

Abstract
Chemical probing represents a very versatile alternative for studying the structure and dynamics of substrates that are intractable by established high-resolution techniques. The implementation of MS-based strategies for the characterization of probing products has not only extended the range of applicability to virtually all types of biopolymers, but has also paved the way for the introduction of new reagents that would not have been viable with traditional analytical platforms. As the availability of probing data is steadily increasing on the wings of the development of dedicated interpretation aids, powerful computational approaches have been explored to enable the effective utilization of such information to generate valid molecular models. This combination of factors has contributed to making the possibility of obtaining actual 3D structures by MS-based technologies (MS3D) a reality. Although approaches for achieving structure determination of unknown substrates or assessing the dynamics of known structures may share similar reagents and development trajectories, they clearly involve distinctive experimental strategies, analytical concerns, and interpretation paradigms. This Perspective offers a commentary on methods aimed at obtaining distance constraints for the modeling of full-fledged structures, while highlighting common elements, salient distinctions, and complementary capabilities exhibited by methods employed in dynamics studies. We discuss critical factors to be addressed for completing effective structural determinations and expose possible pitfalls of chemical methods. We survey programs developed for facilitating the interpretation of experimental data and discuss possible computational strategies for translating sparse spatial constraints into all-atom models. Examples are provided to illustrate how the concerted application of very diverse probing techniques can lead to the solution of actual biological substrates.
Keywords: crosslinking, footprinting, solvent accessibility probes, covalent labeling, structural probing, molecular modeling, structural biology
In the last decade, several factors have reawakened a keen interest in the development of chemical methods for the structural elucidation of biopolymers and their assemblies. Driven by the need to understand the function of new gene products identified at unprecedented rates, the demand for structural information has greatly intensified as a result of the completion of the Human Genome Project1–3 and large scale proteomics initiatives.4, 5 The need for 3D-structure elucidation has been further stimulated by the discovery of riboswitches6, 7 and the realization that sequence information alone is not sufficient to deduce the function of the estimated 98.5% of human genome that does not code for actual proteins.8, 9 In response to the growing demand, large research centers dedicated to structural genomics have been created in the U.S. and abroad to expand the availability of high-resolution structures and to reduce the cost of structural information.10 The weight of these efforts is carried by established high-resolution techniques, such as X-ray crystallography and nuclear magnetic resonance (NMR), which are applicable to a broad range of biopolymers, but present also distinctive restrictions regarding solubility, quantity, size, heterogeneity, and structural flexibility of viable substrates. These intrinsic limitations have been hindering the pursuit of membrane proteins that are marginally soluble in aqueous solvents and highly prone to losing their native fold when removed from the natural lipid bilayer. Size and heterogeneity represent major obstacles to solving larger macromolecular assemblies that are known to support complex cellular functions by clustering together all the necessary components.11 The elucidation of these types of substrates could greatly benefit from the implementation of complementary approaches capable of circumventing such limitations.
Chemical probing represents an excellent source of structural information for substrates that are not directly amenable to established techniques. Different types of reagents can be employed to identify with excellent accuracy any susceptible functional group that may be accessible on the substrate surface, or juxtaposed by its fold.12, 13 In this way, chemical probing is capable of complementing the classic structural approaches and, ever more frequently, compensating for their inability to obtain high-resolution data, thus enabling the comprehensive structural determination of previously intractable substrates. In the case of known structures, the ability to assess the yield of modification can provide very valuable information about their dynamics, thus providing the sought-after insights necessary to understand their function. In recent years, the advances made by mass spectrometry (MS) in the characterization of chemically modified biopolymers have presented an excellent opportunity for revisiting structural probing, for increasing its range of applicability, and for exploring new strategies to tackle biological systems of ever increasing complexity. The concomitant development of powerful computational techniques for generating full-fledged models from sparse experimental constraints14, 15 has greatly increased the value of relatively low-resolution data afforded by chemical probing, thus realizing the possibility of obtaining the actual 3D structure of biopolymers by MS-based technologies (MS3D). In this Perspective, we describe the advances fostered by the implementation of MS detection in chemical probing approaches. We discuss experimental considerations to be taken in account when seeking spatial constraints for actual structure determination and point out critical differences with those addressed when investigating structure dynamics. We highlight the progress made by the development of computational tools to support the acquisition of experimental constraints and to translate this type of information into accurate 3D models.
The evolving trajectory of chemical methods in structural biology
Assessing the susceptibility of functional groups to specific chemical reagents constitutes a very versatile strategy for gaining valuable insights into their spatial situation. This possibility was recognized at a very early stage in the evolution of structural biology, before modern high-resolution techniques were developed. The propensity of exchangeable protons to swap with deuterons in D2O, which can be correlated to their solvent accessibility and structural context, was initially monitored in cellulose as early as 1938,16 in what represents the first example of biopolymer studied by hydrogen-deuterium exchange (HDX). In the mid-50's, HDX was applied to investigate protein conformation in germinal studies that employed gravimetric determinations or direct NMR analysis.17–20 The application of MS to determine the mass changes associated with HDX became possible only decades later, when the introduction of new ionization techniques made the analysis of biomolecules practical.21, 22 The possibility of determining the rate of exchange enables not only the identification of surfaces that are shielded from solvent contact by substrate fold or ligand binding, but also the investigation of substrate stability and conformational dynamics. It is in these types of studies that HDX-MS has grown to assume a prominent role, as discussed in depth by numerous excellent reviews (23–25 and refs. therein).
A similar trajectory was followed by the exploration of other chemical probes that, contrary to the preeminently reversible modification associated with HDX, produce irreversible labeling of surface-exposed regions. The application of selected reagents to introduce specific covalent modifications finds its roots in earlier approaches developed for investigating enzyme structure and mechanism, which took advantage of the inhibitory effects of group reagents to map the composition and architecture of catalytic sites.26–28 In analogous fashion, alkylating agents initially identified for their cytotoxic activity29 were employed to unambiguously identify nucleotides that were not protected by base pairing interactions in complex nucleic acid structures.30 Soon, mono-functional reagents became the basis for footprinting studies of proteins31, 32 and nucleic acids.33, 34 The ability of nucleic acid probes to produce strand scission at the modified site, or to inhibit strand elongation in primer extension reactions, enabled the direct utilization of polyacrylamide gel electrophoresis (PAGE) as a convenient detection platform. However, the absence of protein probes with analogous backbone-cleaving capabilities precluded the utilization of this analytical platform to tackle this type of biopolymer. At the same time, the possible application of Edman degradation to identify probed residues was hindered by intrinsic limitations in the analysis of mixtures and amino acid modifications. In contrast, MS-based approaches (Figure 1) do not rely on probe-directed scission to achieve product characterization and, therefore, allow for the direct application of mono-functional probes to substrates of protein and nucleic acid nature.35–40 The versatility and specificity of such technologies allowed for the utilization of individual reagents, as well as combinations thereof in multiplexed applications that took advantage of their unique mass signatures.41
Figure 1.

General operations and strategies followed to complete MS-based structural probing. In typical MS3D workflows, the substrate of interest is treated with the selected probe. The products can undergo purification/enrichment procedures, or can be analyzed directly by mass spectrometry, according to bottom up or top-down strategies (see text).
Broad-spectrum probes capable of reacting with diverse functional groups were also explored in search of an ideal reagent that would allow for the comprehensive evaluation of all the residues present in any type of biopolymer. The hydroxyl radical (OH˙) has come close to realizing this ideal on the strength of its ability to attack all four nucleotides and a large number of amino acid side-chains.42, 43 Initially, its application was traditionally limited to nucleic acid structures by the utilization of PAGE-based detection, which can take full advantage of the backbone scission induced by this probe. However, the introduction of MS technologies that dispense with the need for probe-induced cleavage has facilitated its recent extension to protein substrates. The possibility of generating hydroxyl radicals directly in situ, either by Fenton reaction,42, 43 water radiolysis,44 or photolysis of hydrogen peroxide,45, 46 has been exploited to accomplish the characterization of interfaces between bound components47 and to pursue detailed analysis of substrate dynamics.48, 49 On the wings of the increasing diffusion of MS-assisted radical footprinting for these types of applications, extensive reviews have been dedicated to the subject over the years.50–52
If MS technologies have contributed to transcend the limits posed by PAGE detection to mono-functional probing, the beneficial effects have been even more significant for the application of bifunctional reagents in structural analysis. These types of probes form irreversible covalent bridges between functional groups that may be distal on the biopolymer sequence, but are juxtaposed or placed within striking reach by the substrate structure. Therefore, the correct identification of modified sites requires the concurrent sequencing of the bridged sections, which is typically difficult by PAGE, but can be accomplished with no ambiguities by MS.53–55 Unlike mono-functional probes that can provide the position of solvent-protected residues, but fail to identify the structure responsible for protection, crosslinking agents are capable of recognizing pairwise residues that were located within the probe span. This characteristic enables the direct evaluation of inter-residue distances in proteins56–58 and nucleic acids,59, 60 which is at the basis of their application to structure determination. At the same time, the possibility of latching onto functional groups that are juxtaposed only temporarily by transient conformations makes them ill-suited for the investigation of dynamics. The ability to obtain actual spatial relationships has been employed to reveal the local arrangement of contiguous secondary structures,61, 62 to determine long-range interactions defining tertiary and quaternary structure,63–65 and to positively identify the different components of large multi-subunit assemblies.66–68 The value of crosslinking information has spurred a great deal of interest in exploring new reagents and analytical approaches for product characterization, which have been extensively reviewed.69–71
Reaping the benefits of MS technologies
The specificity, sensitivity, and speed afforded by MS technologies, which are now capable of accomplishing full characterization of natural and modified biopolymers in unprecedented scale, have contributed to expand significantly the scope of structural probing. At the same time, intrinsic aspects of structural probing make this approach an excellent fit for MS-based analysis. Indeed, the fundamental operations included in these studies parallel those involved in the processing of photographic film. Exposing a substrate to chemical probes creates an "impression" of its structural organization in the form of a permanent modification pattern, which can be subsequently "developed" without worrying about preserving the original fold. This critical feature lends great flexibility to the selection of methods for reading the modification pattern and obtaining the sought-after spatial constraints. With no concerns about denaturing the probed products after the fact (Figure 1), MS-based strategies are directly applicable to virtually all biomolecules and their mixtures, affording numerous advantages over techniques that require chromophores or probe-specific cleavage. The elemental composition of intact molecular ions and corresponding gas-phase fragments is very characteristic for each class of biopolymers and modifiers, alike. Therefore, unambiguous identification can be readily achieved from their unique mass signatures.
Within the broad realm of possibilities stemming from the covalent nature of probing products, the selection of appropriate sample handling and analytical protocols must take in account the intended purpose of a given experiment. Applications aimed at structure determination depend on the unambiguous identification of modified residues, but those aimed at assessing substrate dynamics are based on the ability to obtain accurate adduct quantification. In the latter, the location of susceptible residues may be already known, whereas the amount of modified product, either in absolute terms, or relative to its unmodified counterpart, represents the sought-after information. For this reason, dynamics applications must pay particular attention to any possible source of bias that may lead to inaccurate assessments of product yield, such as adduct instability, inhibition of enzymatic cleavage, modification of chromatographic properties, alteration of ionization efficiency, etc.23–25, 51, 52 The need for accurate quantitative information is somewhat less stringent for structure determination, although its importance will be expected to increase with the development of advanced methods for model validation. Whenever the suspicion of inaccuracy arises, alternative experimental strategies and stricter controls must be considered to stave off possible bias, or at least to estimate its extent.
Typical bottom-up approaches for adduct characterization involve denaturing the probed material by addition of chaotropic and reducing agents, followed by chemical/enzymatic hydrolysis to obtain smaller products that are directly amenable to mass mapping. The masses exhibited by the various species are compared with those predicted from the substrate sequence, taking in account the residue specificity of the hydrolytic step. Probed products are clearly identified by the characteristic deviation corresponding to the modifier's incremental mass. Taking this concept a step forward, the incorporation of ad hoc isotopic labels in the probe structure has been accomplished to facilitate the identification of modified species in complex hydrolytic mixtures.72–76 This characteristic is an intrinsic property of the products of HDX experiments. Unlike other mono-functional reagents, however, transferred deuterons can back-exchange with protons when hydrolysis operations are performed in aqueous medium, thus leading to the possible loss of structural information. For protein targets, this potential problem is minimized by working in acidic environment and by employing proteases that are active at low pH (e.g., pepsin).23–25 New probes have been recently introduced, which undergo partial cleavage under appropriate environmental conditions to produce diagnostic products that facilitate the detection of informative species in complex biological mixtures76, 77 Under typical bottom-up conditions, however, the vast majority of chemical probes produce stable covalent modifications that are not readily reversible in solution, thus minimizing the risk of information loss.
Bottom-up approaches must contend with the possibility that chemical modification may affect the activity of specific proteases employed for product characterization, thus resulting in partial or complete cleavage inhibition. If possible, an absolute match between probe and enzyme specificities should be avoided at the planning stage, where the utilization of alternative cleavage protocols should be carefully considered. Chemical modification can also alter the pKa of the targeted functional group, which may result in unwanted variations of the overall analytical response afforded by the modified product. A case in point is provided by lysine-specific reagents capable of transforming the side-chain amine into either an amide group that prevents protonation, or an ammonium group that exhibits a fixed positive charge. The effects may be felt not only as a variation of enzymatic cleavage efficiency, but also as a reduction of the product's ionization efficiency and gas-phase stability. Further bias between modified and unmodified species may be introduced during possible separation steps, which may hamper the quantification efforts associated with dynamics studies.
Adduct stability in the gas phase is an important consideration for the implementation of tandem mass spectrometry (MS/MS)78 to locate the modified residues within the sequence of hydrolytic products. If only one susceptible residue is present in a given digestion product, then there is no ambiguity about the modification site. However, sequencing data become necessary when two or more residues could be modified by a certain probe. Activating the selected precursor ion with different types of energy leads to the formation of product series that are characteristic for each type of biopolymer and their primary structure.79, 80 When the modification is sufficiently stable to withstand the dissociation process, its unique mass shift can be clearly observed in the characteristic ion series, which unambiguously identifies the modified residue. In some cases, the ability of the probe moiety to undergo partial cleavage has been exploited to either leave readily recognizable remnants attached to the original product, or to produce unique reporter ions that signal the presence of crosslinked species in the mixture.81–86
Probe-associated fragmentation becomes problematic when it represents the preferred dissociation event over the backbone fragmentation that originates informative sequence ions. This situation arises, for example, when specific nucleic acid probes weaken the N-glycosidic bonds of modified nucleotides to induce predominant base-loss and minimal backbone dissociation. As shown in Figure 2a, abundant signals corresponding to facile loss of methyl and methylguanine were observed for an adduct obtained by treating a target RNA construct with the mono-functional probe dimethylsulfate.87 Initially, sparse sequence ions did not allow for the position of the newly created abasic site to be correctly assigned on the oligonucleotide primary structure. The impasse was overcome by isolating and activating the initial fragments in consecutive steps of tandem mass spectrometry (MSn)88 to finally achieve the sequence coverage necessary to locate the modification site (Figure 2b, c).87 Base on this favorable observation, submitting first generation products to further steps of tandem MS should be capable of providing the necessary sequence information for adducts that undergo predominant cleavage of the covalent bridge between crosslinked moieties. Conversely, the opposite case scenario may take place for chemical modifications that increase the stability of precursor ions in the gas-phase, thus limiting the extent of attainable sequence coverage. This situation has been described, for example, for peptides containing oxidized cysteines and methionines produced by radical footprinting.89 In this case, adequate backbone fragmentation was restored by the implementation of electron detachment dissociation (ETD).90 In similar fashion, the increasing diffusion of alternative activation techniques will be expected to facilitate the MS/MS characterization of crosslinked conjugates of protein-protein,81, 91, 92 nucleic acid-nucleic acid,93–96 and protein-nucleic acid nature.97–103
Figure 2.

