric cancers. Focal amplifications were substantially more prevalent in gastric/esophageal adenocarcinomas than colorectal tumors. We identified 64 regions of significant recurrent amplification and deletion, some shared and others unique to the adenocarcinoma types examined. Amplified genes were noted in 37% of gastric/esophageal tumors, including in therapeutically targetable kinases such as ERBB2, FGFR1, FGFR2, EGFR, and MET, suggesting the potential utility of genomic amplifications as biomarkers to guide therapy of gastric and esophageal cancers where targeted therapeutics have been less developed compared to colorectal cancers. Amplified loci implicated genes with known involvement in carcinogenesis but also pointed to regions harboring potentially novel cancer genes, including a recurrent deletion found in 15% of esophageal tumors where the Runt transcription factor subunit RUNX1 was implicated, including by functional experiments in tissue culture. Together, our results defined genomic features that were common and distinct to various gut-derived adenocarcinomas, potentially informing novel opportunities for targeted therapeutic interventions.
Keywords: Esophagus, stomach, colon, adenocarcinoma, copy-number
Introduction
Colorectal, gastric, and esophageal adenocarcinomas collectively account for approximately 180,000 cancer diagnoses and 76,500 deaths each year in the United States and approximately 1.3 million deaths worldwide (1, 2). A better understanding of the somatic genetics of these diseases is a prerequisite for earlier diagnosis and more effective treatment. Colorectal cancer (CRC) genomes have been studied extensively (3, 4); the value of this information is realized by persuasive evidence that KRAS and BRAF mutations in CRC predict lack of response to cetuximab (5, 6). Gastric cancer (GC) and especially esophageal adenocarcinoma (EA) has been subjected to fewer large-scale studies (7-9).
Cancers of the esophagus and stomach commonly arise in a background of intestinal metaplasia, but develop within distinct luminal environments. Nevertheless, they often are treated with identical chemotherapy, and many clinical trials combine patients with these two diseases (10, 11). While the process of intestinal metaplasia preceding GC and EA suggests that these tumors may resemble adenocarcinomas arising from the intestine, they demonstrate distinct clinical behavior from CRC. It is therefore important to define the similarities and differences among digestive tract adenocarcinomas at the genomic and molecular levels. Such a comparison can inform both mechanistic studies and strategies for biomarker-driven therapy.
Two challenges exist in the somatic genetic analysis of cancer: 1) distinguishing ‘driver’ alterations that contribute to tumor development, maintenance, or proliferation from random ‘passenger’ alterations that do not contribute to the neoplastic process, and 2) identifying the specific genes that mediate tumor progression. Both challenges must be confronted in analysis of somatic copy-number alterations (SCNAs) as tumors often harbor many such alterations, each of which often encompasses up to thousands of genes. The study of SCNAs has been greatly enhanced by high-density genomic arrays allowing resolution of individual SCNA boundaries and the ability to study large numbers of tumors. Statistical analysis of SCNAs across many samples can identify regions altered more frequently than expected by chance and also pinpoint the most likely culprit genes in these regions. Pooling data from different but related cancer types can increase both statistical power and the ability to resolve specific gene targets of SCNAs. Given the related origins and documented shared copy-number characteristics of gut-derived adenocarcinomas (12), we hypothesized that evaluating genomic events across these tumors will increase our power to identify common genes active in gut adenocarcinomas and also help uncover differences. Here, we report the largest analysis of SCNAs across gut adenocarcinoma genomes and systematically compare significantly recurrent structural genetic alterations in tumors from distinct regions of the gut. We find multiple known and novel recurrent alterations, including region-specific and shared events.
Materials and Methods
All samples were fresh frozen primary resections from patients not treated with prior chemotherapy or radiation. All cases had diagnoses confirmed by pathologic review and only cases with estimated carcinoma content >70% were selected (Supplementary Table 1). The sample set was not enriched for other features. Tumors annotated as having originated from the gastric-esophageal junction were assigned to the EA collection. DNA was extracted (Supplemental Table 1), quantified with Picogreen dye and hybridized to (214 samples) GeneChip Human Mapping StyI 250K arrays (Affymetrix, Santa Clara, CA) or (271 samples) Genome-Wide Human SNP Array 6.0 (SNP6.0) (Affymetrix) genomic profiling arrays, according to the manufacturer’s instructions. Data from each of the two array platforms were independently normalized and segmented using all data present upon each of the two platforms (12, 13). Regions of known germline copy-number polymorphisms were then removed as previously described (14). Human genome build hg18 was used, and raw data files have been deposited at the Gene Expression Omnibus (GSE36460).
Significantly recurrent SCNAs were identified using GISTIC 2.0. (15) All data from each array were used to generate SCNA profiles for each tumor. To enable probe bound GISTIC analyses across data from two array platforms, the segmented data from each sample was remapped to the 196,800 probes shared by the two platforms. In some cases this remapping modified the position of the probe bounding the transition between two copy-number segments. In these cases, the boundaries were remapped to the nearest probes in the joint set. To remove potentially spurious SCNAs, segments defined by fewer than nine shared probes were removed (16). Additional details are described in Supplemental Methods.
Results
A comprehensive copy-number dataset from digestive tract adenocarcinomas
We analyzed a cohort of 363 new and 123 publically available genome array profiles from primary untreated gut adenocarcinomas including EA (186), GC (110), and CRC (190) (Table S1). We determined genome-wide copy-number profiles using either 250K StyI (238,000 probes) or SNP6.0 genome arrays (1.8 million probes) (Table S1). Copy-number alterations were identified using the full complement of data from each array type. To enable the analysis across platforms, the segmented copy-number data from each sample were remapped to the 196,800 probes common to both arrays. We found no evidence of bias introduced by pooling samples from the two platforms (Supplemental Note 1).
