

Abstract
We here report a study of the intramolecular amination of sp3 C-H bonds via the hydride transfer cyclization of N-tosylimines (HT-amination). In this transformation, 5-aryl-aldehydes are subjected to N-toluenesulfonamide in the presence of BF3•OEt2 to effect imine formation and HT-cyclization, leading to 2-aryl-piperidines and 3-aryl-1,2,3,4-tetrahydroisoquinolines in a one-pot procedure. We examined the reactivity of a range of aldehyde substrates as a function of their conformational flexibility. Substrates of higher conformational rigidity were more reactive, giving higher yields of the desired products. However, a single substituent on the alkyl chain linking the N-tosylimine and the benzylic sp3 C-H bonds was sufficient for HT-cyclization to occur. In addition, an examination of various arenes revealed that the electronic character of the hydridic C-H bonds dramatically affects the efficiency of the reaction. We also found that this transformation is highly stereoselective; 2-substituted aldehydes yield cis-2,5-disubstituted piperidines, while 3-substituted aldehydes afford trans-2,4-disubstituted piperidines. The stereoselectivity is a consequence of thermodynamic control. The pseudo-allylic strain between the arene and tosyl group on the piperidine ring is proposed to rationalize the greater stability of the isomer with the aryl ring in the axial position. This preferential placement of the arene is proposed to affect the observed stereoselectivity.
INTRODUCTION
C-H bond functionalization represents a process of broad synthetic potential owing to the ubiquity of C-H bonds in organic compounds.1 As part of a broad program aimed at the development of new approaches for the direct functionalization of C-H bonds, we have been interested in the design of new methods for the synthesis of arylated pyrrolidines and piperidines.2,3 The wide ranging biological activity of such compounds has granted them an important status in the fields of organic and medicinal chemistry.4
To complement approaches initiated by transition metal insertion into C-H bonds2 and those relying on an external oxidant,3 for preparation of α-aryl-piperidines, we considered an alternative mode of reactivity based on the hydride-transfer cyclization of imines (HT-amination, Scheme 1B).5 This approach is based on a Lewis acid promoted hydride transfer to an activated imine, followed by C-N bond formation. We have previously demonstrated that the HT-cyclization effects the coupling of a range of hydride acceptors, such as aldehydes, enones, enals, vinyl acetals, and unactivated alkynes, with sp3 C-H bonds to deliver a diverse array of complex structural scaffolds, under mild conditions (Figure 1).6 HT-cyclization has since become an active area of research, which has led to the development of a number of interesting transformations.7–12
Scheme 1.

Two Complementary Approaches to α-Arylated Piperidines via C-H Bond Functionalizationa
Figure 1.

Examples of diverse structures prepared by HT-cyclization in our group. The bond formed during the process is highlighted in red. E = CO2Me, E′ = CO2Et, Ar = 4-MeO-C6H4.
In regards to HT-amination reactions, an early report by Reinhoudt et al. describes the thermal-cyclization of ortho-amino benzaldimines of type I to aminals II (Scheme 2A).13 More recently, the research teams of Seidel14a and Akiyama14b showed that triflic acid catalyzed this transformation, leading to expansion of its scope and utility. Both the thermal and the acid catalyzed reactions of aromatic imines can be rationalized by a sigmatropic hydrogen-shift. However, a through-space hydride transfer mechanism is also reasonable. In the context of saturated substrates (where a through-space hydride transfer is the only reasonable mechanistic rationale), Tietze and colleagues have reported the HT-cyclization of imines within conformationally rigid steroidal substrates, giving access to complex isoquinuclidines of type IV (Scheme 2B).15 This reaction has recently been expanded to the corresponding oximes and hydrozones.16
Scheme 2.

Formation of Cyclic Amines via the HT-cyclization of Imines.
In this study we examined the importance of conformational rigidity for the HT-cyclization of imines in a systematic manner. We explored the reactivity of aldehyde substrates with varying degrees of conformational freedom, ranging from rigid aromatic aldehydes to entirely unrestricted 5-aryl-pentanals. We found that a single substituent on the alkyl chain connecting the N-tosylimine (formed in situ from the aldehyde) and the benzylic group is sufficient for an efficient HT-cyclization to occur. Furthermore, we discovered that the HT-cyclization is highly stereoselective and we provide a mechanistic rationale for the observed results.
RESULTS AND DISCUSSION
Initial Investigation of the HT-Amination
One-pot Protocol. We first examined the aromatic N-tosylimine substrate 1 considering the restricted conformational freedom imparted by the aromatic core, which forces the hydride acceptor (N-tosylimine) and the hydride donor (the benzylic C-H bonds) into proximity and thereby should facilitate the hydride transfer (Scheme 3). Moreover, substrates of type 1 are readily accessible from commercially available materials and provide a direct route to an attractive but medicinally under-explored heterocyclic motif, the 3-aryl-1,2,3,4-tetrahydroisoquinoline (also see discussion below).
Scheme 3.

One-pot Condensation/HT-amination Protocol
Substrate 1 was subjected to BF3•OEt2 (2 eq.) in DCE at 50 °C and we were gratified to observe an efficient conversion to the N-tosyl-1,2,3,4-tetrahydroisoquinoline 2 in 92% yield (Scheme 3). As BF3•OEt2 is known to promote the condensation of aldehydes and N-toluenesulfonamide, we subjected the precursor aldehyde 3 to BF3•OEt2 (3 eq.), N-toluenesulfonamide (2 eq.) and 4Å molecular sieves (MS).17 After heating at 80 °C for 2 hours, 1,2,3,4-tetrahydroisoquinoline 2 was isolated in 87% yield. Lewis acid optimization revealed that BF3•OEt2 and TiF4 were the only reagents capable of promoting complete conversion of aldehyde 3 to product 2 (see Table S1 in the Supporting Information for an extensive screen). We also investigated the use of alternative amines in the one-pot procedure with substrate 3. While aniline gave complex product mixtures, 4-nitrobenzenesulfonamide led to a clean reaction mixture. However the rate of reaction was dramatically slower than that with N-toluenesulfonamide and incomplete conversion to the imine was a recurring problem.
With the one-pot procedure in hand, we next directed our attention towards aliphatic aldehyde substrates, which upon HT-amination would yield α-arylated piperidines (Scheme 3). Our investigation began with aldehyde 4, containing gem-dimethyl substitution in the α-position, to promote the intramolecular hydride transfer by increasing the rigidity of the substrate and preventing the potential tautomerization of the imine to the enamine. This substrate gave the 2-aryl-piperidine 5 in 96% yield at room temperature in one hour, the result that stimulated the systematic examination of the aliphatic substrates. Attempts to extend the HT-amination reactivity to ketone substrates, of both the aryl and aliphatic classes were unsuccessful, with complete recovery of the starting ketone observed in all cases.
HT-amination of Aliphatic Aldehydes: Conformational Rigidity of the Substrate Backbone and Electronics of the Benzylic C-H Bonds
Preliminary studies outlined above established that aliphatic aldehyde 4 exhibits high reactivity in the one-pot HT-cyclization protocol. Similarly, the cyclic aldehyde 6 afforded excellent yield of spirocyclic piperidine 7 under mild reaction conditions (Table 1, entry 2). Moving the gem-dimethyl substitution in the β-position had no major effect; substrate 8 furnished the corresponding product 9 in 95% yield under the same conditions (Table 1, entry 3).
Table 1.
HT-amination Scope: Conformational Rigidity and Electronics of Aldehyde Substratesa
| entry | substrate | product | conditions/yield |
|---|---|---|---|
| 1 |
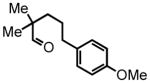 4 |
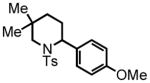 5 |
23 °C, 1 hr 96% |
| 2 |
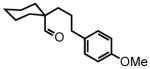 6 |
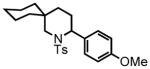 7 |
23 °C, 1 hr 98% |
| 3 |
 8 |
 9 |
23 °C, 1 hr 95% |
| 4 |
 10 |
 11 |
23 °C, 80 min 35%b |
| 5c |
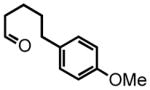 12 |
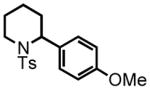 13 |
100 °C, 8 hr 30% |
| 6 |
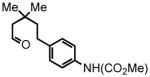 14 |
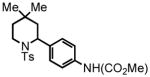 15 |
23 °C, 3 hr 81% |
| 7c |
 16 |
 17 |
100 °C, 18 hr 32% |
Reactions performed at 0.05 M in dry DCE, with 2 eq. TsNH2, 3 eq. BF3•OEt2 and 4Å MS 200% by wt.
Obtained as a 95:5 cis:trans mixture.
Reactions run in a sealed vial.
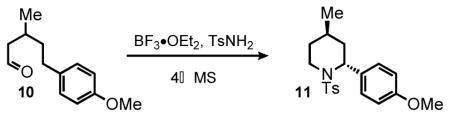
We next examined substrate 10, containing one methyl group in the β-position, and substrate 12, containing no substitution on the alkyl linker (Table 1, entries 4 and 5). Importantly, both substrates afforded the corresponding products, demonstrating the feasibility of HT-amination of aliphatic substrates with a small conformational bias, albeit with significantly lower yield, The efficiency of the HT-cyclization was improved by optimization of reaction conditions as is described in the subsequent section (Table 2). Unfortunately, even under the optimized conditions unsubstituted substrate 12 failed to furnish the desired product in higher yield. We also considered the incorporation of a heteroatom into the alkyl linker, which upon HT-amination would afford morpholine and piperazine products. However, we have found that the presence of a heteroatom in the backbone of various HT-cyclization substrates is generally not tolerated and leads to decomposition of the starting material.
Table 2.
Optimization of the HT-amination protocol for monosubstituted substratesa
| |||||||
|---|---|---|---|---|---|---|---|
| entry | solvent | conc. [M] | BF3•OEt2 | T (°C) | time | yield (%) | trans:cisb |
| 1 | DCE | 0.05 | 3 | 23 | 80 min | 37 | 95:5 |
| 2 | DCE | 0.025 | 3 | 23 | 80 min | 46 | 95:5 |
| 3 | DCE | 0.012 | 3 | 23 | 80 min | 70 | 94:6 |
| 4 | DCE | 0.006 | 3 | 23 | 80 min | 70 | 90:10 |
| 5 | DCM | 0.012 | 3 | 23 | 100 min | 55 | 95:5 |
| 6 | Toluene | 0.012 | 3 | 23 | 100 min | 50 | 95:5 |
| 7 | MeCN | 0.012 | 3 | 23 | 19 hr | 7 | 95:5 |
| 8 | THF | 0.012 | 3 | 23 | 10 hr | 0 | |
|
| |||||||
| 9 | DCE | 0.012 | 3 | 80 | 15 min | 85 | 93:7 |
|
| |||||||
| 10 | DCE | 0.012 | 2 | 80 | 30 min | 80 | 93:7 |
| 11 | DCE | 0.012 | 1 | 80 | 30 min | 81 | 92:8 |
| 12 | DCE | 0.012 | 0.5 | 80 | 30 min | 85 | 92:8 |
| 13c | DCE | 0.012 | 0.5 | 80 | 150 min | 84 | 95:5 |
2 eq. of TsNH2 employed, unless otherwise noted.
Ratio determined by 1H NMR of the crude reaction.
1.1 eq. of TsNH2 employed.
In the context of these substrates, we also explored the effect of electronics of the benzylic C-H bonds and the arene ring. In addition to the methoxy group, the carbamate-containing substrate 14 provided 81% yield of product 15 (Table 1, entry 6). Also, we were glad to find that substrate 16 with an unsubstituted phenyl ring undergoes the cyclization, although with lower efficiency (Table 1, entry 7). As expected, electron-donating substituents are required to achieve high yields of the desired products. This is a limitation pertinent to all HT-cyclization reactions as the stabilization of the carbocation intermediate, formed by the hydride transfer, is required (Scheme 1).
HT-Amination of Aliphatic Aldehydes is Highly Stereoselective
Examining the reactivity of substrate 10, bearing one methyl group in the β-position, revealed that the HT-cyclization is stereoselective (Scheme 4A). Optimization of the reaction conditions revealed that elevating the temperature to 80 °C and decreasing the substrate concentration (to disfavor formation of side products stemming from intermolecular reactions) led to a substantial improvement of the reaction efficiency, from 37% to 85% yield (Table 2). Modification of reaction conditions during optimization had a small effect on the diastereoselectivity.
Scheme 4.

HT-amination is Stereoselective
Under optimized conditions, aldehyde 10 afforded two diastereoisomers in a 93:7 ratio as determined by 1H NMR of the crude mixture (Scheme 4A). The geometry of the major trans-isomer was established by X-ray crystallography (Figure 2A). However, aldehyde 18 with the methyl group in the α-position afforded 2,5-disubstituted piperidine 19 in 65% yield and high stereoselectivity favoring the cis-isomer (d.r. 92:8, Scheme 4B). The structure and geometry of the major isomer was again confirmed by X-ray crystallographic analysis (Figure 2B). As expected, substantial racemization occurs under the HT-amination conditions when optically pure substrate 18 is used (see supporting information).
Figure 2.

Mechanistic Rationale for the Observed Diastereoselectivity. A/The experimental data supports the thermodynamic control of the stereoselectivity. The stereoisomer with the α-aryl group in the axial position is more stable due to the steric bulk of the N-tosyl group and its position on a partially sp2-hybridized nitrogen. ORTEP diagram from the X-ray analysis of the major isomer 11 is shown (right side). B/ The same ratinonale explains the formation of the cis-isomer of 2,5-disubstituted piperidine 19 as the major product. ORTEP diagram of 19 is shown (right side).
While we did observe similar results with a sub-stoichiometric amount of BF3•OEt2 (0.5 equiv., entries 12 & 13, Table 2), these reaction conditions were not general and failed to effect the HT-amination of other substrates in good yield.
Mechanistic Rationale for the Observed Stereoselectivity
To determine whether the diastereoselectivity of the HT-amination is kinetically or thermodynamically driven, a mixture of isomers 11 enriched in the minor cis-isomer (63:37 trans:cis ratio)18 was subjected to the reaction conditions (Figure 2). The result was a shift in the ratio to 90:10 trans:cis, which is very close to the product ratio found in the cyclization reaction. Similar results were obtained for the 2,5-disubstituted piperidines 19 when a 72:28 cis:trans ratio of products was equilibrated under the reaction conditions to a 92:8 cis:trans mixture. These results demonstrate the reversibility of the cyclization step, and support thermodynamic control as the source of the observed high stereoselectivity. In both cases (Figure 2A and B), the aryl ring prefers to adopt an axial position in the chair of the piperidine ring, to avoid the steric interaction with the sulfonamide group. The propensity for axial preference of substituents adjacent to sp2-hybridized nitrogens has been termed “pseudo-allylic strain” and has previously been proposed to explain conformation preferences of N-acyl and N-sulfonyl-piperidines.19 Under circumstances where the reaction would be controlled kinetically, the same pseudo-allylic strain rationale could be invoked in the corresponding transition states.
HT-amination of Monosubstituted Aliphatic Aldehydes: Substrate Scope
As discussed above, substrate 10 containing a methyl group in the β-position gave the trans-isomer of 11 in excellent yield and stereoselectivity (Table 3, entry 1). Similarly, compound 20 presenting a β-phenyl ring also reacted well under the optimized conditions, affording the di-aryl piperidine 21 with excellent stereoselectivity. (d.r. 92:8 favoring the trans-isomer) and modest yield (45%, Table 3, entry 2).
Table 3.
Stereoselective HT-amination: Substrate Scopea
| entry | substrate | product | conditions/yieldb |
|---|---|---|---|
| 1 |
 10 |
 11 |
80 °C, 15 min 85% 93:7 trans:cis |
| 2 |
 20 |
 21 |
80 °C, 2 hr 45% 92:8 trans:cis |
| 3 |
 18 |
 19 |
80 °C, 80 min 65% 92:8 cis:trans |
| 4 |
 22 |
 23 |
80 °C, 6 hr 28% 73:27 cis:trans |
| 5 |
 24 |
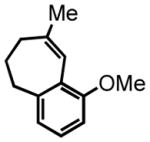 25 |
80 °C, 1 hr 90% |
Reactions performed at 0.012 M in dry DCE, with 2 eq. TsNH2, 3 eq. BF3•OEt2 and 4Å MS 200% by wt.
d.r. determined by 1H NMR of the crude reaction mixture.