Multiple stages of tandem MS for the characterization of mono-methylated adduct of the RNA oligonucleotide SL3: (a) products obtained by isolation/activation of monomethyl SL3 (MS2, precursor ion m/z 1617.02); (b) isolation/activation of the first-generation fragment produced by loss of methylguanine (MS3, m/z 1617.02 → m/z 1576.55 → products); (c) isolation/activation of the second-generation fragment induced by consecutive losses of methylguanine and guanine (MS4, m/z 1617.02 → m/z 1576.55 → m/z 1538.70 → products). All precursor ions selected at each stage had a 4– charge state. Reproduced with permission from reference 87.
Analogous considerations apply also to top-down approaches that dispense with hydrolytic procedures, but seek to obtain direct sequence information by submitting intact substrates to MS/MS.104 A streamlined experimental design and modest sample consumption are rapidly making top-down analysis the approach of choice for structural probing.92, 105, 106 Indeed, eliminating the digestion/mass mapping step may help reduce overall sample demands by minimizing handling losses, while also eliminating any possible bias introduced by specific enzyme activity. On the other hand, energetics considerations may limit the practical size of probed substrates that can be fully characterized. As discussed earlier, popular probes provide adducts that are stable in the gas-phase, or dissociate into recognizable products under typical top-down conditions. The exception is again represented by deuterons transferred by HDX, which can scramble their position with un-exchanged protons during collision-induced dissociation. Recent studies have shown, however, that non-ergodic activation techniques can minimize scrambling and preserve the integrity of the structural information.107–109
Hyphenated techniques combining MS detection with capillary electrophoresis (CE-MS)110, 111 and liquid chromatography (LC-MS)112, 113 have also an important role to play in supporting the characterization of probed products. The implementation of a separation step at the front-end relaxes the requirements imposed on the back-end analysis by eliminating unreacted probes and necessary salt additives, by resolving complex sample mixtures from probing and hydrolysis reactions, and by concentrating the desired adducts in discrete elution bands. On one hand, the concentrating effects typical of these experimental schemes facilitate the determination of samples present in limited amounts. On the other, however, they may introduce unwanted bias between modified and unmodified species, which may hamper dynamics applications. Whenever possible, the selected separation step has taken full advantage of unique physico-chemical properties exhibited by probed products to achieve their selective enrichment over the corresponding unmodified species. For example, the strong interaction between phosphate and TiO2 has been exploited to concentrate peptide-RNA conjugates obtained by digesting photocrosslinked protein-RNA complexes.114 A 2D chromatography system involving both TiO2 and hydrophobic stationary phases was implemented to selectively separate the desired hetero-conjugates from unmodified peptide and oligonucleotide species representing the bulk of the digestion products (Figure 3).
Figure 3.

Schematic overview of a 2D chromatography setup for the automated enrichment of peptide-RNA conjugates derived by complete hydrolysis of protein-RNA complexes irradiated with UV light. Interactions between phosphate groups and TiO2 stationary phase are responsible for the selective retention of nucleic acid components. Hetero-conjugate enrichment is achieved through a multistep process involving the concerted utilization of appropriate C18 traps. Adapted with permission from reference 114.
Selective enrichment of probed species has been also achieved by including specific affinity tags in the reagents structure, which provided the basis for effective capture strategies. Biotinylated crosslinkers have been synthesized to perform affinity purification of probed products by using streptavidin-coated phases.75, 115–118 Concerns about the very high affinity of the biotin-streptavidin interaction, which may hamper the release of captured species for MS analysis, have lead to the introduction of regents in which the tag itself can be readily cleaved off under appropriate environmental conditions.77 Concerns about possible streptavidin contamination of analyte mixtures, especially for those generated by in situ protease treatment of captured material, have lead to the investigation of protein-free retrieval strategies.119 The possibility that the size of the biotin group may somehow affect probe activity has prompted the introduction of strategies in which the desired tag is added only after the probing reaction is complete.120 Considering that the yields of probing reactions are generally low, either by choice or happenstance (vide infra), the ability to enrich the desired products from complex reaction mixtures is often crucial to the success of a probing experiment. For this reason, great efforts are still dedicated to the development of new strategies for increasing the sensitivity and dynamic range achievable in the analysis of conjugated species for assembly characterization and 3D structure determination.121
The perils of chemical methods
Chemical methods tend to be exquisitely sensitive to conformational changes taking place in solution, which may transiently expose susceptible functional groups to probe activity. On one hand, this characteristic has been successfully harnessed to enable the investigation of structure dynamics, which constitutes the prevailing objective of HDX and radical footprinting applications. In this direction, the recent implementation of laser flash photolysis to complete footprinting experiments on a time scale that is faster than typical protein unfolding promises to enable the study of rather short-lived conformational processes.122 On the other hand, however, this characteristic may also represent a possible source of complications when the goal is obtaining unambiguous distance constraints for modeling applications. In the case of bifunctional crosslinkers, for example, a mismatch between substrate dynamics and reaction kinetics can result in the formation of stable covalent bridges across functional groups that are placed only temporarily within crosslinking range. Known as "kinetic trap", this effect inadvertently amplifies the incidence of conformations that are not highly populated in solution, thus providing an inaccurate representation of the structure of interest. In the case of mono-functional reagents, initial modification of susceptible residues can induce unwanted conformational changes that could expose previously protected regions and make secondary sites accessible to the probe. Including these constraints in subsequent modeling operations would provide a distorted view of the structure under investigation. Although, the possibility of incurring in different types of artifacts is intrinsic to probing applications and cannot be completely eliminated, their incidence can be minimized, or at least recognized, by appropriate experimental design.
Possible artifacts can be minimized by operating under ideal "single-hit" conditions, which should allow for each substrate molecule to be modified only once. On one hand, a "single-hit" eliminates the possibility that further modifications may occur at secondary sites exposed by structure distortion. On the other, however, such experimental conditions tend to produce very low reaction yields, which can hamper subsequent steps of adduct characterization. The fact that the single modification may take place in any exposed site in different molecules ensures the ability to obtain a comprehensive view of the structure of interest. Ideal "single-hit" conditions may be achieved by completing the reaction in the shortest possible time, either through rapid quenching or flash photolysis-like implementations, or by keeping the probe to substrate ratio as low as possible in solution. A series of inter-dependent factors should be weighed when calculating the ratio, including the total number of susceptible residues in the substrate sequence regardless their possible protection status; the typical reactivity of the selected probe; the presence of buffers that may affect chemical reactivity; the pH, ionic strength, temperature, and duration of the probing reaction.123–126 Considering that these factors can have widely different effects on different structures, optimal values are very much substrate-dependent and are difficult to predict a priori. For this reason, similar substrates of known behavior constitute very valuable samples for establishing baseline conditions that can be later optimized by using the actual substrate of interest. In this direction, the MS platform enables the implementation of titration schemes, in which the amount of probe is progressively increased and the number of modified sites is monitored by direct analysis of the resulting reaction mixtures (Figure 4). The occurrence of probe-induced conformational changes or kinetic traps is typically revealed by an abrupt jump in the adduct number as a function of reagent concentration.127 Another helpful approach for flagging situations that may be prone to kinetic effects involves the concerted application of nested crosslinkers that present the same reactive groups at the end of modular spacers of increasing span.128 The detection of residue pairs crosslinked by reagents with widely different spacing tends to indicate highly dynamic regions with high risk for trapping effects. Recognizing the conditions at which structural transitions may occur allows for the utilization of higher probe to substrate ratios that are capable of providing better product yields without affecting the accuracy of the structural information.
Figure 4.

Monitoring myoglobin probing with diethylpyrocarbonate (DEPC): (a) representative ESI-MS spectrum; (b) dose response plot. [P]0 and [P] are the initial and final concentration of unmodified protein inferred from the corresponding peak areas. [X]0 and [X] are the DEPC concentrations. The observed deviation from linearity suggests a protein structural change due to the modification. Adapted with permission from reference 127.
Possible artifacts can be ruled out also by designing proper control experiments based on specific properties of the target structure, which can be inferred from earlier probing rounds or other sources. Probing could be repeated after introducing small variations of pH, ionic strength, and temperature, which are expected to have more significant effects on relatively unstable transient conformations than on stable folded structures.87, 129 Site-directed mutagenesis could be employed to severely destabilize a certain fold, which would be expected to result in the elimination of salient crosslinks.65, 130 Mutagenesis is particularly effective for validating long-range tertiary interactions that determine the spatial relationships between contiguous domains and define the global fold of target substrates. Appropriate controls increase the level of confidence in the corresponding spatial constraints and, consequently, in the final structures. Information associated with crosslinking products that are invariably detected under a broad range of conditions should carry higher confidence than products observed only under more narrowly defined conditions, which may not represent the most populated fold. Sensitivity to environmental conditions, reproducibility in repeated experiments, and mutagenesis support are just some of the criteria that should be employed to evaluate the attained experimental constraints. These observations could constitute the basis for scoring algorithms designed to rank the constraints and to guide their selection for subsequent modeling operations.65 In combination with accepted methods for statistical analysis included in current modeling packages, these scoring algorithms could be employed also to assess the confidence level in the final model and to compare the quality of MS3D structures.
Possible ambiguities can also arise from factors capable of influencing the baseline reactivity of specific functional groups. Indeed, the local chemical environment may still affect the reactivity of accessible groups by establishing hydrogen bonding, or other weak interactions, which may not translate into actual steric protection, but may significantly modify the pKa of susceptible residues. The result could be a reduced adduct yield that could be erroneously interpreted as an indication of relevant structural effects. Again, this situation is more likely to affect dynamics applications that need to be sensitive to subtle conformational variations. In some cases, repeating the probing experiment under slightly different conditions to destabilize the putative feature may help confirm its actual existence. In general, a careful examination of the final 3D model is expected to enable the recognition of sites that may be conducive to local effects, which will then become the object of additional control experiments.
Finding a needle in a stack of needles
The characterization of probing products is generally based on mass mapping and MS/MS sequencing data. The possible presence of side reaction products, incomplete “dangling” conjugates, hydrolyzed crosslinkers, probe impurities, and partially digested products can greatly complicate the interpretation of mass spectra obtained from these types of sample mixtures. The majority of software tools for the identification of post-translational modifications in proteins can handle also mono-functional adducts generated by solvent accessibility probes. Programs such as GPMAW,131 MASCOT,132 Protein Prospector,133 and others offer the possibility of adding user-defined modifications, such as those introduced by mono-functional probes. However, typical customization capabilities do not extend to protein-protein crosslinks and do not support the interpretation of their MS/MS data. Only a few programs enable automated interpretation of data obtained from peptide-peptide, peptide-oligonucleotide, and oligonucleotide-oligonucleotide conjugates (summarized in Table 1). Some of these tools are freely available to the public either through dedicated web-servers, or as stand-alone programs. Although they share a similar underlying philosophy, they exhibit different strengths/limitations and have different ranges of applicability, workflows, and I/O file formats. In general, the mass-over-charge ratios (m/z) observed in a certain spectrum are compared with theoretical values predicted from user-defined sequence(s) according to crosslinker identity, residue specificity, and biopolymer cleavage rules. These libraries contain values for all possible combinations of crosslinked, mono-adducted, and unmodified species. Experimental peaklists and theoretical libraries are matched according to a user-defined error accounting for the accuracy of the actual data under consideration, which places a premium on high accuracy determinations to minimize mis-assignments and ambiguous interpretations.
Table 1.
Programs employed to support the MS-based identification of probing products. Summarized are features that determine their performance, such as maximum number of supported sequences, customizability of modifications, integrated spectrum processing, ability to output theoretical mass lists, support for tandem MS analysis, availability of scoring functions to evaluate assignments, and user accessibility. Refer to the text or the provided citation for more details.
| Software | Max # of sequences |
Customizable search |
Spectrum Processing |
Output theoretical mass list |
MS2 data |
Scoring function |
Unique feature | Availability | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| NIH-XL | 2 | limited | offline | - | - | stand alone | 89 | ||
| GPMAW | 2 | limited | offline | - | - | stand alone | 131 | ||
| FindLink | 2 | yes | offline | - | - | - | stand alone | 137 | |
| LINKS/ MS2LINKS |
20 | yes | yes§ | yes | yes | - | customizable MS2 ion search; nucleic acids MS & MS2 data analysis |
CMS3D portal https://ms3d.org/ |
139 |
| X-Link | 2 | yes | offline | no | yes | - | MIX-tagged mass marker peaks for crosslink ID |
stand alone | 135 |
| SEARCHXLINKS | multiple | yes | offline | no | yes | yes | integrated MS & MS2 data analysis; adjustable parameters in MS2 scoring scheme |
webserver* | 136 |
| CLPM | 2 | yes | offline | no | no | - | outputs best match only | webserver http://bioinformatics.ualr.edu/mbc/services/CLPM.html |
138 |
| Pro-CrossLink | 2 | yes | yes¶ | no | yes | yes | streamlined workflow to identify crosslinks |
stand alone http://goodlett.proteomics.washington.edu/helpful_links/ |
155 |
| VIRTUALMSLAB | proteome | yes | offline | yes | no | - | multi-stage experiment editor; integrated output sorting |
stand alone | 134 |
| X!Link | proteome | yes | offline | no | yes | yes | shotgun method | stand alone | 147 |
| MS-Bridge | proteome | yes | offline | yes | no | - | webserver http://prospector.ucsf.edu/mshome.htm |
133 | |
| CrossSearch | 2 | limited | offline | no | no | - | webserver http://talmud.umkc.edu/prot_cross3/ |
144 | |
| iLINK/doLink | multiple | limited ( only bis-NHS ester crosslinkers) |
yes¶ | no | yes | yes | integrated spectrum processing, data viewer and annotation |
stand alone http://tools.proteomecenter.org/wiki/index.php?title=Software:Xlink |
156 |
| XQUEST | proteome | yes | yes¶ | no | yes | yes | search engine with stringent spectrum matching |
stand alone | 145 |
| MSX-3D | 2 | yes | offline | no | no | - | with integrated 3D viewer (PDB file input) |
PBIL portal http://proteomics-pbil.ibcp.fr |
179 |
Webserver does not include data reduction component.
Spectrum processing supports identification of isotope pairs.
URL link is not available.
Several programs have been developed to support specifically the interpretation of data from crosslinking reactions (Table 1). For example, VIRTUALMSLAB,134 X-Link,135 Links/MS2Links,87 MS-Bridge,133 SeachXLinks,136 Findlink,137 CLPM,138 and others offer the ability to handle multiple protein sequences, custom modifications, and peaklists obtained from both MS and LC-MS analysis. Often, these types of programs are made available through portals or suites that offer a broad range of support tools.133, 139 VIRTUALMSLAB presents the unique feature of allowing users to define the sequence by which probing and cleavage reactions are performed, thus replicating the actual wet-lab experimental design. This characteristic enables to minimize possible ambiguities arising from the fact that, for example, the addition of probe before or after proteolysis can lead to different theoretical mass lists, which could greatly affect the matching with actual experimental data. The program is also capable of filtering the theoretical library, as well as the matched peptides, according to different sorting criteria that help evaluate their significance. Links and MS2Links represent recent upgrades of ASAP140 and MS2Assign,141 which afford the unique capability of handling both homo- and hetero-conjugates of the different biopolymers. This added flexibility has greatly facilitated the MS3D investigation of proteins,92, 105, 106 nucleic acids,62, 65 and their functional assemblies.142, 143 In addition to data interpretation, these programs allow for the preview of theoretical mass lists before the actual experiments are performed, which offers the opportunity to perform a risk-free, in silico evaluation of the information content expected from a prospective probing reaction. This functionality enables to streamline the experimental design by identifying the best possible set of reagents capable of maximizing sequence and surface coverage of the target substrate. At the same time, it can help stave off possible ambiguities in the identification of crosslinked products by guiding the selection of more favorable reagents. In similar fashion, the MS-Bridge application of the Protein Prospector suite133 offers the ability of customizing modifications and crosslinks, while also providing the additional capability of performing searches against sequence databases.