Chromosomal instability increases from lower to upper gut adenocarcinomas
Across this set of adenocarcinoma SCNA profiles, visual inspection of the segmented data showed alterations in nearly every part of the genome, and variations in the amount of genomic disruption between different cancers and cancer types (Fig. 1A). To compare levels of genomic disruption, we separately evaluated the frequencies of arm-level (comprising half or more of a chromosome arm) and focal SCNAs in each cancer type. Focal alterations were noted to occur throughout the chromosomes, but showed some predilection for the regions closer to the centromeres and telomeres of each chromosome. (Supplemental Figure 1)
Figure 1. Patterns of chromosomal instability differ across adenocarcinomas of the gastrointestinal tract.
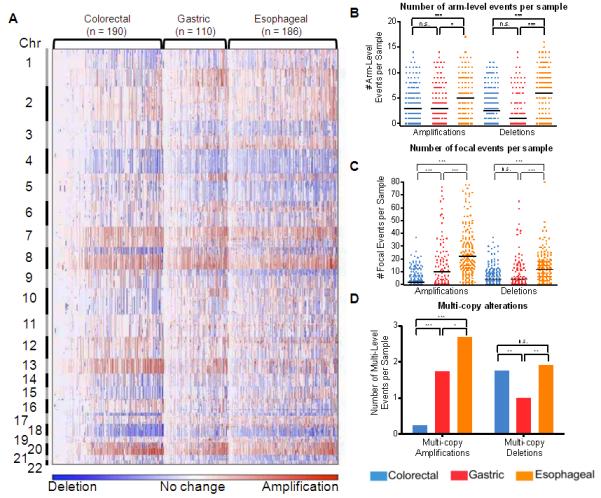
a) Amplifications (red) and deletions (blue) are displayed for 486 gut adenocarcinomas (x-axis) across the genome (y-axis). Quantitation of b) arm-level and c) focal SCNAs. Solid bars represent median numbers of events per sample. Error bars represent S.E.M. d) Quantitation of inferred multi-copy SCNAs (high-level amplifications with inferred copy >4 and presumable homozygous deletions, inferred copy-number <1.3). ***, P < 0.0005; **, P < 0.005; *, P < 0.05; n.s., not significant.
The median number of arm-level gains varied little between types (Fig. 1B), but was significantly increased in focal amplifications in EA and GC compared to CRC (Fig. 1C). Some tumors, particularly among CRC and GC types, demonstrated little apparent genomic disruption, potentially attributable to microsatellite instability or stromal contamination (Supplemental Note 2). The enhanced rate of focal amplification in EA and GC compared to CRC remained valid when these genomically quiet tumors were removed from analysis (Fig. S2). Upper gut adenocarcinomas exhibited an even greater excess of higher-level, multi-copy focal amplifications (Fig. 1D), which remained after we accounted for possible contaminating non-cancer DNA through use of sample-specific thresholds for defining these events (Fig. S3). The higher rates of focal genomic amplifications in EA and GC relative to CRC suggest that underlying mechanisms of genomic instability may differ between upper and lower gastrointestinal adenocarcinomas and that genomic amplification may be a more common means of oncogene activation in GC/EA.
The difference in rates of focal amplifications between upper and lower gastrointestinal adenocarcinomas did not hold for deletions. Fewer arm-level deletions were seen in GC than other gut adenocarcinomas (Fig. 1B). There was a modest increase in focal deletions in EA compared to GC and CRC (Fig. 1B), a trend that persisted when cases without arm-level SCNAs were excluded (Fig. S2). EA and CRC exhibit similar rates of multi-copy deletions that may represent homozygous deletions, rates that are significantly higher than in GC (Fig. 1D). This pattern persisted after exclusion of ‘quiet’ samples and used sample-specific thresholds to identify the multi-copy deletions (Fig. S3). The discordant bias towards focal amplifications but not deletions in upper gastrointestinal cancers suggests that the mechanisms and selective pressures leading to amplification may differ from deletion.
Recurrent amplifications and deletions across the pooled dataset
Arm-level events
We next performed a GISTIC 2.0 analysis to define significantly recurrent SCNAs, starting with arm-level SCNAs. Arm-level amplifications of chromosomes 7p, 8q, 20p, and 20q recurred significantly across all three cancer types (Fig. S4A). Events restricted to specific subtypes included 1q gains in GC and 13q gains in CRC and GC. These significant arm-level gains have been observed previously (17-23). Arm-level deletions were more variable across tumor types. In GC, deletions of 4p and 4q alone were significant reflecting a lower degree of arm-level losses detected earlier in GC (Fig. S4B), but these and other deletions (8p, 18p, 18q) were also detected in EA and CRC. Loss of arms 8p, 14q, and 15q were of higher significance in CRC, whereas loss of 5q, 9p, and 21q were particularly significant among EAs. Loss of 17p (containing TP53) was significant in EA and CRC, but not GC. Unique significant losses of 9p and 21q in EA are notable because these arms respectively contain the known and putative tumor suppressors CDKN2A and RUNX1, both targets of focal deletion in EA (discussed below).
These results are in accordance with previously published data. We compared the frequency of alteration for each cytoband in each cancer type to frequencies determined across 998 CRC, 741 GC, and 71 EA in the Progenetix database curated from cCGH and aCGH data (24, 25). The cytoband-level data are similar for all cancer types, with the exception of lower frequencies of 17q gain in our GC and EA samples (Fig. S5). Notably, the low frequency of deletions in GC data compiled by Progenetix mirrored our data suggesting that result is not unique to the samples in our collection.
Focal alterations
We next evaluated focal SCNAs across all 486 tumors and identified 33 regions subject to significant (q<0.01) focal amplification (Fig. 2 and Table 1). Thirteen of these regions contain known oncogenes, including four genes involved in cell cycle regulation (CCNE1, CCND1, CDK6, MYC) and seven members of tyrosine kinase/MAPK signaling pathways (EGFR, KRAS, MET, ERBB2, FGFR1, FGFR2, and IGF1R). Twenty significant peaks contained no established oncogenes, suggesting potential presence of novel genes or non-coding transcripts that promote intestinal metaplasia and/or gastrointestinal carcinogenesis.
Figure 2. Identification of significant focal SCNAs in a cohort of esophageal, gastric, and colorectal adenocarcinomas.
