

Abstract
Prion propagation involves a templating reaction in which the infectious form of the prion protein (PrPSc) binds to the cellular form (PrPC), generating additional molecules of PrPSc. While several regions of the PrPC molecule have been suggested to play a role in PrPSc formation based on in vitro studies, the contribution of these regions in vivo is unclear. Here, we report that mice expressing PrP deleted for a short, polybasic region at the N terminus (residues 23–31) display a dramatically reduced susceptibility to prion infection and accumulate greatly reduced levels of PrPSc. These results, in combination with biochemical data, demonstrate that residues 23–31 represent a critical site on PrPC that binds to PrPSc and is essential for efficient prion propagation. It may be possible to specifically target this region for treatment of prion diseases as well as other neurodegenerative disorders due to β-sheet-rich oligomers that bind to PrPC.
Introduction
Prion diseases are a class of neurodegenerative disorders caused by the conversion of the cellular form of the prion protein (PrPC), a normal cellular glycoprotein, into PrPSc, a conformationally altered isoform that is infectious (Prusiner, 1998; Collinge, 2001; Weissmann, 2004). Although the three-dimensional structure of PrPSc has not been determined, it is known to have a high content of β-sheets and to be aggregated and protease resistant. PrPSc is thought to propagate by directly interacting with PrPC molecules, triggering their conformational conversion into PrPSc. Direct support for the protein-only model of prion propagation comes from experiments in which PrPSc binds to and converts PrPC substrate molecules into an infectious form in cell-free systems (Kocisko et al., 1994; DebBurman et al., 1997; Horiuchi and Caughey, 1999).
Although it is clear that prion propagation involves a physical interaction between PrPC and PrPSc, the molecular details of the process remain uncertain. Identifying the molecular interfaces that are important in the PrPC–PrPSc conversion process is essential, not only for understanding how this conformational conversion occurs but also for developing small molecules that can interfere with the process for therapeutic purposes.
Several kinds of studies have shed light on regions of the PrPC molecule that are essential for PrPSc formation or that may be involved in binding to PrPSc. Two of these domains (residues 98–110 and 136–158) lie within regions of the protein that are thought to undergo conformational changes during formation of PrPSc and that are therefore likely to form part of the core of the PrPSc structure (Peretz et al., 1997; Morrissey and Shakhnovich, 1999; White et al., 2003; Moroncini et al., 2004; Norstrom and Mastrianni, 2006; Solforosi et al., 2007). Surprisingly, an N-terminal domain (residues 23–31) that has been implicated in the conversion process lies outside the protease-resistant core of PrPSc. This 9 aa region (KKRPKPGGW), encompassing a series of positively charged residues immediately following the N-terminal signal peptide, is of great interest from a cellular and functional standpoint, since it is has been implicated in endocytic trafficking, binding to glycosaminoglycans, and lipid bilayer interactions (Shyng et al., 1995; Pan et al., 2002; Warner et al., 2002; Sunyach et al., 2003; Wadia et al., 2008; Pasupuleti et al., 2009; Taubner et al., 2010).
To directly explore the role of the N-terminal, polybasic domain in the formation of PrPSc, we created transgenic (Tg) mice expressing PrP deleted for residues 23–31. These mice display a dramatically reduced susceptibility to prion infection and accumulate greatly reduced levels of PrPSc in their brains. We demonstrate that residues 23–31 represent a critical site on PrPC that interacts with PrPSc. This information leads to predictions about the nature of the PrPC–PrPSc interface and identifies a novel target site for therapeutic agents that may inhibit formation of PrPSc. Moreover, since the N terminus of PrPC has recently been reported to mediate binding and neurotoxicity of other β-rich oligomers, including those composed of the Alzheimer's Aβ peptide (Chen et al., 2010; Resenberger et al., 2011), our results have relevance to several neurodegenerative disorders due to protein aggregation and misfolding.
Materials and Methods
Generation of Tg mice.
The generation of Tg(Δ23–31) mice has been described previously (Turnbaugh et al., 2011). Transgenic founders were initially crossed to C57BL/6J × CBA hybrid mice before breeding with Prn-p0/0 mice on a pure, C57BL/6J background [European Mouse Mutant Archive (EMMA)]. All Tg(Δ23–31) mice were hemizygous for the transgene array. Both Tga20+/+ and Tga20+/0 mice (Fischer et al., 1996) were used, as noted. Tga20+/0 mice were generated by breeding Tga20+/+ mice to Prn-p0/0 mice (EMMA).
