

Abstract
Stress and anxiety-related behaviors controlled by the basolateral amygdala (BLA) are regulated in vivo by neuropeptide Y (NPY) and corticotrophin-releasing factor (CRF): NPY produces anxiolytic effects, whereas CRF produces anxiogenic effects. These opposing actions are likely mediated via regulation of excitatory output from the BLA to afferent targets. In these studies, we examined mechanisms underlying the effects of NPY and CRF in the BLA using whole-cell patch-clamp electrophysiology in rat brain slices. NPY, even with tetrodotoxin present, caused a dose-dependent membrane hyperpolarization in BLA pyramidal neurons. The hyperpolarization resulted in the inhibition of pyramidal cells, despite arising from a reduction in a voltage-dependent membrane conductance. The Y1 receptor agonist, F7P34 NPY, produced a similar membrane hyperpolarization, whereas the Y1 antagonist, BIBO3304 [(R)-N-[[4-(aminocarbonylaminomethyl)-phenyl]methyl]-N2-(diphenylacetyl)-argininamide trifluoroacetate], blocked the effect of NPY. The NPY-inhibited current was identified as Ih, which is active at and hyperpolarized to rest. Responses to NPY were occluded by either Cs+ or ZD7288 (4-ethylphenylamino-1,2-dimethyl-6-methylaminopyrimidinium chloride), but unaffected by the GIRK-preferring blockers Ba2+ and SCH23390 [(R)-(+)-7-chloro-8-hydroxy-3-methyl-l-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride]. Application of CRF, with or without TTX present, depolarized NPY-sensitive BLA pyramidal neurons, resulting from an increase in Ih. Electrophysiological and immunocytochemical data were consistent with a major role for the HCN1 subunit. Our results indicate that NPY, via Y1 receptors, directly inhibits BLA pyramidal neurons by suppressing a postsynaptic Ih, whereas CRF enhances resting Ih, causing an increased excitability of BLA pyramidal neurons. The opposing actions of these two peptides on the excitability of BLA output cells are consistent with the observed behavioral actions of NPY and CRF in the BLA.
Introduction
Anxiety disorders are among the most prevalent psychiatric disorders (Offord et al., 1996; Kessler et al., 2005; Alonso et al., 2007). Although a number of brain sites are associated with the generation of anxiety, the basolateral amygdala (BLA) is an important mediator of emotional responses (LeDoux, 1992; Davis et al., 1994). The amygdala is critical for assessing the significance and risk of novel situations or objects, with greater perceived risk enhancing the fear response (Amaral, 2002; Sajdyk et al., 2004; Herry et al., 2007; Ghods-Sharifi et al., 2009). Increased amygdala output is associated with fear responses and emotionality, and inappropriately elevated BLA output is associated with an array of anxiety-related disorders (Lorberbaum et al., 2004; Shin et al., 2006; Truitt et al., 2007). Recent efforts toward novel treatments for anxiety disorders have focused on neuropeptides with neuropeptide Y (NPY) being of particular interest as an anxiolytic compound.
BLA activity is governed in part by a balance between excitatory and inhibitory tone with corticotrophin-releasing factor (CRF) and NPY, respectively, playing important roles in governing the activity of this nucleus (Heilig et al., 1994). NPY and CRF have been implicated in modulating the short-term activity of this nucleus as well as inducing longer-term changes in neural activity and behavior (Rainnie et al., 2004; Sajdyk et al., 2008). Administration of NPY into the BLA is anxiolytic and reduces the expression of conditioned fear responses (Broqua et al., 1995; Gutman et al., 2008) primarily via Y1 receptors (Heilig and Widerlöv, 1995; Sajdyk et al., 1999). In contrast to NPY, CRF administration produces anxiogenic-like behavior via CRF R1 receptors, in the BLA (Rainnie et al., 2004; Sajdyk et al., 2004, 2006). These anxiogenic effects of CRF are prevented by pretreatment with NPY (Sajdyk et al., 2004), further underscoring the importance of NPY as a stress buffering system (Heilig et al., 1994).
Cellular responses to NPY have been studied in many other brain regions, including the hippocampus (Colmers and Bleakman, 1994; Pentney et al., 2002), hypothalamus (Cowley et al., 1999; Fu et al., 2004; Melnick et al., 2007), and lateral amygdala (Sosulina et al., 2008). Postsynaptic effects of NPY include activation of inwardly rectifying K+ (GIRK) channels (Sun and Miller, 1999; Sosulina et al., 2008; Chee et al., 2010) causing an inhibition mediated primarily by the Y1 receptor subtype (Zhang et al., 1994; Fu et al., 2004) (but see Ghamari-Langroudi et al., 2005).
In this series of studies, we investigated the mechanism(s) through which NPY mediates anxiolysis by examining its effects on the electrophysiology of pyramidal output neurons in the BLA. In brief, we observed that NPY inhibited pyramidal neurons by suppressing a hyperpolarization-activated, depolarizing current (Ih) that is active at their resting membrane potential. Conversely, CRF enhanced Ih in the same pyramidal neurons in which NPY suppressed Ih. Our findings are consistent with the notion that the fear-related output of the BLA is acutely regulated by the relative states of activation of NPY and CRF receptors converging on Ih in BLA pyramidal cells.
Materials and Methods
Animals.
Male Sprague Dawley rats aged 21–56 d were used for the experiments. The care and use of animals was in accordance with the protocols set by the University of Alberta Animal Care and Use Committee: Health Sciences. Animals were housed in groups of two to four rats, and food and water were supplied ad libitum.
Brain slice preparation.
Rats were decapitated and their brains rapidly submerged in cold (<4°C) artificial CSF (ACSF) that was bubbled with carbogen (95% O2, 5% CO2) and contained the following (in mm): 118 NaCl, 3 KCl, 1.3 MgSO4, 1.4 NaH2PO4, 5.0 MgCl2·6H2O, 10 glucose, 26 NaHCO3, and 1.5 CaCl2. Kynurenic acid (1 mm) was also added to the slicing solution to prevent excitatory damage caused by ionotropic glutamate receptor activation. Coronal brain slices 300 μm thick containing the BLA were prepared using a vibrating slicer (Slicer HR2; Sigmann Elektronik). The brain slices were placed into warm (34 ± 0.5°C) bubbled slicing solution for ∼20–30 min and then were transferred to a room temperature (22°C), carbogenated ACSF solution containing the following (in mm): 124 NaCl, 3 KCl, 1.3 MgSO4, 1.4 NaH2PO4, 10 glucose, 26 NaHCO3, and 2.5 CaCl2. This solution was used as the basis for all remaining experiments. Slices were acclimatized to room temperature for an additional 30 min before being placed into the recording chamber. Slices were held submerged by a platinum and polyester fiber “harp” in a recording chamber that is attached to a fixed stage and viewed with a movable upright microscope (Axioskop FS2; Carl Zeiss). The slices were continuously perfused with warmed (34 ± 0.5°C), carbogenated ACSF at a rate of ∼2 ml/min for ∼20 min before initiating recordings.
Electrophysiology.
Pipettes were pulled from thin-walled borosilicate glass (TW150F; World Precision Instruments) with a two-stage puller (PP-83; Narishige). Tip resistance was 5 MΩ when pipettes were backfilled with an internal solution containing the following (in mm): 5 HEPES, 2 KCl, 136 K+-gluconate, 5 EGTA, 5 Mg·ATP, 8 creatine phosphate, and 0.35 GTP. The pH was adjusted to 7.27 with KOH, and the osmolarity adjusted with distilled water or concentrated K+-gluconate if needed to between 295 and 298 mOsm with an osmometer (3-MO; Advanced Instruments). All recordings were made using either an AxoClamp 2A or a Multiclamp amplifier, and data were acquired using pCLAMP 9.2 via a Digidata 1322 interface (all Molecular Devices). After a recording was completed, we corrected the nominal membrane potential in voltage- and current-clamp recordings for the calculated 15 mV liquid junction potential (Chee et al., 2010). All potential values reported reflect this correction.
The BLA was identified based on the Paxinos and Watson (1986) atlas. Pyramidal neurons within the BLA were identified visually using infrared–differential interference contrast optics. Neurons were selected based on a pyramidal morphology and the presence of large primary dendrite. Gigaohm seals were initially established in current clamp; once a seal was formed, the patch was ruptured with the cell held near the resting potential in the voltage-clamp mode. Once whole-cell recording had been established, neurons were routinely held in voltage clamp at −75 mV except when examining changes in the resting membrane potential and rheobase, which were performed in current clamp. Cells were only studied if they exhibited a stable holding current and access resistance for at least 10 min before experimental manipulations. Not all neurons within the BLA were sensitive to NPY; therefore, only neurons that responded reversibly to an initial application of NPY were used for additional experiments. Concentration–response studies with NPY were conducted beginning with 30 nm and progressively increasing to 3 μm. Recordings of neuronal properties were made immediately before drug application (control), during drug perfusion (lasting ∼3–4 min), and at 2, 5, 10, and 20 min during the washout period. In some cells, the washout was prolonged and recordings were taken until 30 min after washout began. A similar sequence of recordings, including an assessment of access resistance, was acquired for each cell and condition. Changes in the resting membrane potential were assessed by comparison of 120-s-long current-clamp recordings made under each condition (AxoScope 9.2; Molecular Devices). Relatively brief (2–3 min) periods interrupted the recording of membrane potential during which the cell was held in voltage clamp and various experiments were performed.
Rheobase measurements.
In experiments to determine changes in rheobase, neurons were held in current clamp, and families of up to eight depolarizing current ramps were swept from 0 pA initially to 100 pA; the peak current was incremented by 120 pA per successive sweep, with the eighth sweep thus ending at a maximum value of 840 pA. During the peak effects of NPY, this protocol was initially applied from the new, hyperpolarized resting membrane potential. Once this was completed, steady-state depolarizing current was applied to return the membrane to the pre-NPY resting potential and the same current ramp protocol repeated under these conditions. The minimum current necessary to elicit the first action potential during a ramp was designated as the rheobase current of the cell for the condition studied.
Cells were held near rest at −75 mV in voltage-clamp experiments. To assess changes in the steady-state membrane current–voltage (I–V) relationship, we applied a slow (18 mV/s), positive-going, voltage ramp protocol, starting from −135 mV (after initially holding the cell at −135 mV for 2 s), then sweeping to −55 mV before returning to the holding potential. With the exception of Figure 2A, values for mean current (in picoamperes) were taken every 10 mV from the voltage ramp between −135 and −55 mV for each experimental condition. Similar calculations were used throughout for all voltage ramp comparisons. Changes in net current caused by experimental treatment were determined by subtracting the membrane current response during the maximal drug effect from control.
Figure 2.

NPY reduces a steady-state current in BLA pyramidal cells by a postsynaptic action independent of GABAA receptors. A, Raw steady-state current responses to a slow voltage ramp compared in a single BLA neuron in control (black) with 1 μm NPY present (dark gray) and after washout (light gray). B, Comparison of steady-state current–voltage relationships from BLA pyramidal cells at potentials indicated without and with TTX present (n = 12). The dotted line represents zero current. C, Comparison, in the same neurons, of membrane potential change with NPY (1 μm) alone, bicuculline (10 μm) alone, and NPY in the presence of bicuculline (n = 9). D, Net current change (INET) caused by NPY in the absence or presence of bicuculline in the same neurons (n = 9). ns, No significant difference.