Substrate 18 with the methyl group in the α-position exhibited relatively lower reactivity in comparison to the regioisomeric aldehyde 10 (65% yield, Table 3, entry 3), but the stereoselectivy was high and comparable between these two substrates. The transposition of the methoxy group on the arene to the ortho-position resulted in a decrease in yield and diastereoselectivity (28% yield, 73:27 cis:trans; Table 3, entry 4). Positioning the methoxy group in the meta-position disfavored the HT-cyclization and led to bicycle 25, the product of a Friedel-Crafts hydroxyalkylation followed by dehydration (Table 3, entry 5). We attribute this unexpected result to the decreased stabilization of the ensuing carbocation by the methoxy group in the meta-position, which thereby decreases the hydridic character of the benzylic C-H bonds. In addition, this placement of the methoxy group may deactivate the hydridic C-H bonds via a sigma withdrawing effect. With these effects in action the Friedel-Crafts hydroalkylation pathway dominates, leading to cyclization at the electron rich ortho-position of the methoxy group.
HT-amination of Aromatic Aldehydes: Substrate Scope
We revisited the reactivity of ortho-(2-arylethyl)-benzaldehydes in the HT-amination reaction since they afford an interesting and under-explored class of heterocycles, 3-aryl-1,2,3,4-tetrahydroisoquinolines. While there are numerous methods reported in the literature for the synthesis of 1-substituted tetrahydroisoquinolines,20 there are only a few general methods for the incorporation of the aryl substituents at the 3-position.5,21 Following our approach based on the HT-cyclization, 3-aryl-1,2,3,4-tetrahydroisoquinolines can be prepared in four steps from commercially available 2-bromobenzaldehydes.
The introduction of either electron donating (Table 2, entry 2) or withdrawing groups (Table 2, entries 3–5) onto the benzaldehyde ring (hydride acceptor) was well tolerated. Extension of the π-system of the parent substrate 3 to a naphthaldehyde afforded compound 35 in 85% yield, with an extension of the reaction time to 5 hr.
Directing our focus to the hydride donor portion of the substrate, we found that the absence of the para-methoxy group again was not tolerated under the standard reaction conditions. While clean conversion to the imine was observed by 1H NMR, only trace HT-amination product 37 was observed even after heating at 120 °C for 72 hours. However, a phenyl group in the 4-position was tolerated giving the biphenyl-substituted product 39 in 46% yield. The elevated reaction temperature of 120 °C was required in order to promote the HT-cyclization. The isolation of pure crystals of 39 permitted an X-ray analysis, thereby affirming the proposed 3-aryl-1,2,3,4-tetrahydroisoquinoline structure of this and all related products (see supporting information). Substrate 40 was prepared in order to investigate the effects of steric hindrance on the HT-amination, given the potential for the ortho-methyl group to impede cyclization. Such a steric effect was found to be minimal, with product 41 being formed in 80% yield. The methoxy-naphthalene derivative 42 was also reactive furnishing a 71 % yield of product 43 after an extended reaction time of 24 hours.
Deprotection of N-Tosyl Piperidines
Lastly, we investigated the removal of N-tosyl group in the context of the products discussed in this paper, which are all benzylamines. After exploring several known procedures we found that sodium naphthalenide, prepared fresh in dimethoxyethane (DME) at −78 °C was effective. For example, compound 2 was deprotected to afford 3-aryl-1,2,3,4-tetrahydroisoquinoline 44 in 89% (Scheme 5).22 Also, deprotection of substrate 19 was accomplished in 87% yield with minimal impact on the stereochemical integrity of the product.
Scheme 5.

Deprotection of the N-tosyl Group
CONCLUSION
In this study, we examined the formation of α-arylpiperidines via HT-cyclization of N-tosylimines formed in situ from the corresponding aldehydes. We showed that conformationally rigid aromatic aldehydes are readily converted to 3-aryl-1,2,3,4-tetrahydroisoquinolines, an interesting class of heterocycles. In a systematic manner, we examined the reactivity of aliphatic aldehydes (5-aryl-pentanals). We found that a single substituent on the alkyl chain connecting the N-tosylimine and the benzylic group is sufficient for an efficient HT-cyclization to occur. Furthermore, we discovered that the HT-cyclization of these substrates is highly stereoselective affording stereodefined 2,4- and 2,5-disubstituted piperidines. In addition, we have shown that the HT-cyclization of N-tosylimines is a reversible, thermodynamically controlled process and the high stereoselectivity is ascribed to the pseudoallylic strain between the N-tosyl group and the arene ring in the 2-position. This study has served to increase the understanding of the HT-amination reaction and has expanded the scope of HT-transformations in general.
EXPERIMENTAL SECTION
General Considerations
Argon was purified by passage through Drierite. Nuclear Magnetic Resonance spectra were recorded at 300 K on 300, 400 or 500 Fourier transform NMR spectrometers. 1H NMR spectra recorded in CDCl3 solutions were referenced to TMS (0.00 ppm). 13C NMR spectra recorded in CDCl3 were referenced to the residual solvent peak (77.16 ppm). High-resolution mass spectra (HRMS) were obtained on a high resolution sector type double focusing mass spectrometer (ionization mode: FAB+). Flash chromatography was performed on silica gel (230–400 mesh). Reactions were monitored by GC or TLC analysis using hexanes/ethyl acetate and hexanes/diethyl ether mixtures as the eluent and visualized using permanganate stain and/or cerric ammonium molybdate stain and/or UV light. Chloroform-d1 was stored over 4Å molecular sieves. Dichloroethane and triethylamine were freshly distilled from CaH2. Substrates 1223a and 3623b were prepared according to literature procedures.
General HT-amination Protocol
Activated 4Å molecular sieves (200% by wt. relative to substrate) were added to an oven dried flask followed by toluenesulfonamide (2 eq.), the appropriate substrate and a magnetic stir bar. The mixture was sealed under a rubber septum and back filled with argon. DCE, freshly distilled from calcium hydride, was then added via syringe, followed by addition of BF3•OEt2 (3 eq.) via microsyringe. The reaction was placed in an oil bath and heated to 80 °C. The reaction can be monitored by TLC, employing UV and KMnO4 stain visualization, however, the N-tosylimine and cyclized product often exhibit identical Rf values. As such, 1H NMR may be required to ensure the reaction has gone to completion. Following conversion of the starting material, the reaction is filtered through a cotton plug and concentrated in vacuo. The resulting residue was purified by flash column chromatography on silica gel.
Representative Procedure
To a solution of 1-(3-(4-methoxyphenyl)propyl)cyclohexane-carbaldehyde (6) (66 mg, 0.3 mmol) in freshly distilled DCE (6 mL) was added TsNH2 (103 mg, 0.6 mmol) and 4Å molecular sieves (132 mg). The reaction was sealed with a septum under argon. BF3•OEt2 (113 μL, 0.9 mmol) was then added via syringe and the mixture was stirred at room temperature. The reaction was monitored by TLC (30% Et2O:Hex). Upon complete consumption of the starting material, the mixture was filtered through a cotton plug and concentrated in vacuo. The residue was chromatographed on silica gel, eluting with 25% Et2O:Hex to afford 3-(4-methoxyphenyl)-2-tosyl-2-azaspiro[5.5]undecane (7) as a white solid (107 mg, 95%).
General Heck Reaction Protocol
A flame dried Schlenk tube is charged with Pd(OAc)2 (5 mol %), followed by the addition of DMF (0.25 M). The appropriate alkene (1 eq.) and aryl iodide/bromide (1.1 eq.) are subsequently added. Freshly distilled triethylamine (1.1 eq.) is then added to the reaction mixture. The vessel is equipped with a magnetic stir bar and sealed under argon. The reaction is then heated to 80 °C and monitored by TLC. Upon complete consumption of the alkene, the reaction is cooled to room temperature and poured into water (3 × volume of DMF). The resulting solution is extracted with EtOAc (4 × volume of DMF). The combined organic layers are then washed twice with water, once with brine, and dried over MgSO4. The suspension is filtered and the filtrate is concentrated in vacuo. The residue is then chromatographed on silica gel affording the pure product.
General Sonogashira Coupling Protocol
A flame dried flask is charged with PdCl2(PPh3)2 (2.5 mol %) and CuI (2.5 mol %). Freshly distilled triethylamine (0.25 M) is then added followed by the appropriate aryl bromide (1 eq.) and alkyne (1.1 eq.). The reaction is sealed under argon and heated to 50 °C. The reaction is monitored by TLC. Upon consumption of the aryl bromide, the mixture is cooled to room temperature and concentrated in vacuo. The residue is then dissolved in EtOAc and washed with water and NH4Cl (aq. sat.). The organic layer is then dried over MgSO4. The suspension is filtered and the filtrate is concentrated in vacuo. The residue is chromatographed on silica gel affording the pure product.
General Alkene/Alkyne Hydrogenation Protocol
To a solution of the appropriate alkene or alkyne in EtOH (0.4 M) is added 10% by wt. Pd/C (20% by wt. relative to alkyne/alkene). The flask is equipped with a magnetic stir bar and the suspension is sealed with a septum under an atmosphere of H2 supplied via a balloon. The reaction is stirred vigorously and monitored by 1H NMR. Following complete hydrogenation, the suspension is filtered through a pad of celite. The filtrate is concentrated in vacuo to afford the product; the crude product is typically pure by 1H NMR and employed directly in the next step.
General LiAlH4 Reduction Protocol
A flame dried flask is charged with LiAlH4 (1.5 eq.), followed by the addition of THF (0.17 M based on ester). The suspension is then cooled to 0 °C in an ice bath and a solution of the appropriate ester in THF (0.24 M based on ester) is added slowly. The reaction is then allowed to warm to room temperature. The reaction is monitored by TLC. After complete reduction of the starting material, the reaction is cooled to 0 °C and is quenched with water (3 mL/1 g LiAlH4), KOH 20% (9 mL/1 g LiAlH4) and finally water (15 mL/1 g LiAlH4). After stirring for 5 minutes, the suspension is filtered and the filtrate is dried over MgSO4 and concentrated in vacuo. The product alcohol is typically pure by 1H NMR and employed directly in the next step.
General MnO2 Oxidation Protocol
To a solution of alcohol (1 eq.) in DCM (0.3 M) is added MnO2 (5 eq.). The suspension is stirred vigorously and monitored by TLC. Upon complete conversion of the starting material, the mixture is filtered through celite and the filtrate is concentrated in vacuo. The residue is chromatographed on silica gel to afford the pure aldehyde.
General Pyridinium Chlorochromate (PCC) Oxidation Protocol
To a solution of alcohol (1 eq.) in DCM (0.2 M) is added PCC (2.25 eq.). The mixture is stirred and monitored by TLC. Upon conversion of the starting material, the suspension is filtered through a pad of silica gel. The filtrate is concentrated in vacuo and the residue is chromatographed on silica gel to afford the pure aldehyde.
General Swern Oxidation Protocol
A solution of DMSO (2.4 eq.) in dry DCM (1 M) is cooled to −78 °C under an argon atmosphere. Oxalylchloride (1.2 eq., 2.0 M solution in DCM) is added dropwise. The resulting solution is stirred at −78 °C for 30 minutes. A solution of the corresponding alcohol (1 eq.) in dry DCM (5 M) is added dropwise to the reaction mixture. The reaction is stirred at −78 °C for 1 hour. Triethylamine is then added to the solution and the mixture is allowed to warm to room temperature. The reaction mixture is poured into water (3 × total volume of DCM) and extracted four times with DCM. The combined organic layers are then washed sequentially with saturated aqueous CuSO4 solution, and brine, then dried over MgSO4. The suspension is filtered and the filtrate is concentrated in vacuo. The residue is then chromatographed on silica gel affording the pure aldehyde.
N-(2-(4-methoxyphenethyl)benzylidene)-4-methylbenzenesulfonamide (1)
To a solution of 2-(4-methoxyphenethyl)benzaldehyde24 (720 mg, 6.0 mmol) in THF (6 mL) was added TsNH2 (1.03 g, 6.0 mmol) and Ti(OEt)4 (1.9 mL, 9.0 mmol). The reaction was sealed with a septum under argon and stirred for 24 hr. The reaction was then diluted with EtOAc and the resulting solution was poured into brine. The suspension was filtered through Celite and the filtrate was extracted with EtOAc. The combined organic layers were dried over MgSO4. The suspension was filtered and the filtrate was concentrated in vacuo. The residue was then crystallized from Et2O:Hex, affording 1 as a pale yellow solid (611 mg, 52%). 1H NMR (CDCl3, 300 MHz) δ 2.43 (s, 3H); 2.79 (t, J = 8.4 Hz, 2H); 3.19 (d, J = 8.4 Hz, 2H); 3.79 (s, 3H); 6.81 (d, J = 8.7 Hz, 2H); 6.99 (d, J = 8.7 Hz, 2H); 7.34-7.23 (m, 4H); 7.49 (dt, J = 9.0 Hz, J = 1.5 Hz, 1H); 7.85 (d, J = 8.4 Hz, 2H); 7.99 (dd, J = 7.8 Hz, J = 1.2 Hz, 1H); 9.19 (s, 1H); 13C NMR (CDCl3, 75 MHz) δ 21.7, 35.4, 37.7, 55.3, 114.0, 126.9, 128.1, 129.6, 129.9, 130.1, 131.1, 131.2, 132.5, 134.7, 135.4, 144.6, 146.0, 158.2, 168.5. HRMS (FAB+): calculated for C23H24NO3S+ 394.1471 +, measured 394.1483 [M+1].
5-(4-methoxyphenyl)-2,2-dimethylpentanal (4)
Prepared according to the Heck protocol from ethyl 2,2,-dimethylpent-4-enoate25 (1.7 g, 10.9 mmol), 4-iodoanisole (2.63 g, 11.0 mmol) and Pd(OAc)2 (122 mg, 0.55 mmol, 5 mol %), in DMF (50 mL) and TEA (1.54 mL), followed by direct hydrogenation according to the alkene hydrogenation protocol to afford ethyl 2,2-dimethyl-5-phenylpentanoate as a colorless oil (1.39 g, 76% over two steps) 1H NMR (CDCl3, 300 MHz) δ 1.14 (s, 6H); 1.22 (t, J = 7.2 Hz, 3H); 1.54 (apparent d, J = 3.6 Hz, 4H); 2.52 (bt, J = 6.6 Hz, 2H); 3.78 (s, 3H); 4.09 (quart, J = 7.2 Hz, 2H); 6.82 (d, J = 8.4 Hz, 2H); 7.08 (d, J = 8.4 Hz, 2H). The resulting ester (1.39 g, 5.29 mmol) was reduced according to the LiAlH4 reduction protocol employing LiAlH4 (302 mg, 7.93 mmol) in THF (32 + 22 mL) to afford pure product alcohol 2,2-dimethyl-5-phenylpentan-1-ol (1.17 g, 99%) as a colorless oil 1H NMR (CDCl3, 300 MHz) δ 0.86 (s, 6H); 1.30-1.25 (m, 3H); 1.58-1.50 (m, 2H); 2.53 (t, J = 7.8 Hz, 2H); 3.30 (s, 2H); 3.78 (s, 3H); 6.82 (d, J = 8.4 Hz, 2H); 7.10 (d, J = 8.4 Hz, 2H). The resulting alcohol (1.16 g, 5.23 mmol) was oxidized according to the PCC oxidation protocol employing PCC (2.54 g, 11.8 mmol) in DCM (25 mL) to afford aldehyde 4 (888 mg, 77%) as a colorless oil following chromatography on silica gel, eluting with 5% Et2O:Hex. 1H NMR (CDCl3, 300 MHz) δ 1.03 (s, 6H); 1.53-1.45 (bm, 4H); 2.53-2.51 (bm, 2H); 3.78 (s, 3H); 6.82 (d, J = 8.4 Hz, 2H); 7.06 (d, J = 8.7 Hz, 2H); 9.42 (s, 1H); 13C NMR (CDCl3, 75 MHz) δ 21.4, 26.4, 35.5, 36.8, 45.8, 55.3, 113.9, 129.3, 134.1, 157.9, 206.4. HRMS (FAB+): calculated for C14H20O2 220.1463 [M], measured 220.1463 [M].