Mice were genotyped by PCR analysis of tail DNA prepared using the Puregene DNA Isolation Kit (Gentra Systems). Primers P1 and P4 (Chiesa et al., 1998) were used to detect the presence of the Δ23–31 and Tga20+/+ transgenes (Fischer et al., 1996). The Prn-p allele was recognized by primers E2 [referred to as P2 in the study by Chiesa et al. (1998)] and E4 (Li et al., 2007), and the Prn-p0/0 allele was amplified with primers E2 and K4 (GTGAGATGACAGGAGATCCTGCC).
Scrapie inoculation.
A stock of RML scrapie (from Rocky Mountain Laboratory) was prepared by passaging in CD1 mice. Fifty microliters of 1% brain homogenate in PBS from terminally ill CD1 mice was used to intracerebrally inoculate each 4- to 6-week-old recipient mouse of either sex.
Western blotting.
Brain samples (10%, w/v) were homogenized in PBS using plastic pestles (South Jersey Precision Tool and Mold), and membranes were solubilized in 0.5% NP-40/0.5% Na-deoxycholate (DOC), pH 7. Protein levels were quantitated with the BCA kit (Pierce). To deglycosylate PrP, samples containing 20 μg of total protein were treated with N-glycosidase F (PNGase F) (New England Biolabs) according to the manufacturer's guidelines. For proteinase K (PK) treatment, 100 μg of total protein was digested with 20 μg/ml PK (unless otherwise noted) for 30 min at 37°C. Undigested samples were loaded at one-fifth of the amount of PK-treated samples. Samples were subjected to Western blotting using anti-PrP antibody 6D11 (R. Kascsak, New York State Institute for Basic Research in Developmental Disabilities, Staten Island, NY) followed by goat anti-mouse IgG (Pierce), or anti-PrP antibody D18 followed by mouse anti-human IgG (Southern Biotechnology Associates). Blots were developed with ECL Plus (GE Healthcare) and were imaged on the Bio-Rad Chemidoc XRS system.
Immunofluorescence staining.
BHK cells grown in poly-d-lysine (PDL)-coated eight-well chamber slides (BD Biosciences) were transiently transfected with 0.25 μg of DNA and 0.75 μg of Lipofetamine 2000 (Invitrogen) per well. At 24 h after transfection, cells were washed twice with PBS, fixed in 4% paraformaldehyde for 10 min, permeabilized with 0.2% Triton X-100, and blocked in 2% goat serum in PBS. Cells were then stained with 3F4 monoclonal antibody and FLAG polyclonal antibody (Sigma-Aldrich) in blocking solution, followed by incubation with fluorescently conjugated secondary antibodies (Invitrogen), along with DAPI to stain cell nuclei. Cells were viewed on a Nikon Eclipse TE2000-E inverted epifluorescence microscope.
Isolation of detergent-resistant microdomains.
HEK293 cells stably expressing either wild-type (WT) or Δ23–31 PrP were resuspended in lysis buffer containing 25 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5 mm EDTA, 1% Triton X-100, plus protease inhibitors (Roche). Cell lysates were mixed with 60% OptiPrep (Sigma-Aldrich) to achieve a final concentration of 40% Optiprep and placed at the bottom of an ultracentrifuge tube. Thirty percent and 0% OptiPrep layers were made by mixing 60% OptiPrep with lysis buffer (above) and were then added on top of the 40% OptiPrep layer. The gradients were then centrifuged at 99,000 × g for 4 h at 4°C. Fractions were collected, and proteins methanol-precipitated, resuspended in loading buffer, and run on a 12% SDS-PAGE gels. Gels were Western blotted, after which blots were probed with 6D11 antibody, developed with ECL Plus (GE Healthcare), and exposed to autoradiography film. Membranes were then stripped and reprobed with an antibody to flotillin-1 (BD Biosciences).
Detergent insolubility assay.