To study Ih, a family of hyperpolarizing voltage steps was applied from a holding potential of −55 mV (−65 to −125 mV in 10 mV increments; the initial step lasted 1650 ms, and each subsequent step was successively shortened by a 100 ms increment to prevent damage to the membrane). The magnitude of Ih at a given potential step was determined as the difference between the initial peak positive current amplitude and the final, steady-state current for each step. In a series of experiments, the Na+-current blocker, tetrodotoxin (TTX) (0.5 μm) was included in the perfusate to suppress the possible activity-dependent release of other chemical messengers. Drugs were then tested in the presence of TTX after pretreatment with the TTX-containing perfusate alone.
Dual-label immunohistochemistry.
For the immunohistochemical studies, adult male (250–350 g) Sprague Dawley rats (Charles River Laboratories) were housed three to a conventional cage, with ad libitum access to standard laboratory chow and water, located in a temperature (20–22°C)-, humidity (50–55%)-, and illumination (14:10 h light/dark cycle)-controlled, Association for Assessment and Accreditation of Laboratory Animal Care-approved facility. Five days of acclimatization to our facilities were allowed before the animals were included in any experimental procedure. All procedures were approved by the Rosalind Franklin University Institutional Animal Care and Use Committee. Animals were deeply anesthetized with sodium pentobarbital (Sigma-Aldrich; 100 mg/kg, i.p.) and transcardially perfused with 30 ml of PBS (10 mm Na2HPO4, 150 mm NaCl, pH 7.5) containing 0.1% procaine and 100 U/ml heparin at 37°C followed by 60 ml of fixative solution consisting of 4% paraformaldehyde (PFA) in PBS at 4°C. Brains were rapidly dissected out, postfixed for 6 h in 4% PFA solution, followed by an hour-long PBS wash at +4°C. Coronal brain sections were cut in a bath of ice-cold PBS at 40 μm thickness using a vibratome (Vibratome 1000; Ted Pella).
To determine the association of the hyperpolarization-activated, cyclic nucleotide-gated (HCN) subunits with BLA pyramidal cells, we performed double-label immunocytochemistry for CaMKII (a specific marker for BLA projection neurons; mouse monoclonal, clone 6G9; Millipore) (McDonald et al., 2002; Rostkowski et al., 2009) and HCN subunit (1, 2, 3, or 4; NeuroMab) immunoreactivity. On Western blot, the HCN1–4 antibodies detect proteins of the appropriate molecular weight form for each subunit from rat brain and transfected cell lines; no signal for HCN1 was present in tissue from the HCN1 knock-out animal (NeuroMab). Briefly, free-floating sections were rinsed through three changes of PBS over 10 min, followed by a 15 min wash in 1% H2O2 in PBS to diminish endogenous peroxidase activity. Next, tissues were blocked for 3 h in immunocytochemistry (ICC) buffer (0.1 m PBS containing 0.2% gelatin, 0.01% thimerosal, and 0.002% neomycin, pH 7.5) with 5% normal donkey serum (NDS) (Equitech-Bio) to block nonspecific binding. Sections were then incubated at +4°C for 72 h with primary HCN subunit antibody diluted 1:2000 in ICC with 2% NDS. After incubation with primary antibody, sections were washed through five changes of ICC buffer over 50 min and then incubated with biotinylated, affinity-purified donkey anti-rabbit IgG (Jackson ImmunoResearch Laboratories; 1:2000) for 1 h at room temperature. After ICC buffer rinses, sections were incubated in Vectastain Elite ABC (Vector Laboratories; 2 μl/ml) for 30 min. Next, sections were rinsed with PBS and incubated in biotinylated tyramide solution (3 μg/ml biotinylated tyramide and 0.01% H2O2 in PBS) for 10 min. Tissues were then rinsed in ICC buffer and immersed in ICC buffer containing Cy3-conjugated streptavidin (Cy3-SA; Jackson ImmunoResearch Laboratories; 1:250) for 3 h. After washes in four changes of Tris-buffered saline (TBS) (100 mm Tris base, 150 mm NaCl, pH 7.5) over 20 min, sections were then incubated with CaMKII antibody (1:6000; overnight at +4°C in ICC buffer with 2% NDS). Sections were subsequently washed through five changes of ICC buffer over 50 min followed by a 3 h incubation in ICC buffer containing FITC donkey anti-mouse secondary antibody (Jackson ImmunoResearch Laboratories; 1:250) for visualization of CaMKII. After washes in four changes of TBS over 20 min, sections were mounted onto Superfrost Plus slides (Thermo Fisher Scientific), and coverslips were rapidly applied with PVA-DABCO antifade mounting medium.
Confocal microscopy.
Tissues were labeled with multiple fluorescent markers for experiments that required analysis of colocalized immunostained cells. Selected sections of the BLA throughout the rostral-caudal extent of the nucleus [bregma −1.8 to −4.16 mm (Paxinos and Watson, 1986)] were imaged at 60× magnification using an Olympus Fluoview 300 confocal microscope (Olympus; Microscopy Imaging Facility, Rosalind Franklin University) equipped with a motorized x–y–z stage control. The BLA was defined as including the following: posterior subdivision of the basolateral amygdalar nucleus (BLp) and anterior subdivision of the basolateral amygdalar nucleus (BLa); aspects of the lateral amygdala were not included in this analysis. The following excitation wavelengths were used: 488 nm for the secondary fluorophore FITC and 568 nm for Cy3. Colocalization was determined by overlapping signals observed at several focal planes throughout each cell. Omission of either the CaMKII or specific HCN1–4 subunit antibody produced no signal in the appropriate channel, indicating that there was no cross-reactivity of secondary antibodies.
Brightness and contrast of the photomicrographs presented here were adjusted using Adobe Photoshop 6.0 to ensure the highest quality images for publication.
Determination of HCN channel expression in the BLA using reverse transcriptase–PCR.
Adult male Sprague Dawley rats were housed as indicated above for the immunohistochemistry. Rats were anesthetized with 4% isoflurane and killed by decapitation. The BLA was rapidly dissected from 400-μm-thick coronal brain sections under a dissecting microscope. The tissue was immediately placed in RNase-free microcentrifuge tubes on dry ice. The tissue was stored at −80°C until isolation of RNA using Trizol reagent. The amount and integrity of the RNA was determined using spectrophotometic readings at 260 and 280 nm and assessment on agarose gel (1%) electrophoresis. The RNA was treated with DNase I to remove any residual DNA contamination and the resultant RNA (1 μg) was subjected to reverse transcriptase (RT)-PCR (SuperScript III One-Step RT-PCR System; Invitrogen) using primers specific for the HCN1 (forward, 5′-CTCTCTTTGCTAACGCGGAT-3′; reverse, 5′-TTGAAATTGTCCACCGAA-3′; accession number, NM_053375), HCN2 (forward, 5′-GTGGAGCGAACTCTATTCGT-3′; reverse, 5′-GTTCACAATCTCCTCACGCA-3′; accession number, NM_053684), HCN3 (forward, 5′-GCAGCATTTGGTACAACACG-3′; reverse, 5′-AGCGTCTAGCAGATCGAGC-3′; accession number, NM_053685), and HCN4 (forward, 5′-GCAGCGCATCCACGACTAC-3′; reverse, 5′-CGTCACAAAGTTGGGGTCTGC-3′; accession number, NM_021658) subunit mRNA. Appropriate controls (no RNA and no RT) were run to assess specificity. The RT-PCR was run according to manufacturer's guidelines (PCR: 94°C 2 min denaturing; 55–57°C annealing; 68°C 1 min elongation) with modifications of annealing temperatures (HCN1 and 4: 55°C, 40 cycles; HCN2 and 3: 57°C, 34 cycles) to yield optimal signal. This end-point PCR analysis determined the presence of HCN subunit mRNA within the BLA and was not designed to quantitate the amount of mRNA within the tissue. Aliquots of the resultant PCR products were separated using gel electrophoresis; the size of the products was determined using a 100 bp DNA ladder. Images of the gel were obtained using a Kodak Gel Imaging system and the scans were imported into Adobe Photoshop for optimization of contrast and brightness.
Materials.
Human NPY was purchased from Dr. S. St. Pierre (Peptidec Technologies). Kynurenic acid was acquired from either Sigma-Aldrich or Ascent Scientific. KOH was bought from BDH Chemicals. Creatine phosphate and GTP were purchased from Roche Diagnostics. BaCl2, CsCl, K+-gluconate, EGTA, and Mg·ATP were obtained from Sigma-Aldrich. TTX was bought from Alomone Labs. CRF, bicuculline, (R)-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH23390), 4-ethylphenylamíno-1,2-dimethyl-6-methylaminopyrimidinium chloride (ZD7288), and dibutyryl-cAMP (db-cAMP) were purchased from Tocris Bioscience. The Y1 agonist F7P34 NPY ([Phe7, Pro34]NPY) and the Y2 agonist [6-aminohexanoic5-24]NPY ([ahx5-24]NPY) were gifts from Dr. A. G. Beck-Sickinger (Leipzig, Germany). The Y2 antagonist (S)-N2-[[1-[2-[4-[(R,S)-5,11-dihydro-6(6h)-oxodibenz[b,e]azepin-11-yl]-1-piperazinyl]-2-oxoethyl]cylopentyl]acetyl]-N-[2-[1,2-dihydro-3,5(4H)-dioxo-1,2-diphenyl-3H-1,2,4-triazol-4-yl]ethyl]argininamid (BIIE0246) and the Y1 antagonist (R)-N-[[4-(aminocarbonylaminomethyl)-phenyl]methyl]-N2-(diphenylacetyl)-argininamide trifluoroacetate (BIBO3304) were gifts from Dr. H. Doods (Boehringer Ingelheim, Biberach, Germany). The Y5 antagonist trans-naphthalene-2-sulfonic acid 4-((4-(2-dimethylaminopropyl amino)quinazolin-2-ylamino)methyl)cyclohexylmethyl)amide (Novartis 2) was a gift from Dr. P. Hipskind (Lilly Research Laboratories, Indianapolis, IN). All other chemicals were obtained from either Thermo Fisher Scientific or EMD Chemicals. All test compounds were stored as concentrated stock solutions at −20°C and diluted in ACSF immediately before bath application, except BIIE0246, which was dissolved in ethanol as a 1 mm solution and then diluted in ACSF before use.
Data analysis.
Recordings were viewed and analyzed off-line using pClamp 10.2 (Molecular Devices). Figures of electrophysiological traces were composed with Axum 5.0 (Mathsoft). Statistical analyses were performed using GraphPad Prism, version 5.04. Data are expressed as mean ± SEM. In most cases, we used a repeated-measures one-way ANOVA to analyze the effects of a drug (e.g., ZD7288 or Cs+) on neuronal properties characterized by multiple elements (e.g., current–voltage relationships in which current was measured every 10 mV). Such properties were compared in repeated measurements on individual neurons in control conditions, in the presence of a drug, and then either after drug washout, or in the presence of a second drug, such as an antagonist. A Bonferroni test was used for post hoc analysis. The concentration–response curve was fit with a nonlinear regression sigmoidal dose–response equation provided in the software. When comparing the effect of a drug on a single variable in the same neuron, a paired t test was used. Mean differences were considered to be significant at p < 0.05, and the levels of significance are indicated in all figures as follows: *p < 0.05, **p < 0.01, ***p < 0.001.