Increasing the number of subunits and custom modifications considered in the same application results in an exponential increase of the size of the corresponding theoretical libraries, which can affect at different levels the performance of mass matching algorithms. Indeed, the analysis of mono-functional adducts involves calculating the mass of all possible products containing the susceptible residue(s), while crosslinks analysis requires also predicting all possible peptide-peptide, peptide-oligonucleotide and oligonucleotide-oligonucleotide combinations that may be generated by a given substrate. The number of possible combinations is a function of substrate size and total number of residues that may be susceptible to the selected probe. Exhaustive explorations of sizeable libraries place increasing demands on the computational resources and greatly increase the time necessary for completing the task. With the purpose of streamlining this operation, programs such as CrossSearch144 and Links offer the possibility of restricting the search space by eliminating peaks from the input peaklist, which were assigned earlier by control experiments. At the same time, however, larger theoretical libraries increase the sheer probability of incurring false positive matches that are not readily recognizable by the available algorithms. For this reason, resulting outputs tend to require a significant amount of manual interrogation, which can negate any gain of speed and convenience afforded by the utilization of software aids. The pursuit of increasingly complex macromolecular systems will require the development of new algorithms capable of maximizing the benefits of computational power, while minimizing interpretation errors.
In this direction, the program xQuest145 can either generate comprehensive libraries from a user-defined database of up to 100 different proteins, or switch to the so-called "ion-tag mode" for proteome-wide analysis. This approach involves indexing common fragment ions produced by MS/MS of isotopically labeled crosslinked peptides, identifying all peptide sequences (or tags) capable of generating such fragments, and then reconstructing the corresponding MS/MS spectra. The goodness of fit between experimental data and reconstructed spectra is employed for scoring purposes, while Linear Discriminant Analysis (LDA)146 is used to distinguish between true and false positive matches. X!Link was also developed to streamline spectral analysis by direct examination of LC-MS/MS data from crosslinked peptide digests.147, 148 In a characteristic shotgun approach, this program assigns crosslinks according to the accurate mass of parent ions and double filtering of corresponding MS/MS data (i.e., combining the total percentage of sequence ions matched with percentage of precursor ions matched among the top 10 most intense peaks). Discrimination against false positives is achieved by utilizing a statistically optimized filter and a probability-based scoring system. This program is readily applicable to inter-peptide conjugates produced specifically by homo-bifunctional crosslinkers that target amino groups, but does not handle mono-adducts and intra-peptide crosslinks. Similar programs employing database searching and specific scoring schemes have been developed for multiprotein complexes and global crosslinking analysis,120, 149, 150 which enable the assessment of whole proteomes in a single search run. While the shotgun approach provides a seamless and high-throughput way of analyzing crosslinking data, there is still a lot value in examining experimental data to ascertain whether the probing reaction produced structural distortion and artifactual modifications.
The importance of experimental design in facilitating positive identification cannot be discounted. Different approaches have been devised to narrow the focus of analytical and data interpretation procedures to the products of interest, while eliminating from consideration the remaining regions that were not targeted in the probing experiment. The introduction of stable isotope labels either in the probe structure,76, 151, 152 or at the termini of conjugated species during cleavage,153, 154 can provide unique isotopic signatures that represent readily recognizable diagnostics for product identification and quantification. Software packages, such as Pro-Crosslink155 and iLink/doLink,156 support the interpretation of data obtained from isotopically-labeled samples. Pro-Crosslink is an integrated suite of tools for the automated assignment of mass mapping and sequencing data, which can handle substrates digested in the presence of H218O. Peaklists from LC-MS analysis are screened for species bearing 18O atoms that are introduced by hydrolysis at the C-terminus. In this way, the algorithm can readily flag peptide-peptide conjugates, which are recognizable from the characteristic isotopic signature conferred by two labeled C-termini, and designate them for MS/MS sequencing to complete characterization. The masses of each precursor ion and corresponding fragments are employed for mass matching with a theoretical library. The assignments are scored to provide a measure of confidence level. In similar fashion, iXLink/doXLink includes built-in utilities for recognizing the characteristic signatures provided by isotopically-coded (heavy versus light) crosslinkers and 18O end-labels. Also in this case, the products of interest are automatically flagged for MS/MS analysis and the results are scored according to a matching function. The presence of corresponding labeled and unlabeled products facilitates their respective quantification, which is particularly valuable for dynamics applications.
Knowledge of typical fragment ions produced by the various biopolymers in MS/MS experiments has allowed for well-defined dissociation rules to be included in the software tools, which have contributed to enhance the efficiency and accuracy of the automated data interpretation procedures. Knowledge of typical fragments obtained by dissociation of intra- and inter-molecular crosslinks is more limited and has not reached the level where corresponding rules have been hard-coded in interpretation algorithms. Nevertheless, the investigation of the gas-phase behavior of different crosslinked peptides has lead to the identification of distinctive marker ions associated with certain bridged residues.141, 157 To date, these studies have focused on amine-specific homo-bifunctional crosslinkers,123, 158, 159 while other classes of reagents have received little attention. In combination with typical sequence ions, crosslinker-specific markers would be expected to greatly enhance the coverage and accuracy achieved by mass assignment algorithms. These figures of merit could be further enhanced by the inclusion of parameters accounting also for fragment ion intensity and isotopic distribution.
Translating probing data into spatial constraints
Utilizing the information afforded by covalent labeling and crosslinking approaches can be deceptively simple. In reality, a series of considerations need to be properly addressed to translate probing data into spatial constraints that can be safely utilized in molecular modeling operations. The data afforded by HDX and radical footprinting experiments can be readily employed to discriminate regions that are permanently exposed to the solvent from those that are subjected to significant structure dynamics. These regions can be mapped on available Protein Data Bank (PDB)160 structures to identify interfaces between bound components and to highlight flexible domains. Algorithms, such as DSSP,161 NACCESS,162 GETAREA,163 and ASA-VIEW164 calculate the area of accessible surfaces and their gradients, and enable quantitative correlations between reactivity and solvent exposure.35, 39, 165–174 Although this type of information alone is clearly insufficient to support direct model building operations, it can be readily employed to guide the selection among models generated, for example, by homology or course-grained approaches (vide infra).
Bifunctional probes, instead, are capable of determining the actual distance between susceptible functional groups, which can be employed to triangulate their reciprocal positions in 3D space. The feasibility of measuring distances by crosslinking experiments and the accuracy of the corresponding constraints have been evaluated on model proteins of known 3D structure.63, 74, 105, 175 In these studies, the Euclidean distance corresponding to the shortest path between bridged residues was measured in the actual PDB structure and then compared with the reported spacing between the crosslinker's reactive groups. A crosslink measurement was considered valid when the distance between bridged residues was equal or lower than the maximum spacer length. Unfortunately, the latter may not be known with sufficient accuracy, owing to the intrinsic flexibility afforded by typical spacer structures, especially those including series of methylene groups (Figure 5). When all conformational degrees of freedom are taken in account, the vast majority of crosslinkers tend to cover a broad range of permissible distances, as demonstrated by molecular dynamics (MD) simulations of some commercially-available reagents, which found significant deviations from reported values.176 Indeed, the crosslinking literature tends to provide a single value that may represent either the mean or the maximum length of the spacer, while a measure of the possible range is seldom included. A practical consequence of conformational flexibility is that bifunctional reagents with same reactive groups but different spacer lengths (i.e., DSG, DSS, etc., Figure 5) possess widely overlapping span distributions. The concerted application of such analogs could potentially result in conjugation of the same residues pair, which would provide conflicting evaluations of their actual distance. This outcome would be particularly unfavorable in structure dynamics investigations. The introduction of molecular rulers with more rigid spacers (for example consisting of phenyl units)128 would be expected to minimize such ambiguities by narrowing the possible span distributions.
Figure 5.

Structure of common bifunctional N-hydroxysuccinimide esters targeting amino groups: disuccimimidyl tartarate (DST); disuccinimidyl suberate (DSS); disuccinimidyl glutarate (DSG); bis(2 [succinimidooxycarbonyloxy]ethyl)sulfone (BSOCOES); ethyleneglycol bis-(succinimidylsuccinate) (EGS); and bis(sulfosuccinimidyl)adipate (BSSA). *Reported N-N distances were obtained from Pierce reference sheets. **Average N-N distances and N-N distance range distributions were obtained from reference 176.
The intrinsic flexibility of the functional groups targeted by the different probes raises similar issues. For example, the side chains of residues that are placed on the substrate surface and are not constrained by stable interactions can enjoy high levels of conformational freedom, which are not adequately conveyed by the static representations afforded by individual PDB structures. This consideration applies in particular to lysine, arginine, and glutamate residues, which exhibit susceptible functional groups at the end of rather flexible side chains. The 3D space explored by individual side chains could place them within crosslinking distance, even thought their bridging may not have looked possible from a cursory inspection of the PDB. Trajectories obtained from MD simulations would be expected to provide ensembles of conformer structures, which would provide better representations for rationalizing experimental data.61, 177 Only a careful examination of the combined effects of side chain and crosslinker flexibility can provide the confidence necessary to discount improbable crosslinks and to rule out possible structures on the ground of crosslinking information.
The distance between bridged points is not the only piece of information afforded by crosslinking reagents. The fact that a conjugated adduct was positively detected implies not only that the connected residues are laid within proper reciprocal distance, but also that no intervening structure inhibited the closing of the initial "dangling" product. Unlike constraints obtained by Förster resonance energy transfer (FRET) and electron paramagnetic resonance (EPR) spectroscopy, the distances measured by crosslinking reagents are not through-space restraints. Steric effects can directly hinder crosslink formation by blocking the shortest path between susceptible groups. For this reason, alternative methods of evaluating crosslinking data have been developed, which take surface geometry and steric clashes into account. The method proposed by Potluri et al., for example, employs different representations of the substrate surface to find plausible lower and upper bounds of crosslinking distances, which are ranked according to the way the corresponding path intersects with the surface of the substrate.178
Once a product has been observed experimentally, its validity should be evaluated against any structural information that may be already available from initial homology modeling, previous probing rounds, or other structural techniques. Computational resources, such as MSX-3D, 179 enable the inspection of experimental constraints in the context of a target 3D model that can be interactively visualized by Jmol.180 Users can inspect crosslinked positions and look for a clear bridging path that can corroborate or contradict experimental data. The program can be also used beforehand to predict the possible outcome produced by a certain probe based on distance measurements from the available 3D structure. In this case, however, considerations of conformational flexibility should be accounted for by the concerted application of MD simulations, as discussed earlier.
Utilization of spatial constraints from probing data
Different approaches have been explored to take advantage of probing information. Crosslinking data have been successfully employed in the study of complex assemblies to guide docking experiments in which high-resolution structures were available for the individual subunits and their interaction surfaces were modeled according to inter-subunit constraints.181–184 More frequently, however, probing information is employed directly to support the generation of actual models, or indirectly to guide the selection of the most representative structure among a library of plausible models. In either case, the accuracy of the resulting structure depends on the quality of the experimental data and on the accuracy with which such data are translated into spatial constraints.
The direct utilization of probing information in model building operations is based on constraint satisfaction techniques analogous to those employed to solve structures from NMR data. Restrained simulated annealing185 and replica exchange molecular dynamics186 are typically implemented to calculate structure ensembles that satisfy all the experimental distance information. The ability to complete structure determination from sparse constraints, such as those derived by measuring the residual dipolar coupling in NMR, has been successfully demonstrated for the modeling of protein targets.187, 188 Fortunately, software packages developed to support these operations, such as anneal.inp of the CNS suite189 and GROMACS,190 are capable of accepting distance information regardless of the actual source. In this way, crosslinking constraints can be either enforced explicitly during model generation, or incorporated in penalty functions employed for conformational searches.191–194
Additional ways for effectively utilizing the experimental information involve the implementation of different de novo modeling approaches based on reduced representation methods and coarse-grained strategies,195, 196 which have inspired the development of programs, such as ROSETTA,197 ROBETTA,198 CABS,199 and MONSSTER.200 According to these approaches, side-chains and nucleobases are represented as discrete entities, or pseudo-atoms, to simplify the calculation of initial working models. The information afforded by crosslinking constraints can be readily utilized to triangulate the reciprocal position of pseudo-atoms in 3D space, which leads to models that reflect very closely the overall morphology of the target substrate.201–203 The intrinsically low resolution of such representations may preclude the attainment of atomic-level details, such as H-bonding networks, but may still allow for critical functional observations that could prompt otherwise unwarranted hypotheses. Fortunately, low resolution models can be upgraded to molecular-level resolution by the application of homology modeling or other structure prediction techniques.204
Finally, probing constraints can be employed to rank libraries of purely theoretical models and to identify the structure that best fit the available experimental observations.140, 205–207 Computational strategies, such as threading and homology modeling, are typically employed to generate plausible models based on residue/base composition, sequence alignment information, secondary structure prediction, etc. In the case of protein substrates, these strategies can count on the growing availability of protein templates from structural genomics efforts.208, 209 In the case of nucleic acids, a broad range of folding prediction algorithms has been developed, which apply stringent thermodynamics considerations to generate putative structures.204, 210, 211 Once an ensemble of models is generated, the experimental constraints are used as "filters" to accept/discard individual members, or to score them according to statistical criteria expressing best fit. Either way, the selected structure(s) will combine the atomic-level details of the theoretical templates with the actual experimental information to provide the best possible representation of the target substrate.
Regardless the strategy followed to generate them, initial models are typically submitted to energy minimization according to accepted protocols, which is necessary to relax any possible strain introduced by the modeling process. Completing this operation with or without crosslinking restraints can provide helpful information about the model under consideration. Minor deviations between the results observed before and after lifting the restraints should be expected as part of normal relaxation effects striving to correct unnatural angles, geometric violations, clashes, etc. Major variations, however, could indicate the possible manifestation of kinetic traps that might have forced the substrate into a certain fold only when the corresponding crosslinking information was enforced. Furthermore, the final minimized structure should be carefully examined to assess whether it would still allow for the actual formation of the observed crosslinks. Any major discrepancy in terms of distance and steric considerations would raise a flag about the fitness of the model, which should prompt a re-evaluation of the modeling operations.
These considerations underscore the distinctly iterative nature of a typical MS3D project. Indeed, the energy minimized model will be expected to prompt the design of further probing experiments by using reagents capable of targeting very unique features revealed by the model itself. The new set of experimental constraints could be employed to verify the validity of such model, or could be included in an new round of modeling, which should intrinsically result in a more accurate model owing to the greater number of spatial constraints. The iterative process should stop when a new set of information shows no significant discrepancies with the latest structure. It is important to stress also that, at the same time, the model should be expected to prompt other types of experiments aimed at testing its validity, which may involve the preparation of mutants to eliminate a critical interaction, the application of a possible inhibitor ligand, or other biological assays designed to test hypotheses stimulated by the observed structural features. These experiments, however, can provide an assessment based on very general criteria that should be applied to structures obtained by any type of approach, not just to those solved by structural probing, which should find their value and validation in the quality of the new functional insights and the number of testable hypotheses they help generate.
The rewards of structural probing
Recent years have seen a steady increase of examples in which MS3D approaches were not applied only to study well-known substrates with the objective of testing new experimental or computational strategies, but also to tackle poorly-understood systems of biological interest with the goal of achieving actual structural elucidation. In several studies, the tertiary and quaternary structures of protein targets were determined by employing crosslinking data to guide the selection of best-fit models generated by homology modeling.130, 135, 205, 207, 212–216 Targets lacking adequate templates with sufficient sequence similarity, which were not directly amenable to threading or homology modeling, were successfully studied by using 2D structure prediction algorithms to generate low to moderate resolution structures. In 2002, Back et al. first reported a low-resolution model of the prohibitin complex, an unknown protein structure, which was based on computer predictions substantiated by MS3D crosslinking data.137 The final model displayed the structures predicted for the various domains as individual blocks, which were appropriately oriented in 3D space according to the crosslinking information. In similar fashion, Dimova et al. used 2D prediction software to model the individual structures of peptide units constituting Munc 13.217 Six distance constraints from crosslinking experiments were subsequently employed in docking calculations to model the calmodulin-Munc 13 interaction (Figure 6). This model represents a very valuable reference point for studying the role of Munc 13 peptides, which are crucial in the mechanism of synaptic vesicle priming linked with Ca2+ signaling. Based on this structure, for example, candidate Munc 13 mutants could be constructed to evaluate their binding interactions by using the same photo-affinity labeling assay performed to obtain the modeling constraints.