1-(3-(4-methoxyphenyl)propyl)cyclohexanecarbaldehyde (6)
Prepared according to the Heck protocol from methyl 1-allylcyclohexanecarboxylate (1.82 g, 10.0 mmol), 4-iodoanisole (2.58 g, 11.0 mmol) and Pd(OAc)2 (67 mg, 0.30 mmol, 3 mol %), in DMF (50 mL), to afford (E)-methyl 1-(3-(4-methoxyphenyl)allyl)cyclohexanecarboxylate as a pale yellow oil (1.62 g, 56%) 1H NMR (CDCl3, 300 MHz) δ 1.34-1.18 (m, 6H); 1.62-1.54 (m, 2H); 2.12-2.06 (m, 2H); 2.36 (d, J = 7.5 Hz, 2H); 3.67 (s, 3H); 3.79 (s, 3H); 5.89–6.00 (m, 1H); 6.30 (d, J = 15.9 Hz, 1H); 6.82 (d, J = 8.7 Hz, 2H); 7.25 (d, J = 8.7 Hz, 2H). The resulting alkene (1.62 g, 5.60 mmol) was hydrogenated according to the hydrogenation protocol employing 10% by wt. Pd/C (324 mg, 20% by wt.) in EtOH (15 mL) to afford methyl 1-(3-(4-methoxyphenyl)propyl)cyclohexanecarboxylate as a colorless oil (1.53 g, 94%) 1H NMR (CDCl3, 300 MHz) δ 1.33-1.12 (m, 6H); 1.52-1.48 (m, 6H); 2.05 (bd, J = 12.0 Hz, 2H); 2.49 (bs, 2H); 3.65 (s, 3H); 3.78 (s, 3H); 6.81 (d, J = 8.4 Hz, 2H); 7.06 (d, J = 8.4 Hz, 2H). The resulting ester (1.53 g, 5.26 mmol) was reduced according to the LiAlH4 reduction protocol employing LiAlH4 (300 mg, 7.89 mmol) in THF (30 + 20 mL) followed by direct oxidation of the crude product alcohol according to PCC oxidation protocol to afford aldehyde 6 (980 mg, 72% over two steps) as a colorless oil following chromatography on silica gel, eluting with 8% Et2O:Hex. 1H NMR (CDCl3, 400 MHz) δ 1.31-1.21 (m, 4H); 1.53-1.42 (m, 8H); 1.88-1.85 (m, 2H); 2.50 (t, J = 6.8 Hz, 2H); 3.78 (s, 3H); 6.81 (d, J = 8.8 Hz, 2H); 7.05 (d, J = 8.8 Hz, 2H); 9.39 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 22.7, 25.6, 25.9, 31.1, 35.5, 36.1, 49.8, 55.4, 113.9, 129.3, 134.1, 157.9, 207.3. HRMS (FAB+): calculated for C17H24O2 260.1776 [M], measured 260.1767 [M].
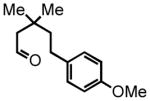
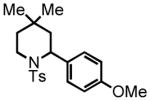
5-(4-methoxyphenyl)-3,3-dimethylpentanal (8)
Prepared according to the Heck protocol from methyl 3,3-dimethylpent-4-enoate (2.13 g, 15.0 mmol), 4-iodoanisole (3.86 g, 16.5 mmol) and Pd(OAc)2 (100 mg, 0.45 mmol, 3 mol %), in DMF (75 mL) and TEA (2.3 mL) to afford (E)-methyl 5-(4-methylphenyl)-3,3-dimethylpent-4-enoate as a colorless oil (2.16 g, 58%). 1H NMR (CDCl3, 300 MHz) δ 1.23 (s, 6H); 2.38 (s, 2H); 3.62 (s, 3H); 3.78 (s, 3H); 6.15 (d, J = 16.2 Hz, 1H); 6.28 (d, J = 16.2 Hz, 1H); 6.83 (d, J = 8.7 Hz, 2H); 7.28 (d, J = 8.7 Hz, 2H). The resulting alkene (2.16 g, 8.70 mmol) was hydrogenated according to the hydrogenation protocol employing 10% by wt. Pd/C (432 mg, 20% by wt.) in EtOH (20 mL) to afford methyl 5-(4-methoxyphenyl)-3,3-dimethylpentanoate as a colorless oil (1.47 g, 67%) 1H NMR (CDCl3, 300 MHz) δ 1.06 (s, 6H); 1.58 (bt, J = 8.1 Hz, 2H); 2.28 (s, 2H); 2.54 (d, J = 8.1 Hz, 2H); 3.66 (s, 3H); 3.78 (s, 3H); 6.82 (d, J = 7.8 Hz, 2H); 7.10 (d, J = 7.5 Hz, 2H). The resulting ester (1.47 g, 5.86 mmol) was reduced according to the LiAlH4 reduction protocol employing LiAlH4 (335 mg, 8.80 mmol) in THF (36 + 24 mL) followed by direct oxidation of the crude product alcohol according to the PCC oxidation protocol to afford aldehyde 8 (733 mg, 57% over two steps) as a colorless oil following chromatography on silica gel, eluting with 8% Et2O:Hex. 1H NMR (CDCl3, 300 MHz) δ 1.12 (s, 6H); 1.58-1.64 (m, 2H); 2.32 (s, 2H); 2.51–2.57 (m, 2H); 3.77 (s, 3H); 6.82 (d, J = 8.7 Hz, 2H); 7.08 (d, J = 8.4 Hz, 2H); 9.86 (t, J = 3.0 Hz, 1H); 13C NMR (CDCl3, 75 MHz) δ 27.6, 29.7, 33.7, 45.2, 54.8, 55.3, 114.0, 129.2, 134.6, 157.9, 203.4. HRMS (FAB+): calculated for C14H21O2+ 221.1536 [M+1], measured 221.1538 [M+1].
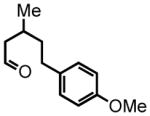
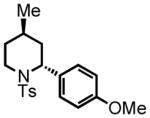
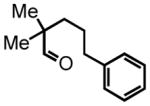
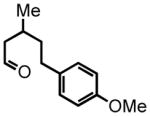
5-(4-methoxyphenyl)-3-methylpentanal (10)
Triethyl phosphonoacetate (0.916 g, 4.08 mmol) is weighed into a flame dried, two-neck round bottom flask equipped with a magnetic bar. Dry THF (3 mL, 2 M) is added under an argon atmosphere. NaH (60% suspension in mineral oil, 120 mg) is washed properly and added to the reaction vessel. The reaction mixture is stirred at room temperature for 30 minutes. 4-(4-methoxyphenyl)butan-2-one (0.73 g, 4.09 mmol) in a solution of THF (2 mL, 2 M) is added dropwise to the reaction mixture. The reaction is monitored by TLC. After 18 hours the mixture is concentrated in vacuo. The residue is then disolved in diethyl ether (30 mL) and washed with saturated NaHCO3 (10 mL). The organic layer is dried over NaSO4. The suspension is filtered and the filtrate is concentrated in vacuo. The residue is chromatographed on silica gel and eluted with EtOAc:Hex (5:95) to afford the corresponding ester (0.64 g, 63%) as a colorless oil. This ester is then subjected to the alkene hydrogenation protocol employing Pd/C 10% by wt. (64 mg) dissolved in EtOH (7 mL) to afford ethyl 5-(4-methoxyphenyl)-3-methylpentanoate (0.511 g, 2.05 mmol, 50% yield over two steps), which is employed directly in the next step; 1H NMR (300 MHz, CDCl3) δ 7.09 (d, J = 8.6 Hz, 2H), 6.82 (d, J = 8.7 Hz, 2H), 4.12 (q, J = 7.2 Hz, 2H), 3.78 (s, 3H), 2.48–2.67 (m, 2H), 2.34 (dd, J = 14.7, 6.2 Hz, 1H), 2.15 (dd, J = 14.8, 8.0 Hz, 1H), 2.00 (sex, J = 6.2 Hz, 1H), 1.59–1.65 (m, 1H), 1.47–1.52 (m, 1H), 1.25 (t, J = 7.1 Hz, 3H), 0.99 (d, J = 6.7 Hz, 3H). The ester (0.492 g, 1.97 mmol) is reduced according to the LiAlH4 reduction protocol employing LiAlH4 (0.112 g, 2.95 mmol) in THF (11 + 8 mL) to afford pure 5-(4-methoxyphenyl)-3-methylpentan-1-ol (0.376 g, 92%) as a colorless oil; 1H NMR (300 MHz, CDCl3) δ 7.10 (d, J = 8.7 Hz, 2H), 6.82 (d, J = 8.7 Hz, 2H), 3.79 (s, 3H), 3.65–3.72 (m, 2H), 2.47–2.67 (m, 2H), 1.56–1.68 (m, 3H), 1.37–1.48 (m, 3H), 0.96 (d, J = 6.3 Hz, 3H). The intermediate alcohol (0.346 g, 1.66 mmol) is then oxidized according to the Swern oxidation protocol employing (COCl)2 (0.99 mL of 2.0 M solution in DCM), DMSO (0.311 g, 3.98 mmol), TEA (0.672 g, 6.65 mmol) in DCM (2 + 4 mL). The crude material is purified by silica gel column chromatography (8% Et2O:Hex) to afford the pure aldehyde 10 (0.250 g, 73%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 9.74 (t, J = 2.2 Hz, 1H), 7.09 (d, J = 8.6 Hz, 2H), 6.83 (d, J = 8.6 Hz, 2H), 3.79 (s, 3H), 2.54–2.63 (m, 2H), 2.44 (ddd, J = 16.0, 5.7, 2.0 Hz, 1H), 2.27 (ddd, J = 16.1, 7.9, 16.1 Hz, 1H), 2.08–2.13 (m, 1H), 1.51–1.66 (m, 2H), 1.02 (d, J = 6.7 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 203.3, 158.2, 134.4; 129.6, 129.6, 114.2, 114.2, 55.7, 51.4, 39.3, 32.7, 28.1, 20.2; HRMS (FAB+): calculated for C13H18O2 207.1307 [M], measured 206.1324 [M].
Methyl 4-(3,3-dimethyl-5-oxopentyl)phenylcarbamate (14)
Prepared according to the Heck protocol from methyl 3,3-dimethylpent-4-enoate (1.30 g, 9.09 mmol), aryl iodide methyl 4-iodophenylcarbamate (2.77 g, 10.0 mmol) and Pd(OAc)2 (102 mg, 0.45 mmol, 5 mol %), in DMF (45 mL) and TEA (2.5 mL) to afford methyl 5-(4-methoxycarbonylamino)phenyl)-3,3-dimethylpentenoate as a colorless oil (1.38 g, 52%); 1H NMR (CDCl3, 300 MHz) δ 1.23 (s, 6H); 2.40 (s, 2H); 3.63 (s, 3H); 3.75 (s, 3H); 6.32-6.18 (m, 2H); 7.08 (bs, 1H); 7.33-7.27 (m, 4H). The resulting alkene (918 mg, 3.15 mmol) was hydrogenated according to the hydrogenation protocol employing 10% by wt. Pd/C (100 mg, 20% by wt.) in EtOH (10 mL) to afford methyl 5-(4-methoxycarbonylamino)phenyl)-3,3-dimethylpentanoate as a colorless oil (877 mg, 95%); 1H NMR (CDCl3, 300 MHz) δ 1.06 (s, 6H); 1.61-1.55 (m, 2H); 2.28 (s, 2H); 2.58-2.52 (m, 2H); 3.66 (s, 3H); 3.76 (s, 3H); 6.65 (bs, 1H); 7.12 (d, J = 8.4 Hz, 2H); 7.28 (d, J = 8.7 Hz, 2H). A flame dried flask is charged with LiBH4 (196 mg, 9.0 mmol, 3 eq.), followed by the addition of THF (7 mL). The suspension is then cooled to 0 °C in an ice bath and a solution of methyl 5-(4-methoxycarbonylamino)phenyl)-3,3-dimethylpentanoate in THF (3 mL) is added slowly. The reaction is then allowed to warm to room temperature and is stirred overnight. After complete reduction of the starting material, the mixture is diluted with water (50 mL), followed by the slow addition of 10% HCl (aq.). The solution is then neutralized with saturated NaHCO3 and the resulting solution is extracted with EtOAc. The combined organic layers are washed with brine, dried over MgSO4 and concentrated in vacuo. The crude intermediate alcohol is employed directly in the next step. The alcohol is the subjected to Swern oxidation protocol employing DMSO (0.51 mL, 7.20 mmol), ox-alyl chloride 2 M in DCM (1.80 mL, 3.60 mmol), and triethylamine (1.70 mL, 12 mmol) in DCM (10 mL). to afford aldehyde 14 (603 mg, 76% over two steps) as a colorless oil following chromatography on silica gel, eluting with 30% Et2O:Hex. 1H NMR (CDCl3, 300 MHz) δ 1.13 (s, 6H); 1.64-1.58 (m, 2H); 2.34 (d, J = 3.0 Hz, 2H); 2.58-2.53 (m, 2H); 3.76 (s, 3H); 6.63 (bs, 1H); 7.10 (d, J = 8.4 Hz, 2H); 7.29 (d, J = 8.4 Hz, 2H); 9.87 (t, J = 3.0 Hz, 1H). HRMS (FAB+): calculated for C15H21NO3 263.1521 [M], measured 263.1528 [M].
2,2-dimethyl-5-phenylpentanal (16)
Prepared according to the Heck protocol from ethyl 2,2,-dimethylpent-4-enoate25 (1.56 g, 10.0 mmol), 4-iodobenzene (1.3 mL, 11.0 mmol) and Pd(OAc)2 (67 mg, 0.30 mmol, 3 mol %) in DMF (50 mL) to afford (E)-ethyl 3,3-dimethyl-5-phenylpent-4-enoate as a pale yellow oil (1.85 g, 80%); 1H NMR (CDCl3, 300 MHz) δ 1.22 (s, 6H); 1.25 (t, J = 7.2 Hz, 3H); 2.43 (dd, J = 7.5 Hz, J = 0.9 Hz, 2H); 4.13 (quart, J = 7.2 Hz, 2H); 6.20-6.09 (m, 1H); 6.40 (d, J = 15.9 Hz, 1H); 7.34-7.20 (m, 5H). The resulting alkene (1.85 g, 7.97 mmol) is hydrogenated according to the hydrogenation protocol employing 10% by wt. Pd/C (370 mg, 20% by wt.) in EtOH (20 mL) to afford ethyl 3,3-dimethyl-5-phenylpentanoate as a colorless oil (1.70 g, 91%); 1H NMR (CDCl3, 300 MHz) δ 1.15 (s, 6H); 1.22 (t, J = 7.2 Hz, 3H); 1.56 (bs, 4H); 2.59 (bs, 2H); 4.09 (quart, J = 7.2 Hz, 2H); 7.27-7.15 (m, 5H). The intermediate ester (1.70 g, 7.25 mmol) is reduced according to the LiAlH4 reduction protocol employing LiAlH4 (413 mg, 10.88 mmol) in THF (42 + 28 mL) followed by direct oxidation of the crude product alcohol according to the PCC oxidation protocol to afford aldehyde 16 (1.11 g, 81% over two steps) as a colorless oil following chromatography on silica gel, eluting with 5% Et2O:Hex; 1H NMR (CDCl3, 300 MHz) δ 1.03 (s, 6H); 1.55-1.51 (bm, 4H); 2.60 (t, J = 6.9 Hz, 2H); 7.30-7.14 (m, 5H); 9.42 (s, 1H); 13C NMR (CDCl3, 75 MHz) δ 21.4, 26.2, 36.5, 36.9, 45.9, 126.0, 128.5, 128.7, 142.1, 206.4. HRMS (FAB+): calculated for C13H18O 190.1358 [M], measured 190.1371 [M].