Results
Identification of pyramidal neurons in the BLA
Pyramidal neurons are the most abundant cell type within the BLA (McDonald, 1982). They were identified based on their characteristic pyramid-shaped soma, by their electrophysiological characteristics, and by exclusion of surrounding interneurons based on their smaller size, and rounder appearance, and differing electrical properties (Rainnie et al., 1993). The average resting membrane potential of a representative sample (n = 231) of BLA pyramidal neurons was −75.9 ± 0.57 mV. More than 300 BLA pyramidal neurons were examined for the effects of NPY alone. Approximately 50% of these neurons were sensitive to NPY, although we could determine no other characteristics, such as resting membrane potential or input resistance that helped distinguish NPY-sensitive versus NPY-insensitive neurons. A neuron was deemed to have responded to NPY when we observed a consistent hyperpolarization of ≥2 mV after NPY application, and which reversed on washout. All mechanistic experiments were performed in cells first demonstrating clear responses to NPY application.
NPY inhibits BLA pyramidal neurons via Y1 receptors
Application of NPY (1 μm) to neurons in current clamp caused a reversible membrane hyperpolarization from rest of 7.1 ± 1.3 mV (n = 93; p < 0.0001) (Fig. 1A1). Although in some cases there appeared to be an effect on spontaneous synaptic events in the BLA neurons (Fig. 1A1), the actions of NPY on the resting membrane potential of pyramidal cells did not depend on the activity of other neurons, because 1 μm NPY hyperpolarized them by −7.05 ± 0.31 mV with 500 nm TTX present (n = 128; p > 0.9 vs NPY alone) (Fig. 1A2). When applied at concentrations between 30 nm to 3 μm, NPY hyperpolarized pyramidal neuron membrane potentials in a concentration-dependent manner, with an EC50 of 404 nm.
Figure 1.
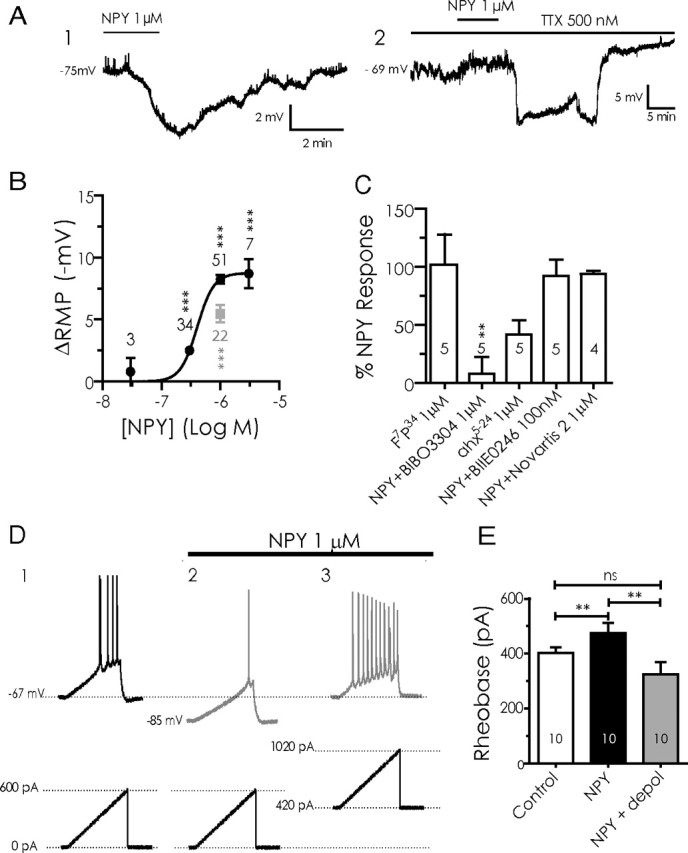
NPY inhibits BLA pyramidal neurons. A1, Current-clamp voltage recording (10 min continuous trace) of the hyperpolarization caused in a single BLA pyramidal neuron by application of NPY (1 μm). A2, NPY has a similar effect when applied in the presence of 500 nm TTX in a different BLA neuron. B, Concentration–response curve for the hyperpolarizing effect of NPY on membrane potential (ΔRMP). Concentrations ranged from 30 nm to 3 μm, with NPY (1 μm) having a near-maximal effect. Values next to each point denote the number of cells observed under that condition; significance values represent differences from zero effect. Effect of the Y1 agonist F7P34 NPY (1 μm) is shown as a gray square. C, Summary bar graph comparing the response to NPY receptor-selective agonists or the effect of NPY in the presence of receptor-selective antagonists against the response to NPY itself in BLA pyramidal cells. Values within each bar represent number of cells per condition. Error bars represent mean ± SEM. D, Effect of NPY (1 μm) on BLA neuron excitability. Membrane potential responses to current ramps (bottom traces) in control (D1) and in NPY (D2). D3, Increased firing response in the presence of NPY to current ramp in D1 and D2 superimposed on steady depolarizing current (bottom trace) returning cell to control membrane potential. E, Rheobase determined for BLA pyramidal cells in control, in the presence of 1 μm NPY, and depolarized to the control membrane potential with NPY present. Error bars in this and all subsequent figures represent mean ± SEM. Numbers of neurons tested under each condition for this and all other bar graphs are indicated in the bar. Significance levels for statistical tests in this and all other figures as described are denoted as follows: *p < 0.05, **p < 0.01, ***p < 0.001.
Because 1 μm was close to the maximal effective concentration of NPY (Fig. 1B), we used this concentration in the following mechanistic experiments. The Y1 receptor-selective agonist, F7P34 NPY (1 μm) (Soll et al., 2001; El Bahh et al., 2005), elicited a hyperpolarization similar in magnitude (5.5 ± 0.8 mV; n = 5) (Fig. 1B,C) to that seen in the same neurons with NPY itself (6.3 ± 1.3 mV; n = 5; p > 0.5). Furthermore, pretreatment with the Y1-selective antagonist BIBO3304 (1 μm) (Wieland et al., 1998) significantly reduced the magnitude of hyperpolarization by NPY from 8.7 ± 0.69 mV (n = 5) to 0.95 ± 1.3 mV (n = 5; p < 0.01). The Y2 receptor agonist [ahx5-24]NPY (1 μm) (Rist et al., 1995; El Bahh et al., 2002) produced a slight but not significant hyperpolarization (3.2 ± 1.2 mV; n = 5) that was much less than that caused by NPY in the same cells (6.9 ± 1.5 mV; n = 5; p < 0.01). In addition, the actions of NPY on membrane potential before (5.8 ± 1.2 mV; n = 5) or after pretreatment with the Y2-selective antagonist BIIE0246 (100 nm) (El Bahh et al., 2002) did not differ significantly (4.9 ± 0.60 mV; n = 5; p > 0.1). Finally, the actions of NPY on membrane potential before (8.8 ± 1.1 mV; n = 4) or after application of the Y5 receptor antagonist Novartis 2 (1 μm) (Pronchuk et al., 2002) were not significantly different (8.1 ± 0.8 mV; n = 4; p > 0.1). A comparison of the magnitude of hyperpolarization induced under each condition as a percentage of the NPY response in the same cells is shown in Figure 1C.
NPY reduces excitability in BLA pyramidal cells
To determine whether the hyperpolarization resulting from Y1 receptor activation in BLA pyramidal cells also made the neurons less readily excitable, we determined the rheobase in these neurons under control conditions and at the peak NPY effect. In the absence of NPY, the rheobase determined in this manner (Chee et al., 2010) was 402 ± 22 pA (n = 10) (Fig. 1D). In the presence of NPY, these 10 neurons hyperpolarized from −64.7 ± 1.1 to −72.5 ± 1.8 mV (p < 0.01). Rheobase, when measured from the hyperpolarized membrane potential caused by NPY application, was increased to 475 ± 37 pA (p < 0.02; n = 10) (Fig. 1D). Interestingly, when steady-state current was used to depolarize the cell to the original membrane potential with NPY present, the rheobase as determined with current ramps was only 324 ± 45 pA, significantly smaller than in control (p < 0.05; n = 10) (Fig. 1E). All effects of NPY reversed on washout. Thus, when NPY hyperpolarized these neurons, more current was needed to achieve threshold from their new resting potential, but when the cells were returned with depolarizing current to their original resting potential with NPY present, they appeared more excitable.
The effect of NPY on steady-state membrane current
To determine the mechanism of the actions of NPY, we studied its effects on the steady-state current–voltage relationship of BLA pyramidal neurons using a voltage ramp protocol (Fig. 2A) (Kombian and Colmers, 1992; Pentney et al., 2002). NPY (1 μm) application caused a prominent reduction in inward current at potentials at or negative to rest (Fig. 2A). Near the resting membrane potential (−75 mV), this resulted in a net outward shift in membrane current (156 ± 13 pA; n = 77).
As above, we used the Na+ channel blocker, TTX (500 nm), to determine whether activity in other neurons was necessary for the actions of NPY on membrane current. Addition of TTX to control ACSF did not significantly change the membrane current–voltage relationship (n = 12) (Fig. 2B), consistent with NPY acting predominantly at a postsynaptic site. Furthermore, neither the NPY-mediated membrane hyperpolarization nor underlying change in membrane current were affected by the presence of the GABAA receptor antagonist, bicuculline (10 μm), suggesting that the actions of NPY did not require intact GABAA transmission (n = 9) (Fig. 2C,D).
The actions of NPY do not involve GIRK activation
In other neurons, hyperpolarizations caused by NPY receptor activation have been shown to result from the activation of a GIRK (Sun and Miller, 1999; Sun et al., 2001; Paredes et al., 2003; Fu et al., 2004), including in pyramidal neurons of the lateral amygdala (Sosulina et al., 2008). Therefore, we tested the hypothesis that NPY modulates a GIRK in BLA neurons. Initially, we used Ba2+ to determine whether GIRK activation mediated the actions of NPY, as Ba2+ is commonly used to block potassium currents, including GIRK currents (Sodickson and Bean, 1996, 1998; Fernandez-Fernandez et al., 1999; Takigawa and Alzheimer, 1999; Slesinger, 2001). By calculating the net difference in the net NPY response at −120 mV in the absence or presence of Ba2+ at different concentrations (10, 30, and 100 μm), we determined that Ba2+ (10 μm) does not occlude the NPY-sensitive current (130 ± 12%; n = 4) (Fig. 3A). Even in 100 μm Ba2+, a concentration usually quite effective at blocking GIRK responses (Fernandez-Fernandez et al., 1999), the NPY-sensitive current was reduced only to 75 ± 22% (n = 4; p > 0.05; not significant) of the control current.
Figure 3.
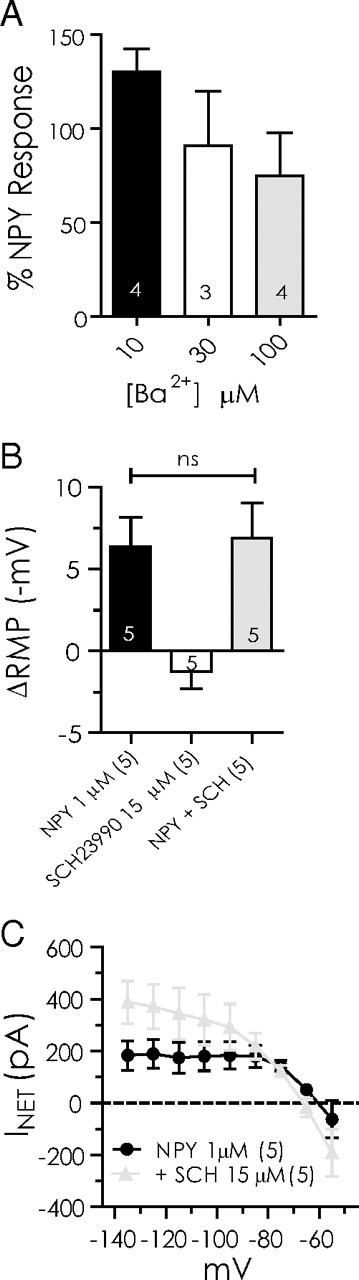
The inhibitory actions of NPY do not involve a GIRK. A, Response to NPY compared at −120 mV before and after treatment with different concentrations of Ba2+ (10, 30, and 100 μm). B, Comparison of hyperpolarization by NPY (1 μm) alone, SCH23390 (10 μm) alone, and NPY after SCH23390 pretreatment. C, Comparison of net current blocked by NPY before and after SCH23390 pretreatment. Differences were not significant (n = 5).