Figure 6.

Model of the calmodulin-Munc13 peptide complex based on crosslinking and photoaffintiy labeling (PAL).217 Munc13 (13-1) peptide (blue) structure was predicted and modeled as an α-helix, and calmodulin (CaM, grey) was modeled after multiple CaM structures in the PDB. The complex was created using PatchDock and refined using ROSETTADock based on PAL of CaM residues M122, M124 (green) and 13-1 residue W7 (green), and crosslinks between13-1 residue K13 (purple) with CaM lysines (yellow, and red). Adapted with permission from reference 217.
Utilizing solved high-resolution subunits to assemble full-fledged structures through docking or simulated annealing is rapidly emerging as one of the strategies of choice for maximizing the benefits of structural probing.181–184 This strategy is capable of producing results that are comparable to those obtained by established structural methods, as demonstrated by the elucidation of the Ffh-FtsY complexes of E. coli and T. aquaticus, which were tackled by using the amine-specific crosslinkers DSS and BSSA (Figure 5). A total of nine intermolecular crosslinks identified by tandem MS was utilized to determine the binding interface and relative orientation of the different components of the Ffh-FtsY NG complex.218 In particular, the high-resolution structures of the NG domains of Ffh and FtsY were treated as “pseudo” rigid bodies by the program MODELLER219 to generate an ensemble of 1000 complexes that maximally satisfied all nine distance constraints. Inspecting the ensemble revealed that the models ranked in the top 10% were highly clustered and shared a tight binding interface and single interacting orientation. The selected models were further refined by rounds of docking and energy minimization to obtain the final MS3D structure (Figure 7a). Remarkably, when crystal structures were finally obtained for the complex,220, 221 they displayed a backbone that deviated by only 3.8 Å from that of the corresponding MS3D model. More importantly, however, the latter included also the M domain that was conspicuously absent from the crystal structure. The crosslinking data placed this domain in close proximity to the rest of the Ffh-FtsY complex (Figure 7b), which contradicted previous information and suggested possible dynamic roles in the targeting and trafficking processes performed by this substrate.
Figure 7.

(a) The model of T. aquaticus Ffh-FtsY NG complex was created by docking the apo-NG domains of Ffh (green) and FtsY (blue) according to crosslinking constraints.218 The MS3D model overlays perfectly with the crystal structure (grey) of the complex. (b) The structure of S. solfataricus SRP54 (gray) was superimposed with T. aquaticus Ffh (green) to generate a model for the Ffh_FtsY complex including the M domain. Relative positions of T. aquaticus lysine residues in the SRP54 M domain are mapped (green). These residues formed crosslinks with residues G(−3) and K62 of FtsY (magenta line), suggesting a close proximity of the M domain to the Ffh-FtsY complex interface. Adapted with permission from reference 218.
The potential for the elucidation of large membrane-bound complexes was demonstrated by Schulz et al., who employed ROSETTA with crosslinking data to elucidate the 3D structure of the full-fledged A2t complex.222 The high-resolution structures available for the N-terminal truncated annexin A2 (ANXA2) and p11-ANXA2 hetero-tetramer, which represent the essential components of this ~100 kDa octameric complex, were docked according to information provided by isotope-labeled amine crosslinkers of different spacer lengths. More specifically, three independent distance constraints were employed to appropriately place the C-terminal ANXA2 protein near the p11 dimer, whereas one constraint was used to position the N-terminal ANXA2 in relation to the rest of the complex (Figure 8). The resulting models were further refined by employing an additional set of unique distance constraints. The information afforded by "dangling" mono-functional products was not discarded, but was instead utilized to generate a surface topology map that confirmed the quaternary organization of the A2t complex.
Figure 8.
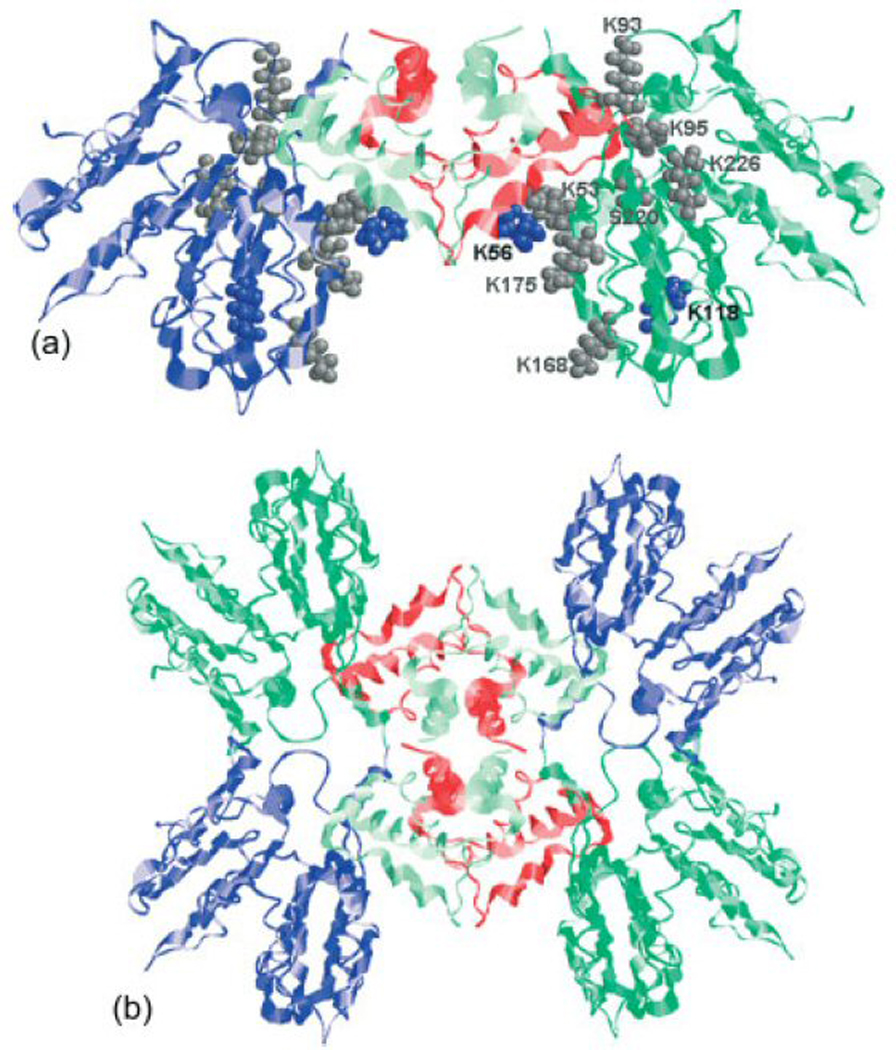
(a) MS3D model of the ~100 kDa ANXA2/P11 complex.222 The location of lysine residues involved in intra- and inter-protein crosslinking of the p11 dimer and full-length ANXA2 are marked as blue and grey spheres. (b) These crosslinks were used for docking calculations that provided a hetero-octameric A2t complex consisting of four ANXA2 (dark green and blue) located on the periphery connected by two p11 dimers (red and light green) in the center. Adapted with permission from reference 222.
Successful MS3D investigations have not been limited only to protein substrates, but have covered also complex nucleic acid structures. The reliance of these biopolymers on base-pairing interactions to define their higher-order structure has paved the way for the extensive utilization of solvent-accessibility reagents to reveal their specific pairing arrangements, which can be readily translated into very accurate 3D models by different computational strategies. For example, a combination of distance information from crosslinking probes and footprinting data from mono-functional reagents was employed to solve the 3D structures of two ribosomal frameshifting pseudoknots of retroviral origin.62 In this case, initial models were generated by the constraint-satisfaction algorithm MC-SYM223 and then energy minimized by CNS.189 In the final MS3D model of the mouse mammary tumor virus pseudoknot, the coordinates of atoms present in the double-stranded regions afforded an average root mean square deviation (rmsd) of only ~3 Å from the coordinates of the corresponding atoms of the actual NMR structure available in the PDB.224
The highly hierarchical nature of the RNA folding processes is particularly conducive to the divide-and-conquer strategy described earlier for protein complexes. In fact, such processes tend to proceed from the initial formation of discrete elements of secondary structure (e.g., stem-loop hairpins), which are stabilized by the annealing of contiguous sequences, to the formation of long-range tertiary interactions between them, which define the global fold of the RNA substrates.225 The size and crystallization properties of discrete domains frequently enable their solution by the established structural techniques, which can thus afford the desired high-resolution building blocks. In the case of the elucidation of the HIV-1 packaging signal (Ψ-RNA), initial footprinting experiments confirmed the presence of four stem-loop domains in the context of the full-length construct,65 which had been already solved separately by NMR and crystallography. This observation allowed for the utilization of inter-domain distances provided by crosslinking probes to guide the assembly of the individual high-resolution components into an all-atom 3D model of full-length Ψ-RNA (Figure 9a). The resulting structure exhibited a tight cloverleaf architecture stabilized by a distinctive GNRA-tetraloop receptor interaction between distal domains, which was modeled by homology with other known interactions of similar type (Figure 9b).65 The structure was confirmed by site-directed mutagenesis and paved the way for the examination of the roles played by Ψ-RNA in the context of the entire 5’-untranslated region of the HIV-1 genome.
Figure 9.

(a) MS3D model of the full-length Ψ-RNA generated using footprinting and crosslinking data.65 High-resolution structures for the discrete stem-loop domains SL1 (red), SL2 (green), SL3 (blue) and SL4(yellow) were employed as building blocks. Linker regions (orange) were generated de novo. (b) Details of H-bonding and interactions between SL1 and SL4 domains in the observed GNRA-tetraloop interaction. Adapted with permission from reference 65.
In conclusion, these few examples clearly demonstrate that the time has never been more ripe for the effective utilization of chemical methods for actual structure elucidation. The recent technological advances in the MS field have stimulated a revival of structural probing as a viable tool for pursuing the elucidation of biomolecules that elude the established high-resolution techniques. In particular, the implementation of MS-based approaches has greatly expanded the wealth of protein- and nucleic acid-specific chemistries that could be explored for probing applications. The advancements of instrumentation and experimental techniques for the characterization of probed biopolymers have been matched by the concomitant development of software tools for supporting the interpretation of data of increasing complexity and for translating such data into spatial constraints for molecular modeling. At the same time, the refinement of computational approaches for model generation and new strategies for utilizing sparse experimental constraints have increased the ability of successfully concluding probing projects with the attainment of high quality, valid 3D models that represent the substrate structure with excellent accuracy. In addition to applications to progressively larger multi-subunit assemblies, the flexibility afforded by the MS platform will be expected to enable the investigation of biopolymers of increasing complexity, such as glycoproteins implicated in immunological host-pathogen responses and lipoprotein complexes involved in membrane trafficking and signaling. The ability of probe molecules to cross cell membranes, combined with the sensitivity and specificity of MS-based detection, will be expected to result in increasing interest for in vivo probing applications, which could help reconcile high-resolution information obtained in vitro with the actual structure assumed by the substrate of interest in its natural environment. In this way, the integration of MS3D approaches with other structural biology techniques will lead to a better understanding of the structure-function relationship of substrates of great biological interest.
Acknowledgments
This publication was funded by the National Institutes of Health (GM643208 and RR019864) and by the National Science Foundation (Chem 043967). Sandia is a multiprogram laboratory operated by Sandia Corporation, a Lockheed Martin Company, for the United States Department of Energy’s National Nuclear Security Administration under contract DE-AC04-94AL85000.