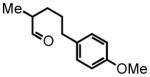
5-(4-methoxyphenyl)-2-methylpentanal (18)
Prepared according to the Heck protocol from ethyl 2-methylpent-4-enoate (1.00 g, 7.03 mmol), 4-iodoanisole (1.81 g, 7.74 mmol) and PdCl2(PPh3)2 (24 mg, 5 mol %) in DMF (28 mL) and TEA (0.78 g, 7.7 mmol) to afford (E)-ethyl-5-(4-methoxyphenyl)-2-methylpent-4-enoate as a pale yellow oil. The resulting alkene is hydrogenated according to the hydrogenation protocol employing Pd/C 10% by wt. (99.0 mg) in EtOH (10.0 mL) to afford ethyl 5-(4-methoxyphenyl)-2-methylpentanoate (1.12 g, 4.5 mmol, 64% over two steps) which was employed directly in the next step; 1H NMR (300 MHz, CDCl3) δ 7.08 (d, J = 8.6 Hz, 2H), 6.82 (d, J = 8.7 Hz, 2H), 4.12 (q, J = 8.7 Hz, 2H), 3.79 (s, 3H), 2.55 (t, J = 7.4 Hz, 2H), 2.43 (t, J = 7.4 Hz, 1H), 1.54–1.73 (m, 3H), 1.43–1.49 (m, 1H), 1.24 (t, J = 7.3 Hz, 3H), 1.13 (d, J = 6.9 Hz, 3H). The crude ester (0.975 g, 3.90 mmol) was reduced according to the LiAlH4 reduction protocol employing LiAlH4 (0.222 g, 5.85 mmol) in THF (22 + 16 mL) to afford pure 5-(4-methoxyphenyl)-2-methylpentan-1-ol (0.722 g, 3.71 mmol, 95%) as a colorless oil following silica gel column chromatography, eluting with 20% EtOAc:Hex; 1H NMR (300 MHz, CDCl3) δ 7.03 (d, J = 8.4 Hz, 2H), 6.77 (d, J = 8.5 Hz, 2H), 3.72 (s, 3H), 3.33–3.48 (m, 2H), 2.49 (ddd, J = 8.8, 8.8, 2.6 Hz, 2H), 1.50–1.63 (m, 3H), 1.22–1.44 (m, 1H), 1.15 (bs, 1H), 1.03–1.10 (m, 1H), 0.85 (d, J = 6.7 Hz, 3H). The resulting alcohol (0.70 g, 3.36 mmol) was oxidized according to the Swern oxidation protocol employing (COCl)2 (2.02 mL of 2.0 M solution in DCM), DMSO (0.630 g, 8.07 mmol), TEA (1.36 g, 13.4 mmol) in DCM (2 + 4 mL). The crude material is purified by silica gel column chromatography eluting with 5% EtOAc:Hex to afford the pure aldehyde 18 (0.506 g, 2.45 mmol, 73%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 9.58 (d, J = 1.9 Hz, 1H), 7.07 (d, J = 8.5 Hz, 2H), 6.82 (d, J = 8.5 Hz, 2H), 3.77 (s, 3H), 2.56 (t, J = 7.3 Hz, 2H), 2.33 (dsex, J = 6.9; 1.9 Hz, 1H), 1.57–1.76 (m, 3H), 1.34–1.43 (m, 1H), 1.08 (d, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 205.5, 158.2, 134.4; 129.7, 129.6, 114.2, 114.2, 55.6, 46.6, 35.3, 30.4, 29.3, 13.7. HRMS (FAB+): calculated for C13H18O2 206.1307 [M], measured 206.1321 [M].
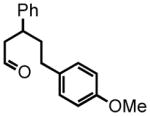
5-(4-methoxyphenyl)-3-phenylpentanal (20)
Prepared according to the procedure for compound 10 from triethyl phosphonoacetate (0.821 g, 2.66 mmol), NaH from 60 % mineral oil suspension (0.140 g) and 3-(4-methoxyphenyl)-1-phenylpropan-1-one (0.80 g, 3.33 mmol) in THF (4 + 4 mL). After purification using silica gel column chormatography (5% EtOAc: Hex) the corresponding ester is submitted to direct hydrogenation according to the hydrogenation protocol using Pd/C 10% by wt. (80 mg) in EtOH (10 mL) to afford ethyl 5-(4-methoxyphenyl)-3-phenylpentanoate (0.52 g, 1.66 mmol, 50 % over two steps) which is employed directly in the next step; 1H NMR (300 MHz, CDCl3) δ 7.31–7.34 (m, 2H), 7.19–7.28 (m, 3H), 7.01 (d, J = 8.4 Hz, 2H), 6.79 (d, J = 8.7 Hz, 2H), 4.02 (q, J = 7.1 Hz, 2H), 3.77 (s, 3H), 3.12–3.23 (m, 1H), 2.60 (t, J = 6.1 Hz, 2H), 2.40 (t, J = 8.4 Hz, 2H), 1.86–1.97 (m, 2H), 1.12 (t, J = 6.9 Hz, 3H). The crude ester (0.75 g, 2.40 mmol) is reduced according to the LiAlH4 reduction protocol employing LiAlH4 (0.14 g, 3.61 mmol) in THF (14 + 10 mL) to afford pure 5-(4-methoxyphenyl)-3-phenylpentan-1-ol (0.616 g, 2.28 mmol, 95%) as a colorless oil; 1H NMR (300 MHz, CDCl3) δ 7.32 (t, J = 7.3 Hz, 2H), 7.20 (dd, J = 5.5, 4.1 Hz, 3H), 7.01 (d, J = 8.5 Hz, 2H), 6.79 (d, J = 8.5 Hz, 2H), 3.77 (s, 3H), 3.43–3.56 (m, 2H), 2.72 (t, J = 7.9 Hz, 1H), 2.40 (t, J = 7.9 Hz, 2H), 1.86–1.99 (m, 4H), 1.56 (brs, 1H). The intermediate alcohol (0.53 g, 1.95 mmol) is oxidized according to the Swern oxidation protocol employing (COCl)2 (1.17 mL of 2.0 M solution in DCM), DMSO (0.37 g, 4.68 mmol), TEA (0.79 g, 7.80 mmol) in DCM (2 + 4 mL). The crude product is purified by silica gel column chromatography 5% EtOAc:Hex to afford the pure aldehyde 20 (0.37 g, 1.36 mmol, 70%) as a colorless oil; 1H NMR (300 MHz, CDCl3) δ 9.63 (t, J = 2.1 Hz, 1H), 7.33 (t, J = 6.5 Hz, 2H), 7.21–7.23 (m, 3H) 7.01 (d, J = 8.8 Hz, 2H), 6.80 (d, J = 8.7 Hz, 2H), 3.77 (s, 3H), 3.19 (quintet, J = 7.4 Hz, 1H), 2.72 (ddd J = 7.4, 2.1, 1.3 Hz, 2H), 2.42 (ddd, J = 9.0, 9.0, 1.8 Hz, 2H), 1.90–1.99 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 202.1, 158.2, 143.8; 134.1, 129.6, 129.6, 129.1, 129.1, 128.0, 128.0, 127.1, 114.2, 114.2, 55.6, 51.1, 39.9, 38.7, 32.0. HRMS (FAB+): calculated for C18H20O2 268.1463 [M], measured 268.1476 [M].
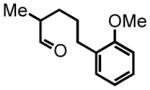
5-(2-methoxyphenyl)-2-methylpentanal (22)
Prepared according to the Heck protocol from ethyl 2-methylpent-4-enoate (1.00 g, 7.03 mmol), 2-iodoanisole (1.81 g, 7.74 mmol) and PdCl2(PPh3)2 (24 mg, 5 mol %) in DMF (28 mL) and TEA (0.78 g, 7.7 mmol). The resulting alkene is submitted to hydrogenation protocol using EtOH (10mL) and Pd/C 10% by wt. (100 mg) to afford ester ethyl 5-(2-methoxyphenyl)-2-methylpentanoate (0.88 g, 3.52 mmol, 50% over two steps) which was used employed directly in the next step; 1H NMR (300 MHz, CDCl3) δ 7.16 (ddd, J = 9.6, 7.8, 1.8 Hz, 1H), 7.11 (dd, J = 7.4, 1.6 Hz, 1H), 6.82–6.89 (m, 2H), 4.12 (q, J = 7.2 Hz, 2H), 3.81 (s, 3H), 2.60 (t, J = 7.5 Hz, 2H), 2.45 (sex, J = 7.0 Hz, 1H), 1.65–1.73 (m, 1H), 1.56–1.61 (m, 2H), 1.40–1.50 (m, 1H), 1.24 (t, J = 7.1 Hz, 3H), 1.14 (d, J = 6.9 Hz, 3H). The crude ester (0.87 g, 3.46 mmol) was reduced according to the LiAlH4 reduction protocol employing LiAlH4 (0.197 g, 5.19 mmol) in THF (17 + 14 mL) to afford pure 5-(2-methoxyphenyl)-2-methylpentan-1-ol (0.633 g, 3.04 mmol, 88%) as a colorless oil; 1H NMR (300 MHz, CDCl3) δ 7.12–7.20 (m, 2H), 6.83–6.93 (m, 2H), 3.82 (s, 3H), 3.51 (dd, J = 10.4, 6.0 Hz, 1H), 3.42 (dd, J = 10.2, 6.6 Hz, 1H), 2.60 (ddd, J = 8.8, 6.9, 2.3 Hz, 2H), 1.41–1.70 (m, 4H), 1.30 (brs, 1H), 1.17–1.23 (m, 1H), 0.93 (d, J = 6.7 Hz, 3H). The intermediate alcohol (0.77 g, 3.70 mmol) is oxidized according to the Swern oxidation protocol employing (COCl)2 (2.21 mL of 2.0 M solution in DCM), DMSO (0.693 g, 8.90 mmol), TEA (1.50 g, 14.7 mmol) in DCM (3 + 6 mL). The crude product is purified by silica gel column chromatography eluting with 10% EtOAc:Hex to afford the pure aldehyde 22 (0.412 g, 2.0 mmol, 54%) as a colorless oil; 1H NMR (300 MHz, CDCl3) δ 9.57 (d, J = 2.0 Hz, 1H), 7.06–7.16 (m, 2H), 6.78–6.87 (m, 2H), 3.76 (s, 3H), 2.62 (t, J = 7.4 Hz, 2H), 2.33 (dsex, J = 7.0, 1.9 Hz, 1H), 1.59–1.73 (m, 3H), 1.36–1.42 (m, 1), 1.05 (d, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 205.6, 157.8, 130.7; 130.2, 127.6, 120.8, 110.7, 55.6, 46.6, 30.6, 30.6, 27.6, 13.7. HRMS (FAB+): calculated for C13H18O2 206.1307 [M], measured 206.1311 [M].
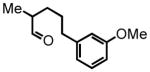
5-(3-methoxyphenyl)-2-methylpentanal (24)
Prepared according to the Heck protocol from ethyl 2-methylpent-4-enoate (1.00 g, 7.03 mmol), 2-iodoanisole (1.81 g, 7.74 mmol) and PdCl2(PPh3)2 (24 mg, 5 mol %) in DMF (28 mL) and TEA (0.78 g, 7.7 mmol). The resulting alkene is submitted to the hydrogenation protocol using EtOH (10mL) and Pd/C 10% by wt. (100 mg) to afford ethyl 5-(e-methoxyphenyl)-2-methylpentanoate (0.65 g, 2.60 mmol, 37% over two steps) which was used employed directly in the next step; 1H NMR (300 MHz, CDCl3) δ 7.19 (ddd, J = 7.4, 7.4, 2.0 Hz, 1H), 6.72–6.77 (m, 4H), 4.12 (q, J = 7.1 Hz, 2H), 3.79 (s, 3H), 2.59 (t, J = 7.4 Hz, 2H), 2.43 (sex, J = 6.8 Hz, 1H), 1.56–1.72 (m, 3H), 1.42–1.50 (m, 1H), 1.24 (t, J = 7.0 Hz, 3H), 1.14 (d, J = 7.0 Hz, 3H). The crude ester (0.65 g, 2.60 mmol) was reduced according to the LiAlH4 reduction protocol employing LiAlH4 (0.150 g, 3.95 mmol) in THF (13 + 11 mL) to afford pure 5-(3-methoxyphenyl)-2-methylpentan-1-ol (0.285 g, 1.37 mmol, 53%) as a colorless oil; 1H NMR (300 MHz, CDCl3) δ 7.17–7.22 (m, 1H), 6.72–6.79 (m, 3H), 3.80 (s, 3H), 3.42–3.49 (m, 2H), 2.59 (td, J = 7.9, 2.7 Hz, 2H), 1.60–1.68 (m, 2H), 1.57 (s, 1H), 1.42–1.46 (m, 1H), 1.18–1.25 (m, 2H), 0.92 (d, J = 6.7 Hz, 3H). The intermediate alcohol (0.510 g, 2.45 mmol) is oxidized according to the Swern oxidation protocol employing (COCl)2 (1.50 mL of 2.0 M solution in DCM), DMSO (0.459 g, 5.87 mmol), TEA (0.99 g, 9.80 mmol) in DCM (2.5 + 1.5 mL). The crude product is purified by silica gel column chromatography eluting with 10% EtOAc:Hex to afford the pure aldehyde 24 (0.430 g, 2.08 mmol, 85%) as a colorless oil; 1H NMR (300 MHz, CDCl3) δ 9.60 (dd, J = 1.9, 0.8 Hz, 1H), 7.20 (t, J = 7.5 Hz, 1H), 6.72–6.78 (m, 3H), 3.80 (s, 3H), 2.61 (t, J = 7.3 Hz, 2H), 2.35 (dsex, J = 7.0, 1.9 Hz, 1H), 1.63–1.71 (m, 3H), 1.48–1.41 (m, 1H), 1.09 (d, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 204.9, 159.7, 143.6; 129.3, 120.8, 114.2, 111.1, 55.0, 46.2, 35.9, 30.0, 28.6, 13.3. HRMS (FAB+): calculated for C13H18O2 206.1307 [M], measured 206.1311 [M].
5-fluoro-2-(4-methoxyphenethyl)benzaldehyde (28)
Prepared according to the Sonogashira conditions from 2-bromo-5-fluorobenzaldehyde (1.40 g, 6.88 mmol), 4-ethynylanisole (1.0 g, 7.57 mmol), PdCl2(PPh3)2 (91 mg, 0.13 mmol, 2 mol %) and CuI (25 mg, 0.13 mmol, 2 mol %) in freshly distilled triethylamine (27 mL) to afford 5-fluoro-2-((4-methoxyphenyl)ethynyl)benzaldehyde as a white solid (1.63 g, 93%) following chromatography on silica gel, eluting with 10% Et2O:Hex; 1H NMR (CDCl3, 300 MHz) δ 3.84 (s, 3H); 6.90 (d, J = 8.7 Hz, 2H); 7.31-7.24 (m, 1H); 7.49 (d, J = 8.7 Hz, 2H); 7.63-7.58 (m, 2H); 10.58 (s, 1H). The intermediate alkyne (1.53 g, 6.00 mmol) was hydrogenated according to the hydrogenation protocol employing 10% by wt. Pd/C (300 mg, 20% by wt.) in EtOH (15 mL), followed by direct oxidation of the crude product alcohol according to the manganese dioxide oxidation protocol to afford aldehyde 28 (1.21 g, 78% over two steps) as a white solid following chromatography on silica gel, eluting with 5% Et2O:Hex; 1H NMR (CDCl3, 400 MHz) δ 2.82 (t, J = 8.4 Hz, 2H); 3.24 (t, J = 8.0 Hz, 2H); 3.76 (s, 3H); 6.80 (d, J = 8.4 Hz, 2H); 7.02 (d, J = 8.8 Hz, 2H); 7.17-7.15 (m, 2H); 7.49 (d, J = 8.4 Hz, 1H); 10.09 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 34.0, 37.7, 55.3, 113.9, 116.9 (d, J = 21.6 Hz), 120.9 (d, J = 21.1 Hz), 129.6, 132.7, 133.1 (d, J = 6.9 Hz), 135.3 (d, J = 5.5 Hz), 140.2, 158.2, 161.5 (d, J = 245.8 Hz), 190.5. HRMS (FAB+): calculated for C16H15FO2 258.1056 [M], measured 258.1054 [M].
4-fluoro-2-(4-methoxyphenethyl)benzaldehyde (30)
Prepared according to the Sonogashira conditions from methyl 2-bromo-4-fluorobenzoate (1.6 mL, 6.88 mmol), 4-ethynylanisole (1.0 g, 7.57 mmol), PdCl2(PPh3)2 (91 mg, 0.13 mmol, 2 mol %) and CuI (25 mg, 0.13 mmol, 2 mol %) in freshly distilled triethylamine (23 mL) to afford methyl 4-fluoro-2-((4-methoxyphenyl)ethynyl)benzoate as a yellow solid (1.81 g, 93%) following chromatography on silica gel, eluting with 20% Et2O:Hex; 1H NMR (CDCl3, 300 MHz) δ 3.81 (s, 3H); 3.94 (s, 3H); 6.88 (d, J = 9.0 Hz, 2H); 7.03 (dt, J = 7.8 Hz, J = 2.4 Hz, 1H); 7.30 (d, J = 2.4 Hz, 1H); 7.51 (d, J = 8.7 Hz, 2H); 7.98 (dd, J = 6.0 Hz, J = 3.0 Hz, 1H). The intermediate alkyne (1.81 g, 6.37 mmol) was hydrogenated according to the hydrogenation protocol employing 10% by wt. Pd/C (362 mg, 20% by wt.) in EtOH (22 mL) to afford methyl 4-fluoro-2-(4-methoxyphenylethyl)benzoate as a colorless oil (1.77 g, 96%); 1H NMR (CDCl3, 300 MHz) δ 2.84 (dd, J = 8.4 Hz, J = 5.7 Hz, 2H); 3.22 (dd, J = 8.4 Hz, J = 5.7 Hz, 2H); 3.79 (s, 3H); 3.89 (s, 3H); 6.83 (d, J = 8.4 Hz, 2H); 6.95-6.84 (m, 2H); 7.13 (d, J = 8.4 Hz, 2H); 7.94 (dd, J = 8.7 Hz, J = 6.0 Hz, 1H). The resulting ester (1.75 g, 6.06 mmol) is reduced according to the LiAlH4 reduction protocol employing LiAlH4 (345 mg, 9.09 mmol) in THF (36 + 24 mL) followed by direct oxidation of the crude product alcohol according to the manganese dioxide oxidation protocol to afford aldehyde 30 (1.19 g, 76% over two steps) as a white solid following chromatography on silica gel, eluting with 15% EtOAc:Hex; 1H NMR (CDCl3, 300 MHz) δ 2.83 (t, J = 8.1 Hz, 2H); 3.27 (t, J = 8.1 Hz, 2H); 3.75 (s, 3H); 6.80 (d, J = 8.1 Hz, 2H); 6.89 (d, J = 9.3 Hz, 1H); 7.09-6.99 (m, 3H); 7.81 (t, J = 7.8 Hz, 1H); 10.08 (s, 1H); 13C NMR (CDCl3, 75 MHz) δ 34.9, 37.0, 55.2, 113.9 (d, J = 21.5 Hz), 113.9, 118.0 (d, J = 21.4 Hz), 129.5, 130.5, 132.8, 135.1 (d, J = 9.9 Hz), 147.9 (d, J = 8.8 Hz), 158.2, 165.7 (d, J = 254.9 Hz), 190.5. HRMS (FAB+): calculated for C16H15FO2 258.1056 [M], measured 258.1049 [M].