Because Ba2+ is not uniquely selective for blocking GIRKS, we next used SCH23990, a dopamine D1 receptor antagonist that also selectively blocks GIRK channels (Kuzhikandathil and Oxford, 2002; Sosulina et al., 2008; Chee et al., 2010) to determine whether GIRK channels mediated the NPY response. Application of SCH23390 (15 μm; 5 min) did not significantly affect resting membrane potential (Fig. 3B). The hyperpolarization caused by NPY was unaffected by pretreatment or cotreatment with SCH23990 (6.4 ± 1.8 vs 6.9 ± 2.2 mV; n = 5; p > 0.6) (Fig. 3B). The net current blocked by NPY between −135 and −75 mV was also unaffected by the inclusion of SCH23390 in the perfusate (Fig. 3C). Based on the above, we concluded that it is unlikely that modulation of a GIRK plays a significant role in mediating the effects of NPY.
NPY reduces the amplitude of Ih
Because blockade of GIRK channels did not greatly affect the actions of NPY, we next examined whether modulation of another inwardly rectifying current, Ih, mediated the effect of NPY on BLA neurons. Ih is a hyperpolarization-activated, nonselective cation current that conducts both Na+ and K+ (for review, see Pape, 1996). Previous studies in the BLA have identified the presence of a Cs+-sensitive Ih specifically in pyramidal neurons (Womble and Moises, 1993; Park et al., 2007). We therefore applied Cs+ extracellularly to block Ih (DiFrancesco, 1982; Pape, 1996) to determine whether it affected the postsynaptic response to NPY. In the absence of Cs+, NPY (1 μm) hyperpolarized BLA neurons by 5.9 ± 1.4 mV (n = 5; p < 0.05) (Fig. 4A). After NPY washout, bath application of 1 mm Cs+ for 10 min produced a slight, nonsignificant membrane hyperpolarization (1.6 ± 1.2 mV; n = 5). In voltage clamp, the overall membrane conductance was reduced by Cs+ (1 mm), most notably in the voltage region negative to Ek (Fig. 4B). One millimolar Cs+ blocked ∼60% of the control membrane conductance (n = 5) at −135 mV. More significantly, in the presence of Cs+, NPY no longer had any hyperpolarizing effect on the resting membrane potential (−1.7 ± 1.2 mV; n = 5; p > 0.02 vs NPY alone) (Fig. 4A), instead causing a small depolarization. Cs+ also occluded the NPY-sensitive current at all potentials (Fig. 4C).
Figure 4.
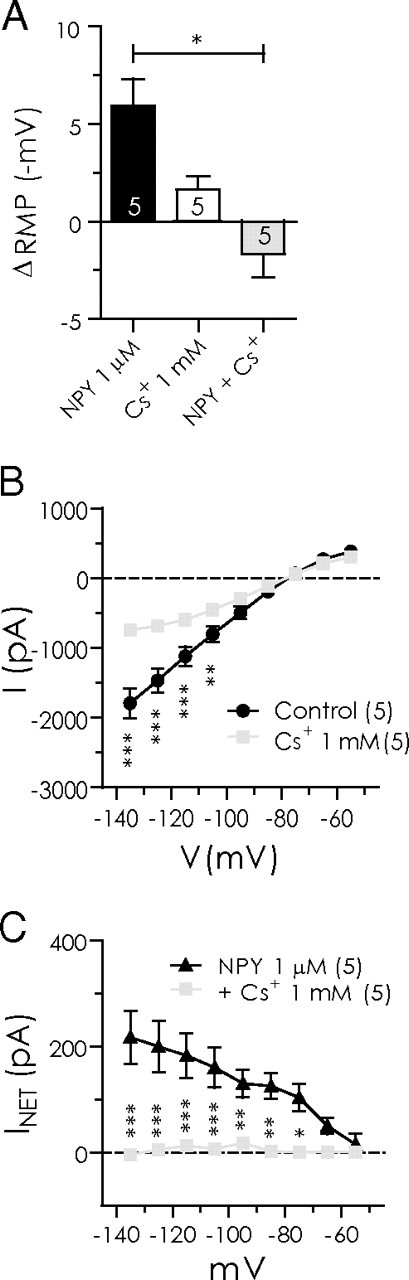
The response to NPY in BLA pyramidal cells is occluded by Cs+. A, Magnitude of resting membrane potential change with NPY (1 μm) alone, Cs+ (1 mm) alone, and NPY after Cs+ pretreatment. B, Steady-state membrane current response to voltage ramp (n = 5) is significantly reduced in presence of Cs+ alone. C, Voltage ramp summary comparing net current blocked by NPY in the absence and presence of Cs+. Cs+ completely occluded the effect of NPY (n = 5).
In experiments using a hyperpolarizing voltage step protocol from a (corrected) holding potential of −55 mV, we observed BLA pyramidal neurons to exhibit a robust Ih (Fig. 5A). The threshold for activation of Ih was approximately −65 mV and the current had an average apparent reversal potential near −57 mV. NPY (1 μm) significantly reduced the amplitude of Ih between −75 and −125 mV (Fig. 5B,C). The average amount of Ih reduced by NPY remained relatively steady between −95 and −125 mV, ranging from −130 ± 7 pA at −105 mV to −132 ± 8 pA at −115 mV (n = 77). NPY produced a small negative shift to the voltage dependence of activation of Ih from −101 to −105 mV (Fig. 5D). Consistent with an involvement of Ih in the actions of NPY, Cs+ (1 mm) alone also significantly affected the amplitude of Ih at potentials equal or negative to −95 mV, with the largest change to Ih amplitude at −125 mV (n = 5) (Fig. 5E). Interestingly, virtually all BLA pyramidal cells examined in voltage clamp demonstrated a clear Ih, whether or not they were sensitive to NPY or CRH.
Figure 5.
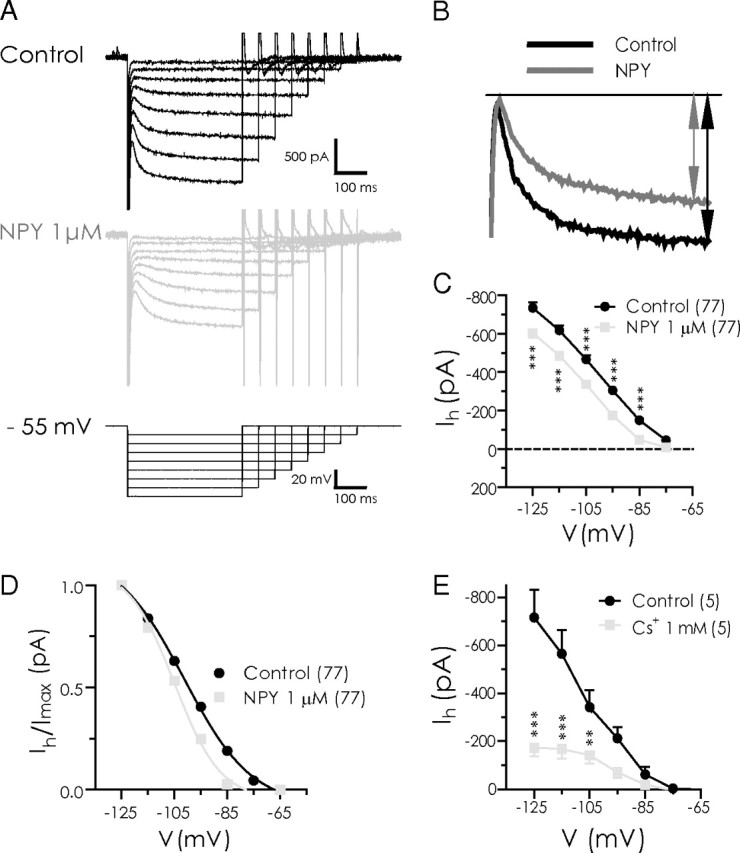
NPY reduces the amplitude of Ih in BLA pyramidal cells. A, Ih isolated (between −65 and −125 mV) with hyperpolarizing voltage steps in control (top) and with 1 μm NPY (middle). Cells were held at −55 mV and hyperpolarizing steps applied (bottom traces). B, Current responses to voltage step from −55 to −125 mV illustrating reduction in Ih amplitude with NPY (1 μm). C, Comparison of mean Ih amplitude at potentials between −125 and −75 mV in control and with NPY application (n = 77). Ih amplitude for each cell and condition was determined as illustrated in B. D, NPY induced a small leftward shift in the activation curve for Ih (n = 77). Data were normalized to the maximum Ih available for each condition, transformed to absolute values, and fitted to a nonlinear regression. Voltage shift for Ih was determined at 50% I/Imax. E, Cs+ strongly suppresses Ih in BLA pyramidal cells, determined using the same hyperpolarizing voltage step protocol as described in A (n = 5).
Effect of ZD7288 on the NPY-sensitive current
Given the above evidence that the response of NPY was related to a suppression of Ih, we tested the effect of the selective Ih blocker ZD7288 (Gasparini and DiFrancesco, 1997; Pentney et al., 2002). Perfusion of ZD7288 (10 μm) onto NPY-sensitive neurons for 20 min significantly hyperpolarized the neurons (7.5 ± 2.2 mV; n = 5; p < 0.05) (Fig. 6A) and blocked ∼60% of the total steady-state membrane current at −135 mV (Fig. 6B), thus closely resembling the effects of Cs+. As expected, the effect of ZD7288 on the Ih amplitude in the voltage step protocol was also similar to that observed with Cs+ (Fig. 6C), both in the magnitude of change and the voltage region affected, with the greatest difference observed at −125 mV (−657 ± 96 to −34 ± 20 pA; n = 5; p < 0.05). In control, NPY hyperpolarized BLA pyramidal neurons by 7.3 ± 2.4 mV (n = 5), whereas subsequent to a 20 min perfusion of ZD7288, NPY application actually modestly depolarized the membrane (by 2.1 ± 0.3 mV; p < 0.005; n = 5) (Fig. 6A). At rest, ZD7288 occluded the NPY-sensitive current from −75 ± 18 to 31 ± 31 pA (n = 5; p < 0.05) (Fig. 6D). Thus, blocking Ih with ZD7288 almost completely occluded the inhibitory effect of NPY across all potentials measured. These data provided stronger evidence that the postsynaptic actions of NPY are mediated by a suppression of Ih.
Figure 6.

The Ih blocker ZD7288 hyperpolarizes BLA pyramidal neurons and occludes the actions of NPY. A, Net membrane potential hyperpolarization compared in the same neurons with NPY (1 μm), ZD7288 (10 μm), and NPY after ZD7288 pretreatment (n = 5). B, Total steady-state membrane current is reduced by ZD7288 in BLA pyramidal cells (n = 5). C, ZD7288 sharply reduces Ih amplitude (n = 5). D, Net current caused by NPY in the absence and the presence of ZD7288 (n = 5).