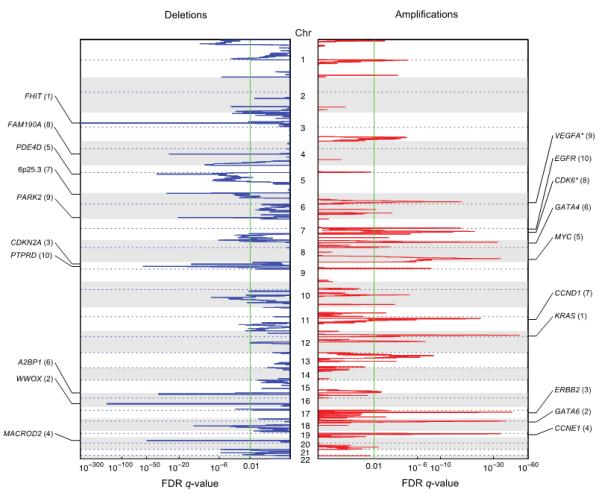
GISTIC q-values (x-axis) for deletions (left, blue) and amplifications (right, red) are plotted across the genome (y-axis). The 10 most significant peaks are labeled with known or putative gene targets. Values in parentheses represent the number of genes in the peak region. Asterisks indicate genes immediately adjacent to the peak.
Table 1.
Significantly amplified focal SCNAs in a cohort of esophageal, gastric, and colorectal adenocarcinomas (q-value <0.01).
| Cytoband | Residual q-value |
Peak Boundaries (Mb) |
Number of Genes in Peak |
Candidate Target(s) (Bold designates therapeutic target) |
Gut Adenocarcinoma Types Represented |
|---|---|---|---|---|---|
| Gut Adenocarcinomas | |||||
| 12p12.1 | 8.04E-51 | 25.23-25.34 | 3 | KRAS | E, G |
| 18q11.2 | 5.05E-37 | 17.95-18.05 | 1 | GATA6 | E, G, C |
| 17q12 | 4.34E-35 | 34.97-35.27 | 10 | ERBB2 | E, G, C |
| 19q12 | 5.97E-34 | 34.95-35.10 | 1 | CCNE1 | E, G |
| 8q24.21 | 8.23E-34 | 128.50- 128.83 |
2 | MYC | E, G, C |
| 8p23.1 | 2.10E-32 | 11.41-11.71 | 4 | GATA4 | E, G |
| 11q13.2 | 5.26E-22 | 68.97-69.49 | 5 |
CCND1, FGF3, FGF4, FGF19 |
E, G |
| 7q21.2 | 3.36E-17 | 92.32-92.50 | 1 | CDK6* | E |
| 6p21.1 | 2.12E-16 | 43.79-43.99 | 2 | VEGFA | E, G |
| 7p11.2 | 6.71E-16 | 54.92-55.28 | 1 | EGFR | E, G |
| 17q21.2 | 2.23E-11 | 37.02-37.21 | 7 | E | |
| 9p13.3 | 7.03E-09 | 35.52-35.93 | 24 | E | |
| 12q15 | 6.23E-08 | 67-09-68.25 | 10 | MDM2, FRS2 | E, G |
| 7q22.1 | 1.41E-07 | 98.41-99.02 | 16 | E | |
| 13q13.1 | 2.04E-06 | 32.18-33.33 | 4 | E | |
| 10q22.2 | 1.10E-05 | 75.00-75.80 | 16 | E | |
| 7q31.2 | 1.32E-05 | 115.98- 116.42 |
6 | MET | E |
| 1q21.3 | 1.69E-05 | 146.23- 150.89 |
106 | MCL1 | E |
| 1q42.3 | 1.71E-04 | 233.02- 233.42 |
4 | G | |
| 10q26.12 | 3.17E-04 | 122.75- 123.37 |
1 | FGFR2 | E |
| 13q14.11 | 3.67E-04 | 40.27-40.83 | 10 | E | |
| 13q22.1 | 3.90E-04 | 72.47-73.00 | 2 | KLF5 | None |
| 6q23.3 | 8.14E-04 | 135.32- 135.87 |
5 | MYB | E |
| 7q34 | 1.03E-03 | 141.92- 142.26 |
4 | EPHB6 | E |
| 8p12 | 1.28E-03 | 38.23-38.46 | 5 | FGFR1 | C |
| 3q26.2 | 1.55E-03 | 169.95- 175.23 |
32 | PRKCl | E |
| 11p14.2 | 1.81E-03 | 26.94-27.57 | 6 | E | |
| 1p36.22 | 1.99E-03 | 103.10- 110.18 |
10 | G | |
| 15q26.1 | 3.52E-03 | 86.15-100.34 | 94 | IGF1R | None |
| 17q24.3 | 4.18E-03 | 67.82-68.05 | 1 | SOX9 | None |
| 11q14.1 | 4.19E-03 | 75.12-79.48 | 31 | GAB2 | None |
| 20q13.2 | 6.89E-03 | 51.55-52.11 | 3 | None | |
| 3q27.1 | 8.69E-03 | 185.72- 186.17 |
3 | EPHB3 | G |
The 18q11.2 amplification peak is the second most significant peak after KRAS and contains only the endodermal transcription factor GATA6. Coupled with functional data suggesting a role for GATA6 in esophageal carcinogenesis (26, 27), these results implicate GATA6 as an important contributor to gastrointestinal neoplasia. The sixth most significant peak in the composite dataset, located at 8p23.1, contains 4 genes, including the related transcription factor GATA4, a candidate target noted previously (28).
We also identified 30 regions of significant focal deletion (Fig. 2 and Table 2). Eight of these regions include genes such as FHIT and WWOX, with exons spread over large genomic loci (in excess of 1 Mb). Prior studies suggest that such regions often lie in “fragile sites” or areas of low gene density where deletion may be tolerated, and may not harbor functional tumor suppressors (12, 29). An additional eight regions contain the known tumor suppressors CDKN2A, SMAD4, PTEN, APC, RUNX1, ARID1A, and ATM and the putative tumor suppressor PARD3B (30). Fourteen regions did not contain known tumor suppressors or large-footprint genes, but could contain novel factors whose loss contributes to intestinal metaplasia or cancer. Our analysis would not have detected regions of loss of heterozygosity that did not lead to copy-number loss.