References
- 1.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Raymond C, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blocker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowski J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C, Yao A, Ye J, Zhan M, Zhang W, Zhang H, Zhao Q, Zheng L, Zhong F, Zhong W, Zhu S, Zhao S, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An H, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigo R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi H, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Mays A, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu X, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen M, Wu D, Wu M, Xia A, Zandieh A, Zhu X. The sequence of the human genome. Science. 2001;291(5507):1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 3.Consortium IHGS. Finishing the euchromatic sequence of the human genome. Nature. 2004;431(7011):931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 4.Mann M, Hendrickson RC, Pandey A. Analysis of proteins and proteomes by mass spectrometry. Annu Rev Biochem. 2001;70:437–473. doi: 10.1146/annurev.biochem.70.1.437. [DOI] [PubMed] [Google Scholar]
- 5.Yates JR, Ruse CI, Nakorchevsky A. Proteomics by mass spectrometry: approaches, advances, and applications. Annu Rev Biomed Eng. 2009;11:49–79. doi: 10.1146/annurev-bioeng-061008-124934. [DOI] [PubMed] [Google Scholar]
- 6.Nahvi A, Sudarsan N, Ebert MS, Zou X, Brown KL, Breaker RR. Genetic control by a metabolite binding mRNA. Chem Biol. 2002;9(9):1043–1049. doi: 10.1016/s1074-5521(02)00224-7. [DOI] [PubMed] [Google Scholar]
- 7.Nudler E, Mironov AS. The riboswitch control of bacterial metabolism. Trends Biochem. Sci. 2004;29(1):11–17. doi: 10.1016/j.tibs.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 8.Pheasant M, Mattick JS. Raising the estimate of functional human sequences. Genome Res. 2007;17(9):1245–1253. doi: 10.1101/gr.6406307. [DOI] [PubMed] [Google Scholar]
- 9.Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, Kuehn MS, Taylor CM, Neph S, Koch CM, Asthana S, Malhotra A, Adzhubei I, Greenbaum JA, Andrews RM, Flicek P, Boyle PJ, Cao H, Carter NP, Clelland GK, Davis S, Day N, Dhami P, Dillon SC, Dorschner MO, Fiegler H, Giresi PG, Goldy J, Hawrylycz M, Haydock A, Humbert R, James KD, Johnson BE, Johnson EM, Frum TT, Rosenzweig ER, Karnani N, Lee K, Lefebvre GC, Navas PA, Neri F, Parker SC, Sabo PJ, Sandstrom R, Shafer A, Vetrie D, Weaver M, Wilcox S, Yu M, Collins FS, Dekker J, Lieb JD, Tullius TD, Crawford GE, Sunyaev S, Noble WS, Dunham I, Denoeud F, Reymond A, Kapranov P, Rozowsky J, Zheng D, Castelo R, Frankish A, Harrow J, Ghosh S, Sandelin A, Hofacker IL, Baertsch R, Keefe D, Dike S, Cheng J, Hirsch HA, Sekinger EA, Lagarde J, Abril JF, Shahab A, Flamm C, Fried C, Hackermuller J, Hertel J, Lindemeyer M, Missal K, Tanzer A, Washietl S, Korbel J, Emanuelsson O, Pedersen JS, Holroyd N, Taylor R, Swarbreck D, Matthews N, Dickson MC, Thomas DJ, Weirauch MT, Gilbert J, Drenkow J, Bell I, Zhao X, Srinivasan KG, Sung WK, Ooi HS, Chiu KP, Foissac S, Alioto T, Brent M, Pachter L, Tress ML, Valencia A, Choo SW, Choo CY, Ucla C, Manzano C, Wyss C, Cheung E, Clark TG, Brown JB, Ganesh M, Patel S, Tammana H, Chrast J, Henrichsen CN, Kai C, Kawai J, Nagalakshmi U, Wu J, Lian Z, Lian J, Newburger P, Zhang X, Bickel P, Mattick JS, Carninci P, Hayashizaki Y, Weissman S, Hubbard T, Myers RM, Rogers J, Stadler PF, Lowe TM, Wei CL, Ruan Y, Struhl K, Gerstein M, Antonarakis SE, Fu Y, Green ED, Karaoz U, Siepel A, Taylor J, Liefer LA, Wetterstrand KA, Good PJ, Feingold EA, Guyer MS, Cooper GM, Asimenos G, Dewey CN, Hou M, Nikolaev S, Montoya-Burgos JI, Loytynoja A, Whelan S, Pardi F, Massingham T, Huang H, Zhang NR, Holmes I, Mullikin JC, Ureta-Vidal A, Paten B, Seringhaus M, Church D, Rosenbloom K, Kent WJ, Stone EA, Batzoglou S, Goldman N, Hardison RC, Haussler D, Miller W, Sidow A, Trinklein ND, Zhang ZD, Barrera L, Stuart R, King DC, Ameur A, Enroth S, Bieda MC, Kim J, Bhinge AA, Jiang N, Liu J, Yao F, Vega VB, Lee CW, Ng P, Shahab A, Yang A, Moqtaderi Z, Zhu Z, Xu X, Squazzo S, Oberley MJ, Inman D, Singer MA, Richmond TA, Munn KJ, Rada-Iglesias A, Wallerman O, Komorowski J, Fowler JC, Couttet P, Bruce AW, Dovey OM, Ellis PD, Langford CF, Nix DA, Euskirchen G, Hartman S, Urban AE, Kraus P, Van Calcar S, Heintzman N, Kim TH, Wang K, Qu C, Hon G, Luna R, Glass CK, Rosenfeld MG, Aldred SF, Cooper SJ, Halees A, Lin JM, Shulha HP, Zhang X, Xu M, Haidar JN, Yu Y, Ruan Y, Iyer VR, Green RD, Wadelius C, Farnham PJ, Ren B, Harte RA, Hinrichs AS, Trumbower H, Clawson H, Hillman-Jackson J, Zweig AS, Smith K, Thakkapallayil A, Barber G, Kuhn RM, Karolchik D, Armengol L, Bird CP, de Bakker PI, Kern AD, Lopez-Bigas N, Martin JD, Stranger BE, Woodroffe A, Davydov E, Dimas A, Eyras E, Hallgrimsdottir IB, Huppert J, Zody MC, Abecasis GR, Estivill X, Bouffard GG, Guan X, Hansen NF, Idol JR, Maduro VV, Maskeri B, McDowell JC, Park M, Thomas PJ, Young AC, Blakesley RW, Muzny DM, Sodergren E, Wheeler DA, Worley KC, Jiang H, Weinstock GM, Gibbs RA, Graves T, Fulton R, Mardis ER, Wilson RK, Clamp M, Cuff J, Gnerre S, Jaffe DB, Chang JL, Lindblad-Toh K, Lander ES, Koriabine M, Nefedov M, Osoegawa K, Yoshinaga Y, Zhu B, de Jong PJ. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chandonia JM, Brenner SE. The impact of structural genomics: expectations and outcomes. Science. 2006;311(5759):347–351. doi: 10.1126/science.1121018. [DOI] [PubMed] [Google Scholar]
- 11.Trinkle-Mulcahy L, Lamond AI. Toward a high-resolution view of nuclear dynamics. Science. 2007;318(5855):1402–1407. doi: 10.1126/science.1142033. [DOI] [PubMed] [Google Scholar]
- 12.Sergiev PV, Dontsova OA, Bogdanov AA. [Study of ribosome structure using the biochemical methods: judgment day] Mol Biol (Mosk) 2001;35(4):559–583. [PubMed] [Google Scholar]
- 13.Whirl-Carrillo M, Gabashvili IS, Bada M, Banatao DR, Altman RB. Mining biochemical information: lessons taught by the ribosome. Rna. 2002;8(3):279–289. doi: 10.1017/s135583820202407x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barth P, Wallner B, Baker D. Prediction of membrane protein structures with complex topologies using limited constraints. Proc Natl Acad Sci U S A. 2009;106(5):1409–1414. doi: 10.1073/pnas.0808323106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forster F, Webb B, Krukenberg KA, Tsuruta H, Agard DA, Sali A. Integration of small-angle X-ray scattering data into structural modeling of proteins and their assemblies. J Mol Biol. 2008;382(4):1089–1106. doi: 10.1016/j.jmb.2008.07.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Champetier G, Vaillard R. Réaction d'éxchange de la cellulose et de l'eau lourde. Bull. Soc. Chim. Fr. 1938;33:1042. [Google Scholar]
- 17.Hvidt A, Linderstrøm-Lang K. Exchange of hydrogen atoms in insulin with deuterium atoms in aqueous solutions. Biochim Biophys Acta. 1954;14(4):574–575. doi: 10.1016/0006-3002(54)90241-3. [DOI] [PubMed] [Google Scholar]
- 18.Hvidt A, Linderstrøm-Lang K. The kinetics of the deuterium exchange of insulin with D2O; an amendment. Biochim Biophys Acta. 1955;16(1):168–169. doi: 10.1016/0006-3002(55)90200-6. [DOI] [PubMed] [Google Scholar]
- 19.Burley RW, Nicholls CH, Speakman JB. The crystalline/amorphous ratio of keratin fibers. The hydrogen-deuterium exchange reaction. J. Text. Inst. 1955;47:T427–T432. [Google Scholar]
- 20.Saunders M, Wishnia A, Kirkwood JC. Thee nuclear magnetic resonance spectrum of ribonuclease. J. Am. Chem. Soc. 1957;79:3289–3290. [Google Scholar]
- 21.Sethi SK, Smith DL, McCloskey JA. Determination of active hydrogen content by fast atom bombardment mass spectrometry following hydrogen-deuterium exchange. Biochem Biophys Res Commun. 1983;112(1):126–131. doi: 10.1016/0006-291x(83)91806-5. [DOI] [PubMed] [Google Scholar]
- 22.Katta V, Chait B. Conformational changes in proteins probed by hydrogen-deuterium exchange electrospray ionization mass spectometry. Rapid Commun. Mass Spectrom. 1991;5:214–217. doi: 10.1002/rcm.1290050415. [DOI] [PubMed] [Google Scholar]
- 23.Englander SW. Hydrogen exchange and mass spectrometry: A historical perspective. J Am Soc Mass Spectrom. 2006;17(11):1481–1489. doi: 10.1016/j.jasms.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 25.Konermann L, Tong X, Pan Y. Protein structure and dynamics studied by mass spectrometry: H/D exchange, hydroxyl radical labeling, and related approaches. J Mass Spectrom. 2008;43(8):1021–1036. doi: 10.1002/jms.1435. [DOI] [PubMed] [Google Scholar]
- 26.Sizer IW. Chemical aspects of enzyme inhibition. Science. 1957;125(3237):54–59. doi: 10.1126/science.125.3237.54. [DOI] [PubMed] [Google Scholar]
- 27.Fraenkel-Conrat H. Methods for investigating the essential groups for enzyme activity. Methods Enzymol. 1957;IV:247–269. [Google Scholar]
- 28.Singer TP. On the mechanism of enzyme inhibition by sulfhydryl reagents. J. Biol. Chem. 1948;174:11–21. [PubMed] [Google Scholar]
- 29.Staehelin M. Inactivation of virus nucleic acids with glyoxal derivatives. Biochim. Biophys. Acta. 1959;31:448–454. doi: 10.1016/0006-3002(59)90019-8. [DOI] [PubMed] [Google Scholar]
- 30.Litt M, Hancock V. Kethoxal--a potentially useful reagent for the determination of nucleotide sequences in single-stranded regions of transfer ribonucleic acid. Biochemistry. 1967;6(6):1848–1854. doi: 10.1021/bi00858a036. [DOI] [PubMed] [Google Scholar]
- 31.Simpson RT. Modification of chromatin with acetic anhydride. Biochemistry. 1971;10(24):4466–4470. doi: 10.1021/bi00800a018. [DOI] [PubMed] [Google Scholar]
- 32.Biroc SL, Reeder RH. Iodination of Xenopus laevis histone F2a1 in chromatin. Biochemistry. 1976;15(7):1440–1448. doi: 10.1021/bi00652a014. [DOI] [PubMed] [Google Scholar]
- 33.Peattie DA, Gilbert W. Chemical probes for higher-order structure in RNA. Proc. Natl. Acad. Sci. U.S.A. 1980;77(8):4679–4682. doi: 10.1073/pnas.77.8.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ehresmann C, Baudin F, Mougel M, Romby P, Ebel J-P, Ehresmann B. Probing the structure of RNAs in solution. Nucleic Acids Res. 1987;12(22):9109–9128. doi: 10.1093/nar/15.22.9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suckau D, Mak M, Przybylski M. Protein surface topology-probing by selective chemical modification and mass spectrometric peptide mapping. Proc Natl Acad Sci U S A. 1992;89(12):5630–5634. doi: 10.1073/pnas.89.12.5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hahner S, Ludemann HC, Kirpekar F, Nordhoff E, Roepstorff P, Galla HJ, Hillenkamp F. Matrix-assisted laser desorption/ionization mass spectrometry (MALDI) of endonuclease digests of RNA. Nucleic Acids Res. 1997;25(10):1957–1964. doi: 10.1093/nar/25.10.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kowalak JA, Pomerantz SC, Crain PF, McCloskey JA. A novel method for the determination of post-transcriptional modification in RNA by mass spectrometry. Nucleic Acids Res. 1993;21(19):4577–4585. doi: 10.1093/nar/21.19.4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kvaratskhelia M, Miller JT, Budihas SR, Pannell LK, Le Grice SF. Identification of specific HIV-1 reverse transcriptase contacts to the viral RNA:tRNA complex by mass spectrometry and a primary amine selective reagent. Proc Natl Acad Sci U S A. 2002;99(25):15988–15993. doi: 10.1073/pnas.252550199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leitner A, Lindner W. Functional probing of arginine residues in proteins using mass spectrometry and an arginine-specific covalent tagging concept. Anal Chem. 2005;77(14):4481–4488. doi: 10.1021/ac050217h. [DOI] [PubMed] [Google Scholar]
- 40.Pomerantz SC, McCloskey JA. Analysis of RNA hydrolyzates by liquid chromatography-mass spectrometry. Methods Enzymol. 1990;193:796–824. doi: 10.1016/0076-6879(90)93452-q. [DOI] [PubMed] [Google Scholar]
- 41.Yu E, Fabris D. Toward multiplexing the application of solvent accessibility probes for the investigation of RNA three-dimensional structures by electrospray ionization-Fourier transform mass spectrometry. Anal Biochem. 2004;334(2):356–366. doi: 10.1016/j.ab.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 42.Heyduk T, Baichoo N, Heyduk E. Hydroxyl radical footprinting of proteins using metal ion complexes. Met Ions Biol Syst. 2001;38:255–287. [PubMed] [Google Scholar]
- 43.Tullius TD, Dombroski BA, Churchill ME, Kam L. Hydroxyl radical footprinting: a high-resolution method for mapping protein-DNA contacts. Methods Enzymol. 1987;155:537–558. doi: 10.1016/0076-6879(87)55035-2. [DOI] [PubMed] [Google Scholar]
- 44.Sclavi B, Woodson S, Sullivan M, Chance MR, Brenowitz M. Time-resolved synchrotron X-ray "footprinting", a new approach to the study of nucleic acid structure and function: application to protein-DNA interactions and RNA folding. J Mol Biol. 1997;266(1):144–159. doi: 10.1006/jmbi.1996.0775. [DOI] [PubMed] [Google Scholar]
- 45.Hambly DM, Gross ML. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J Am Soc Mass Spectrom. 2005;16(12):2057–2063. doi: 10.1016/j.jasms.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 46.Aye TT, Low TY, Sze SK. Nanosecond laser-induced photochemical oxidation method for protein surface mapping with mass spectrometry. Anal Chem. 2005;77(18):5814–5922. doi: 10.1021/ac050353m. [DOI] [PubMed] [Google Scholar]
- 47.Goldsmith SC, Guan JQ, Almo S, Chance M. Synchrotron protein footprinting: a technique to investigate protein-protein interactions. J Biomol Struct Dyn. 2001;19(3):405–418. doi: 10.1080/07391102.2001.10506750. [DOI] [PubMed] [Google Scholar]
- 48.Brenowitz M, Chance MR, Dhavan G, Takamoto K. Probing the structural dynamics of nucleic acids by quantitative time-resolved and equilibrium hydroxyl radical "footprinting". Curr Opin Struct Biol. 2002;12(5):648–653. doi: 10.1016/s0959-440x(02)00366-4. [DOI] [PubMed] [Google Scholar]
- 49.Kiselar JG, Janmey PA, Almo SC, Chance MR. Visualizing the Ca2+-dependent activation of gelsolin by using synchrotron footprinting. Proc Natl Acad Sci U S A. 2003;100(7):3942–3947. doi: 10.1073/pnas.0736004100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guan JQ, Chance MR. Structural proteomics of macromolecular assemblies using oxidative footprinting and mass spectrometry. Trends Biochem Sci. 2005;30(10):583–592. doi: 10.1016/j.tibs.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 51.Maleknia SD, Downard K. Radical approaches to probe protein structure, folding, and interactions by mass spectrometry. Mass Spectrom Rev. 2001;20(6):388–401. doi: 10.1002/mas.10013. [DOI] [PubMed] [Google Scholar]