4,5-difluoro-2-(4-methoxyphenethyl)benzaldehyde (32)
Prepared according to the Sonogashira conditions from methyl 2-bromo-4,5-difluorobenzoate (1.72 mL, 6.88 mmol), 4-ethynylanisole (1.0 g, 7.57 mmol), PdCl2(PPh3)2 (91 mg, 0.13 mmol, 2 mol %) and CuI (25 mg, 0.13 mmol, 2 mol %) in freshly distilled triethylamine (23 mL) to afford methyl 4,5-difluoro-2-((4-methoxyphenyl)ethynyl)benzoate as a yellow solid (1.68 g, 81%) following chromatography on silica gel, eluting with 8% EtOAc:Hex; 1H NMR (CDCl3, 300 MHz) δ 3.83 (s, 3H); 3.95 (s, 3H); 6.89 (d, J = 8.7 Hz, 2H); 7.40 (dd, J = 10.8 Hz, J = 7.8 Hz, 1H); 7.50 (d, J = 8.7 Hz, 2H); 7.81 (dd, J = 10.8 Hz, J = 8.4 Hz, 1H). The intermediate alkyne (1.68 g, 5.57 mmol) is hydrogenated according to the hydrogenation protocol employing 10% by wt. Pd/C (337 mg, 20% by wt.) in EtOH (19 mL) to afford methyl 4,5-difluoro-2-(4-methoxyphenethyl)benzoate as a white solid (1.70 g, 99%); 1H NMR (CDCl3, 300 MHz) δ 2.81 (dd, J = 8.4 Hz, J = 5.7 Hz, 2H); 3.20 (dd, J = 8.4 Hz, J = 5.7 Hz, 2H); 3.79 (s, 3H); 3.89 (s, 3H); 6.83 (d, J = 8.4 Hz, 2H); 6.96 (dd, J = 11.1 Hz, J = 7.8 Hz, 1H); 7.10 (d, J = 8.4 Hz, 2H); 7.77 (dd, J = 11.1 Hz, J = 8.1 Hz, 1H). The resulting ester (1.70 g, 5.57 mmol) is reduced according to the LiAlH4 reduction protocol employing LiAlH4 (320 mg, 8.36 mmol) in THF (33 + 22 mL) followed by direct oxidation of the crude product alcohol according to the manganese dioxide oxidation protocol to afford aldehyde 32 (842 mg, 55% over two steps) as a white solid following recrystallization from Et2O:Hex; 1H NMR (CDCl3, 300 MHz) δ 2.84 (t, J = 8.4 Hz, 2H); 3.24 (t, J = 8.1 Hz, 2H); 3.78 (s, 3H); 6.80 (d, J = 8.7 Hz, 2H); 7.04-6.97 (m, 3H); 7.64 (dd, J = 10.5 Hz, J = 8.4 Hz, 1H); 10.03 (d, J = 2.1 Hz, 1H); 13C NMR (CDCl3, 75 MHz) δ 33.9, 37.4, 55.4, 114.1, 119.6 (d, J = 17.3 Hz), 120.0 (d, J = 17.3 Hz), 129.6, 130.8, 132.2, 142.5, 149.8 (dd, J = 332.1 Hz, J = 13.1 Hz), 153.2 (dd, J = 340.5 Hz, J = 12.9 Hz), 158.4, 189.0. HRMS (FAB+): calculated for C16H14F2O2 276.0962 [M], measured 276.0949 [M].
1-(4-methoxyphenethyl)naphthalene-2-carbaldehyde (34)
Prepared according to the Sonogashira conditions from 1-bromonaphthalene-2-carboxaldehyde (1.35 g, 5.73 mmol), 4-ethynylanisole (832 mg, 6.30 mmol), PdCl2(PPh3)2 (97 mg, 0.14 mmol, 2.5 mol %) and CuI (27 mg, 0.14 mmol, 2.5 mol %) in freshly distilled triethylamine (23 mL) to afford 1-((4-methoxyphenyl)ethynyl)naphthalene-2-carboxaldehyde as a yellow solid (1.18 g, 72%) following chromatography on silica gel, eluting with 10% Et2O:Hex; 1H NMR (CDCl3, 300 MHz) δ 3.88 (s, 3H); 6.97 (dd, J = 6.9 Hz, J = 2.1 Hz, 2H); 7.69-7.62 (m, 4H); 7.91-7.84 (m, 2H); 7.98 (d, J = 8.7 Hz, 1H); 8.61 (dd, J = 6.3 Hz, J = 3.6 Hz, 1H); 10.89 (s, 1H). The intermediate alkyne (1.18 g, 4.12 mmol) was hydrogenated according to the hydrogenation protocol employing 10% by wt. Pd/C (236 mg, 20% by wt.) in EtOH (15 mL), followed by direct oxidation of the crude alcohol according to the manganese dioxide oxidation protocol to afford 34 (464 mg, 39%, across two steps) as a white solid following chromatography on silica gel, eluting with 15% Et2O:Hex; 1H NMR (CDCl3, 300 MHz) δ 2.99 (t, J = 6.3 Hz, 2H); 3.82-3.77 (m, 2H, eclipsed by singlet at 3.79); 3.79 (s, 3H); 6.85 (dd, J = 6.6 Hz, J = 1.8 Hz, 2H); 7.12 (d, J = 8.7 Hz, 2H); 7.65-7.62 (m, 2H); 7.81 (d, J = 8.7 Hz, 1H); 7.93-7.88 (m, 2H); 8.32-8.28 (m, 1H); 10.41 (s, 1H); 13C NMR (CDCl3, 75 MHz) δ 29.0, 37.2, 55.4, 114.1, 124.6, 125.0, 127.1, 127.4, 128.6, 129.1, 129.5, 131.0, 131.9, 132.0, 136.4, 143.6, 158.3, 191.6. HRMS (FAB+): calculated for C20H19O2+ 291.1380 [M+1], measured 291.1385 [M+1].
2-(2-(biphenyl-4-yl)ethyl)benzaldehyde (38)
To a flame dried flask equipped with a magnetic stir bar was added 95% NaH (288 mg, 12.0 mmol) followed by DMF (40 mL). The resulting suspension was cooled to 0 °C and triphenyl-(2-carbomethoxybenzyl)phosphonium bromide26 (5.88 g, 12 mmol) was added portionwise. Upon complete addition, the mixture was warmed to room temperature and stirred for 30 minutes. 4-Biphenyl carboxaldehyde (1.82 g, 10 mmol) was then added in one portion and the mixture was stirred at room temperature for 12 hr. Upon complete consumption of the aldehyde, the reaction mixture was poured into water (200 mL) and extracted with EtOAc. The combined organic layers were then washed twice with water and once with brine. The organic layer was dried over MgSO4 and the resulting suspension was filtered. The filtrate was concentrated in vacuo and the residue was then chromatographed on silica gel, eluting with 20% EtOAc:Hex, to afford (E)-methyl 2-(2-(biphenyl-4-yl)vinyl)benzoate as a white solid (3.14 g, 99%); 1H NMR (CDCl3, 400 MHz) δ 3.93 (s, 3H); 7.04 (d, J = 16.4 Hz, 1H); 7.36-7.29 (m, 2H); 7.44 (t, J = 7.2 Hz, 2H); 7.51 (t, J = 7.2 Hz, 1H); 7.63-7.58 (m, 6H); 7.73 (d, J = 8.0 Hz, 1H); 7.93 (d, J = 7.6 Hz, 1H); 8.04 (d, J = 16.4 Hz, 1H). The intermediate alkene (3.14 g, 9.99 mmol) was hydrogenated according to the hydrogenation protocol employing 10% by wt. Pd/C (640 mg, 20% by wt.) in EtOH (25 mL), followed by direct reduction of the crude product ester according to the LiAlH4 reduction protocol. The resulting crude alcohol was subjected to the manganese dioxide oxidation protocol to afford aldehyde 38 (1.74 g, 61% over three steps) as a white solid following chromatography on silica gel, eluting with 15% Et2O:Hex; 1H NMR (CDCl3, 300 MHz) δ 2.93 (dd, J = 8.4 Hz, J = 6.0 Hz, 2H); 3.34 (dd, J = 8.4 Hz, J = 5.7 Hz, 2H); 7.52-7.20 (m, 10H); 7.58 (d, J = 8.7 Hz, 2H); 7.82 (dd, J = 7.8 Hz, J = 1.2 Hz, 1H); 10.21 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 35.1, 38.0, 126.9, 127.1, 127.2, 127.2, 128.8, 129.1, 131.4, 132.9, 133.9, 139.2, 140.5, 141.1, 144.3, 192.6. HRMS (FAB+): calculated for C21H18O 286.1358 [M], measured 286.1367 [M].
2-(4-methoxy-2-methylphenethyl)benzaldehyde (40)
Prepared according to the hydrogenation protocol from alkyne 2-((4-methoxy-2-methylphenyl)ethynyl)benzaldehyde27 (1.51 g, 6.00 mmol) and 10 % by wt. Pd/C (302 mg, 20% by wt.) in EtOH (15 mL) followed by direct oxidation of the crude alcohol via the manganese dioxide oxidation protocol to afford aldehyde 40 (813 mg, 53% over two steps) following chromatography on silica gel, eluting with 8% Et2O:Hex; 1H NMR (CDCl3, 400 MHz) δ 2.25 (s, 3H); 2.84 (dd, J = 8.4 Hz, J = 6.0 Hz, 2H); 3.25 (dd, J = 8.4 Hz, J = 6.0 Hz, 2H); 3.77 (s, 3H); 6.70-6.65 (m, 2H); 7.01 (d, J = 8.4 Hz, 1H); 7.22 (d, J = 7.6 Hz, 1H); 7.38 (dt, J = 7.6 Hz, J = 1.2 Hz, 1H); 7.49 (dt, J = 7.6 Hz, J = 1.6 Hz, 1H); 7.82 (dd, J = 7.6 Hz, J = 1.6 Hz, 1H); 10.17 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 19.6, 33.9, 34.9, 55.3, 111.3, 115.9, 126.8, 130.3, 131.4, 131.6, 132.1, 133.9, 134.0, 137.4, 144.7, 158.1, 192.3. HRMS (FAB+): calculated for C17H18O2 254.1307 [M], measured 254.1317 [M].
2-(2-(6-methoxynaphthalen-2-yl)ethyl)benzaldehyde (42)
Prepared according to the Sonogashira conditions from 2-bromobenzaldehyde (1.17 mL, 10.0 mmol), 2-ethynyl-6-methoxynaphthalene (2 g, 11.0 mmol), PdCl2(PPh3)2 (140 mg, 0.20 mmol, 2 mol %) and CuI (38 mg, 0.20 mmol, 2 mol %) in freshly distilled triethylamine (40 mL) to afford 2-((6-methoxynaphthalen-2-yl)ethynyl)benzaldehyde as a white solid (1.90 g, 66%) following chromatography on silica gel, eluting with 20% Et2O:Hex; 1H NMR (CDCl3, 400 MHz) δ 3.92 (s, 3H); 7.11 (d, J = 2.0 Hz, 1H); 7.18 (dd, J = 9.2 Hz, J = 2.4 Hz, 1H); 7.43 (dt, J = 8.0 Hz, J = 0.8 Hz, 1H); 7.59-7.53 (m, 2H); 7.66 (d, J = 8.0 Hz, 1H); 7.71 (dd, J = 8.8 Hz, J = 2.4, 2H); 7.96 (d, J = 0.8 Hz, 1H); 8.00 (s, 1H); 10.70 (s, 1H). The intermediate alkyne (1.85 g, 6.74 mmol) was hydrogenated according to the hydrogenation protocol employing 10 % by wt. Pd/C (370 mg, 20% by wt.) in EtOH (17 mL) followed by direct oxidation of the crude alcohol the via manganese dioxide oxidation protocol to afford aldehyde 42 (1.40 g, 75% over two steps) following chromatography on silica gel, eluting with 15% Et2O:Hex; 1H NMR (CDCl3, 300 MHz) δ 3.01 (t, J = 8.4 Hz, 2H); 3.37 (t, J = 8.4 Hz, 2H); 3.89, (s, 3H); 7.13-7.10 (m, 2H); 7.22 (d, J = 7.2 Hz, 1H); 7.39-7.31 (m, 2H); 7.48-7.43 (m, 1H); 7.52 (s, 1H); 7.68-7.63 (m, 2H); 7.82 (dd, J = 7.5 Hz, J = 0.9, 1H); 10.22 (s, 1H); 13C NMR (CDCl3, 75 MHz) δ 35.1, 38.3, 55.4, 105.7, 118.8, 126.6, 126.8, 126.9, 127.9, 129.0, 129.1, 131.4, 132.7, 133.2, 133.9, 136.5, 144.4, 157.3, 192.6. HRMS (FAB+): calculated for C20H18O2 290.1307 [M], measured 290.1312 [M].
3-(4-methoxyphenyl)-2-tosyl-1,2,3,4-tetrahydroisoquinoline (2)
Purification by flash column chromatography on silica gel afforded 2 as a white solid, eluting with 15% EtOAc:Hex. 1H NMR (CDCl3, 500 MHz) δ 2.36 (s, 3H); 3.05-3.03 (m, 2H); 3.73 (s, 3H); 4.14 (d, J = 17 Hz, 1H); 4.68 (d, J = 16.5 Hz, 1H); 5.37 (bs, 1H); 6.72 (d, J = 8.5 Hz, 2H); 6.99-6.97 (m, 1H); 7.05-7.03 (m, 1H); 7.13-7.10 (m, 4H); 7.16 (d, J = 8.0 Hz, 2H); 7.63 (d, J = 8.0 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 21.6, 32.2, 43.9, 54.1, 55.3, 113.8, 126.0, 126.5, 127.1, 127.2, 128.6, 128.8, 129.6, 131.8, 132.5, 132.7, 137.4, 143.3, 158.9. HRMS (FAB+): calculated for C23H24NO3S 394.1471 [M+1], measured 394.1483 [M+1].
2-(4-methoxyphenyl)-5,5-dimethyl-1-tosylpiperidine (5)
Purification by flash column chromatography on silica gel afforded 5 as a white solid, eluting with 25% Et2O:Hex. 1H NMR (CDCl3, 300 MHz) δ 0.79 (s, 6H); 1.28-1.22 (bm, 2H); 2.10-2.03 (bm, 2H); 2.39 (s, 3H); 2.84 (d, J = 13.5 Hz, 1H); 3.37 (d, J = 13.2 Hz, 1H); 3.77 (s, 3H); 5.16 (s, 1H); 6.77 (d, J = 8.7 Hz, 2H); 7.07 (d, J = 8.4 Hz, 2H); 7.22 (d, J = 8.1 Hz, 2H); 7.65 (d, J = 8.1 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 21.6, 24.2, 25.7, 28.7, 30.4, 32.6, 52.5, 55.0, 55.3, 113.8, 127.1, 128.2, 129.5, 130.8, 138.7, 142.8, 158.4. HRMS (FAB+): calculated for C21H28NO3S+ 374.1784 [M+1], measured 374.1798 [M+1].