Table 2.
Significantly deleted focal SCNAs in a cohort of esophageal, gastric, and colorectal adenocarcinomas (q-value <0.01).
| Cytoband | Residual q-value |
Peak Boundaries (Mb) |
Number of Genes in Peak |
Candidate Target(s) |
Gut Adenocarcinoma Types Represented |
|---|---|---|---|---|---|
| Gut Adenocarcinomas | |||||
| 3p14.2 | 1.207E- 313 |
58.98-61.52 | 1 | FHIT | E, G, C |
| 16q23.1 | 2.96E-151 | 76.69-78.21 | 2 | WWOX | E, G, C |
| 9p21.3 | 5.08E-56 | 21.98-22.01 | 1 | CDKN2A | E |
| 20p12.1 | 3.22E-50 | 14.21-15.99 | 2 | MACROD2 | E, G, C |
| 5q11.2 | 9.09E-38 | 58.30-59.84 | 3 | PDE4D | E, G, C |
| 16p13.3 | 8.41E-36 | 50.63-77.09 | 2 | A2BP1 | C |
| 6p25.3 | 2.09E-28 | 1.54-2.57 | 2 | E, G | |
| 4q22.1 | 2.60E-27 | 91.37-93.49 | 2 | FAM190A | E, G, C |
| 6q26 | 3.61E-21 | 161.61- 163.13 |
1 | PARK2 | E, G, C |
| 9p24.1 | 1.32E-14 | 7.77-12.71 | 3 | PTPRD | E, G |
| 18q21.2 | 7.52E-14 | 46.77-46.97 | 1 | SMAD4 | E, C |
| 1p36.11 | 1.36E-11 | 26.77-27.31 | 11 | ARID1A | E, C |
| 21q11.2 | 1.61E-06 | 1.00-1.64 | 17 | E | |
| 1q44 | 2.64E-06 | 24.39-24.48 | 1 | E | |
| 8p23.3 | 2.64E-06 | 1.00-1.71 | 7 | E | |
| 10q23.31 | 2.64E-06 | 89.44-90.07 | 4 | PTEN | C |
| 21q22.12 | 5.76E-06 | 35.03-35.35 | 2 | RUNX1 | E |
| 4q35.1 | 1.80E-05 | 179.06- 187.23 |
31 | E | |
| 7q36.3 | 3.01E-05 | 156.49- 158.82 |
12 | E | |
| 5q22.2 | 5.22E-05 | 110.88- 113.74 |
12 | APC | C |
| 1p13.2 | 8.53E-05 | 111.01- 119.86 |
79 | C | |
| 2q33.2 | 1.15E-04 | 204.53- 206.27 |
1 | PARD3B | E |
| 3p26.2 | 1.24E-04 | 4.32-4.51 | 1 | None | |
| 4q35.2 | 4.15E-04 | 185.37- 191.27 |
36 | CASP3 | C |
| 17p12 | 1.38E-03 | 11.40-12.51 | 5 | None | |
| 11q22.3 | 2.26E-03 | 103.49- 112.70 |
62 | ATM | E |
| 5q15 | 3.85E-03 | 83.69-106.74 | 56 | None | |
| 21q22.3 | 8.17E-03 | 38.21-46.94 | 116 | None | |
| 12q21.2 | 8.88E-03 | 71.30-86.91 | 39 | None | |
| 10q11.23 | 9.24E-03 | 52.31-53.77 | 5 | C | |
The combined analysis across three tumor types enabled identification of less common SCNAs. Five amplification peaks were significant in the composite set but not in any individual cancer type. One of these, 13q22.1, contains only two genes, including the proliferative transcription factor KLF5. The combined dataset also enabled more precise identification of the likely targets of focal SCNAs. For example, the 1p36.11 deletion narrowed from 89 genes in the EA set to only 11 genes in the combined dataset, including the chromatin-modifying enzyme ARID1A, a recently identified target of frequent mutation in clear cell ovarian and gastric adenocarcinomas (31, 32). However, combining data across platforms also entailed some loss of resolution for the SNP6 data. We therefore performed a separate analysis of the SNP6 data, which yielded similar results to the composite analysis, though with fewer peaks (Supplemental Note 3 and Table S6).
To identify relationships between genes targeted by focal alterations, we evaluated the co-occurrence or exclusivity of focal alterations at all GISTIC peak regions. After correcting for multiple hypothesis testing and tissue type, the only significant (Bonferroni-corrected p-value ≥0.05) findings were correlations of amplifications of CCNE1 with each of two peaks with unknown targets: a deletion peak at 6p25.3 and amplification of 1q42.3. These findings may suggest cooperativity between these novel events and amplification of CCNE1, or reflect subsets of tumors that for other reasons tend to share alterations in these regions.
Comparison of focal alterations across gut adenocarcinoma and other cancer types
Comparison of focal amplifications
We analyzed focal alterations in each tumor type separately and identified five, 14 and 25 amplification peaks in CRC, GC, and EA, respectively (Figs. 3A and S6 and Table S2). Highlighting the similarities and differences among digestive tract cancers, only three amplifications were significant in all tumor types: 8q24.21 (containing MYC), 17q12 (containing ERBB2), and 18q11.2 (containing GATA6). Among these peaks, ERBB2 is amplified more commonly in esophageal (17%) and gastric (13%) than in colorectal (6%) tumors (Fig. S7).
Figure 3. Venn diagram of focal events common across gut adenocarcinomas.

Significantly amplified (a) or deleted (b) peaks (or gene if likely candidate available) as determined by GISTIC analysis of focal alterations in each adenocarcinoma subtype. The color code denotes peaks that are unique to a given tumor type or shared between two or three of the adenocarcinoma subtypes. Bolded genes represent genes significantly altered and common across esophageal, gastric, and colorectal tumors. Asterisks represent significant SCNAs in a dataset containing 3,131 cancers not including CRC, EA or GC (12).