- 52.Takamoto K, Chance MR. Radiolytic protein footprinting with mass spectrometry to probe the structure of macromolecular complexes. Annu Rev Biophys Biomol Struct. 2006;35:251–276. doi: 10.1146/annurev.biophys.35.040405.102050. [DOI] [PubMed] [Google Scholar]
- 53.Jensen ON, Barofsky DF, Young MC, von Hippel PH, Swenson S, Seifried SE. Direct observation of UV-crosslinked protein-nucleic acid complexes by matrix-assisted laser desorption ionization mass spectrometry. Rapid Commun Mass Spectrom. 1993;7(6):496–501. doi: 10.1002/rcm.1290070619. [DOI] [PubMed] [Google Scholar]
- 54.Urlaub H, Hartmuth K, Luhrmann R. A two-tracked approach to analyze RNA-protein crosslinking sites in native, nonlabeled small nuclear ribonucleoprotein particles. Methods. 2002;26(2):170–181. doi: 10.1016/S1046-2023(02)00020-8. [DOI] [PubMed] [Google Scholar]
- 55.Wang Y, Zhang Q, Wang Y. Tandem mass spectrometry for the determination of the sites of DNA interstrand cross-link. J Am Soc Mass Spectrom. 2004;15(11):1565–1571. doi: 10.1016/j.jasms.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 56.Zahn H, Meienhofer J. Reaktionen von 1,5-difluor-2,4,-dinitrobenzol mit insulin. 2. Mitt. versuche mit insulin. Makromol. Chem. 1958;26:153–166. [Google Scholar]
- 57.Fasold H. [On chemical studies of the tertiary structure of proteins. II. Analysis of azoglobin, separation and identification of individual bridged peptides] Biochem Z. 1965;342(3):295–302. [PubMed] [Google Scholar]
- 58.Hartman FC, Wold F. Cross-linking of bovine pancreatic ribonuclease A with dimethyl adipimidate. Biochemistry. 1967;6(8):2439–2448. doi: 10.1021/bi00860a021. [DOI] [PubMed] [Google Scholar]
- 59.Golinska B, Millon R, Backendorf C, Olomucki M, Ebel JP, Ehresmann B. Identification of a 16-S RNA fragment crosslinked to protein S1 within Escherichia coli ribosomal 30-S subunits by the use of a crosslinking reagent: ethyl 4-azidobenzoylaminoacetimidate. Eur J Biochem. 1981;115(3):479–484. doi: 10.1111/j.1432-1033.1981.tb06227.x. [DOI] [PubMed] [Google Scholar]
- 60.Summerton J, Bartlett PA. Sequence-specific crosslinking agents for nucleic acids. Use of 6-bromo-5,5-dimethoxyhexanohydrazide for crosslinking cytidine to guanosine and crosslinking RNA to complementary sequences of DNA. J Mol Biol. 1978;122(2):145–162. doi: 10.1016/0022-2836(78)90032-3. [DOI] [PubMed] [Google Scholar]
- 61.Jacobsen RB, Sale KL, Ayson MJ, Novak P, Hong J, Lane P, Wood NL, Kruppa GH, Young MM, Schoeniger JS. Structure and dynamics of dark-state bovine rhodopsin revealed by chemical cross-linking and high-resolution mass spectrometry. Protein Sci. 2006;15(6):1303–1317. doi: 10.1110/ps.052040406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu ET, Zhang Q, Fabris D. Untying the FIV frameshifting pseudoknot structure by MS3D. J. Mol. Biol. 2005;345:69–80. doi: 10.1016/j.jmb.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 63.Huang BX, Kim HY, Dass C. Probing three-dimensional structure of bovine serum albumin by chemical cross-linking and mass spectrometry. J Am Soc Mass Spectrom. 2004;15(8):1237–1247. doi: 10.1016/j.jasms.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 64.Onisko B, Fernandez EG, Freire ML, Schwarz A, Baier M, Camina F, Garcia JR, Rodriguez-Segade Villamarin S, Requena JR. Probing PrPSc structure using chemical cross-linking and mass spectrometry: evidence of the proximity of Gly90 amino termini in the PrP 27–30 aggregate. Biochemistry. 2005;44(30):10100–10109. doi: 10.1021/bi0501582. [DOI] [PubMed] [Google Scholar]
- 65.Yu ET, Hawkins AE, Eaton J, Fabris D. MS3D structural elucidation of the HIV-1 packaging signal. Proc Natl Acad Sci U S A. 2008;105:12248–12253. doi: 10.1073/pnas.0800509105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chang Z, Kuchar J, Hausinger RP. Chemical cross-linking and mass spectrometric identification of sites of interaction for UreD, UreF, and urease. J Biol Chem. 2004;279(15):15305–15313. doi: 10.1074/jbc.M312979200. [DOI] [PubMed] [Google Scholar]
- 67.Guerrero C, Tagwerker C, Kaiser P, Huang L. An integrated mass spectrometry-based proteomic approach: quantitative analysis of tandem affinity-purified in vivo cross-linked protein complexes (QTAX) to decipher the 26 S proteasome-interacting network. Mol Cell Proteomics. 2006;5(2):366–378. doi: 10.1074/mcp.M500303-MCP200. [DOI] [PubMed] [Google Scholar]
- 68.Vasilescu J, Guo X, Kast J. Identification of protein-protein interactions using in vivo cross-linking and mass spectrometry. Proteomics. 2004;4(12):3845–3854. doi: 10.1002/pmic.200400856. [DOI] [PubMed] [Google Scholar]
- 69.Friedhoff P. Mapping protein-protein interactions by bioinformatics and cross-linking. Anal Bioanal Chem. 2005;381(1):78–80. doi: 10.1007/s00216-004-2892-7. [DOI] [PubMed] [Google Scholar]
- 70.Sinz A. Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein-protein interactions. Mass Spectrom Rev. 2006;25(4):663–682. doi: 10.1002/mas.20082. [DOI] [PubMed] [Google Scholar]
- 71.Trakselis MA, Alley SC, Ishmael FT. Identification and mapping of protein-protein interactions by a combination of cross-linking, cleavage, and proteomics. Bioconjug Chem. 2005;16(4):741–750. doi: 10.1021/bc050043a. [DOI] [PubMed] [Google Scholar]
- 72.Chen X, Chen YH, Anderson VE. Protein cross-links: universal isolation and characterization by isotopic derivatization and electrospray ionization mass spectrometry. Anal Biochem. 1999;273(2):192–203. doi: 10.1006/abio.1999.4243. [DOI] [PubMed] [Google Scholar]
- 73.Muller DR, Schindler P, Towbin H, Wirth U, Voshol H, Hoving S, Steinmetz MO. Isotope-tagged cross-linking reagents. A new tool in mass spectrometric protein interaction analysis. Anal Chem. 2001;73(9):1927–1934. doi: 10.1021/ac001379a. [DOI] [PubMed] [Google Scholar]
- 74.Pearson KM, Pannell LK, Fales HM. Intramolecular Cross-Linking Experiments on Cytochrome C and Ribonuclease a Using an Isotope Multiplet Method. Rapid Commun. Mass Spectrom. 2002;16:149–159. doi: 10.1002/rcm.554. [DOI] [PubMed] [Google Scholar]
- 75.Trester-Zedlitz M, Kamada K, Burley SK, Fenyö D, Chait BT, Muir TW. A modular cross-linking approach for exploring protein interactions. J. Am. Chem. Soc. 2003:2416–2425. doi: 10.1021/ja026917a. [DOI] [PubMed] [Google Scholar]
- 76.Petrotchenko EV, Olkhovik VK, Borchers CH. Isotopically coded cleavable cross-linker for studying protein-protein interaction and protein complexes. Mol Cell Proteomics. 2005;4(8):1167–1179. doi: 10.1074/mcp.T400016-MCP200. [DOI] [PubMed] [Google Scholar]
- 77.Fonovic M, Verhelst SH, Sorum MT, Bogyo M. Proteomics evaluation of chemically cleavable activity-based probes. Mol Cell Proteomics. 2007;6(10):1761–1770. doi: 10.1074/mcp.M700124-MCP200. [DOI] [PubMed] [Google Scholar]
- 78.McLafferty FW. Tandem mass spectrometry. Science. 1981;214:280–287. doi: 10.1126/science.7280693. [DOI] [PubMed] [Google Scholar]
- 79.Roepstorff P, Fohlman J. Proposal for a common nomenclature for sequence ions in mass spectra of peptides. Biol Mass Spectrom. 1984;11:601. doi: 10.1002/bms.1200111109. [DOI] [PubMed] [Google Scholar]
- 80.McLuckey SA, Habibi-Goudarzi S. Decompositions of multiply charged oligonucleotide anions. J. Am. Chem. Soc. 1993;115:12085–12095. [Google Scholar]
- 81.Back JW, Hartog AF, Dekker HL, Muijsers AO, de Koning LJ, de Jong L. A new crosslinker for mass spectrometric analysis of the quaternary structure of protein complexes. J Am Soc Mass Spectrom. 2001;12(2):222–227. doi: 10.1016/S1044-0305(00)00212-9. [DOI] [PubMed] [Google Scholar]
- 82.Tang X, Munske GR, Siems WF, Bruce JE. Mass spectrometry identifiable cross-linking strategy for studying protein-protein interactions. Anal Chem. 2005;77(1):311–318. doi: 10.1021/ac0488762. [DOI] [PubMed] [Google Scholar]
- 83.Chowdhury SM, Munske GR, Tang X, Bruce JE. Collisionally activated dissociation and electron capture dissociation of several mass spectrometry-identifiable chemical cross-linkers. Anal Chem. 2006;78(24):8183–8193. doi: 10.1021/ac060789h. [DOI] [PubMed] [Google Scholar]
- 84.Soderblom EJ, Goshe MB. Collision-induced dissociative chemical cross-linking reagents and methodology: Applications to protein structural characterization using tandem mass spectrometry analysis. Anal Chem. 2006;78(23):8059–8068. doi: 10.1021/ac0613840. [DOI] [PubMed] [Google Scholar]
- 85.Lu Y, Tanasova M, Borhan B, Reid GE. Ionic reagent for controlling the gas-phase fragmentation reactions of cross-linked peptides. Anal Chem. 2008;80(23):9279–9287. doi: 10.1021/ac801625e. [DOI] [PubMed] [Google Scholar]
- 86.Gardner MW, Vasicek LA, Shabbir S, Anslyn EV, Brodbelt JS. Chromogenic cross-linker for the characterization of protein structure by infrared multiphoton dissociation mass spectrometry. Anal Chem. 2008;80(13):4807–4819. doi: 10.1021/ac800625x. [DOI] [PubMed] [Google Scholar]
- 87.Kellersberger KA, Yu E, Kruppa GH, Young MM, Fabris D. Top-down characterization of nucleic acids modified by structural probes using high-resolution tandem mass spectrometry and automated data interpretation. Anal. Chem. 2004;76(9):2438–2445. doi: 10.1021/ac0355045. [DOI] [PubMed] [Google Scholar]
- 88.Solouki T, Pasa-Tolic L, Jackson GS, Guan S, Marshall AG. High-resolution multistage MS, MS2, and MS3 matrix-assisted laser desorption/ionization FT-ICR mass spectra of peptides from a single laser shot. Anal. Chem. 1996;68:3718–3725. doi: 10.1021/ac960312d. [DOI] [PubMed] [Google Scholar]
- 89.Srikanth R, Wilson J, Bridgewater JD, Numbers JR, Lim J, Olbris MR, Kettani A, Vachet RW. Improved sequencing of oxidized cysteine and methionine containing peptides using electron transfer dissociation. J Am Soc Mass Spectrom. 2007;18(8):1499–1506. doi: 10.1016/j.jasms.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 90.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Nat. Acad. Sci. USA. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sinz A, Wang K. Mapping protein interfaces with a fluorogenic cross-linker and mass spectrometry: application to nebulin-calmodulin complexes. Biochemistry. 2001;40(26):7903–7913. doi: 10.1021/bi010259+. [DOI] [PubMed] [Google Scholar]
- 92.Novak P, Haskins WE, Ayson MJ, Jacobsen RB, Schoeniger JS, Leavell MD, Young MM, Kruppa GH. Unambiguous assignment of intramolecular chemical cross-links in modified mammalian membrane proteins by Fourier transform-tandem mass spectrometry. Anal Chem. 2005;77(16):5101–5106. doi: 10.1021/ac040194r. [DOI] [PubMed] [Google Scholar]
- 93.Zhang Q, Yu ET, Kellersberger KA, Crosland E, Fabris D. Toward building a database of bifunctional probes for the MS3D investigation of nucleic acids structures. J Am Soc Mass Spectrom. 2006;17:1570–1581. doi: 10.1016/j.jasms.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 94.Gao Y, Wang Y. The photochemistry of 5-BrdCpdC, dCp5-BrdCpdA, and dCp5-BrdCdT in aqueous solution. Nucleosides Nucleotides Nucleic Acids. 2006;25(3):279–287. doi: 10.1080/15257770500446972. [DOI] [PubMed] [Google Scholar]
- 95.Gupta R, Beck JL, Sheil MM, Ralph SF. Identification of bifunctional GA and AG intrastrand crosslinks formed between cisplatin and DNA. J Inorg Biochem. 2005;99(2):552–559. doi: 10.1016/j.jinorgbio.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 96.Park S, Tretyakova N. Structural characterization of the major DNA-DNA cross-link of 1,2,3,4-diepoxybutane. Chem Res Toxicol. 2004;17(2):129–136. doi: 10.1021/tx0342058. [DOI] [PubMed] [Google Scholar]
- 97.Jensen ON, Kulkarni S, Aldrich JV, Barofsky DF. Characterization of peptide-oligonucleotide heteroconjugates by mass spectrometry. Nucleic Acids. Res. 1996;24(19):3866–3872. doi: 10.1093/nar/24.19.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Steen H, Jensen ON. Analysis of protein-nucleic acid interactions by photochemical cross-linking and mass spectrometry. Mass Spectrom. Rev. 2002;21(3):163–182. doi: 10.1002/mas.10024. [DOI] [PubMed] [Google Scholar]
- 99.Steen H, Petersen J, Mann M, Jensen ON. Mass spectrometric analysis of a UV-cross-linked protein-DNA complex: tryptophans 54 and 88 of E. coli SSB cross-link to DNA. Protein Sci. 2001;10(10):1989–2001. doi: 10.1110/ps.07601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tiss A, Barre O, Michaud-Soret I, Forest E. Characterization of the DNA-binding site in the ferric uptake regulator protein from Escherichia coli by UV crosslinking and mass spectrometry. FEBS Lett. 2005;579(25):5454–5460. doi: 10.1016/j.febslet.2005.08.067. [DOI] [PubMed] [Google Scholar]
- 101.Pingoud V, Geyer H, Geyer R, Kubareva E, Bujnicki JM, Pingoud A. Identification of base-specific contacts in protein-DNA complexes by photocrosslinking and mass spectrometry: a case study using the restriction endonuclease SsoII. Mol Biosyst. 2005;1(2):135–141. doi: 10.1039/b503091a. [DOI] [PubMed] [Google Scholar]
- 102.Luo X, Hsiao HH, Bubunenko M, Weber G, Court DL, Gottesman ME, Urlaub H, Wahl MC. Structural and functional analysis of the E. coli NusB-S10 transcription antitermination complex. Mol Cell. 2008;32(6):791–802. doi: 10.1016/j.molcel.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Thiede B, Urlaub H, Neubauer H, Grelle G, Wittmann-Liebold B. Precise determination of RNA-protein contact sites in the 50 S ribosomal subunit of Escherichia coli. Biochem J. 1998;334(Pt 1):39–42. doi: 10.1042/bj3340039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kelleher NL, Lin HY, Valaskovic GA, Aaserud DJ, Fridriksson EK, McLafferty FW. Top down versus bottom up protein characterization by tandem high-resolution mass spectrometry. J. Am. Chem. Soc. 1999;121:806–812. [Google Scholar]
- 105.Novak P, Young M, Schoeniger J, Kruppa GH. A top down approach to protein structure studies using chemical cross-linking and Fourier transform mass spectrometry. Eur. J. Mass Spectrom. 2003;9:623–631. doi: 10.1255/ejms.590. [DOI] [PubMed] [Google Scholar]
- 106.Novak P, Kruppa GH, Young MM, Schoeniger J. A top down method for the determination of residue specific solvent accessibility in proteins. J. Mass Spectrom. 2004;39:322–328. doi: 10.1002/jms.587. [DOI] [PubMed] [Google Scholar]
- 107.Pan J, Han J, Borchers CH, Konermann L. Electron capture dissociation of electrosprayed protein ions for spatially resolved hydrogen exchange measurements. J Am Chem Soc. 2008;130(35):11574–11575. doi: 10.1021/ja802871c. [DOI] [PubMed] [Google Scholar]