3-(4-methoxyphenyl)-2-tosyl-2-azaspiro[5.5]undecane (7)
Purification by flash column chromatography on silica gel afforded 7 as a white solid, eluting with 15% Et2O:Hex. 1H NMR (CDCl3, 400 MHz) δ 1.21-1.07 (m, 4H); 1.43-1.31 (m, 8H); 2.03-1.97 (m, 2H); 2.38 (s, 3H); 2.76 (d, J = 13.6 Hz, 1H); 3.69 (d, J = 13.6 Hz, 1H); 5.14 (t, J = 3.6 Hz, 1H); 6.76 (d, J = 8.8 Hz, 2H); 7.08 (d, J = 8.0 Hz, 2H); 7.20 (d, J = 8.0 Hz, 2H); 7.64 (d, J = 8.0 Hz, 2H); 13C NMR (CDCl3, 100 MHz) δ 21.5, 21.6, 25.0, 26.5, 30.9, 31.9, 32.7, 37.8, 50.0, 55.3, 113.8, 127.0, 128.2, 129.4, 130.9, 138.6, 142.7, 158.4. HRMS (FAB+): calculated for C24H32NO3S+ 414.2097 [M+1], measured 414.2092 [M+1].
2-(4-methoxyphenyl)-4,4-dimethyl-1-tosylpiperidine (9)
Purification by flash column chromatography on silica gel afforded 9 as a white solid, eluting with 15% Et2O:Hex. 1H NMR (CDCl3, 300 MHz) δ 0.58 (s, 3H); 0.80 (s, 3H); 1.25 (t, J = 3.6 Hz, 2H); 1.49 (dd, J = 14.1 Hz, J = 5.7 Hz, 1H); 1.91 (dd, J = 14.1 Hz, J = 4.8 Hz, 1H); 2.41 (s, 3H); 3.46-3.37 (m, 1H); 3.69-3.63 (m, 1H); 3.77 (s, 3H); 4.93 (t, J = 4.8 Hz, 1H); 6.78 (d, J = 8.4 Hz, 2H); 7.16 (d, J = 8.7 Hz, 2H); 7.24 (d, J = 8.1 Hz, 2H); 7.64 (d, J = 8.1 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 21.6, 27.3, 28.9, 31.1, 37.3, 40.2, 42.7, 55.3, 113.6, 127.2, 127.5, 129.5, 132.9, 138.0, 142.9, 158.3. HRMS (FAB+): calculated for C21H28NO3S+ 374.1784 [M+1], measured 374.1798 [M+1].
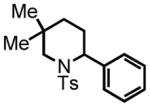
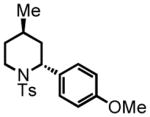
trans-2-(4-methoxyphenyl)-4-methyl-1-tosylpiperidine (11)
Purification by flash column chromatography on silica gel, eluting with 5 % EtOAc:Hex afforded 11 as a 93:7 trans:cis mixture. The major diastereomer was isolated by recrystallization of the mixture from diethyl ether with slow vapor diffusion of pentane. This material was used for characterization purposes. 1H NMR (300 MHz, CDCl3) δ 7.75 (d, J = 8.2 Hz, 2H), 7.29 (d, J = 8.0 Hz, 2H), 7.23 (d, J = 8.3 Hz, 2H), 6.84 (d, J = 8.2 Hz, 2H), 5.27 (d, J = 5.0 Hz, 1H), 3.86 (d, J = 13.9 Hz, 1H), 3.79 (s, 3H), 2.97 (ddd, J = 13.5, 13.5, 2.6 Hz, 1H), 2.43 (s, 3H), 2.14 (d, J = 13.8 Hz, 1H), 1.61-1.48 (m, 1H), 1.37 (d, J = 13.1 Hz, 1H), 1.25 (ddd, J = 12.6, 12.6, 5.4 Hz, 1H), 0.90 (ddd, J = 16.8, 12.6, 4.5 Hz, 1H), 0.79 (d, J = 6.4 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 158.8, 143.3, 139.2, 131.4, 130.0, 130.0, 128.4, 128.4, 127.4, 127.4, 114.3, 114.3, 55.7, 55.3, 42.8, 36.0, 33.3, 25.5, 22.6, 21.9. HRMS (FAB+): calculated for C20H26NO3S+ 360.1628 [M+1], measured 360.1630 [M+1]. X-ray structure was also obtained, see supporting information.
2-(4-methoxyphenyl)-1-tosylpiperidine (13)
Purification by flash column chromatography on silica gel afforded 13 as a colorless oil, eluting with 10% Et2O:Hex. 1H NMR (CDCl3, 300 MHz) δ 1.48-1.29 (m, 5H); 2.16 (bd, J = 12.6 Hz, 1H); 2.44 (s, 3H); 2.99 (dt, J = 14.7 Hz, J = 3.3 Hz, 1H); 3.83-3.77 (m, 4H); 5.21 (s, 1H); 6.86 (d, J = 8.7 Hz, 2H); 7.31-7.24 (m, 4H); 7.75 (d, J = 8.4 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 19.1, 21.7, 24.5, 27.4, 41.9, 55.0, 55.4, 114.1, 127.2, 128.4, 129.8, 131.0, 139.0, 143.0, 158.6. HRMS (FAB+): calculated for C19H24NO3S+ 346.1471 [M+1], measured 346.1482 [M+1].
Methyl 4-(4,4-dimethyl-1-tosylpiperidin-2-yl)phenylcarbamate (15)
Purification by flash column chromatography on silica gel afforded 15 as a white solid, eluting with 40% Et2O:Hex. 1H NMR (CDCl3, 300 MHz) δ 0.57 (s, 3H); 0.80 (s, 3H); 1.28-1.23 (m, 2H); 1.52 (dd, J = 10.5 Hz, J = 4.2 Hz, 1H); 1.92 (dd, J = 10.5 Hz, J = 3.3 Hz, 1H); 2.42 (s, 3H); 3.42-3.35 (m, 1H); 3.68 (dt, J = 10.5 Hz, J = 3.3 Hz, 1H); 3.77 (s, 3H); 4.96 (t, J = 3.6 Hz, 1H); 6.56 (bs, 1H); 7.19 (d, J = 6.6 Hz, 2H); 7.29-7.25 (m, 4H); 7.65 (d, J = 6.3 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 21.6, 27.2, 29.0, 31.3, 37.3, 40.2, 42.6, 52.4, 55.4, 118.6, 127.1, 127.3, 129.6, 136.2, 136.5, 138.0, 143.1, 154.1. HRMS (FAB+): calculated for C22H29N2O4S+ 417.1843 [M+1], measured 417.1849 [M+1].
5,5-dimethyl-2-phenyl-1-tosylpiperidine (17)
Purification by flash column chromatography on silica gel afforded 17 as a golden oil, eluting with 5% Et2O:Hex. 1H NMR (CDCl3, 300 MHz) δ 0.80 (s, 6H); 1.26-1.21 (m, 2H); 2.12-2.08 (m, 2H); 2.40 (s, 3H); 2.86 (d, J = 13.5 Hz, 1H); 3.41 (d, J = 13.5 Hz, 1H); 5.23 (bs, 1H); 7.27-7.14 (m, 7H); 7.65 (d, J = 8.4 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 21.6, 24.2, 25.8, 28.8, 30.5, 32.6, 52.7, 55.5, 126.8, 127.1, 127.2, 128.6, 129.5, 138.7, 139.0, 142.9. HRMS (FAB+): calculated for C20H25NO2S 343.1606 [M], measured 343.1610 [M].
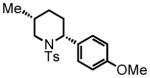
cis-2-(4-methoxyphenyl)-5-methyl-1-tosylpiperidine (19)
Purification by flash column chromatography on silica gel, eluting with 10 % EtOAc:Hex afforded 19 as a 92:8 cis:trans mixture. The major diastereomer was isolated by recrystallization of the mixture from diethyl ether with slow vapor diffusion of pentane. This material was used for characterization purposes. 1H NMR (300 MHz, CDCl3) δ 7.75 (d, J = 8.4 Hz, 2H), 7.28 (d, J = 8.2 Hz, 2H), 7.23 (d, J = 8.7 Hz, 2H), 6.85 (d, J = 8.8 Hz, 2H), 5.21 (d, J = 4.6 Hz, 1H), 3.80 (s, 3H), 3.79-3.75 (m, 1H), 2.53 (dd, J = 14.3, 11.7 Hz, 1H), 2.43 (s, 3H), 2.18 (ddd, J = 14.0, 5.2, 3.1 Hz, 1H), 1.64 (dddd, J = 14.0, 14.0, 5.4, 3.7 Hz, 1H), 1.50-1.37 (m, 2H), 1.07 (ddd, J = 13.5, 12.0, 3.3 Hz, 1H), 0.70 (d, J = 6.3 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 158.9, 143.3, 139.2, 131.0, 130.0, 130.0, 128.5, 128.5, 127.4, 127.4, 114.3, 114.3, 55.6, 54.3, 48.5, 30.3, 28.1, 19.3. 27.5, 21.9, HRMS (FAB+): calculated for C20H26NO3S+ 360.1628 [M+1], measured 360.1637 [M+1]. X-ray structure was also obtained, see supporting information.
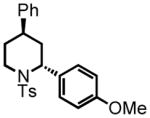
trans-2-(4-methoxyphenyl)-4-phenyl-1-tosylpiperidine (21)
Purification by flash column chromatography on silica gel afforded 21 as a white solid, eluting with 10% EtOAc:Hex. 1H NMR (300 MHz, CDCl3) δ 7.82 (d, J = 8.3 Hz, 2H), 7.34 (d, J = 7.9 Hz, 2H), 7.29 (d, J = 9.0 Hz, 2H), 7.25 (t, J = 7.0 Hz, 2H), 7.18 (ddd, J = 7.3, 1.3, 1.3 Hz, 1H), 6.97 (d, J = 7.8 Hz, 2H), 6.89 (d, J = 8.8 Hz, 2H), 5.40 (d, J = 4.6 Hz, 1H), 4.00 (d, J = 14.2 Hz, 1H), 3.81 (s, 3H), 3.12 (ddd, J = 14.7, 12.9, 3.1 Hz, 1H), 2.71 (ddd, J = 12.6, 3.4, 3.4 Hz, 1H), 2.46 (s, 3H), 2.35 (d, J = 13.9 Hz, 1H), 1.79 (ddd, J = 13.4, 13.4, 5.3 Hz, 1H), 1.57-1.55 (m, 1H), 1.45 (ddd, J = 17.3, 12.8, 4.5 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 159.0, 145.6, 143.6, 139.2, 130.8, 130.2, 130.2, 129.0, 129.0, 128.4, 128.4, 127.5, 127.5, 127.0, 127.0, 126.9, 114.6, 114.6, 55.7, 55.4, 42.1, 36.8, 34.6, 32.2, 21.9. HRMS (FAB+): calculated for C25H28NO3S+ 422.1784 [M+1], measured 422.1790 [M+1].
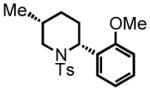
cis-2-(2-methoxyphenyl)-5-methyl-1-tosylpiperidine (major diastereomer cis-23)
Purification by flash column chromatography on silica gel, eluting with 10 % EtOAc:Hex afforded 23 as a 73:27 trans:cis mixture. The major diastereomer cis-23 was isolated by recrystallization of the mixture from diethyl ether with slow vapor diffusion of pentane. This material was used for characterization purposes. 1H NMR (300 MHz, CDCl3) δ 7.61 (d, J = 7.6 Hz, 2H), 7.20-7.06 (m, 4H), 6.81 (d, J = 8.3 Hz, 1H), 6.74 (t, J = 7.5 Hz, 1H), 5.50 (d, J = 5.8 Hz, 1H), 3.95 (dd, J = 13.3, 4.5 Hz, 1H), 3.79 (s, 3H), 2.96 (dd, J = 13.2, 11.7 Hz, 1H), 2.38 (s, 3H), 2.17-2.12 (m, 1H), 1.75-1.64 (m, 2H), 1.55-1-.42 (m, 2H), 0.95-0.91 (m, 1H), 0.83 (d, J = 6.4 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 156.6, 143.0, 138.7, 129.9, 129.7, 129.7, 128.2, 128.1, 127.3, 127.3, 120.3, 110.9, 55.5, 51.7, 50.3, 30.8, 29.7, 28.1, 21.8, 19.5. HRMS (FAB+): calculated for C20H26NO3S+ 360.1628 [M+1], measured 360.1647 [M+1].
trans-2-(2-methoxyphenyl)-5-methyl-1-tosylpiperidine (minor diastereomer trans -23)
Purification by flash column chromatography on silica gel afforded trans-23 as a white solid, eluting with 10% EtOAc:Hex. The minor diasteromer was isolated after several silica gel chromatography columns. This material was used for characterization purposes. 1H NMR (300 MHz, CDCl3) δ 7.41 (d, J = 8.3 Hz, 2H), 7.27 (d, J = 8.3 Hz, 1H), 7.12 (m, 3H), 6.80 (ddd, J = 7.7, 7.7, 1.0 Hz, 1H), 6.68 (d, J = 8.5 Hz, 1H), 4.75 (dd, J = 7.9, 4.6 Hz, 1H), 3.88 (dd, J = 12.5, 4.2 Hz, 1H), 3.70 (s, 3H), 2.84 (dd, J = 12.7, 7.7 Hz, 1H), 2.37 (s, 3H), 1.98-1.95 (m, 2H), 1.85-1.83 (m, 1H), 1.73-1.68 (m, 1H), 1.10-1.07 (m, 1H), 0.94 (d, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 156.1, 142.4, 136.4, 129.3, 129.1, 128.9, 128.9, 128.1, 127.5, 127.5, 119.9, 110.0, 55.0, 54.6, 52.1, 29.5, 29.5, 29.5, 21.5, 18.9. HRMS (FAB+): calculated for C20H26NO3S+ 360.1628 [M+1], measured 360.1637 [M+1].
(Z)-1-methoxy-8-methyl-6,7-dihydro-5H-benzo[7]annulene (25)
Purification by flash column chromatography on silica gel afforded 25, eluting with 5% EtOAc:Hex. 1H NMR (300 MHz, CDCl3) δ 7.01 (d, J = 8.3 Hz, 1H), 6.69-6.63 (m, 2H), 6.21 (s, 1H), 3.79 (s, 3H), 2.78-2.75 (m, 2H), 2.30 (t, J = 6.8 Hz, 2H), 1.98-1.94 (m, 2H), 1.91 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 157.6, 142.7, 137.7; 131.5, 129.6, 125.4, 114.6, 110.7, 55.2, 36.6, 35.8, 27.1, 27.0. HRMS (FAB+): calculated for C13H17O+ 189.1274 [M+1], measured 189.1290 [M+1].
7-fluoro-3-(4-methoxyphenyl)-2-tosyl-1,2,3,4-tetrahydroisoquinoline (29)
Purification by flash column chromatography on silica gel afforded 29 as a white solid, eluting with 15% EtOAc:Hex. 1H NMR (CDCl3, 400 MHz) δ 2.34 (s, 3H); 2.99 (bd, J = 4.0 Hz, 2H); 3.71 (s, 3H); 4.08 (d, J = 16.8 Hz, 1H); 4.66 (d, J = 16.8 Hz, 1H); 5.35 (t, J = 4.4 Hz, 1H); 6.68 (dd, J = 8.8 Hz, J = 2.4 Hz, 1H); 6.72 (dd, J = 6.8 Hz, J = 2.0 Hz, 2H); 6.82 (dt, J = 8.8 Hz, J = 2.8 Hz, 1H); 7.00 (dd, J = 8.4 Hz, J = 6.0 Hz, 1H); 7.09 (d, J = 8.4 Hz, 2H); 7.17 (d, J = 8.0 Hz, 2H); 7.63 (d, J = 8.4 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 21.5, 31.2, 43.7, 54.0, 55.3, 122.6 (d, J = 21.7 Hz), 113.8, 114.3 (d, J = 21.3 Hz), 127.1, 128.3, 128.5, 129.6, 130.2 (d, J = 7.8 Hz), 131.3, 134.2 (d, J = 7.0 Hz), 137.3, 143.4, 159.0, 161.2 (d, J = 243.7 Hz). HRMS (FAB+): calculated for C23H23FNO3S+ 412.1377 [M+1], measured 412.1375 [M+1].