Two amplification peaks were restricted to colorectal cancers. One amplicon contains the RTK, FGFR1, not previously reported in this disease, but noted to be overexpressed (33). The other unique peak is adjacent to the CRC oncogene CDK8 at 13q12.2 (34). Esophageal and gastric adenocarcinomas shared seven amplicons, containing VEGFA, EGFR, GATA4, CCND1, MDM2, CCNE1, and KRAS. The most significantly amplified gene across our dataset, KRAS, showed a strong foregut preference. Only 5% of CRCs carried focal KRAS amplification, compared to 21% of upper GI cancers. Conversely, CRCs have substantially higher rates of KRAS mutation (Fig. S8) demonstrating how upper and lower gastrointestinal cancers show distinct ways of altering the same oncogene (35).
An additional 14 regions of amplification were specific to EA. Six of these peaks contained genes known to contribute to cancer (CDK6, MCL1, PRKCI, MYB, MET, and FGFR2), while the others contain no previously described oncogenes. To evaluate how the large sample size enabled identification of relevant targets, we compared our analysis to the SCNA analyses of the largest previously published EA datasets, comprising 42 and 56 tumors (8, 36). Amplifications at CCNE1, MET, FGFR2 and MYB were not identified in previous datasets, but these genes have been noted to be overexpressed in EA (37-40). Many peaks that lack known oncogenes were also not noted in earlier reports. Although MET and CCNE1 amplifications had been detected previously by focused gene inquiry, MYB and FGFR2 amplifications were not noted in prior data. Our sample numbers also afforded greater resolution to identify targets of previously identified SCNAs. For example, an amplicon at 6p21.1 was reported in studies of 42 and 56 EAs to contain between 50 and 70 genes (8, 36). We narrowed this region to only 2 genes, including the vascular endothelial growth factor, VEGFA, the target of the therapeutic antibody bevacizumab. A prior study of EA also identified a 94-gene region of amplification on 3q and attributed this event to PIK3CA (36). Our analysis narrows this peak to a region containing PRKCI, >5 Mb away from PIK3CA.
The presence of FGFR2 amplifications in EA suggests a potential new therapeutic target for these tumors, similar to in GC (9). We confirmed the presence of FGFR2 amplification in individual EA cases through quantitative RT-PCR (Fig. S9). These results indicate FGFR2 amplification may serve as a biomarker for the use of FGFR2-directed therapy in EA in addition to GC.
Four amplification peaks were restricted to gastric adenocarcinoma. Among these, only 3q27.1, containing the RTK gene, EPHB3, has been suggested to play a role in cancer progression (41). Compared to prior studies, we not only confirmed recurrent focal amplifications involving GATA4 and GATA6, but also identified novel peaks on 6p21.1 (VEGFA), 3q27.1 (EPHB3), 1p36.22, 12q15, and 1q42.3 (9).
The most significant focally amplified tyrosine kinases, ERBB2, EGFR, MET, FGFR1, and FGFR2, are known oncogenes and targets of therapeutic agents in current use or development. We detected amplifications involving one or more RTKs in 42% of EA, 28% of GC, and 14% of CRC samples (Fig. 4). Only 10% of tumors had concurrent amplifications of RTKs and KRAS.
Figure 4. Amplified therapeutic determinants in gut adenocarcinomas.
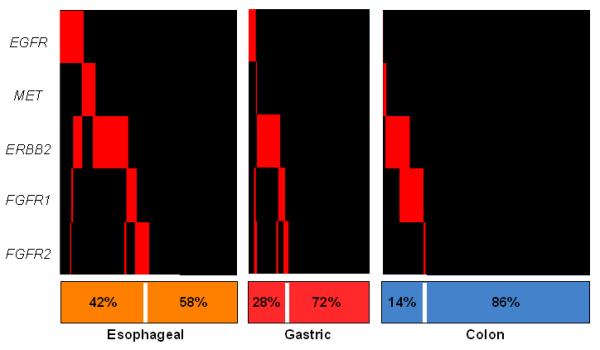
The presence of focal amplifications (inferred copy-number > 2.3) is shown in red for recurrently amplified receptor tyrosine kinase genes across all tumor specimens (vertical bars).
Finally, we compared amplifications in GI adenocarcinomas to those found in a study of 2,311 diverse cancers (12). Among the 33 focal amplifications in GI cancers, 42% overlapped with peak regions in other cancers (Fig. S10), including 11 regions containing the known oncogenes MCL1, MYB, EGFR, FGFR1, MYC, CCND1, KRAS, MDM2, ERBB2, and CCNE1 (marked with asterisks in Fig 3). Peak regions present in gut, but absent from non-gut adenocarcinomas, encompassed genes encoding the tissue-specific transcription factors GATA4 and GATA6 and the known or putative oncogenes CDK6, FGFR2, MET, EPHB3, and EPHB6. Some of these genes are occasionally amplified in other cancer types, but did not show statistical significance. HMGA2 amplifications on chromosome 12 are notably absent in GI tumors, despite the presence of chromosome 12 amplification at MDM2 in both GI and non-GI carcinomas. Similarly, amplification of the G1/S cell cycle dependent-kinase CDK6 is restricted to GI tumors while CDK4 amplification is significant in all other tumors further suggesting that many cancer utilize similar pathways with multiple inputs to reach the same output (Table S3).
Comparison of focal deletions
We identified 16, 8, and 21 peaks of significant deletion in CRC, GC, and EA, respectively (Figs. 3B and S11 and Table S4). Six peaks were shared across the three cancer types, all containing genes spanning large genomic loci (including FHIT, WWOX, and MACROD2) (12, 42).
Seven deletion peaks were unique to CRC, including two peaks that encompass known tumor suppressors (APC and PTEN), and five that do not. Among the latter, a previously unidentified peak at 10q25 contains the apoptosis effector caspase gene CASP7, suggesting a mechanism to evade apoptosis (Table S4A).
Eleven deletion peaks were unique to EA while none were unique to GC (Fig. 3B). Among the EA peaks, two contain known esophageal tumor suppressors (CDKN2A and ATM; Table S4B and Fig. S7D). One peak contains RUNX1, which we consider further below. Two peaks were seen in both esophageal and gastric tumors, including one large gene (PTPRD) and one region (6p25.3) with no known tumor suppressor.