Ten percent brain homogenates in PBS were diluted in a buffer containing 10 mm Tris-HCl, pH 7.4, 0.5% DOC, 0.5% TX-100, and 150 mm NaCl, supplemented with protease inhibitors (Roche), and protein concentration was determined using the BCA kit (Pierce). A total of 100 μg of total protein was further diluted to a final concentration of 0.5 μg/μl in 400 μl of 0.5% DOC/0.5% NP-40 plus protease inhibitors. Samples were mixed for 20 min at 4°C, and then centrifuged for 5 min at 10,000 × g at 4°C to pellet any unsolubilized debris. The clarified supernatant was then ultracentrifuged at 186,000 × g at 4°C for 1 h. Proteins in the resulting supernatant were methanol-precipitated, and both the pellet (insoluble) and supernatant (soluble) fractions were immunoblotted for PrP.
Endocytosis assay.
N2a cells were grown in PDL-coated, eight-well chamber slides (BD Biosciences) and were transiently transfected with 0.25 μg of DNA [pCDNA3.1(+)Hygro vector, or vector encoding WT (3F4) or Δ23–31 (3F4) PrPs] plus 0.75 μg of Lipofetamine 2000 (Invitrogen) per well. At 24 h after transfection, cells were surface stained with 3F4 antibody on ice, and then incubated in OptiMem in the presence of 250 μm CuSO4 (Pauly and Harris, 1998) at 37°C for 30 min to initiate endocytosis. Cells were then incubated in the presence or absence of phosphatidylinositol-specific phospholipase C (PIPLC) (0.5 U/ml) for 2 h at 37°C before being fixed, permeabilized with 0.5% Triton X-100 in PBS, and incubated with a fluorescently tagged secondary antibody before DAPI staining.
Scrapie-infected neuroblastoma cells.
Chronically infected ScN2a.3 cells (a subclone of N2a cells that has been infected with RML) were transiently transfected with pCDNA3.1+(Hygro) vector, or with vector encoding WT (3F4) or Δ23–31 (3F4) PrPs plus Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). At 48 h after transfection, cells were lysed in 0.5% NP-40/0.5% DOC, pH 7. Proteins in 20% of the cell lysate were methanol-precipitated and boiled in sample buffer. The remaining 80% of the lysate was digested with 20 μg/ml PK for 40 min at 37°C. The reaction was stopped by the addition of 2 mm PMSF, followed by ultracentrifugation at 186,000 × g at 4°C for 1 h. The resulting pellet was boiled in sample buffer in the presence of PMSF, and then all samples were subjected to Western blotting. Blots were probed with 3F4 antibody (1:5000) followed by goat anti-mouse IgG (Pierce), and were developed with ECL (GE Healthcare) before exposure to autoradiography film.
Histology and immunohistochemistry.
After killing by CO2, brains were immediately immersion-fixed in 4% paraformaldehyde at 4°C for 24 h. Brains were then rinsed with PBS, dehydrated, and embedded in paraffin. Immunohistochemistry for PrPSc was performed on 4 μm deparaffinized sections according to a previous protocol (Bell et al., 1997). Sections were subjected to a three-step pretreatment procedure, including hydrated autoclaving at 121°C for 10 min, incubation in 98% formic acid for 5 min at room temperature, and incubation in 4 m guanidine thiocyanate at 4°C for 2 h. After blocking in 5% goat serum for 30 min at room temperature, monoclonal antibody 6H4 (Prionics; 1:10,000 in 5% goat serum) was applied overnight at 4°C. An EnVision+ Dual Link System-HRP (DAB+) kit (Dako) was used to visualize the primary antibody. Analysis of histological and immunohistochemical specimens was performed on a Nikon Eclipse TE2000-E inverted microscope.
PMCA reactions.
Aliquots of 10% brain homogenate from infected mice were serially diluted into 10% normal brain homogenates and loaded into 0.2 ml PCR tubes. Tubes were positioned on an adaptor placed on the plate holder of a microsonicator (Misonix model 3000) that was programmed to perform cycles of 30 min incubation at 37°C followed by a 20 s pulse of sonication at a power setting of 9. During amplification, samples were immersed in the water of the sonicator bath without shaking. Nonamplified samples were kept frozen and not subjected to sonication.
To detect PK-resistant PrPSc, samples were incubated in the presence of PK (20 μg/ml) for 60 min at 56°C with constant shaking at 450 rpm. The digestion was stopped by adding NuPAGE LDS sample buffer (Invitrogen), and proteins were fractionated on an SDS-PAGE gel, electroblotted onto a PVDF membrane (Millipore), and probed with D18 antibody followed by a mouse anti-human IgG antibody (Southern Biotechnology Associates). Immunoreactive bands were visualized by incubation with West Dura (Pierce) and imaged using a Chemidoc imaging system (Bio-Rad).