- 108.Abzalimov RR, Kaplan DA, Easterling ML, Kaltashov IA. Protein conformations can be probed in top-down HDX MS experiments utilizing electron transfer dissociation of protein ions without hydrogen scrambling. J Am Soc Mass Spectrom. 2009;20(8):1514–1517. doi: 10.1016/j.jasms.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kaltashov IA, Bobst CE, Abzalimov RR. H/D exchange and mass spectrometry in the studies of protein conformation and dynamics: is there a need for a top-down approach? Anal Chem. 2009;81(19):7892–7899. doi: 10.1021/ac901366n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hofstadler SA, Wahl JH, Bakhtiar R, Anderson GA, Bruce JE, Smith RD. Capillary electrophoresis Fourier-transform ion-cyclotron resonance mass speectrometry with sustained off-resonance irradiation for the characterization of protein and peptide mixtures. J. Am. Soc. Mass Spectrom. 1994;5:894–899. doi: 10.1016/1044-0305(94)87014-4. [DOI] [PubMed] [Google Scholar]
- 111.Valaskovic GA, Kelleher NL, McLafferty FW. Attomole protein characterization by capillary electrophoresis-mass spectrometry. Science. 1996;273:1199–1202. doi: 10.1126/science.273.5279.1199. [DOI] [PubMed] [Google Scholar]
- 112.Bruins AP, Covey TR, Henion JD. Ion spray interface for combined liquid chromatography-atmospheric pressure ionization mass spectrometry. Anal Chem. 1987;59:2642–2646. [Google Scholar]
- 113.Emmett MR, Caprioli RM. Micro-electrospray mass spectrometry: ultra-high-sensitivity analysis of peptides and proteins. J. Am. Soc. Mass Spectrom. 1994;5:605–613. doi: 10.1016/1044-0305(94)85001-1. [DOI] [PubMed] [Google Scholar]
- 114.Richter FM, Hsiao H-H, Plessmann U, Urlaub H. Enrichment of protein-RNA crosslinks from crude UV-irradiated mixtures for MS analysis by on-line chromatography using titanium oxide columns. Biopolymers. 2009;91(4):297–309. doi: 10.1002/bip.21139. [DOI] [PubMed] [Google Scholar]
- 115.Gabant G, Augier J, Armengaud J. Assessment of solvent residues accessibility using three Sulfo-NHS-biotin reagents in parallel: application to footprint changes of a methyltransferase upon binding its substrate. J Mass Spectrom. 2008;43(3):360–370. doi: 10.1002/jms.1328. [DOI] [PubMed] [Google Scholar]
- 116.Tomohiro T, Hashimoto M, Hatanaka Y. Cross-linking chemistry and biology: development of multifunctional photoaffinity probes. Chem Rec. 2005;5(6):385–395. doi: 10.1002/tcr.20058. [DOI] [PubMed] [Google Scholar]
- 117.Azim-Zadeh O, Hillebrecht A, Linne U, Marahiel MA, Klebe G, Lingelbach K, Nyalwidhe J. Use of biotin derivatives to probe conformational changes in proteins. J Biol Chem. 2007;282(30):21609–21617. doi: 10.1074/jbc.M610921200. [DOI] [PubMed] [Google Scholar]
- 118.Kang S, Mou L, Lanman J, Velu S, Brouillette WJ, Prevelige PE., Jr Synthesis of biotin-tagged chemical cross-linkers and their applications for mass spectrometry. Rapid Commun Mass Spectrom. 2009;23(11):1719–1726. doi: 10.1002/rcm.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yan F, Che FY, Rykunov D, Nieves E, Fiser A, Weiss LM, Hogue Angeletti R. Nonprotein based enrichment method to analyze peptide cross-linking in protein complexes. Anal Chem. 2009;81(17):7149–7159. doi: 10.1021/ac900360b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chowdhury SM, Du X, Tolic N, Wu S, Moore RJ, Mayer MU, Smith RD, Adkins JN. Identification of cross-linked peptides after click-based enrichment using sequential collision-induced dissociation and electron transfer dissociation tandem mass spectrometry. Anal Chem. 2009;81(13):5524–5532. doi: 10.1021/ac900853k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Paramelle D, Cantel S, Enjalbal C, Amblard M, Forest E, Heymann M, Geourjon C, Martinez J, Subra G. A new generation of cross-linkers for selective detection by MALDI MS. Proteomics. 2009;9(23):5384–5388. doi: 10.1002/pmic.200900562. [DOI] [PubMed] [Google Scholar]
- 122.Gau BC, Sharp JS, Rempel DL, Gross ML. Fast photochemical oxidation of protein footprints faster than protein unfolding. Anal Chem. 2009;81(16):6563–6571. doi: 10.1021/ac901054w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Madler S, Bich C, Touboul D, Zenobi R. Chemical cross-linking with NHS esters: a systematic study on amino acid reactivities. J Mass Spectrom. 2009;44(5):694–706. doi: 10.1002/jms.1544. [DOI] [PubMed] [Google Scholar]
- 124.Swaim CL, Smith JB, Smith DL. Unexpected products from the reaction of the synthetic cross-linker 3,3'-dithiobis(sulfosuccinimidyl propionate), DTSSP with peptides. J Am Soc Mass Spectrom. 2004;15(5):736–749. doi: 10.1016/j.jasms.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 125.Leavell MD, Novak P, Behrens CR, Schoeniger JS, Kruppa GH. Strategy for selective chemical cross-linking of tyrosine and lysine residues. J Am Soc Mass Spectrom. 2004;15(11):1604–1611. doi: 10.1016/j.jasms.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 126.Richter S, Fabris D, Binaschi M, Gatto B, Capranico G, Palumbo M. Effects of common buffer systems on drug activity: the case of clerocidin. Chem. Res. Toxicol. 2004;17(4):492–501. doi: 10.1021/tx034210b. [DOI] [PubMed] [Google Scholar]
- 127.Mendoza VL, Vachet RW. Protein surface mapping using diethylpyrocarbonate with mass spectrometric detection. Anal Chem. 2008;80(8):2895–2904. doi: 10.1021/ac701999b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang Q, Crosland E, Fabris D. Nested Arg-specific bifunctional crosslinkers for MS-based structural analysis of proteins and protein assemblies. Anal. Chim. Acta. 2008;627:117–128. doi: 10.1016/j.aca.2008.05.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yu E, Fabris D. Direct probing of RNA structures and RNA-protein interactions in the HIV-1 packaging signal by chemical modification and electrospray ionization Fourier transform mass spectrometry. J. Mol. Biol. 2003;330(2):211–223. doi: 10.1016/s0022-2836(03)00589-8. [DOI] [PubMed] [Google Scholar]
- 130.Gao Q, Doneanu CE, Shaffer SA, Adman ET, Goodlett DR, Nelson SD. Identification of the interactions between cytochrome P450 2E1 and cytochrome b5 by mass spectrometry and site-directed mutagenesis. J Biol Chem. 2006;281(29):20404–20417. doi: 10.1074/jbc.M601785200. [DOI] [PubMed] [Google Scholar]
- 131.Peri S, Steen H, Pandey A. GPMAW--a software tool for analyzing proteins and peptides. Trends Biochem Sci. 2001;26(11):687–689. doi: 10.1016/s0968-0004(01)01954-5. [DOI] [PubMed] [Google Scholar]
- 132.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 133.Clauser KR, Baker P, Burlingame AL. Role of accurate mass measurement (+/− 10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal Chem. 1999;71(14):2871–2882. doi: 10.1021/ac9810516. [DOI] [PubMed] [Google Scholar]
- 134.de Koning LJ, Kasper PT, Back JW, Nessen MA, Vanrobaeys F, Van Beeumen J, Gherardi E, de Koster CG, de Jong L. Computer-assisted mass spectrometric analysis of naturally occurring and artificially introduced cross-links in proteins and protein complexes. Febs J. 2006;273(2):281–291. doi: 10.1111/j.1742-4658.2005.05053.x. [DOI] [PubMed] [Google Scholar]
- 135.Taverner T, Hall NE, O'Hair RA, Simpson RJ. Characterization of an antagonist interleukin-6 dimer by stable isotope labeling, cross-linking, and mass spectrometry. J Biol Chem. 2002;277(48):46487–46492. doi: 10.1074/jbc.M207370200. [DOI] [PubMed] [Google Scholar]
- 136.Wefing S, Schnaible V, Hoffmann D. SearchXLinks. A program for the identification of disulfide bonds in proteins from mass spectra. Anal Chem. 2006;78(4):1235–1241. doi: 10.1021/ac051634x. [DOI] [PubMed] [Google Scholar]
- 137.Back JW, Sanz MA, De Jong L, De Koning Koning, Nijtmans LG, De Koster CG, Grivell LA, Van Der Spek H, Muijsers AO. A structure for the yeast prohibitin complex: Structure prediction and evidence from chemical crosslinking and mass spectrometry. Protein Sci. 2002;11(10):2471–2478. doi: 10.1110/ps.0212602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tang Y, Chen Y, Lichti CF, Hall RA, Raney KD, Jennings SF. CLPM: a cross-linked peptide mapping algorithm for mass spectrometric analysis. BMC Bioinformatics. 2005;(6 Suppl 2):S9. doi: 10.1186/1471-2105-6-S2-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yu ET, Hawkins A, Kuntz ID, Rahn LA, Rothfuss A, Sale K, Young MM, Yang CL, Pancerella CM, Fabris D. The collaboratory for MS3D: a new cyberinfrastructure for the structural elucidation of biological macromolecules and their assemblies using mass spectrometry-based approaches. J Proteome Res. 2008;7(11):4848–4857. doi: 10.1021/pr800443f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Young MM, Tang N, Hempel JC, Oshiro CM, Taylor EW, Kuntz ID, Gibson BW, Dollinger G. High throughput protein fold identification by using experimental constraints derived from intramolecular cross-links and mass spectrometry. Proc Natl Acad Sci U S A. 2000;97(11):5802–5806. doi: 10.1073/pnas.090099097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Schilling B, Row RH, Gibson BW, Guo X, Young MM. MS2Assign, automated assignment and nomenclature of tandem mass spectra of chemically crosslinked peptides. J Am Soc Mass Spectrom. 2003;14(8):834–850. doi: 10.1016/S1044-0305(03)00327-1. [DOI] [PubMed] [Google Scholar]
- 142.Fabris D, Chaudhari P, Hagan N, Turner K. Functional investigations of retroviral proteinribonucleic acid complexes by nanospray Fourier transform ion cyclotron resonance mass spectrometry. Eur J Mass Spectrom (Chichester, Eng) 2007;13(1):29–33. doi: 10.1255/ejms.839. [DOI] [PubMed] [Google Scholar]
- 143.Yu E, Gaucher SP, Sale K, Hadi MZ. DNA-mediated Ref1 Conformational Changes: Possible Mechanism for Protein Recruitment in Base Excision Repair (BER) Pathway; Denver, CO. 56th ASMS Conference on Mass Spectrometry and Allied Topics.Denver, CO: 2008. [Google Scholar]
- 144.Nadeau OW, Wyckoff GJ, Paschall JE, Artigues A, Sage J, Villar MT, Carlson GM. CrossSearch, a user-friendly search engine for detecting chemically cross-linked peptides in conjugated proteins. Mol Cell Proteomics. 2008;7(4):739–749. doi: 10.1074/mcp.M800020-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rinner O, Seebacher J, Walzthoeni T, Mueller LN, Beck M, Schmidt A, Mueller M, Aebersold R. Identification of cross-linked peptides from large sequence databases. Nat Methods. 2008;5(4):315–318. doi: 10.1038/nmeth.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Selvin S. Practical biostatistical methods. Duxbury Press; 1995. p. 503. [Google Scholar]
- 147.Lee YJ, Lackner LL, Nunnari JM, Phinney BS. Shotgun cross-linking analysis for studying quaternary and tertiary protein structures. J Proteome Res. 2007;6(10):3908–3917. doi: 10.1021/pr070234i. [DOI] [PubMed] [Google Scholar]
- 148.Lee YJ. Probability-based shotgun cross-linking sites analysis. J Am Soc Mass Spectrom. 2009;20(10):1896–1899. doi: 10.1016/j.jasms.2009.06.020. [DOI] [PubMed] [Google Scholar]
- 149.Maiolica A, Cittaro D, Borsotti D, Sennels L, Ciferri C, Tarricone C, Musacchio A, Rappsilber J. Structural analysis of multiprotein complexes by cross-linking, mass spectrometry, and database searching. Mol Cell Proteomics. 2007;6(12):2200–2211. doi: 10.1074/mcp.M700274-MCP200. [DOI] [PubMed] [Google Scholar]
- 150.Singh P, Shaffer SA, Scherl A, Freeman TL, Holman C, Pfuetzner RA, Miller SI, Hernandez P, Appel RA, Goodlett DRIn. Identification of Crosslinked Peptides by an "open-modification" search tool; Denver, CO. 56th ASMS Conference on Mass Spectrometry and Allied Topics.Denver, CO: 2008. [Google Scholar]
- 151.Petrotchenko EV, Xiao K, Cable J, Chen Y, Dokholyan NV, Borchers CH. BiPS, a photocleavable, isotopically coded, fluorescent cross-linker for structural proteomics. Mol Cell Proteomics. 2009;8(2):273–286. doi: 10.1074/mcp.M800265-MCP200. [DOI] [PubMed] [Google Scholar]
- 152.Petrotchenko EV, Serpa JJ, Borchers CH. Use of a combination of isotopically coded cross-linkers and isotopically coded N-terminal modification reagents for selective identification of inter-peptide crosslinks. Anal Chem. 2010;82(3):817–823. doi: 10.1021/ac901637v. [DOI] [PubMed] [Google Scholar]
- 153.Yao X, Freas A, Ramirez J, Demirev PA, Fenselau C. Proteolytic 18O labeling for comparative proteomics: model studies with two serotypes of adenovirus. Anal Chem. 2001;73(13):2836–2842. doi: 10.1021/ac001404c. [DOI] [PubMed] [Google Scholar]
- 154.Meng Z, Limbach PA. Quantitation of ribonucleic acids using 18O labeling and mass spectrometry. Anal Chem. 2005;77(6):1891–1895. doi: 10.1021/ac048801y. [DOI] [PubMed] [Google Scholar]
- 155.Gao Q, Xue S, Doneanu CE, Shaffer SA, Goodlett DR, Nelson SD. Pro-CrossLink. Software tool for protein cross-linking and mass spectrometry. Anal Chem. 2006;78(7):2145–2149. doi: 10.1021/ac051339c. [DOI] [PubMed] [Google Scholar]
- 156.Seebacher J, Mallick P, Zhang N, Eddes JS, Aebersold R, Gelb MH. Protein cross-linking analysis using mass spectrometry, isotope-coded cross-linkers, and integrated computational data processing. J Proteome Res. 2006;5(9):2270–2282. doi: 10.1021/pr060154z. [DOI] [PubMed] [Google Scholar]
- 157.Raftery MJ, Geczy CL. Electrospray low energy CID and MALDI PSD fragmentations of protonated sulfinamide cross-linked peptides. J Am Soc Mass Spectrom. 2002;13(6):709–718. doi: 10.1016/S1044-0305(02)00359-8. [DOI] [PubMed] [Google Scholar]
- 158.Gaucher SP, Hadi MZ, Young MM. Influence of crosslinker identity and position on gas-phase dissociation of Lys-Lys crosslinked peptides. J Am Soc Mass Spectrom. 2006;17(3):395–405. doi: 10.1016/j.jasms.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 159.Iglesias AH, Santos LF, Gozzo FC. Collision-induced dissociation of Lys-Lys intramolecular crosslinked peptides. J Am Soc Mass Spectrom. 2009;20(4):557–566. doi: 10.1016/j.jasms.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 160.Berman HM, Westbrook J, Feng Z, Gilliland GT, Bhat N, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucl. Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22(12):2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 162.Hubbard S, Thornton J. NACCESS. :1992–1996. [Google Scholar]
- 163.Franczkiewicz R, Braun W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comp. Chem. 1998;19:319–333. [Google Scholar]
- 164.Ahmad S, Gromiha M, Fawareh H, Sarai A. ASAView: database and tool for solvent accessibility representation in proteins. BMC Bioinformatics. 2004;5:51. doi: 10.1186/1471-2105-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Akinsiku OT, Yu ET, Fabris D. Mass spectrometric investigation of protein alkylation by the RNA footprinting probe kethoxal. J. Mass Spectrom. 2005;40:1372–1381. doi: 10.1002/jms.932. [DOI] [PubMed] [Google Scholar]
- 166.Yan X, Broderick D, Leid ME, Schimerlik MI, Deinzer ML. Dynamics and ligand-induced solvent accessibility changes in human retinoid X receptor homodimer determined by hydrogen deuterium exchange and mass spectrometry. Biochemistry. 2004;43(4):909–917. doi: 10.1021/bi030183c. [DOI] [PubMed] [Google Scholar]
- 167.Sharp JS, Becker JM, Hettich RL. Analysis of protein solvent accessible surfaces by photochemical oxidation and mass spectrometry. Anal Chem. 2004;76(3):672–683. doi: 10.1021/ac0302004. [DOI] [PubMed] [Google Scholar]