Among the 30 deletion peaks in the composite tumor set (Table 2), 15 were also significant in the non-GI adenocarcinoma study (Fig. S10). The common sites included nine peaks encompassing large genes, four peaks containing known deleted tumor suppressors (APC, CDKN2A, ATM, and PTEN), and two peaks without either. One of the latter peaks contains the effector caspase gene CASP3, suggesting that deletion of an effector caspase, CASP3 or CASP7, may mark GI adenocarcinomas more generally and not CRC alone. Focal loss of RB1 and TP53 were identified in non-GI but not in digestive tract adenocarcinomas, although 17p deletion, containing TP53, was significant in CRC and EAC. The 15 deletion peaks restricted to GI adenocarcinomas include 13 that lack known tumor suppressors (Table S5). The other two contain the known tumor suppressors SMAD4 and RUNX1.
Functional significance of RUNX1 deletions in esophageal adenocarcinoma
We observed highly focal RUNX1 deletions at 21q22.12 in 15% of EAs, also noted in a recent report (Fig. 5A) (36). RUNX1 behaves as a tumor suppressor in leukemia, where translocations and mutations disrupt gene function (43). We therefore evaluated a possible tumor suppressor function for RUNX1 in EA by reintroducing it into the EA cell line OE33, which carries a focal RUNX1 deletion (Fig. 5A). We observed a 69% reduction in anchorage-independent growth relative to GFP-infected cells (Fig. 5B-C). As we did not possess an EA cell line without deletion at the RUNX1 locus, we ectopically expressed RUNX1 in A549 lung cancer cells, which have no focal RUNX1 deletion, to evaluate for generalized cellular toxicity due to overexpression of this gene. In contrast to OE33 cells, RUNX1 expression did not significantly affect A549 colony formation (Fig. 5B-C). These results are consistent with a potential role for RUNX1 as a tumor suppressor in EA.
Figure 5. RUNX1 re-expression can inhibit anchorage-independent growth in an esophageal adenocarcinoma cell model.

a) Focal deletion of locus 21q22.12 containing RUNX1 in esophageal adenocarcinomas, OE33 (EA), and A549 (non-small cell lung cancer) cell lines. b) OE33 and A549 cell lines were stably transduced with retrovirus containing RUNX1. Immunoblotting confirmed positive expression of RUNX1. c) Soft-agar colony formation for OE33 and A549 cells with ectopic GFP or RUNX1 expression. Representative images of colony formation at 6.3X magnification. Quantitation of three independent experiments; error bars represent S.E.M. **, P < 0.005; n.s., not significant.
Discussion
These data provide the most comprehensive, high-resolution analysis to date of SCNA patterns across the three most common forms of gut adenocarcinoma. Our cohort size enhanced the ability to detect significant SCNAs, and we identified several focused areas of recurrent genomic alteration pointing toward genes that may contribute to cancer. Notably, the genomes of EA and GC cancers contain alterations selected for their contributions to the process of both intestinal metaplasia and malignant transformation to adenocarcinoma.
We observed more focal amplifications in upper GI adenocarcinomas compared to CRC. Unlike CRC, EA and GC emerge in a setting of bile and acid injury, which may generate DNA strand breaks and contribute to the high rates of SCNA (44). Alternatively, distinct DNA repair pathways or selection for differing stimuli may account for these differences. It is unclear why the enhanced rates of focal amplifications in EA/GC were not matched by a similarly increased rate of focal deletions nor why the rates of deletion in GC fell below those in EA or CRC. Although the loci of the most significant amplifications peaks fell at known or plausible oncogenes, many focal deletions peaks lie in potential fragile sites. Thus, the mechanisms and selection pressures underlying deletions may differ from those responsible for amplification.
The GATA6 and GATA4 transcription factor genes lie in the second and sixth most significant amplification peaks across the full dataset, respectively. The developmental role of these transcription factors and selective amplification in GI cancers suggests that they may add to the growing number of lineage-survival transcription factor oncogenes (45). A parallel phenomenon is amplification of SOX2 in squamous esophageal and lung carcinomas (46), an event absent in EA.
A key clinical observation emerging from these data is that focally amplified RTKs were observed most prominently in EA and GC, suggesting that genomic amplifications will be more important biomarkers in upper gastrointestinal cancers than in CRC. Recent clinical trials reveal benefit when the HER2-directed antibody trastuzumab is combined with chemotherapy in treating ERBB2-amplified or – overexpressing GC/EA (11). The presence of ERBB2 amplifications in 6% of CRCs suggests that ERBB2-directed therapy may benefit select CRC patients (47). Additionally, based on evidence that KRAS mutation negatively predicts cetuximab response, highly prevalent KRAS amplification in upper GI tumors may similarly impact clinical decisions. When KRAS is evaluated as a biomarker in upper GI cancers, it will be important to examine both SCNAs and point mutations. Our data also point to other genomic amplifications of clinical relevance. CDK6 (48) and VEGFA focal amplifications may serve as a marker of response to targeted inhibitors. More broadly, these data point to the inclusion of gene amplifications to guide therapy in upper gastrointestinal adenocarcinomas.
While these data suggest that many patients with EA or GC may benefit from treatment targeting an amplified RTK, it is unlikely that therapies directed against these targets alone will lead to durable responses. The progression-free survival provided by single-agent therapy with trastuzumab in GC/EA has been modest (11). The presence of complex SCNA profiles in these tumors suggest there could be co-occurring alterations that confer primary resistance (49) or enhanced genomic instability that speeds acquired resistance. In our cohort, the rates of co-occurrence between RTK-associated and other events did not significantly deviate from what would be expected for statistically independent events. However, co-occurrences were detected, and the rates with which individual pairs of events co-occur are likely to inform combinational treatment strategies. While evaluation of RTK-targeted agents is needed, the genomic complexity and variability of these cancers suggests that combination inhibitor strategies will ultimately be essential.