CRF increases the amplitude of Ih
Based on behavioral evidence that CRF receptor activation is anxiogenic (Rainnie et al., 2004; Sajdyk et al., 2004), we hypothesized that CRF will influence Ih, in the direction opposite to that of NPY. We first determined the effects of CRF (30 nm) on BLA neurons by applying and washing out CRF, and then applying NPY (1 μm). In all five neurons, CRF produced a reversible depolarization (9.2 ± 0.98 mV; n = 5) (Fig. 7A1). After CRF washed out, NPY hyperpolarized the membrane of these neurons by a similar amount (8.7 ± 0.82 mV; n = 5; not illustrated). In separate experiments, we determined that the actions of CRF on these neurons were similar in the absence or presence of TTX (10.6 ± 2.8 mV, n = 9, vs 7.8 ± 1.5 mV, n = 10; p > 0.39) (Fig. 7A2).
Figure 7.
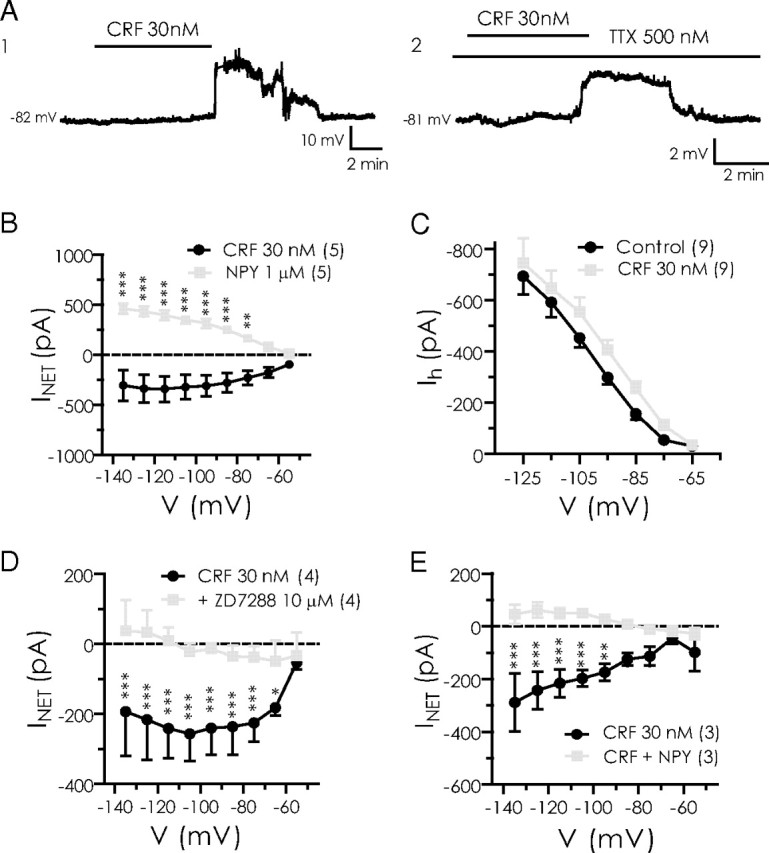
CRF depolarizes NPY-sensitive BLA neurons by enhancement of Ih. A1, A2, Change in membrane potential caused by 30 nm CRF in control (A1) or in the presence of TTX (A2) (recordings are from different neurons). B, Changes in steady-state current caused by CRF and NPY application in the same neurons (n = 5). C, Increase in Ih amplitude caused by CRF. D, CRF-sensitive current is blocked by ZD7288 (n = 4). E, NPY blocks CRF effects in CRF-sensitive neurons (n = 3).
Using the voltage ramp protocol, we determined that CRF increased an inward current while decreasing the outward current at potentials at or depolarized to rest (n = 5) (Fig. 7B). At −75 mV, CRF increased the inward current from −28 ± 20 to −270 ± 46 pA (n = 5; p < 0.05). By calculating the net current after either CRF or NPY treatment, we found that, whereas NPY reduced an inward current as discussed above, CRF appeared to enhance a current over the same potential range (Fig. 7B). Using the voltage step protocol, we determined that CRF increased Ih across the entire voltage region examined, and significantly enhanced it over control values at potentials between −75 and −105 mV (Fig. 7C). Near rest (−75 mV), CRF increased Ih from −55 ± 14 to −111 ± 20 pA (n = 9; p < 0.01). To confirm that CRF was indeed acting on Ih, we examined the effect of CRF in the absence and presence of ZD7288 (10 μm). In four pyramidal neurons, the CRF-mediated depolarization (9.4 ± 1.7 mV) was blocked when retested in the presence of 10 μm ZD7288 (0.36 ± 0.16 mV; n = 4; p < 0.05 vs CRF alone; not illustrated). Consistent with this, the net current induced by CRF was significantly blocked by ZD7288 between −75 and −115 mV (Fig. 7D). At −75 mV, the current produced by CRF was reduced from 225 ± 27 pA to only 38 ± 16 pA with ZD7288 present (n = 4; p < 0.01). In the presence of ZD7288, the CRF-mediated response was not significant (p > 0.25). Interestingly, when NPY was coapplied with CRF to CRF-responsive neurons, the Ih response to CRF was also abolished (Fig. 7E).
HCN channel subtypes carrying Ih in NPY-sensitive BLA cells
Four different isoforms of the HCN-gated channels mediate Ih (Santoro et al., 1997, 1998; Ludwig et al., 1998; Santoro and Tibbs, 1999). The four subunits, HCN1–HCN4, are differentially distributed throughout the CNS and vary in their responses both to voltage and cAMP (Ludwig et al., 1998, 1999; Santoro et al., 1998, 2000; Seifert et al., 1999; Chen et al., 2001). We tested the sensitivity of NPY-responsive neurons to elevations of cyclic nucleotides by adding the membrane-permeant analog, db-cAMP, to the bath perfusate at 30 μm, for at least 5 min before NPY reapplication (n = 5). Application of db-cAMP alone at 30 μm produced no significant effect on resting membrane potential (Fig. 8A), nor did the presence of db-cAMP alter the membrane hyperpolarization produced by NPY (1 μm) in the same neurons (7.5 ± 0.94 vs 7.7 ± 2.1 mV; n = 5; p > 0.9). In the presence of db-cAMP, the effect of NPY on net current blocked was not different from that seen before db-cAMP treatment (Fig. 8B). The only measurable effect was a modest increase in the amount of NPY-sensitive current measured at −135 mV in the presence of 30 μm db-cAMP. These results are consistent with the primary HCN subunit gating Ih in BLA pyramidal neurons being the virtually cAMP-insensitive HCN1 subunit (Santoro et al., 1998, 2000; Chen et al., 2001).
Figure 8.
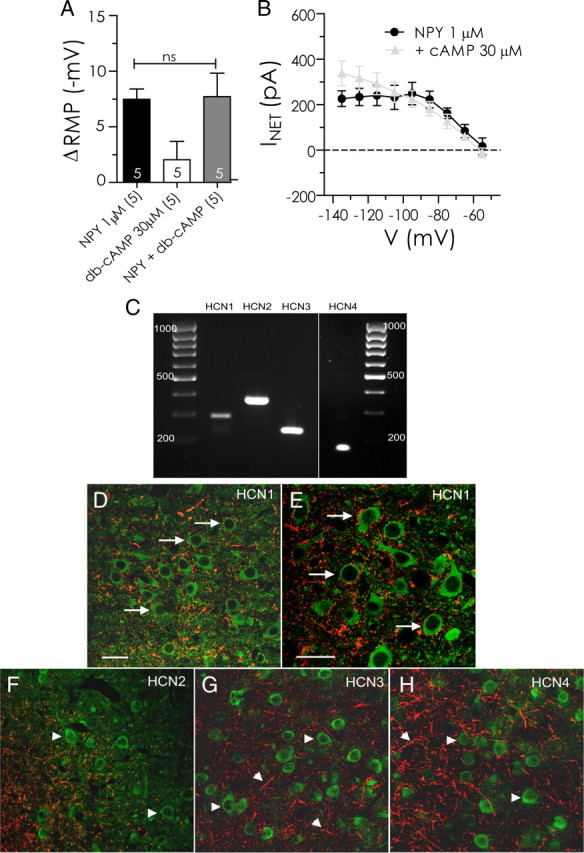
NPY-sensitive H-current is likely mediated by HCN1 subunits. A, NPY-mediated change in resting membrane potential is unaltered by pretreatment with the membrane-permeant cAMP analog, db-cAMP (30 μm). B, Net current caused by NPY is unaltered in the presence of db-cAMP (cAMP, 30 μm; n = 5). C, Gel electrophoresis of RT-PCR products showing specific bands for HCN1 (288 bp), HCN2 (370 bp), HCN3 (233 bp), and HCN4 (172 bp) mRNA expression in BLA tissue. cDNA sizes were determined via elution against a 100 bp DNA ladder. D–H, Photomicrographs of CaMKII ir (green) and, in red, HCN1 ir (D, E), HCN2 ir (F), HCN3 ir (G), and HCN4 ir (H) in the BLA (bregma, −2.56 mm). The arrows indicate CaMKII-immunoreactive cells that are closely associated with HCN expression; arrowheads indicate single labeled CaMKII- or HCN subunit-immunoreactive structures. Scale bars: D, F, G, H (all with 60× objective), E (with 100× objective), 40 μm.
The expression and distribution of the different HCN channel subunits and their association with projection neurons within the BLA was examined using RT-PCR and dual-label immunohistochemistry. CaMKII immunoreactivity (ir) was used as a selective marker to identify pyramidal cells within the BLA (McDonald et al., 2002; Rostkowski et al., 2009). Gene expression (Fig. 8C) and immunoreactivity for all HCN subunits (HCN1–4) was detected within the BLA and each subunit displayed a characteristic staining pattern, although all exhibited punctate staining patterns. RT-PCR demonstrated bands for each of the HCN subunits at the predicted size; no signal was present in the controls. HCN1 ir was strong in the BLA, but not lateral amygdala, and staining was evident in short fibers (Fig. 8D). Higher magnification demonstrated that HCN1-immunoreactive punctae were identified around the perimeter of a relatively large proportion of CaMKII-immunoreactive cells (Fig. 8E). By contrast, HCN2 (Fig. 8F) and HCN 3 ir (Fig. 8G) were much less abundant in the basolateral/lateral complex, whereas numerous HCN4-immunoreactive fibers were present throughout both the basolateral complex (Fig. 8H). Except for HCN1, no HCN subunit demonstrated appreciable association with the CaMKII-immunoreactive (pyramidal) cell population, consistent with it being the predominant component of Ih in BLA pyramidal neurons.
Discussion
NPY is potently anxiolytic when injected into the BLA. Although the acute anxiolytic actions of NPY in the BLA have been recognized for some time, to our knowledge this is the first study of its mechanism of action there. Activation of Y1 receptors in a large number of BLA pyramidal cells suppresses an Ih current that is active at rest, thereby hyperpolarizing and inhibiting them. Conversely, these same neurons are depolarized and excited by activation of CRF receptors that increase membrane Ih. These acute cellular actions of NPY or CRF would be expected to reduce or increase the output of BLA projection neurons, respectively. As BLA output is correlated with anxiogenic behaviors, increases being anxiogenic and decreases anxiolytic, respectively (Rainnie et al., 2004; Sajdyk et al., 2008), our results are consistent with the hypothesis that the acute behavioral actions of NPY and CRF result from their convergent actions at Ih in BLA pyramidal neurons.