6-fluoro-3-(4-methoxyphenyl)-2-tosyl-1,2,3,4-tetrahydroisoquinoline (31)
Purification by flash column chromatography on silica gel afforded 31 as a white solid, eluting with 20% EtOAc:Hex. 1H NMR (CDCl3, 300 MHz) δ 2.35 (s, 3H); 3.00 (s, 2H); 3.71 (s, 3H); 4.09 (d, J = 16.5 Hz, 1H); 4.66 (d, J = 16.5 Hz, 1H); 5.35 (t, J = 3.9 Hz, 1H); 6.73 (d, J = 8.4 Hz, 2H); 6.84-6.77 (m, 2H); 6.96-6.92 (m, 1H); 7.10 (d, J = 8.7 Hz, 2H); 7.16 (d, J = 8.1 Hz, 2H); 7.63 (d, J = 8.1 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 21.5, 32.1, 43.3, 53.6, 55.2, 113.6 (d, J = 21.1 Hz), 113.8, 115.2 (d, J = 21.7 Hz), 127.1, 127.4 (d, J = 8.2 Hz), 128.4, 129.4, 129.6, 131.3, 135.0 (d, J = 7.7 Hz), 137.3, 143.3, 159.0, 161.6 (d, J = 243.9 Hz). HRMS (FAB+): calculated for C23H23FNO3S+ 412.1377 [M+1], measured 412.1375 [M+1].
6,7-fluoro-3-(4-methoxyphenyl)-2-tosyl-1,2,3,4-tetrahydroisoquinoline (33)
Purification by flash column chromatography on silica gel afforded 33 as a white solid, eluting with 10% EtOAc:Hex. 1H NMR (CDCl3, 300 MHz) δ 2.38 (s, 3H); 2.97 (bd, J = 3.6 Hz, 2H); 3.74 (s, 3H); 4.00 (d, J = 16.8 Hz, 1H); 4.62 (d, J = 16.8 Hz, 1H); 5.35 (t, J = 4.2 Hz, 1H); 6.74 (d, J = 8.4 Hz, 2H); 6.90-6.78 (m, 2H); 7.08 (d, J = 8.4 Hz, 2H); 7.20 (d, J = 8.1 Hz, 2H); 7.63 (d, J = 8.1 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 21.5, 31.1, 43.0, 53.5, 55.3, 114.0, 114.7 (d, J = 17.8 Hz), 117.3 (d, J = 17.0 Hz), 127.2, 128.4, 128.8, 129.3, 129.7, 130.9, 137.3, 143.6, 148.8 (dd, J = 244.7 Hz, J = 11.5 Hz), 149.4 (dd, J = 243.0, Hz, J = 9.4 Hz), 159.1. HRMS (FAB+): calculated for C23H21F2NO3S 429.1210 [M], measured 429.1214 [M].
3-(4-methoxyphenyl)-2-tosyl-1,2,3,4-tetrahydrobenzo[f]isoquinoline (35)
Purification by flash column chromatography on silica gel afforded 35 as a white solid, eluting with 30% Et2O:Hex. 1H NMR (CDCl3, 300 MHz) δ 2.25 (s, 3H); 3.20 (dd, J = 17.1 Hz, J = 6.3 Hz, 1H); 3.57 (d, J = 17.1 Hz, 1H); 3.67 (s, 3H); 4.18 (d, J = 17.4 Hz, 1H); 4.81 (d, J = 17.7 Hz, 1H); 5.61 (d, J = 6.3 Hz, 1H); 6.66 (d, J = 8.4 Hz, 2H); 7.11-7.03 (m, 5H); 7.53-7.44 (m, 2H); 7.67-7.62 (m, 3H); 7.87-7.88 (m, 2H); 13C NMR (CDCl3, 75 MHz) δ 21.5, 27.1, 43.6, 53.3, 55.3, 113.8, 122.5, 124.2, 125.7, 126.6, 126.9, 127.1, 127.6, 128.8, 129.6, 131.0, 131.8, 132.7, 137.7, 143.3, 159.0. HRMS (FAB+): calculated for C27H26NO3S+ 444.1628 [M+1], measured 444.1616 [M+1].
3-(biphenyl-4-yl)-2-tosyl-1,2,3,4-tetrahydroisoquinoline (39)
Purification by flash column chromatography on silica gel afforded 39 as a white solid, eluting with 20% Et2O:Hex. 1H NMR (CDCl3, 300 MHz) δ 2.36 (s, 3H); 3.10 (d, J = 4.2 Hz, 2H); 4.25 (d, J = 16.5 Hz, 1H); 4.72 (d, J = 16.5 Hz, 1H); 5.43 (t, J = 4.5 Hz, 1H); 7.03-7.00 (m, 1H); 7.08-7.05 (m, 1H); 7.18-7.13 (m, 4H); 7.28-7.25 (m, 2H); 7.33 (d, J = 7.2 Hz, 1H); 7.44-7.38 (m, 4H); 7.51 (d, J = 6.9 Hz, 2H); 7.65 (d, J = 8.1 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 21.6, 32.6, 44.3, 54.7, 126.0, 126.6, 127.1, 127.2, 127.3, 127.4, 127.8, 128.8, 128.9, 129.7, 132.6, 132.7, 137.4, 139.0, 140.4, 140.8, 143.3. HRMS (FAB+): calculated for C28H26NO2S+ 440.1679 [M+1], measured 440.1680 [M+1]. X-ray structure was also obtained, see supporting information.
3-(4-methoxy-2-methylphenyl)-2-tosyl-1,2,3,4-tetrahydroisoquinoline (41)
Purification by flash column chromatography on silica gel afforded 41 as a white solid, eluting with 20% EtOAc:Hex. The product was isolated as a 3:1 mixture of rotomers. Elevated temperature 1H NMR failed to result in coalescence of the signals. For the purposes of spectral characterization the major and minor rotomers designated as a and b, respectively. 1H NMR (CDCl3, 300 MHz) δ 2.19 (s, 0.91Hb); 2.30 (s, 3Ha); 2.41 (s, 0.97Hb); 2.50 (s, 3Ha); 2.85-2.77 (m, 1Ha + 0.44Hb); 3.07-3.01 (m, 1Ha + 0.67Hb); 3.17 (dd, J = 11.1 Hz, J = 4.8 Hz, 0.32Hb); 3.53 (s, 0.96Hb); 3.72 (s, 3Ha); 4.13 (d, J = 12.0 Hz, 0.32Hb); 4.21 (d, J = 12.3 Hz, 1Ha); 4.63 (d, J = 12.6 Hz, 1Ha); 4.76 (d, J = 11.7 Hz, 0.30Hb); 5.43-5.41 (m, 1Ha + 0.31Hb); 6.04 (d, J = 1.5 Hz, 0.29Hb); 6.44 (dd, J = 6.3 Hz, J = 2.1 Hz, 0.98Hb); 6.51 (d, J = 1.8 Hz, 0.35Hb); 6.70 (d, J = 2.1 Hz, 1Ha); 6.77 (d, J = 6.6 Hz, 1Ha); 6.93-6.91 (m, 1H + 0.39Hb); 7.19-7.00 (m, 6Ha + 1.5Hb); 7.29 (d, J = 6.0 Hz, 0.69Hb); 7.53 (d, J = 6.0 Hz, 2Ha); 7.78 (d, J = 6.0 Hz, 0.63Hb); 13C NMR (CDCl3, 75 MHz) δ18.6, 20.0, 21.5, 32.4, 41.7, 45.0, 50.4, 52.4, 52.7, 55.2, 105.1, 110.8, 116.4, 116.9, 125.5, 125.9, 126.5, 126.6, 126.8, 127.3, 127.4, 127.7, 127.8, 128.1, 128.3, 128.7, 129.2, 129.9, 131.2, 131.6, 133.2, 133.4, 134.8, 136.0, 136.8, 137.7, 138.0, 140.2, 143.1, 143.5, 158.8, 159.5. HRMS (FAB+): calculated for C24H26NO3S+ 408.1628 [M+1], measured 408.1652 [M+1].
3-(6-methoxynaphthalen-2-yl)-2-tosyl-1,2,3,4-tetrahydroisoquinoline (43)
Purification by flash column chromatography on silica gel afforded 43 as a white solid, eluting with 20% Et2O:Hex. 1H NMR (CDCl3, 300 MHz) δ 2.33 (s, 3H); 3.19-3.06 (m, 2H); 3.88 (s, 3H); 4.19 (d, J = 16.5 Hz, 1H); 4.73 (d, J = 16.8 Hz, 1H); 5.53-5.50 (m, 1H); 6.99-6.96 (m, 1H); 7.15-7.05 (m, 7H); 7.36 (dd, J = 8.7 Hz, J = 1.8 Hz, 1H); 7.44, (s, 1H); 7.47 (d, J = 16.2 Hz, 1H); 7.60 (d, J = 8.7 Hz, 1H); 7.65 (d, J = 8.4 Hz, 2H); 13C NMR (CDCl3, 100 MHz) δ 21.6, 32.1, 44.2, 54.8, 55.4, 105.6, 118.9, 126.0, 126.1, 126.5, 127.2, 128.5, 128.8, 129.6, 132.5, 132.7, 133.9, 134.9, 137.4, 143.3, 157.9. HRMS (FAB+): calculated for C27H25NO3S 443.1555 [M], measured 443.1557 [M].
3-(4-methoxyphenyl)-1,2,3,4-tetrahydroisoquinoline (44)
Napthalene (0.274 g, 2.14 mmol) was dissolved in dry DME (1.5 M) and sodium (0.039 g, 1.69 mmol) was added in portions. The mixture was stirred at room temperature for one hour. It was then added dropwise to a solution of the tosylamine 2 (0.060 g, 0.152 mmol) in DME (0.09 M) at −78 °C until a green color persisted. Saturated NaHCO3 was added and the mixture was extracted with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4 and concentrated under reduce pressure. The resulting residue was purified by silica gel column chromatography (80 % EtOAc:MeOH) to afford 44 (0.039 g, 0.135 mmol, 89%) as pale yellow oil. 1H NMR (300 MHz, CDCl3) δ 7.36 (d, J = 8.4 Hz, 2H), 7.16-7.10 (m, 4H), 6.91 (d, J = 8.7 Hz, 2H), 3.97 (t, J = 7.2 Hz, 1H), 3.82 (s, 3H), 2.96 (d, J = 7.1 Hz, 2H), 1.99 (brs, 1H); 13C NMR (75 MHz, CDCl3) δ 159.0, 136.3, 135.0, 134.9, 129.2, 127.8, 127.8, 126.4, 126.3, 114.1, 114.1, 58.0, 55.4, 49.3, 37.7, 29.8. HRMS (FAB+): calculated for C16H17NO 239.1310 [M], measured 239.1315 [M].
cis-2-(4-methoxyphenyl)-5-methylpiperidine (45)
Napthalene (0.464 g, 3.62 mmol) was dissolved in dry DME (1.5 M) and sodium (0.065 g, 2.83 mmol) was added in portions. The mixture was stirred at room temperature for one hour. It was added dropwise to a solution of the tosylamine 19 (0.093 g, 0.260 mmol) in DME (0.09 M) at −78 °C until a green color persisted. Saturated NaHCO3 was added and the mixture was extracted with EtOAc. The combined organic layers were extracted with HCl 10%. The resulting aqueous layers were combined and washed with ethyl acetate. The aqueous layer was then neutralized with NaOH 1N and subsequently extracted with EtOAc. The organic layers were dried over Na2SO4 and concentrated under reduced pressure to afford 45 (0.046 g, 0.226 mmol, 87%) as a pale yellow oil that was pure by NMR analysis (mixture of diasteromers 90:10 cis:trans). 1H NMR (300 MHz, CDCl3) δ 7.30 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.6 Hz, 2H), 3.79 (s, 3H), 3.58 (d, J = 9.2 Hz, 1H), 2.98 (dd, J = 11.7, 3.3 Hz, 1H), 2.84 (d, J = 11.7 Hz, 1H), 1.79-1.73 (m, 4H), 1.58 (d, J = 10.1 Hz, 2H), 1.14 (d, J = 6.9 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 158.9, 137.9, 128.1, 128.1, 114.1, 114.1, 61.3, 55.6, 52.7, 31.1, 29.8, 28.1, 17.6. HRMS (FAB+): calculated for C13H20NO+ 206.1539 [M+1], measured 206.1544 [M+1].
Chiral substrate (S)-5-(4-methoxyphenyl)-2-methylpentanal (S5)
To a suspension of (4S)-4-benzyl-oxazolidin-2-one (S1) (0.131 g, 0.739 mmol), DMAP (0.012 g, 0.098 mmol) and 5-(4-methoxyphenyl)pentanoic acid23a (0.20 g, 0.96 mmol) in DCM (0.75 M in oxazolidinone) at 0 °C under an argon atmosphere, is added DCC (0.200 g, 1.00 mmol) in one portion. The reaction is warmed to room temperature after 10 min and stirring is continued until no starting material was detected by TLC. The suspension is filtered through celite and the precipitate is washed with DCM. The filtrate is washed with sat. NaHCO3, dried over Na2SO4 and concentrated at reduced pressure. The resulting residue is purified by silica gel chromatography, eluting with 20% EtOAc:Hex to afford (S)-4-benzyl-3-(5-(4-methoxyphenyl)pentanoyl)oxazolidin-2-one (S2) (0.243 g, 0.662 mmol, 69%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.36-7.33 (m, 3H), 7.23 (d, J = 7.1 Hz, 2H), 7.14 (d, 8.7 Hz, 2H), 6.86 (d, J = 8.3 Hz, 2H), 4.68 (oct, J = 8.3 Hz, 1H), 4.19-4.18 (m, 2H), 3.81 (s, 3H), 3.31 (dd, J = 13.4, 3.1 Hz, 1H), 3.01-2.98 (m, 2H), 2.79 (dd, J = 13.4, 9.6 Hz, 1H), 2.65 (t, J = 7.2 Hz, 2H), 1.76-1.74 (m, 4H); 13C NMR (75 MHz, CDCl3) δ 173.2, 157.8, 153.5, 135.4, 134.2, 129.5, 129.5, 129.3, 129.3, 129.0, 129.0, 127.3, 114.8, 113.8, 113.8 66.2, 55.3, 55.1, 37.9, 35.4, 34.7, 31.1, 23.9. To a stirred solution of N,N-diisopropylamine (0.275, 2.72 mmol) in anhydrous THF (0.4 M) at −78°C was added a 2.5 M solution of n-BuLi in hexanes (1.8 mL). After stirring for 15 min at −78°C, S2 (0.833 g, 2.26 mmol) in THF (0.8 M) was added dropwise. The mixture was stirred for 30 min after which methyl iodide (1.29 g, 0.09 mmol) was added. The reaction mixture was stirred overnight at room temperature. The reaction was then quenched with 0.1 M aqueous HCl solution and extracted four times with Et2O. The extract was washed once with brine, dried over Na2SO4 and concentrated under reduced pressure to furnish the crude product, which was purified by silica gel chromatography, eluting with 15% EtOAc:Hex to afford (S)-4-benzyl-3-((S)-5-(4-methoxyphenyl)-2-methylpentanoyl)oxazolidin-2-one (S3) (0.632 g, 1.66 mmol, 73%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.34-7.24 (m, 3H), 7.20 (d, J = 7.5 Hz, 2H), 7.08 (d, J = 8.5 Hz, 2H), 6.81 (d, J = 8.6 Hz, 2H), 4.68-4.59 (m, 1H), 4.15-4.13 (m, 2H), 3.77 (s, 3H), 3.74 (q, J = 6.7 Hz, 1H), 3.24 (dd, J = 13.4, 3.1 Hz, 1H), 2.76 (dd, J = 13.4, 9.6 Hz, 1H), 2.65 (dd, J = 12.9, 7.1 Hz, 2H), 1.83-1.76 (m, 1H), 1.66-1.56 (m, 2H), 1.47-1.45 (m, 1H), 1.21 (d, J = 6.7 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 177.5, 158.2, 153.4, 135.7, 134.7, 129.8, 129.8, 129.7, 129.3, 129.3, 127.7, 114.1, 114.1, 66.4, 55.7, 55.6, 38.3, 38.0, 35.3, 33.4, 29.6, 17.9; HRMS (FAB+): calculated for C23H28NO4+ 382.2013 [M+1], measured 382.2029 [M+1]. (S)-4-benzyl-3-((S)-5-(4-methoxyphenyl)-2-methylpentanoyl)oxazolidin-2-one (S3) (0.60 g, 1.57 mmol) was dissolved in dry THF (0.3 M in ester) under an argon atmosphere and cooled to 0 °C. EtOH was added (101 uL), followed by LiBH4 (0.041 g, 1.88 mmol). The mixture was then stirred overnight. Following the disappearence of the starting material as detected by TLC, EtOAc was added followed by the careful addition of saturated NH4Cl. The aqueous layer was extratcted with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure to yield (S)-5-(4-methoxyphenyl)-2-methylpentan-1-ol (S4) (0.274 g, 1.31 mmol) as a colorless oil following purification by silica gel column chromatography, eluting with 20% EtOAc:Hex. 1H NMR (300 MHz, CDCl3) δ 7.09 (d, J = 8.4 Hz, 2H), 6.82 (d, J = 8.5 Hz, 2H), 3.78 (s, 3H), 3.45 (s, 2H), 2.55 (ddd, J = 8.8, 8.8, 2.6 Hz, 2H), 1.63-1.50 (m, 3H), 1.44-1.32 (m, 1H), 1.22 (bs, 1H), 1.15-1.03 (m, 1H), 0.91 (d, J = 6.7 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 158.1, 135.2, 129.6, 129.6, 114.1, 114.1, 68.6, 55.6, 36.1, 35.7, 33.2, 29.5, 17.0. Oxidation was carried out according to the Swern oxidation protocol from (S)-5-(4-methoxyphenyl)-2-methylpentan-1-ol (S4) (0.439 g, 2.11 mmol) and (COCl)2 (1.26 mL of 2.0 M solution in DCM), DMSO (0.395 g, 5.06 mmol), TEA (0.853 g, 8.44 mmol) in DCM (2 + 4 mL). The crude reaction residue was purified by silica gel column chromatography 5% EtOAc:Hex to afford the pure (S)-5-(4-methoxyphenyl)-2-methylpentanal (S5) (0.305 g, 1.49 mmol, 70%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 9.58 (d, J = 1.9 Hz, 1H), 7.07 (d, J = 8.5 Hz, 2H), 6.82 (d, J = 8.5 Hz, 2H), 3.77 (s, 3H), 2.56 (t, J = 7.3 Hz, 2H), 2.33 (dsex, J = 6.9; 1.9 Hz, 1H), 1.76-1.57 (m, 3H), 1.43-1.34 (m, 1H), 1.08 (d, J = 7.1 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 205.5, 158.2, 134.4; 129.7, 129.6, 114.2, 114.2, 55.6, 46.6, 35.3, 30.4, 29.3, 13.7; HRMS (FAB+): calculated for C13H19O2+ 207.1380 [M+1], measured 207.1390 [M+1].