Beyond these immediately clinically relevant targets, these data provide enhanced insight into specific genes responsible for different subtypes of gastrointestinal adenocarcinoma. Despite a shared intestinal origin, upper gastrointestinal cancers exhibit many distinct events from those seen in CRC, such as alteration of cell cycle regulators (CCND1, CCNE1, CDK6, CDKN2A). Moreover, as evidenced by the selective deletion of RUNX1 in EA, clear genomic differences exist between EA and GC. While our analysis has helped identify loci of recurrent alteration and potential targets of these events, many recurrent regions of SCNA do not contain known oncogenes or tumor suppressors. One challenge moving forward will be to determine which of these regions harbor additional genes or non-coding elements that contribute toward intestinal metaplasia or transformation to cancer. High-resolution genome analyses of large sample numbers, as presented here, can guide future studies and inform the development of strategies to reverse the effects of the somatic genetic events that drive these cancers.
Supplementary Material
Acknowledgements
We thank the members of the Broad Institute Biological Samples Platform and Genomic Analysis Platform for their work enabling this project and also thank the physicians and hospital staff whose efforts in collecting these samples is essential to this research.
Grant Support: A.J.B is supported by the National Cancer Institute (K08CA134931) and GI SPORE Developmental Project award - P50CA127003) and the DeGregorio Family Foundation. R.B. is supported by the National Cancer Institute (K08CA122833 and U54CA143798) and a V Foundation Scholarship. F.R. is supported by a grant from Istituto Toscano Tumori.
Footnotes
Conflicts of interest: R.B., M.M., and R.A.S are consultants for Novartis. M.M. is a founder of Foundation Medicine.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: a cancer journal for clinicians. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.American Cancer Society Vol. 1 http://www.cancer.org/docroot/STT/stt_0asp. Copyright 2011. [Google Scholar]
- 3.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 4.Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 5.Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26:5705–12. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 6.Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. The New England journal of medicine. 2008;359:1757–65. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 7.Rossi E, Klersy C, Manca R, Zuffardi O, Solcia E. Correlation between genomic alterations assessed by array comparative genomic hybridization, prognostically informative histologic subtype, stage, and patient survival in gastric cancer. Human pathology. 2011 doi: 10.1016/j.humpath.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 8.Goh XY, Rees JR, Paterson AL, Chin SF, Marioni JC, Save V, et al. Integrative analysis of array-comparative genomic hybridisation and matched gene expression profiling data reveals novel genes with prognostic significance in oesophageal adenocarcinoma. Gut. 2011;60:1317–26. doi: 10.1136/gut.2010.234179. [DOI] [PubMed] [Google Scholar]
- 9.Deng N, Goh LK, Wang H, Das K, Tao J, Tan IB, et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut. 2012 doi: 10.1136/gutjnl-2011-301839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wainberg ZA, Lin LS, DiCarlo B, Dao KM, Patel R, Park DJ, et al. Phase II trial of modified FOLFOX6 and erlotinib in patients with metastatic or advanced adenocarcinoma of the oesophagus and gastro-oesophageal junction. British journal of cancer. 2011;105:760–5. doi: 10.1038/bjc.2011.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–97. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 12.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome biology. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A. 2007;104:20007–12. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bardi G, Sukhikh T, Pandis N, Fenger C, Kronborg O, Heim S. Karyotypic characterization of colorectal adenocarcinomas. Genes Chromosomes Cancer. 1995;12:97–109. doi: 10.1002/gcc.2870120204. [DOI] [PubMed] [Google Scholar]
- 18.Buffart TE, Louw M, van Grieken NC, Tijssen M, Carvalho B, Ylstra B, et al. Gastric cancers of Western European and African patients show different patterns of genomic instability. BMC Med Genomics. 2011;4:7. doi: 10.1186/1755-8794-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He QJ, Zeng WF, Sham JS, Xie D, Yang XW, Lin HL, et al. Recurrent genetic alterations in 26 colorectal carcinomas and 21 adenomas from Chinese patients. Cancer Genet Cytogenet. 2003;144:112–8. doi: 10.1016/s0165-4608(02)00959-7. [DOI] [PubMed] [Google Scholar]
- 20.Molkentin JD. The zinc finger-containing transcription factors GATA-4, -5, and - 6. Ubiquitously expressed regulators of tissue-specific gene expression. J Biol Chem. 2000;275:38949–52. doi: 10.1074/jbc.R000029200. [DOI] [PubMed] [Google Scholar]
- 21.Muleris M, Dutrillaux AM, Olschwang S, Salmon RJ, Dutrillaux B. Predominance of normal karyotype in colorectal tumors from hereditary non-polyposis colorectal cancer patients. Genes Chromosomes Cancer. 1995;14:223–6. doi: 10.1002/gcc.2870140312. [DOI] [PubMed] [Google Scholar]
- 22.Nancarrow DJ, Handoko HY, Smithers BM, Gotley DC, Drew PA, Watson DI, et al. Genome-wide copy number analysis in esophageal adenocarcinoma using high-density single-nucleotide polymorphism arrays. Cancer Res. 2008;68:4163–72. doi: 10.1158/0008-5472.CAN-07-6710. [DOI] [PubMed] [Google Scholar]
- 23.Tsafrir D, Bacolod M, Selvanayagam Z, Tsafrir I, Shia J, Zeng Z, et al. Relationship of gene expression and chromosomal abnormalities in colorectal cancer. Cancer Res. 2006;66:2129–37. doi: 10.1158/0008-5472.CAN-05-2569. [DOI] [PubMed] [Google Scholar]
- 24.Baudis M. Progenetix oncogenomic online resource. 2009 doi: 10.1093/database/baab043. http://www.progenetix.net. [DOI] [PMC free article] [PubMed]