Just as the anxiolytic behavioral actions of NPY in the BLA are primarily mediated via activation of Y1 receptors (Wahlestedt et al., 1993; Karlsson et al., 2008), so too are the actions of NPY on Ih. Thus, the Y1 receptor agonist, F7P34 NPY, mimicked the actions of NPY and the selective Y1 antagonist, BIBO3304, prevented the postsynaptic actions of subsequent NPY applications. Neither Y2 nor Y5 receptor antagonists were effective in blocking NPY effects. Although behavioral experiments on Y2 receptor knock-out mice have reported an anxiolytic phenotype (Tschenett et al., 2003; Tasan et al., 2009, 2010), this appears to result from loss of presynaptic Y2 receptors on fibers originating in the central amygdala. Given the apparently reciprocal actions of Y1 and Y2 receptors on anxiety-like behaviors, we were somewhat surprised that the Y2-preferring agonist [ahx5-24]NPY had no significant actions on BLA pyramidal cells. This suggests that, at least in our preparation, synaptic activity from inputs regulated by Y2 receptors is at low levels. We also note that the receptor antagonists alone did not affect membrane potential (data not shown) consistent with low ambient NPY levels in the BLA.
The EC50 for the actions of NPY was ∼400 nm in BLA pyramidal neurons, which is higher than previously reported for hypothalamus (EC50 = 28 nm) (Pronchuk et al., 2002), but not inconsistent with values from the hippocampus (136 nm) (El Bahh et al., 2002). An EC50 near 400 nm is somewhat surprising, as NPY is locally produced within GABAergic neurons in the BLA (McDonald, 1985). Although it is possible that NPY itself, which will also activate the anxiogenic Y2 receptor, could diminish its own Y1-mediated actions (Kask et al., 1996; Nakajima et al., 1998; Sajdyk et al., 1999, 2002a,b), the effect of NPY on BLA neurons is unaltered in the presence of the Y2 antagonist, BIIE0246, making this unlikely. Another possibility could be that there is a low abundance of Y1 receptors coupled to Ih, thus requiring a greater level of receptor activation to produce the suppression of Ih.
Mechanisms underlying the NPY-mediated response
Our results do not support roles for either GABAA receptors or GIRK channels in mediating NPY-induced hyperpolarization of BLA pyramidal neurons. Thus, NPY is equally effective in the absence or the presence of the GABAA antagonist, bicuculline, unlike the actions of another anxiety-related peptide, Neuropeptide S (Jüngling et al., 2008). Furthermore, although Y1 receptors activate GIRK channels in lateral amygdala projection neurons (Sosulina et al., 2008), here, blockers of GIRK channels did not significantly affect the actions of NPY in BLA.
Our data are entirely consistent with NPY suppressing an Ih that is active at the resting membrane potential in BLA pyramidal cells. First, NPY selectively reduced the gradually increasing current responses in voltage step protocols designed to isolate Ih (Fig. 5A). Second, the actions of NPY were occluded by either Cs+ or ZD7288, both known blockers of Ih (DiFrancesco, 1982, 1995; Pentney et al., 2002). Third, the Ih-specific blocker, ZD7288, not only mimics the hyperpolarizing actions of NPY but also occludes them. Finally, the inhibition of neuronal firing caused by NPY is mediated by the hyperpolarization it elicits. Thus, neurons depolarized to their original resting potential in the presence of NPY are more sensitive to the depolarizing effects of current, consistent with reduction of a resting depolarizing conductance by NPY.
Ih is a mixed cation conductance activated by hyperpolarization, but which depolarizes cells (Pape, 1996; Accili et al., 2002). A number of roles have been postulated for Ih in neurons, including protection from excessive hyperpolarization (Womble and Moises, 1993; Pape, 1996), depolarizing bursting neurons after the strong hyperpolarization at the termination of a burst (Accili et al., 2002), and mediating dendritic integration (Magee, 1999; Berger et al., 2001). A recent report has implicated the physiological loss of postsynaptic Ih to result in an increased excitability subsequent to the induction of long-term depression in CA1 pyramidal neurons (Brager and Johnston, 2007). However, with NPY, we have observed the opposite effect, in which a receptor-mediated loss of postsynaptic Ih results in a hyperpolarization accompanied by an inhibition. As this response occurs in the presence of TTX, it appears to be independent of changes in synaptic properties.
With the single exception above (Brager and Johnston, 2007), reports of receptor-mediated effects on Ih have linked increased Ih activity with increases in excitability. Agonists at dopamine (Rosenkranz and Johnston, 2006; Chen and Yang, 2007), NMDA (Fan et al., 2005), κ-opioid (Pan, 2003), and CRF (Qiu et al., 2005; Wanat et al., 2008) receptors all cause increased Ih activity. Within the BLA, we observed that CRF augments a resting Ih, to depolarize and excite pyramidal neurons. Ih inhibition, by contrast, is reported to result from different mechanisms entirely. The norepinephrine (via dopamine D2 receptors)-mediated inhibition of ventral tegmental area dopaminergic cells is indirect, in which inhibition of Ih results from an increase in an outward K+ conductance (Arencibia-Albite et al., 2007). Propofol or loperamide reportedly both inhibit Ih by directly binding to the extracellular region of the HCN channel in cortical or dorsal root ganglion cells, respectively (Chen et al., 2005; Vasilyev et al., 2007). Thus, to our knowledge, the only other reported instance of G-protein-coupled receptor-mediated inhibition of Ih as with NPY here is mediated by activation of α2-adrenoreceptors in dorsal root ganglion neurons (Yagi and Sumino, 1998).
The Ih current can be carried by homotetramers or heterotetramers of the four known HCN subunits, HCN1–HCN4. Although they are distinguished by voltage sensitivity and kinetics, the HCN1 subunit is quite insensitive to cAMP (Santoro et al., 1999; Chen et al., 2001). In the BLA, elevation of intracellular cAMP with a membrane-permeant analog affected neither resting membrane potential nor the hyperpolarizing response to NPY, suggesting that the cAMP-insensitive channel, HCN1, is the predominant form mediating these responses (Biel et al., 2009). This is further supported by the close association of HCN1 channel subunit immunoreactivity with pyramidal cell bodies. Although our histological analysis does not allow us to state conclusively that these channels are inserted within the cell membrane, when considered together with the cellular pharmacological studies and molecular studies demonstrating the presence of HCN1 mRNA within the BLA, we believe it constitutes strong evidence that the HCN1 channel is important in regulating the activity of BLA pyramidal neurons.
Behavioral relevance of Ih modulation
NPY and CRF act in a reciprocal manner to balance emotional responses (Heilig et al., 1994; Britton et al., 2000). A mechanism by which NPY inhibits depolarization of BLA pyramidal cells in vitro is consistent with its acute anxiolytic actions in vivo. Acute injections of NPY into the BLA block the generation of behavioral stress responses induced by either CRF injections in the BLA or restraint stress (Sajdyk et al., 2006). Moreover, repeated injections of either NPY or CRF into the BLA have profound effects on behavior that greatly outlast their acute actions and result in the induction of a long-term behavioral “stress resilience” (Sajdyk et al., 2008), or “stress vulnerability,” respectively (Rainnie et al., 2004). In agreement with behavioral studies, we determined that the opposing actions of these two neuropeptides converge on the same target, Ih, within the BLA. NPY acts by suppressing a tonically active Ih to decrease excitability, whereas CRF enhances the same current, resulting in membrane depolarization and increased neuronal excitability. Although the enhancement of Ih has been described previously (Qiu et al., 2005; Wanat et al., 2008), this is to our knowledge the first example of reciprocal regulation of Ih to result in opposing behavioral outputs.
Conclusion
NPY acutely inhibits a significant subset of BLA pyramidal neurons, hyperpolarizing the postsynaptic membrane via the Y1 receptor. The NPY-mediated inhibition resulted from the suppression of an Ih that is tonically active at rest. Inhibition of Ih is a novel modulatory mechanism for NPY receptors. In contrast, CRF acutely depolarizes the membrane of pyramidal neurons by enhancing Ih at rest. Understanding the mechanism of the anxiolytic effects of NPY within the BLA, and how this relates to the opposing actions of CRF, will provide valuable information for developing pharmaceuticals that can reduce the anxious behaviors characterizing anxiety spectrum disorders.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants MH62121 (J.H.U.) and MH081152 (J.H.U., W.F.C.), and Canadian Institutes of Health Research (CIHR) Grant MOP 10250 (W.F.C.). C.J.G. was supported by a Canada Graduate Scholarship (M) from CIHR. J.P.M. was supported by a Graduate Student Recruitment Studentship, Faculty of Medicine and Dentistry, University of Alberta. W.F.C. is a Medical Scientist of the Alberta Heritage Foundation for Medical Research. The monoclonal antibodies HCN1, HCN2, HCN3, and HCN4 were obtained from the University of California, Davis/NIH NeuroMab Facility, supported by NIH Grant U24NS050606 (Department of Neurobiology, Physiology, and Behavior, College of Biological Sciences, University of California, Davis, Davis, CA). We gratefully acknowledge Gina DeJoseph for her excellent technical assistance.