- 168.Scholten A, Visser NF, van den Heuvel RH, Heck AJ. Analysis of protein-protein interaction surfaces using a combination of efficient lysine acetylation and nanoLC-MALDI-MS/MS applied to the E9:Im9 bacteriotoxin--immunity protein complex. J Am Soc Mass Spectrom. 2006;17(7):983–994. doi: 10.1016/j.jasms.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 169.Charvatova O, Foley BL, Bern MW, Sharp JS, Orlando R, Woods RJ. Quantifying protein interface footprinting by hydroxyl radical oxidation and molecular dynamics simulation: application to galectin-1. J Am Soc Mass Spectrom. 2008;19(11):1692–1705. doi: 10.1016/j.jasms.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sharp JS, Guo JT, Uchiki T, Xu Y, Dealwis C, Hettich RL. Photochemical surface mapping of C14S-Sml1p for constrained computational modeling of protein structure. Anal Biochem. 2005;340(2):201–212. doi: 10.1016/j.ab.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 171.Obungu VH, Gelfanova V, Rathnachalam R, Bailey A, Sloan-Lancaster J, Huang L. Determination of the mechanism of action of anti-FasL antibody by epitope mapping and homology modeling. Biochemistry. 2009;48(30):7251–7260. doi: 10.1021/bi900296g. [DOI] [PubMed] [Google Scholar]
- 172.Li J, Lim MS, Li S, Brock M, Pique ME, Woods VL, Jr, Craig L. Vibrio cholerae toxin-coregulated pilus structure analyzed by hydrogen/deuterium exchange mass spectrometry. Structure. 2008;16(1):137–148. doi: 10.1016/j.str.2007.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Brock M, Fan F, Mei FC, Li S, Gessner C, Woods VL, Jr, Cheng X. Conformational analysis of Epac activation using amide hydrogen/deuterium exchange mass spectrometry. J Biol Chem. 2007;282(44):32256–32263. doi: 10.1074/jbc.M706231200. [DOI] [PubMed] [Google Scholar]
- 174.Mandell JG, Falick AM, Komives EA. Identification of protein-protein interfaces by decreased amide proton solvent accessibility. Proc Natl Acad Sci U S A. 1998;95(25):14705–14710. doi: 10.1073/pnas.95.25.14705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Dihazi GH, Sinz A. Mapping low-resolution three-dimensional protein structures using chemical cross-linking and Fourier transform ion-cyclotron resonance mass spectrometry. Rapid Commun Mass Spectrom. 2003;17(17):2005–2014. doi: 10.1002/rcm.1144. [DOI] [PubMed] [Google Scholar]
- 176.Green NS, Reisler E, Houk KN. Quantitative evaluation of the lengths of homobifunctional protein cross-linking reagents used as molecular rulers. Protein Sci. 2001;10(7):1293–1304. doi: 10.1110/ps.51201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Schoeniger J, Jacobsen RB, Sale KIn. Protein Structure and Conformation Revealed Using Sequential Cross-linker Rate Measurements (SCRaM): Direct Measurement of Inter-Residue Distances and Secondary Structure; Denver, CO. 55th ASMS Conference on Mass Spectrometry and Allied Topics.Denver, CO: 2007. [Google Scholar]
- 178.Potluri S, Khan AA, Kuzminykh A, Bujnicki JM, Friedman AM, Bailey-Kellogg C. Geometric analysis of cross-linkability for protein fold discrimination. Pac Symp Biocomput. 2004:447–458. doi: 10.1142/9789812704856_0042. [DOI] [PubMed] [Google Scholar]
- 179.Heymann M, Paramelle D, Subra G, Forest E, Martinez J, Geourjon C, Deleage G. MSX-3D: a tool to validate 3D protein models using mass spectrometry. Bioinformatics. 2008;24(23):2782–2783. doi: 10.1093/bioinformatics/btn510. [DOI] [PubMed] [Google Scholar]
- 180.Gorelick RJ, Chabod DJ, Rein A, Henderson LE, Arthur LO. The two zinc fingers in the human immunodeficiency virus type 1 nucleocapsid protein are not functionally equivalent. J. Virol. 1993;67(7):4027–4036. doi: 10.1128/jvi.67.7.4027-4036.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Giron-Monzon L, Manelyte L, Ahrends R, Kirsch D, Spengler B, Friedhoff P. Mapping protein-protein interactions between MutL and MutH by cross-linking. J Biol Chem. 2004;279(47):49338–49345. doi: 10.1074/jbc.M409307200. [DOI] [PubMed] [Google Scholar]
- 182.Grintsevich EE, Benchaar SA, Warshaviak D, Boontheung P, Halgand F, Whitelegge JP, Faull KF, Loo RR, Sept D, Loo JA, Reisler E. Mapping the cofilin binding site on yeast G-actin by chemical cross-linking. J Mol Biol. 2008;377(2):395–409. doi: 10.1016/j.jmb.2007.12.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Mouradov D, Craven A, Forwood JK, Flanagan JU, Garcia-Castellanos R, Gomis-Ruth FX, Hume DA, Martin JL, Kobe B, Huber T. Modelling the structure of latexin-carboxypeptidase A complex based on chemical cross-linking and molecular docking. Protein Eng Des Sel. 2006;19(1):9–16. doi: 10.1093/protein/gzi070. [DOI] [PubMed] [Google Scholar]
- 184.Carlsohn E, Angstrom J, Emmett MR, Marshall AG, Nilsson CL. Chemical cross-linking of the urease complex from Helicobacter pylori and analysis by Fourier transform ion cyclotron resonance mass spectrometry and molecular modeling. International Journal of Mass Spectrometry. 2004;234(1–3):137–144. [Google Scholar]
- 185.Nilges M, Clore GM, Gronenborn AM. Determination of three-dimensional structures of proteins from interproton distance data by hybrid distance geometry-dynamical simulated annealing calculations. FEBS Lett. 1988;229(2):317–324. doi: 10.1016/0014-5793(88)81148-7. [DOI] [PubMed] [Google Scholar]
- 186.Geyer CJ. Markov chain Monte Carlo maximum likelihood; Computing Science and Statistics, 23rd Symposium on the Interface, 1991.1991. [Google Scholar]
- 187.Aszodi A, Gradwell MJ, Taylor WR. Global fold determination from a small number of distance restraints. J Mol Biol. 1995;251(2):308–326. doi: 10.1006/jmbi.1995.0436. [DOI] [PubMed] [Google Scholar]
- 188.Lin M, Lu HM, Chen R, Liang J. Generating properly weighted ensemble of conformations of proteins from sparse or indirect distance constraints. J Chem Phys. 2008;129(9):094101. doi: 10.1063/1.2968605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstelve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR System: a new software suite for macromolecular structure determination. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 190.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 191.DeWitte RS, Michnick SW, Shakhnovich EI. Exhaustive enumeration of protein conformations using experimental restraints. Protein Sci. 1995;4(9):1780–1791. doi: 10.1002/pro.5560040913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Faulon JL, Sale K, Young M. Exploring the conformational space of membrane protein folds matching distance constraints. Protein Sci. 2003;12(8):1750–1761. doi: 10.1110/ps.0305003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Sale K, Faulon JL, Gray GA, Schoeniger JS, Young MM. Optimal bundling of transmembrane helices using sparse distance constraints. Protein Sci. 2004;13(10):2613–2627. doi: 10.1110/ps.04781504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zheng W, Brooks BR. Modeling protein conformational changes by iterative fitting of distance constraints using reoriented normal modes. Biophys J. 2006;90(12):4327–4336. doi: 10.1529/biophysj.105.076836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Levitt M. A simplified representation of protein conformations for rapid simulation of protein folding. J Mol Biol. 1976;104(1):59–107. doi: 10.1016/0022-2836(76)90004-8. [DOI] [PubMed] [Google Scholar]
- 196.Levitt M, Warshel A. Computer simulation of protein folding. Nature. 1975;253(5494):694–698. doi: 10.1038/253694a0. [DOI] [PubMed] [Google Scholar]
- 197.Simons KT, Bonneau R, Ruczinski I, Baker D. Ab initio protein structure prediction of CASP III targets using ROSETTA. Proteins. 1999;(Suppl 3):171–176. doi: 10.1002/(sici)1097-0134(1999)37:3+<171::aid-prot21>3.3.co;2-q. [DOI] [PubMed] [Google Scholar]
- 198.Chivian D, Kim DE, Malmstrom L, Bradley P, Robertson T, Murphy P, Strauss CE, Bonneau R, Rohl CA, Baker D. Automated prediction of CASP-5 structures using the Robetta server. Proteins. 2003;(53 Suppl 6):524–533. doi: 10.1002/prot.10529. [DOI] [PubMed] [Google Scholar]
- 199.Latek D, Ekonomiuk D, Kolinski A. Protein structure prediction: combining de novo modeling with sparse experimental data. J Comput Chem. 2007;28(10):1668–1676. doi: 10.1002/jcc.20657. [DOI] [PubMed] [Google Scholar]
- 200.Skolnick J, Kolinski A, Ortiz AR. MONSSTER: a method for folding globular proteins with a small number of distance restraints. J Mol Biol. 1997;265(2):217–241. doi: 10.1006/jmbi.1996.0720. [DOI] [PubMed] [Google Scholar]
- 201.Duarte CM, Wadley LM, Pyle AM. RNA structure comparison, motif search and discovery using a reduced representation of RNA conformational space. Nucleic Acids Res. 2003;31(16):4755–4761. doi: 10.1093/nar/gkg682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kolinski A. Protein modeling and structure prediction with a reduced representation. Acta Biochim Pol. 2004;51(2):349–371. [PubMed] [Google Scholar]
- 203.Kolinski A, Skolnick J. Assembly of protein structure from sparse experimental data: an efficient Monte Carlo model. Proteins. 1998;32(4):475–494. [PubMed] [Google Scholar]
- 204.Jonikas MA, Radmer RJ, Altman RB. Knowledge-based instantiation of full atomic detail into coarse-grain RNA 3D structural models. Bioinformatics. 2009;25(24):3259–3266. doi: 10.1093/bioinformatics/btp576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.D'Ambrosio C, Talamo F, Vitale RM, Amodeo P, Tell G, Ferrara L, Scaloni A. Probing the dimeric structure of porcine aminoacylase 1 by mass spectrometric and modeling procedures. Biochemistry. 2003;42(15):4430–4443. doi: 10.1021/bi0206715. [DOI] [PubMed] [Google Scholar]
- 206.Silva RA, Hilliard GM, Fang J, Macha S, Davidson WS. A three-dimensional molecular model of lipid-free apolipoprotein A–I determined by cross-linking/mass spectrometry and sequence threading. Biochemistry. 2005;44(8):2759–2769. doi: 10.1021/bi047717+. [DOI] [PubMed] [Google Scholar]
- 207.Tubb MR, Silva RA, Fang J, Tso P, Davidson WS. A three-dimensional homology model of lipid-free apolipoprotein A-IV using cross-linking and mass spectrometry. J Biol Chem. 2008;283(25):17314–17323. doi: 10.1074/jbc.M800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hasegawa H, Holm L. Advances and pitfalls of protein structural alignment. Curr Opin Struct Biol. 2009;19(3):341–348. doi: 10.1016/j.sbi.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 209.Qu X, Swanson R, Day R, Tsai J. A guide to template based structure prediction. Curr Protein Pept Sci. 2009;10(3):270–285. doi: 10.2174/138920309788452182. [DOI] [PubMed] [Google Scholar]
- 210.Jabbari H, Condon A, Zhao S. Novel and efficient RNA secondary structure prediction using hierarchical folding. J Comput Biol. 2008;15(2):139–163. doi: 10.1089/cmb.2007.0198. [DOI] [PubMed] [Google Scholar]
- 211.Wu JC, Gardner DP, Ozer S, Gutell RR, Ren P. Correlation of RNA secondary structure statistics with thermodynamic stability and applications to folding. J Mol Biol. 2009;391(4):769–783. doi: 10.1016/j.jmb.2009.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Chyou DT, Rawle VL, Trotman CN. Quaternary structure of Artemia haemoglobin II: analysis of T and C polymer alignment and interpolymer interface. BMC Struct Biol. 2007;7:26. doi: 10.1186/1472-6807-7-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Fuzesi M, Gottschalk KE, Lindzen M, Shainskaya A, Kuster B, Garty H, Karlish SJ. Covalent cross-links between the gamma subunit (FXYD2) and alpha and beta subunits of Na,K-ATPase: modeling the alpha-gamma interaction. J Biol Chem. 2005;280(18):18291–18301. doi: 10.1074/jbc.M500080200. [DOI] [PubMed] [Google Scholar]
- 214.Lubben M, Portmann R, Kock G, Stoll R, Young MM, Solioz M. Structural model of the CopA copper ATPase of Enterococcus hirae based on chemical cross-linking. Biometals. 2009;22(2):363–375. doi: 10.1007/s10534-008-9173-4. [DOI] [PubMed] [Google Scholar]
- 215.Taverner T, Hernandez H, Sharon M, Ruotolo BT, Matak-Vinkovic D, Devos D, Russell RB, Robinson CV. Subunit architecture of intact protein complexes from mass spectrometry and homology modeling. Acc Chem Res. 2008;41(5):617–627. doi: 10.1021/ar700218q. [DOI] [PubMed] [Google Scholar]
- 216.Dietrich KA, Sindelar CV, Brewer PD, Downing KH, Cremo CR, Rice SE. The kinesin-1 motor protein is regulated by a direct interaction of its head and tail. Proc Natl Acad Sci U S A. 2008;105(26):8938–8943. doi: 10.1073/pnas.0803575105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Dimova K, Kalkhof S, Pottratz I, Ihling C, Rodriguez-Castaneda F, Liepold T, Griesinger C, Brose N, Sinz A, Jahn O. Structural insights into the calmodulin-Munc13 interaction obtained by cross-linking and mass spectrometry. Biochemistry. 2009;48(25):5908–5921. doi: 10.1021/bi900300r. [DOI] [PubMed] [Google Scholar]
- 218.Chu F, Shan SO, Moustakas DT, Alber F, Egea PF, Stroud RM, Walter P, Burlingame AL. Unraveling the interface of signal recognition particle and its receptor by using chemical cross-linking and tandem mass spectrometry. Proc Natl Acad Sci U S A. 2004;101(47):16454–16459. doi: 10.1073/pnas.0407456101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Sali A, Potterton L, Yuan F, van Vlijmen H, Karplus M. Evaluation of comparative protein modeling by MODELLER. Proteins. 1995;23(3):318–326. doi: 10.1002/prot.340230306. [DOI] [PubMed] [Google Scholar]
- 220.Egea PF, Shan SO, Napetschnig J, Savage DF, Walter P, Stroud RM. Substrate twinning activates the signal recognition particle and its receptor. Nature. 2004;427(6971):215–221. doi: 10.1038/nature02250. [DOI] [PubMed] [Google Scholar]
- 221.Focia PJ, Shepotinovskaya IV, Seidler JA, Freymann DM. Heterodimeric GTPase core of the SRP targeting complex. Science. 2004;303(5656):373–377. doi: 10.1126/science.1090827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Schulz DM, Kalkhof S, Schmidt A, Ihling C, Stingl C, Mechtler K, Zschornig O, Sinz A. Annexin A2/P11 interaction: new insights into annexin A2 tetramer structure by chemical crosslinking, high-resolution mass spectrometry, and computational modeling. Proteins. 2007;69(2):254–269. doi: 10.1002/prot.21445. [DOI] [PubMed] [Google Scholar]
- 223.Gautheret D, Major F, Cedergren R. Modeling the three-dimensional structure of RNA using discrete nucleotide conformational sets. J Mol Biol. 1993;229(4):1049–1064. doi: 10.1006/jmbi.1993.1104. [DOI] [PubMed] [Google Scholar]
- 224.Shen LX, Tinoco I., Jr The structure of an RNA pseudoknot that causes efficient frameshifting in mouse mammary tumor virus. J Mol Biol. 1995;247(5):963–978. doi: 10.1006/jmbi.1995.0193. [DOI] [PubMed] [Google Scholar]
- 225.Tinoco I, Bustamante C. How RNA folds. J. Mol. Biol. 1999;293(2):271–281. doi: 10.1006/jmbi.1999.3001. [DOI] [PubMed] [Google Scholar]