- 25.Baudis M, Cleary ML. Progenetix.net: an online repository for molecular cytogenetic aberration data. Bioinformatics. 2001;17:1228–9. doi: 10.1093/bioinformatics/17.12.1228. [DOI] [PubMed] [Google Scholar]
- 26.Alvarez H, Opalinska J, Zhou L, Sohal D, Fazzari MJ, Yu Y, et al. Widespread hypomethylation occurs early and synergizes with gene amplification during esophageal carcinogenesis. PLoS genetics. 2011;7:e1001356. doi: 10.1371/journal.pgen.1001356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin L, Bass AJ, Lockwood WW, Wang Z, Silvers AL, Thomas DG, et al. Activation of GATA binding protein 6 (GATA6) sustains oncogenic lineage-survival in esophageal adenocarcinoma. Proc Natl Acad Sci U S A. 2012;109:4251–6. doi: 10.1073/pnas.1011989109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin L, Aggarwal S, Glover TW, Orringer MB, Hanash S, Beer DG. A minimal critical region of the 8p22-23 amplicon in esophageal adenocarcinomas defined using sequence tagged site-amplification mapping and quantitative polymerase chain reaction includes the GATA-4 gene. Cancer Res. 2000;60:1341–7. [PubMed] [Google Scholar]
- 29.Smith DI, Zhu Y, McAvoy S, Kuhn R. Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett. 2006;232:48–57. doi: 10.1016/j.canlet.2005.06.049. [DOI] [PubMed] [Google Scholar]
- 30.Rothenberg SM, Mohapatra G, Rivera MN, Winokur D, Greninger P, Nitta M, et al. A genome-wide screen for microdeletions reveals disruption of polarity complex genes in diverse human cancers. Cancer Res. 2010;70:2158–64. doi: 10.1158/0008-5472.CAN-09-3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones S, Wang TL, Shih Ie M, Mao TL, Nakayama K, Roden R, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–31. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011 doi: 10.1038/ng.982. [DOI] [PubMed] [Google Scholar]
- 33.Jang JH. Reciprocal relationship in gene expression between FGFR1 and FGFR3: implication for tumorigenesis. Oncogene. 2005;24:945–8. doi: 10.1038/sj.onc.1208254. [DOI] [PubMed] [Google Scholar]
- 34.Firestein R, Bass AJ, Kim SY, Dunn IF, Silver SJ, Guney I, et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature. 2008;455:547–51. doi: 10.1038/nature07179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacConaill LE, Campbell CD, Kehoe SM, Bass AJ, Hatton C, Niu L, et al. Profiling critical cancer gene mutations in clinical tumor samples. PLoS One. 2009;4:e7887. doi: 10.1371/journal.pone.0007887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gu J, Ajani JA, Hawk ET, Ye Y, Lee JH, Bhutani MS, et al. Genome-wide catalogue of chromosomal aberrations in barrett’s esophagus and esophageal adenocarcinoma: a high-density single nucleotide polymorphism array analysis. Cancer Prev Res (Phila) 2010;3:1176–86. doi: 10.1158/1940-6207.CAPR-09-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brabender J, Lord RV, Danenberg KD, Metzger R, Schneider PM, Park JM, et al. Increased c-myb mRNA expression in Barrett’s esophagus and Barrett’s-associated adenocarcinoma. The Journal of surgical research. 2001;99:301–6. doi: 10.1006/jsre.2001.6186. [DOI] [PubMed] [Google Scholar]
- 38.Herrera LJ, El-Hefnawy T, Queiroz de Oliveira PE, Raja S, Finkelstein S, Gooding W, et al. The HGF receptor c-Met is overexpressed in esophageal adenocarcinoma. Neoplasia. 2005;7:75–84. doi: 10.1593/neo.04367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iida S, Katoh O, Tokunaga A, Terada M. Expression of fibroblast growth factor gene family and its receptor gene family in the human upper gastrointestinal tract. Biochemical and biophysical research communications. 1994;199:1113–9. doi: 10.1006/bbrc.1994.1346. [DOI] [PubMed] [Google Scholar]
- 40.Lin L, Prescott MS, Zhu Z, Singh P, Chun SY, Kuick RD, et al. Identification and characterization of a 19q12 amplicon in esophageal adenocarcinomas reveals cyclin E as the best candidate gene for this amplicon. Cancer Res. 2000;60:7021–7. [PubMed] [Google Scholar]
- 41.Ji XD, Li G, Feng YX, Zhao JS, Li JJ, Sun ZJ, et al. EphB3 is overexpressed in non-small-cell lung cancer and promotes tumor metastasis by enhancing cell survival and migration. Cancer Res. 2011;71:1156–66. doi: 10.1158/0008-5472.CAN-10-0717. [DOI] [PubMed] [Google Scholar]
- 42.Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463:893–8. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cohen M., Jr Perspectives on RUNX genes: An update. Am J Med Genet Part A. 2009:2629–46. doi: 10.1002/ajmg.a.33021. [DOI] [PubMed] [Google Scholar]
- 44.Bernstein H, Bernstein C, Payne CM, Dvorakova K, Garewal H. Bile acids as carcinogens in human gastrointestinal cancers. Mutation research. 2005;589:47–65. doi: 10.1016/j.mrrev.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 45.Garraway LA, Sellers WR. Lineage dependency and lineage-survival oncogenes in human cancer. Nature reviews Cancer. 2006;6:593–602. doi: 10.1038/nrc1947. [DOI] [PubMed] [Google Scholar]
- 46.Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009;41:1238–42. doi: 10.1038/ng.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, et al. A Molecularly Annotated Platform of Patient-Derived Xenografts (“Xenopatients”) Identifies HER2 as an Effective Therapeutic Target in Cetuximab-Resistant Colorectal Cancer. Cancer Discovery. 2011 doi: 10.1158/2159-8290.CD-11-0109. [DOI] [PubMed] [Google Scholar]
- 48.Ismail A, Bandla S, Reveiller M, Toia L, Zhou Z, Gooding WE, et al. Early G cyclin-dependent kinases as prognostic markers and potential therapeutic targets in esophageal adenocarcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:4513–22. doi: 10.1158/1078-0432.CCR-11-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maher EA, Brennan C, Wen PY, Durso L, Ligon KL, Richardson A, et al. Marked genomic differences characterize primary and secondary glioblastoma subtypes and identify two distinct molecular and clinical secondary glioblastoma entities. Cancer Res. 2006;66:11502–13. doi: 10.1158/0008-5472.CAN-06-2072. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.