References
- Accili EA, Proenza C, Baruscotti M, DiFrancesco D. From funny current to HCN channels: 20 years of excitation. News Physiol Sci. 2002;17:32–37. doi: 10.1152/physiologyonline.2002.17.1.32. [DOI] [PubMed] [Google Scholar]
- Alonso J, Lépine JP ESEMeD/MHEDEA 2000 Scientific Committee. Overview of key data from the European Study of the Epidemiology of Mental Disorders (ESEMeD) J Clin Psychiatry. 2007;68(Suppl 2):3–9. [PubMed] [Google Scholar]
- Amaral DG. The primate amygdala and the neurobiology of social behavior: implications for understanding social anxiety. Biol Psychiatry. 2002;51:11–17. doi: 10.1016/s0006-3223(01)01307-5. [DOI] [PubMed] [Google Scholar]
- Arencibia-Albite F, Paladini C, Williams JT, Jiménez-Rivera CA. Noradrenergic modulation of the hyperpolarization-activated cation current (Ih) in dopamine neurons of the ventral tegmental area. Neuroscience. 2007;149:303–314. doi: 10.1016/j.neuroscience.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger T, Larkum ME, Lüscher HR. High Ih channel density in the distal apical dendrite of layer V pyramidal cells increases bidirectional attenuation of EPSPs. J Neurophysiol. 2001;85:855–868. doi: 10.1152/jn.2001.85.2.855. [DOI] [PubMed] [Google Scholar]
- Biel M, Wahl-Schott C, Michalakis S, Zong X. Hyperpolarization-activated cation channels: from genes to function. Physiol Rev. 2009;89:847–885. doi: 10.1152/physrev.00029.2008. [DOI] [PubMed] [Google Scholar]
- Brager DH, Johnston D. Plasticity of intrinsic excitability during long-term depression is mediated through mGluR-dependent changes in Ih in hippocampal CA1 pyramidal neurons. J Neurosci. 2007;27:13926–13937. doi: 10.1523/JNEUROSCI.3520-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton KT, Akwa Y, Spina MG, Koob GF. Neuropeptide Y blocks anxiogenic-like behavioral action of corticotropin-releasing factor in an operant conflict test and elevated plus maze. Peptides. 2000;21:37–44. doi: 10.1016/s0196-9781(99)00169-2. [DOI] [PubMed] [Google Scholar]
- Broqua P, Wettstein JG, Rocher MN, Gauthier-Martin B, Junien JL. Behavioral effects of neuropeptide Y receptor agonists in the elevated plus-maze and fear-potentiated startle procedures. Behav Pharmacol. 1995;6:215–222. [PubMed] [Google Scholar]
- Chee MJ, Myers MG, Jr, Price CJ, Colmers WF. Neuropeptide Y suppresses anorexigenic output from the ventromedial nucleus of the hypothalamus. J Neurosci. 2010;30:3380–3390. doi: 10.1523/JNEUROSCI.4031-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Yang XL. Hyperpolarization-activated cation current is involved in modulation of the excitability of rat retinal ganglion cells by dopamine. Neuroscience. 2007;150:299–308. doi: 10.1016/j.neuroscience.2007.09.015. [DOI] [PubMed] [Google Scholar]
- Chen S, Wang J, Siegelbaum SA. Properties of hyperpolarization-activated pacemaker current defined by coassembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. J Gen Physiol. 2001;117:491–504. doi: 10.1085/jgp.117.5.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Shu S, Bayliss DA. Suppression of Ih contributes to propofol-induced inhibition of mouse cortical pyramidal neurons. J Neurophysiol. 2005;94:3872–3883. doi: 10.1152/jn.00389.2005. [DOI] [PubMed] [Google Scholar]
- Colmers WF, Bleakman D. Effects of neuropeptide Y on the electrical properties of neurons. Trends Neurosci. 1994;17:373–379. doi: 10.1016/0166-2236(94)90046-9. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Pronchuk N, Fan W, Dinulescu DM, Colmers WF, Cone RD. Integration of NPY, AGRP, and melanocortin signals in the hypothalamic paraventricular nucleus: evidence of a cellular basis for the adipostat. Neuron. 1999;24:155–163. doi: 10.1016/s0896-6273(00)80829-6. [DOI] [PubMed] [Google Scholar]
- Davis M, Rainnie D, Cassell M. Neurotransmission in the rat amygdala related to fear and anxiety. Trends Neurosci. 1994;17:208–214. doi: 10.1016/0166-2236(94)90106-6. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D. Block and activation of the pace-maker channel in calf Purkinje fibres: effects of potassium, caesium and rubidium. J Physiol. 1982;329:485–507. doi: 10.1113/jphysiol.1982.sp014315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFrancesco D. Cesium and the pacemaker current. J Cardiovasc Electrophysiol. 1995;6:1152–1155. doi: 10.1111/j.1540-8167.1995.tb00394.x. [DOI] [PubMed] [Google Scholar]
- El Bahh B, Cao JQ, Beck-Sickinger AG, Colmers WF. Blockade of neuropeptide Y2 receptors and suppression of NPY's anti-epileptic actions in the rat hippocampal slice by BIIE0246. Br J Pharmacol. 2002;136:502–509. doi: 10.1038/sj.bjp.0704751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Bahh B, Balosso S, Hamilton T, Herzog H, Beck-Sickinger AG, Sperk G, Gehlert DR, Vezzani A, Colmers WF. The anti-epileptic actions of neuropeptide Y in the hippocampus are mediated by Y and not Y receptors. Eur J Neurosci. 2005;22:1417–1430. doi: 10.1111/j.1460-9568.2005.04338.x. [DOI] [PubMed] [Google Scholar]
- Fan Y, Fricker D, Brager DH, Chen X, Lu HC, Chitwood RA, Johnston D. Activity-dependent decrease of excitability in rat hippocampal neurons through increases in Ih. Nat Neurosci. 2005;8:1542–1551. doi: 10.1038/nn1568. [DOI] [PubMed] [Google Scholar]
- Fernandez-Fernandez JM, Wanaverbecq N, Halley P, Caulfield MP, Brown DA. Selective activation of heterologously expressed G protein-gated K+ channels by M2 muscarinic receptors in rat sympathetic neurones. J Physiol. 1999;515:631–637. doi: 10.1111/j.1469-7793.1999.631ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu LY, Acuna-Goycolea C, van den Pol AN. Neuropeptide Y inhibits hypocretin/orexin neurons by multiple presynaptic and postsynaptic mechanisms: tonic depression of the hypothalamic arousal system. J Neurosci. 2004;24:8741–8751. doi: 10.1523/JNEUROSCI.2268-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini S, DiFrancesco D. Action of the hyperpolarization-activated current (Ih) blocker ZD 7288 in hippocampal CA1 neurons. Pflugers Arch. 1997;435:99–106. doi: 10.1007/s004240050488. [DOI] [PubMed] [Google Scholar]
- Ghamari-Langroudi M, Colmers WF, Cone RD. PYY3-36 inhibits the action potential firing activity of POMC neurons of arcuate nucleus through postsynaptic Y2 receptors. Cell Metab. 2005;2:191–199. doi: 10.1016/j.cmet.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Ghods-Sharifi S, St Onge JR, Floresco SB. Fundamental contribution by the basolateral amygdala to different forms of decision making. J Neurosci. 2009;29:5251–5259. doi: 10.1523/JNEUROSCI.0315-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutman AR, Yang Y, Ressler KJ, Davis M. The role of neuropeptide Y in the expression and extinction of fear-potentiated startle. J Neurosci. 2008;28:12682–12690. doi: 10.1523/JNEUROSCI.2305-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig M, Widerlöv E. Neurobiology and clinical aspects of neuropeptide Y. Crit Rev Neurobiol. 1995;9:115–136. [PubMed] [Google Scholar]
- Heilig M, Koob GF, Ekman R, Britton KT. Corticotropin-releasing factor and neuropeptide Y: role in emotional integration. Trends Neurosci. 1994;17:80–85. doi: 10.1016/0166-2236(94)90079-5. [DOI] [PubMed] [Google Scholar]
- Herry C, Bach DR, Esposito F, Di Salle F, Perrig WJ, Scheffler K, Lüthi A, Seifritz E. Processing of temporal unpredictability in human and animal amygdala. J Neurosci. 2007;27:5958–5966. doi: 10.1523/JNEUROSCI.5218-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jüngling K, Seidenbecher T, Sosulina L, Lesting J, Sangha S, Clark SD, Okamura N, Duangdao DM, Xu YL, Reinscheid RK, Pape HC. Neuropeptide S-mediated control of fear expression and extinction: role of intercalated GABAergic neurons in the amygdala. Neuron. 2008;59:298–310. doi: 10.1016/j.neuron.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson RM, Choe JS, Cameron HA, Thorsell A, Crawley JN, Holmes A, Heilig M. The neuropeptide Y Y1 receptor subtype is necessary for the anxiolytic-like effects of neuropeptide Y, but not the antidepressant-like effects of fluoxetine, in mice. Psychopharmacology. 2008;195:547–557. doi: 10.1007/s00213-007-0945-2. [DOI] [PubMed] [Google Scholar]
- Kask A, Rägo L, Harro J. Anxiogenic-like effect of the neuropeptide Y Y1 receptor antagonist BIBP3226: antagonism with diazepam. Eur J Pharmacol. 1996;317:R3–R4. doi: 10.1016/s0014-2999(96)00838-2. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:593–602. doi: 10.1001/archpsyc.62.6.593. [DOI] [PubMed] [Google Scholar]
- Kombian SB, Colmers WF. Neuropeptide Y selectively inhibits slow synaptic potentials in rat dorsal raphe nucleus in vitro by a presynaptic action. J Neurosci. 1992;12:1086–1093. doi: 10.1523/JNEUROSCI.12-03-01086.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzhikandathil EV, Oxford GS. Classic D1 dopamine receptor antagonist R-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH23390) directly inhibits G protein-coupled inwardly rectifying potassium channels. Mol Pharmacol. 2002;62:119–126. doi: 10.1124/mol.62.1.119. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. Emotion and the amygdala. In: Aggleton JP, editor. The amygdala: neurobiological aspects of emotion, memory, and mental dysfunction. Wilmington, DE: Wiley; 1992. pp. 339–351. [Google Scholar]
- Lorberbaum JP, Kose S, Johnson MR, Arana GW, Sullivan LK, Hamner MB, Ballenger JC, Lydiard RB, Brodrick PS, Bohning DE, George MS. Neural correlates of speech anticipatory anxiety in generalized social phobia. Neuroreport. 2004;15:2701–2705. [PubMed] [Google Scholar]
- Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M. A family of hyperpolarization-activated mammalian cation channels. Nature. 1998;393:587–591. doi: 10.1038/31255. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Zong X, Stieber J, Hullin R, Hofmann F, Biel M. Two pacemaker channels from human heart with profoundly different activation kinetics. EMBO J. 1999;18:2323–2329. doi: 10.1093/emboj/18.9.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC. Dendritic Ih normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci. 1999;2:508–514. doi: 10.1038/12229. [DOI] [PubMed] [Google Scholar]
- McDonald AJ. Neurons of the lateral and basolateral amygdaloid nuclei: a Golgi study in the rat. J Comp Neurol. 1982;212:293–312. doi: 10.1002/cne.902120307. [DOI] [PubMed] [Google Scholar]
- McDonald AJ. Immunohistochemical identification of gamma-aminobutyric acid-containing neurons in the rat basolateral amygdala. Neurosci Lett. 1985;53:203–207. doi: 10.1016/0304-3940(85)90186-7. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Muller JF, Mascagni F. GABAergic innervation of alpha type II calcium/calmodulin-dependent protein kinase immunoreactive pyramidal neurons in the rat basolateral amygdala. J Comp Neurol. 2002;446:199–218. doi: 10.1002/cne.10204. [DOI] [PubMed] [Google Scholar]
- Melnick I, Pronchuk N, Cowley MA, Grove KL, Colmers WF. Developmental switch in neuropeptide Y and melanocortin effects in the paraventricular nucleus of the hypothalamus. Neuron. 2007;56:1103–1115. doi: 10.1016/j.neuron.2007.10.034. [DOI] [PubMed] [Google Scholar]
- Nakajima M, Inui A, Asakawa A, Momose K, Ueno N, Teranishi A, Baba S, Kasuga M. Neuropeptide Y produces anxiety via Y2-type receptors. Peptides. 1998;19:359–363. doi: 10.1016/s0196-9781(97)00298-2. [DOI] [PubMed] [Google Scholar]
- Offord DR, Boyle MH, Campbell D, Goering P, Lin E, Wong M, Racine YA. One-year prevalence of psychiatric disorder in Ontarians 15 to 64 years of age. Can J Psychiatry. 1996;41:559–563. doi: 10.1177/070674379604100904. [DOI] [PubMed] [Google Scholar]