(S)-2-(methoxymethyl)-N-((S)-5-(4-methoxyphenyl)-2-methylpentylidene)pyrrolidin-1-amine (S6)
The enantiomeric excess of aldehyde S5 was determined by conversion of the aldehyde to the 1-amino-2-(methoxymethyl)pyrrolidine (SAMP) imine. SAMP (0.054g, 0.415 mmol) was added to S5 (0.103 g, 0.500 mmol) at 0 °C in the absence of solvent. After 1 hour the reaction mixture was dissolved in DCM, dried over Na2SO4 and concentrated under reduced pressure. The resulting residue was analyzed by 1H NMR, which indicated a diasteromeric ratio of >99:1. The residue was purified by silica gel column chromatography eluting with 20% EtOAc:Hex to afford (S)-2-(methoxymethyl)-N-((S)-5-(4-methoxyphenyl)-2-methylpentylidene)pyrrolidin-1-amine (S6) (0.137 g, 0.430 mmol, 86 %) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 7.08 (d, J = 7.1 Hz, 2H), 6.81 (d, J = 8.7 Hz, 2H), 6.47 (d, J = 6.5 Hz, 1H), 3.77 (s, 3H), 3.57 (ddd, J = 8.9, 3.5, 1.7 Hz, 1H), 3.44-3.32 (m, 6H), 2.69 (quintet, J = 8.7 Hz, 1H), 2.54 (t, J = 7.0 Hz, 2H), 2.33 (septet, J = 6.8 Hz, 1H), 1.93-1.88 (m, 4H), 1.61 (quintet, J = 7.6 Hz, 2H), 1.45-1.37 (m, 2H), 1.03 (d, J = 6.9 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 157.6, 144.2, 144.2, 134.8, 129.3, 129.3, 113.7, 113.7, 74.8, 63.6, 59.2, 55.2, 50.5, 37.0, 35.1, 35.1, 29.3, 22.1, 19.1; HRMS (FAB+): calculated for C19H31N2O2+ 319.2386 [M+1], measured 319.2379 [M+1].
Supplementary Material
Table 4.
Synthesis of 3-Aryl-1,2,3,4-tetrahydroisoquinolines via HT-aminationa
| entry | substrate | product | conditions/yield |
|---|---|---|---|
| 1 |
 3 |
 2 |
80 °C, 2 hr 87% |
| 2 |
 26 |
 27 |
80 °C, 2 hr 75% |
| 3 |
 28 |
 29 |
80 °C, 6 hr 88% |
| 4 |
 30 |
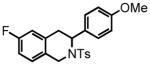 31 |
80 °C, 6 hr 88% |
| 5 |
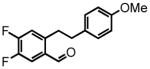 32 |
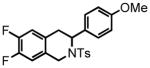 33 |
80 °C, 6 hr 88% |
| 6 |
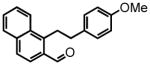 34 |
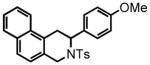 35 |
80 °C, 5 hr 85% |
| 7b |
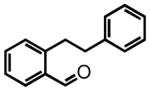 36 |
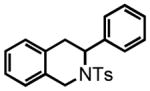 37 |
120 °C, 72 hr trace |
| 8b |
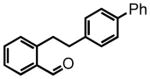 38 |
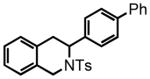 39 |
120 °C, 72 hr 46% |
| 9 |
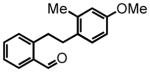 40 |
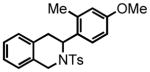 41 |
80 °C, 3 hr 80% |
| 10 |
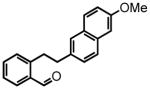 42 |
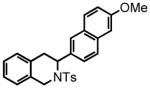 43 |
80 °C, 24 hr 71% |
Reactions performed at 0.05 M in dry DCE, with 2 eq. TsNH2, 3 eq. BF3•OEt2 and 4Å MS 200% by wt.
Reactions run in a sealed vial.
Acknowledgments
This work was supported by the National Institute of General Medical Sciences (GM60326). We would also like to thank the Parkin research group for X-ray crystal analysis. The National Science Foundation (CHE-0619638) is thanked for acquisition of an X-ray diffractometer. Authors Paul A. Vadola and Igna-cio Carrera contributed equally to this work.
Footnotes
Supporting Information Available. 1H and 13C NMR spectra of HT-amination substrates and products, X-ray data for compounds 11, 19 and 39 and discussion of the reactivity of chiral substrates. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Godula K, Sames D. Science. 2006;312:67. doi: 10.1126/science.1114731. [DOI] [PubMed] [Google Scholar]
- 2.Pastine SJ, Gribkov DV, Sames D. J Am Chem Soc. 2006;128:14220–14221. doi: 10.1021/ja064481j.Gribkov DV, Pastine SJ, Schnurch M, Sames D. J Am Chem Soc. 2007;129:11750–11755. doi: 10.1021/ja072577n.Review on transition metal-catalyzed α-functionalization of amines: Campos KR. Chem Soc Rev. 2007;36:1069–1084. doi: 10.1039/b607547a.
- 3.For examples of oxidative a-arylation of cyclic amines: Baslé O, Li C-J. Org Lett. 2008;10:3661–3663. doi: 10.1021/ol8012588.and references therein.
- 4.During the preparation of this manuscript a report concerning the HT-amination of aromatic aldehydes was published in Organic Letters: Mori K, Kawasaki T, Akiyama T. Org Lett. 2012;14:1436–1439. doi: 10.1021/ol300180w.
- 5.(a) Horton DA, Bourne GT, Smythe ML. Chem Rev. 2003;103:893–930. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]; (b) Scott JD, Williams RM. Chem Rev. 2002;102:1669–1730. doi: 10.1021/cr010212u. [DOI] [PubMed] [Google Scholar]; (c) Stevenson GI, Huscroft I, MacLeod AM, Swain CJ, Cascier MA, Chicci GC, Grahm MI, Harrison T, Kelleher FJ, Kurtz M, Ladduwahetty T, Merchant KJ, Metzger JM, Macintyre DE, Sadowski S, Sohal B, Owens AP. J Med Chem. 1998;41:4623–4635. doi: 10.1021/jm980376b. [DOI] [PubMed] [Google Scholar]
- 6.(a) Pastine SJ, McQuaid KM, Sames D. J Am Chem Soc. 2005;127:12180–12181. doi: 10.1021/ja053337f. [DOI] [PubMed] [Google Scholar]; (b) Pastine SJ, Sames D. Org Lett. 2005;7:5429–5431. doi: 10.1021/ol0522283. [DOI] [PubMed] [Google Scholar]; (c) McQuaid KM, Sames D. J Am Chem Soc. 2009;131:402–403. doi: 10.1021/ja806068h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) McQuaid KM, Long JZ, Sames D. Org Lett. 2009;11:2971–2975. doi: 10.1021/ol900915p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Vadola PA, Sames D. J Am Chem Soc. 2009;131:16525–16528. doi: 10.1021/ja906480w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Examples of Asymmetric HT-cyclization Muraka S, Deb Indubhusan D, Zhang C, Seidel D. J Am Chem Soc. 2009;131:13236–13227. doi: 10.1021/ja905213f.Kang YK, Kim SM, Kim DY. J Am Chem Soc. 2010;132:11847–11849. doi: 10.1021/ja103786c.Cao W, Liu X, Wang W, Lin L, Feng X. Org Lett. 2011;13:600–603. doi: 10.1021/ol1028282.Zhou G, Liu F, Zhang J. Chem Eur J. 2011;17:3101–3104. doi: 10.1002/chem.201100019.Mori K, Ehara K, Kurihara K, Akiyama T. J Am Chem Soc. 2011;133:6166–6169. doi: 10.1021/ja2014955.
- 8.Recent examples of HT-cyclization of tertiary amines: Matyus P, Elias O, Tapolcsanyi P, Polonka-Balint A, Halasz-Dajka H. Synthesis. 2006:2625–2639.Ruble JC, Hurd AR, Johnson TA, Sherry DA, Barbachyn MR, Toogood PL, Bundy GL, Graber DR, Kamilar GM. J Am Chem Soc. 2009;131:3991–3997. doi: 10.1021/ja808014h.Murarka S, Zhang C, Konieczynska MD, Seidel D. Org Lett. 2009;11:129–132. doi: 10.1021/ol802519r.Zhou G, Zhang J. Chem Commun. 2010;46:6593–6595. doi: 10.1039/c0cc01946a.Haibach MC, Deb I, Kanta De C, Seidel D. J Am Chem Soc. 2011;133:2100–2103. doi: 10.1021/ja110713k.
- 9.Examples of HT-cyclization induced by alkyne activation: Bajracharya GB, Pahadi NK, Gridnev ID, Yamamoto Y. J Org Chem. 2006;71:6204–6210. doi: 10.1021/jo060951g.Barluenga J, Fañanás-Mastral M, Aznar F, Valdés C. Angew Chem Int Ed. 2008;47:6594–6597. doi: 10.1002/anie.200802268.Shikanai D, Murase H, Hata T, Urabe H. J Am Chem Soc. 2009;131:3166–3167. doi: 10.1021/ja809826a.Yang S, Li Z, Jian X, He C. Angew Chem Int Ed. 2009;48:3999–4001. doi: 10.1002/anie.200900368.Tobisu M, Hiromi N, Chatani N. J Org Chem. 2009;74:5471–5475. doi: 10.1021/jo901045g.Jurberg ID, Odabachian Y, Gagosz F. J Am Chem Soc. 2010;132:3543–3552. doi: 10.1021/ja9100134.
- 10.Example of HT-cyclization with ketenimine hydride acceptors: Alajarin M, Bonillo B, Ortin M-M, Sanchez-Andrada P, Vidal A, Orenes R-A. Org Biomol Chem. 2010;8:4690–4700. doi: 10.1039/c0ob00193g.
- 11.Preparation of carbocycles using HT-cyclization: Mahoney SJ, Moon DT, Hollinger J, Fillion E. Tet Lett. 2009;50:4706–4709.Jin T, Himuro M, Yamamoto Y. J Am Chem Soc. 2010;132:5590–5591. doi: 10.1021/ja101633x.Mori K, Sueoka S, Akiyama T. J Am Chem Soc. 2011;133:2424–2426. doi: 10.1021/ja110520p.
- 12.Example of preparation of benzopyrans using HT-cyclization: Mori K, Kawasaki T, Sueoka S, Akiyama T. Org Lett. 2010;12:1732–1735. doi: 10.1021/ol100316k.
- 13.Verboom W, Hamzink MRJ, Reinhoudt DN, Visser R. Tetrahedron Lett. 1984;25:4309–4312. [Google Scholar]
- 14.(a) Zhang C, Murarka S, Seidel D. J Org Chem. 2009;74:419–422. doi: 10.1021/jo802325x. [DOI] [PubMed] [Google Scholar]; (b) Mori K, Ohshima Y, Ehara K, Akiyama T. Chem Lett. 2009;38:524–525. [Google Scholar]
- 15.(a) Wölfing J, Frank É, Schneider G, Tietze LF. Angew Chem Int Ed. 1999;38:200–201. [Google Scholar]; (b) Wölfing J, Frank É, Schneider G, Tietze LF. Eur J Org Chem. 1999:3013–3020. [Google Scholar]; (c) Wölfing J, Frank É, Schneider G, Tietze LF. Eur J Org Chem. 2004:90–100. [Google Scholar]
- 16.For the HT-amination of hydrazones and oximes see: Frank É, Schneider G, Kádár Z, Wölfing J. Eur J Org Chem. 2009:3544–3553.
- 17.(a) McKay WR, Proctor GR. J Chem Soc, Perkin Trans. 1981;1:2435–2442. [Google Scholar]; (b) Duguet N, Campbell CD, Slawin AMZ, Smith AD. Org Biomol Chem. 2008;6:1108– 1113. doi: 10.1039/b800857b. [DOI] [PubMed] [Google Scholar]
- 18.The 63:37 trans:cis ratio of isomers of product 11 was obtained by concentrating a single fraction from the column chromatography of 11 which contained both diastereomers. The same procedure was used to obtain the 72:28 cis:trans ratio of product 19.
- 19.(a) Paulsen H, Todt K. Angew Chem Int Ed. 1966;5:899–900. [Google Scholar]; (b) Johnson RA. J Org Chem. 1968;33:3627–3632. [Google Scholar]; (c) Watson PS, Jiang B, Scott B. Org Lett. 2000;2:3679–3681. doi: 10.1021/ol006589o. [DOI] [PubMed] [Google Scholar]; (d) Seel S, Thaler T, Takatsu K, Zhang C, Zipset H, Straub BF, Mayer P, Knochel P. J Am Chem Soc. 2011;133:4774–4777. doi: 10.1021/ja201008e. [DOI] [PubMed] [Google Scholar]
- 20.(a) Chrzanowska M, Rozwadowska MD. Chem Rev. 2004;104:3341–3370. doi: 10.1021/cr030692k. [DOI] [PubMed] [Google Scholar]; (b) Li CJ. Acc Chem Res. 2009;42:335–344. doi: 10.1021/ar800164n. [DOI] [PubMed] [Google Scholar]
- 21.(a) Vicario JL, Badia D, Carillo L, Anakabe E. Tetrahedron: Asymmetry. 2003;14:347. [Google Scholar]; (b) Allen CP, Benkovics T, Turek AK, Yoon TP. J Am Chem Soc. 2009;131:12560–12561. doi: 10.1021/ja906183g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ueno S, Ohtsubo M, Kuwano R. J Am Chem Soc. 2009;131:12904–12905. doi: 10.1021/ja905988e. [DOI] [PubMed] [Google Scholar]
- 22.Wuts PGM, Greene TW. Greene’s Protecting Groups in Organic Synthesis. 4. John Wiley & Sons, Inc; Hoboken, New Jersey: 2007. pp. 855–859. [Google Scholar]
- 23.(a) McCalmont WF, Patterson JR, Lindenmuth MA, Heady TN, Haverstick DM, Gray LS, Macdonald TL. Bioorg Med Chem. 2005;13:3821–3839. doi: 10.1016/j.bmc.2005.03.004. [DOI] [PubMed] [Google Scholar]; (b) Kirmse W, Konrad W, Özkir IS. Tetrahedron. 1997;53:9935–9964. [Google Scholar]
- 24.Mahoney SJ, Moon DT, Hollinger J, Fillion E. Tetrahedron Lett. 2009;50:4706–4709. [Google Scholar]
- 25.Brace NO. J Org Chem. 1964;29:1247–1249. [Google Scholar]
- 26.Laufer SA, Ahrens GM, Karcher SC, Hering JS, Niess R. J Med Chem. 2006;49:7912–7915. doi: 10.1021/jm061072p. [DOI] [PubMed] [Google Scholar]
- 27.Alfonsi M, Dell’Acqua M, Facoetti D, Arcadi A, Abbiati G, Rossi E. Eur J Org Chem. 2009;17:2852–2862. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.