- Pan ZZ. Kappa-opioid receptor-mediated enhancement of the hyperpolarization-activated current (Ih) through mobilization of intracellular calcium in rat nucleus raphe magnus. J Physiol. 2003;548:765–775. doi: 10.1113/jphysiol.2002.037622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC. Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu Rev Physiol. 1996;58:299–327. doi: 10.1146/annurev.ph.58.030196.001503. [DOI] [PubMed] [Google Scholar]
- Paredes MF, Greenwood J, Baraban SC. Neuropeptide Y modulates a G protein-coupled inwardly rectifying potassium current in the mouse hippocampus. Neurosci Lett. 2003;340:9–12. doi: 10.1016/s0304-3940(03)00036-3. [DOI] [PubMed] [Google Scholar]
- Park K, Lee S, Kang SJ, Choi S, Shin KS. Hyperpolarization-activated currents control the excitability of principal neurons in the basolateral amygdala. Biochem Biophys Res Commun. 2007;361:718–724. doi: 10.1016/j.bbrc.2007.07.064. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. Ed 2. New York: Academic; 1986. The rat brain in stereotaxic coordinates. [DOI] [PubMed] [Google Scholar]
- Pentney AR, Baraban SC, Colmers WF. NPY sensitivity and postsynaptic properties of heterotopic neurons in the MAM model of malformation-associated epilepsy. J Neurophysiol. 2002;88:2745–2754. doi: 10.1152/jn.00500.2002. [DOI] [PubMed] [Google Scholar]
- Pronchuk N, Beck-Sickinger AG, Colmers WF. Multiple NPY receptors inhibit GABAA synaptic responses of rat medial parvocellular effector neurons in the hypothalamic paraventricular nucleus. Endocrinology. 2002;143:535–543. doi: 10.1210/endo.143.2.8655. [DOI] [PubMed] [Google Scholar]
- Qiu DL, Chu CP, Shirasaka T, Tsukino H, Nakao H, Kato K, Kunitake T, Katoh T, Kannan H. Corticotrophin-releasing factor augments the IH in rat hypothalamic paraventricular nucleus parvocellular neurons in vitro. J Neurophysiol. 2005;94:226–234. doi: 10.1152/jn.01325.2004. [DOI] [PubMed] [Google Scholar]
- Rainnie DG, Asprodini EK, Shinnick-Gallagher P. Intracellular recordings from morphologically identified neurons of the basolateral amygdala. J Neurophysiol. 1993;69:1350–1362. doi: 10.1152/jn.1993.69.4.1350. [DOI] [PubMed] [Google Scholar]
- Rainnie DG, Bergeron R, Sajdyk TJ, Patil M, Gehlert DR, Shekhar A. Corticotrophin releasing factor-induced synaptic plasticity in the amygdala translates stress into emotional disorders. J Neurosci. 2004;24:3471–3479. doi: 10.1523/JNEUROSCI.5740-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rist B, Wieland HA, Willim KD, Beck-Sickinger AG. A rational approach for the development of reduced-size analogues of neuropeptide Y with high affinity to the Y1 receptor. J Pept Sci. 1995;1:341–348. doi: 10.1002/psc.310010509. [DOI] [PubMed] [Google Scholar]
- Rosenkranz JA, Johnston D. Dopaminergic regulation of neuronal excitability through modulation of Ih in layer V entorhinal cortex. J Neurosci. 2006;26:3229–3244. doi: 10.1523/JNEUROSCI.4333-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostkowski AB, Teppen TL, Peterson DA, Urban JH. Cell-specific expression of Neuropeptide Y Y1 receptor immunoreactivity in the rat basolateral amygdala. J Comp Neurol. 2009;517:166–176. doi: 10.1002/cne.22143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajdyk TJ, Vandergriff MG, Gehlert DR. Amygdalar neuropeptide Y Y1 receptors mediate the anxiolytic-like actions of neuropeptide Y in the social interaction test. Eur J Pharmacol. 1999;368:143–147. doi: 10.1016/s0014-2999(99)00018-7. [DOI] [PubMed] [Google Scholar]
- Sajdyk TJ, Schober DA, Smiley DL, Gehlert DR. Neuropeptide Y-Y2 receptors mediate anxiety in the amygdala. Pharmacol Biochem Behav. 2002a;71:419–423. doi: 10.1016/s0091-3057(01)00679-7. [DOI] [PubMed] [Google Scholar]
- Sajdyk TJ, Schober DA, Gehlert DR. Neuropeptide Y receptor subtypes in the basolateral nucleus of the amygdala modulate anxiogenic responses in rats. Neuropharmacology. 2002b;43:1165–1172. doi: 10.1016/s0028-3908(02)00234-4. [DOI] [PubMed] [Google Scholar]
- Sajdyk TJ, Shekhar A, Gehlert DR. Interactions between NPY and CRF in the amygdala to regulate emotionality. Neuropeptides. 2004;38:225–234. doi: 10.1016/j.npep.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Sajdyk TJ, Fitz SD, Shekhar A. The role of neuropeptide Y in the amygdala on corticotropin-releasing factor receptor-mediated behavioral stress responses in the rat. Stress. 2006;9:21–28. doi: 10.1080/10253890600557315. [DOI] [PubMed] [Google Scholar]
- Sajdyk TJ, Johnson PL, Leitermann RJ, Fitz SD, Dietrich A, Morin M, Gehlert DR, Urban JH, Shekhar A. Neuropeptide Y in the amygdala induces long-term resilience to stress-induced reductions in social responses but not hypothalamic-adrenal-pituitary axis activity or hyperthermia. J Neurosci. 2008;28:893–903. doi: 10.1523/JNEUROSCI.0659-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Tibbs GR. The HCN gene family: molecular basis of the hyperpolarization-activated pacemaker channels. Ann N Y Acad Sci. 1999;868:741–764. doi: 10.1111/j.1749-6632.1999.tb11353.x. [DOI] [PubMed] [Google Scholar]
- Santoro B, Grant SG, Bartsch D, Kandel ER. Interactive cloning with the SH3 domain of N-src identifies a new brain specific ion channel protein, with homology to eag and cyclic nucleotide-gated channels. Proc Natl Acad Sci U S A. 1997;94:14815–14820. doi: 10.1073/pnas.94.26.14815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Liu DT, Yao H, Bartsch D, Kandel ER, Siegelbaum SA, Tibbs GR. Identification of a gene encoding a hyperpolarization-activated pacemaker channel of brain. Cell. 1998;93:717–729. doi: 10.1016/s0092-8674(00)81434-8. [DOI] [PubMed] [Google Scholar]
- Santoro B, Chen S, Luthi A, Pavlidis P, Shumyatsky GP, Tibbs GR, Siegelbaum SA. Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. J Neurosci. 2000;20:5264–5275. doi: 10.1523/JNEUROSCI.20-14-05264.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert R, Scholten A, Gauss R, Mincheva A, Lichter P, Kaupp UB. Molecular characterization of a slowly gating human hyperpolarization-activated channel predominantly expressed in thalamus, heart, and testis. Proc Natl Acad Sci U S A. 1999;96:9391–9396. doi: 10.1073/pnas.96.16.9391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin LM, Rauch SL, Pitman RK. Amygdala, medial prefrontal cortex, and hippocampal function in PTSD. Ann N Y Acad Sci. 2006;1071:67–79. doi: 10.1196/annals.1364.007. [DOI] [PubMed] [Google Scholar]
- Slesinger PA. Ion selectivity filter regulates local anesthetic inhibition of G-protein-gated inwardly rectifying K+ channels. Biophys J. 2001;80:707–718. doi: 10.1016/S0006-3495(01)76050-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodickson DL, Bean BP. GABAB receptor-activated inwardly rectifying potassium current in dissociated hippocampal CA3 neurons. J Neurosci. 1996;16:6374–6385. doi: 10.1523/JNEUROSCI.16-20-06374.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodickson DL, Bean BP. Neurotransmitter activation of inwardly rectifying potassium current in dissociated hippocampal CA3 neurons: interactions among multiple receptors. J Neurosci. 1998;18:8153–8162. doi: 10.1523/JNEUROSCI.18-20-08153.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soll RM, Dinger MC, Lundell I, Larhammer D, Beck-Sickinger AG. Novel analogues of neuropeptide Y with a preference for the Y1-receptor. FEBS J. 2001;268:2828–2837. doi: 10.1046/j.1432-1327.2001.02161.x. [DOI] [PubMed] [Google Scholar]
- Sosulina L, Schwesig G, Seifert G, Pape HC. Neuropeptide Y activates a G-protein-coupled inwardly rectifying potassium current and dampens excitability in the lateral amygdala. Mol Cell Neurosci. 2008;39:491–498. doi: 10.1016/j.mcn.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Sun L, Miller RJ. Multiple neuropeptide Y receptors regulate K+ and Ca2+ channels in acutely isolated neurons from the rat arcuate nucleus. J Neurophysiol. 1999;81:1391–1403. doi: 10.1152/jn.1999.81.3.1391. [DOI] [PubMed] [Google Scholar]
- Sun QQ, Huguenard JR, Prince DA. Neuropeptide Y receptors differentially modulate G-protein-activated inwardly rectifying K+ channels and high-voltage-activated Ca2+ channels in rat thalamic neurons. J Physiol. 2001;531:67–79. doi: 10.1111/j.1469-7793.2001.0067j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takigawa T, Alzheimer C. G protein-activated inwardly rectifying K+ (GIRK) currents in dendrites of rat neocortical pyramidal cells. J Physiol. 1999;517:385–390. doi: 10.1111/j.1469-7793.1999.0385t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasan RO, Lin S, Hetzenauer A, Singewald N, Herzog H, Sperk G. Increased novelty-induced motor activity and reduced depression-like behavior in neuropeptide Y (NPY)-Y4 receptor knockout mice. Neuroscience. 2009;158:1717–1730. doi: 10.1016/j.neuroscience.2008.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasan RO, Nguyen NK, Weger S, Sartori SB, Singewald N, Heilbronn R, Herzog H, Sperk G. The central and basolateral amygdala are critical sites of neuropeptide Y/Y2 receptor-mediated regulation of anxiety and depression. J Neurosci. 2010;30:6282–6290. doi: 10.1523/JNEUROSCI.0430-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truitt WA, Sajdyk TJ, Dietrich AD, Oberlin B, McDougle CJ, Shekhar A. From anxiety to autism: spectrum of abnormal social behaviors modeled by progressive disruption of inhibitory neuronal function in the basolateral amygdala in Wistar rats. Psychopharmacology. 2007;191:107–118. doi: 10.1007/s00213-006-0674-y. [DOI] [PubMed] [Google Scholar]
- Tschenett A, Singewald N, Carli M, Balducci C, Salchner P, Vezzani A, Herzog H, Sperk G. Reduced anxiety and improved stress coping ability in mice lacking NPY-Y2 receptors. Eur J Neurosci. 2003;18:143–148. doi: 10.1046/j.1460-9568.2003.02725.x. [DOI] [PubMed] [Google Scholar]
- Vasilyev DV, Shan Q, Lee Y, Mayer SC, Bowlby MR, Strassle BW, Kaftan EJ, Rogers KE, Dunlop J. Direct inhibition of Ih by analgesic loperamide in rat DRG neurons. J Neurophysiol. 2007;97:3713–3721. doi: 10.1152/jn.00841.2006. [DOI] [PubMed] [Google Scholar]
- Wahlestedt C, Pich EM, Koob GF, Yee F, Heilig M. Modulation of anxiety and neuropeptide Y-Y1 receptors by antisense oligodeoxynucleotides. Science. 1993;259:528–531. doi: 10.1126/science.8380941. [DOI] [PubMed] [Google Scholar]
- Wanat MJ, Hopf FW, Stuber GD, Phillips PE, Bonci A. Corticotropin-releasing factor increases mouse ventral tegmental area dopamine neuron firing through a protein kinase C-dependent enhancement of Ih. J Physiol. 2008;586:2157–2170. doi: 10.1113/jphysiol.2007.150078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland HA, Engel W, Eberlein W, Rudolf K, Doods HN. Subtype selectivity of the novel nonpeptide neuropeptide Y Y1 receptor antagonist BIBO 3304 and its effect on feeding in rodents. Br J Pharmacol. 1998;125:549–555. doi: 10.1038/sj.bjp.0702084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womble MD, Moises HC. Hyperpolarization-activated currents in neurons of the rat basolateral amygdala. J Neurophysiol. 1993;70:2056–2065. doi: 10.1152/jn.1993.70.5.2056. [DOI] [PubMed] [Google Scholar]
- Yagi J, Sumino R. Inhibition of a hyperpolarization-activated current by clonidine in rat dorsal root ganglion neurons. J Neurophysiol. 1998;80:1094–1104. doi: 10.1152/jn.1998.80.3.1094. [DOI] [PubMed] [Google Scholar]
- Zhang X, Bao L, Xu ZQ, Kopp J, Arvidsson U, Elde R, Hökfelt T. Localization of neuropeptide Y Y1 receptors in the rat nervous system with special reference to somatic receptors on small dorsal root ganglion neurons. Proc Natl Acad Sci U S A. 1994;91:11738–11742. doi: 10.1073/pnas.91.24.11738. [DOI] [PMC free article] [PubMed] [Google Scholar]