PrPC–PrPSc binding assay.
The binding assay was performed as described previously (Miller et al., 2011). Briefly, 5% brain homogenate was made in Tris-buffered saline (50 mm Tris-HCl, 200 mm NaCl, pH 7.5), vortexed briefly, sonicated, and clarified by centrifugation at 470 × g for 15 min at 4°C. A volume of 12.4 μl of clarified RML brain homogenate was diluted into 250 μl of binding buffer (50 mm Tris, 200 mm NaCl, 1% Triton X-100, 1% Tween 20, pH 7.5) in siliconized 1.5 ml tubes (Thermo Fisher Scientific). A total of 3.5 μg of recombinant Δ23–28 or WT PrP was added, and samples were incubated for 1 h at 4°C with end-over-end rotation. Protein A beads (Invitrogen) linked to anti-myc antibody 9E10 (Santa Cruz Biotechnology) were incubated with the PrPC–PrPSc complex for 3 h at 4°C, collected with a magnetic separator, and rinsed with binding buffer. Ten percent of the bound sample was kept as an undigested control. The remainder was resuspended in 25 μg/ml PK in 0.5% DOC/0.5% NP-40, transferred to clean tubes, and digested for 30 min at 37°C. Proteins were eluted with sample buffer, and analyzed by Western blotting using anti-PrP antibody D18.
Results
Δ23–31 PrP is posttranslationally processed and localized like WT PrP
To investigate the role of the N-terminal polybasic region in PrPSc conversion, we generated a mutant PrP deleted for amino acids 23–31 in the mouse PrP sequence (Fig. 1A). To confirm that Δ23–31 PrP is correctly processed and localized, this molecule was expressed in several different cell lines, and its biochemical properties and subcellular distribution were characterized.
Figure 1.

Schematic of Δ23–31 PrP, and expression in transfected cells. A, PrP residues 23–31, shown in WT PrP, were deleted to make Δ23–31 PrP. SP, N-terminal signal peptide; OR, octapeptide repeats; GPI, C-terminal GPI addition signal. B, Vector (lanes 1, 4), WT PrP (lanes 2, 5), or Δ23–31 PrP (lanes 3, 6) were expressed by transient transfection of N2a cells. Both PrP constructs carried an epitope tag for monoclonal antibody 3F4. Equal amounts of total protein were either left untreated (lanes 1–3) or were treated with PNGase F (lanes 4–6) before blotting with 3F4 antibody. Molecular size markers are shown in kilodaltons. C, WT PrP (FLAG-tagged) and Δ23–31 PrP (3F4-tagged) were coexpressed in BHK cells. Staining for FLAG (red) (i) and 3F4 (green) (ii) completely overlap in the merged image (yellow) (iii). DAPI staining of the nucleus is shown in blue. Scale bar, 25 μm.
When expressed in N2a neuroblastoma cells, Δ23–31 PrP showed a glycosylation pattern similar to that of WT PrP, including unglycosylated, monoglycosylated, and diglycosylated forms (Fig. 1B, lanes 2, 3). Treatment with PNGase F caused these species to collapse into a single band corresponding to the unglycosylated form, which was ∼1 kDa smaller for the deleted protein (Fig. 1B, lanes 5, 6). We noticed that, in transfected N2a cells, as well as in other cell lines, the Δ23–31 mutant was generally expressed at higher levels than WT PrP (Fig. 1B, compare lanes 3, 6, with lanes 2, 5). This difference is likely due to a lower rate of internalization for Δ23–31 PrP (see below), which has been shown to correlate with an extended half-life for other N-terminal deletion mutants (Nunziante et al., 2003).
To analyze the localization patterns of Δ23–31 and WT PrPs within the same cell, BHK cells were cotransfected with plasmids encoding FLAG-tagged WT PrP and 3F4-tagged Δ23–31 PrP (Fig. 1Ci,ii). Using antibodies that selectively recognize the two proteins, we found that their subcellular distribution patterns completely overlapped (Fig. 1Ciii). Additional experiments in BHK cells demonstrated that, like WT, Δ23–31 PrP is primarily localized to the plasma membrane, where it is tethered by a PIPLC-cleavable glycosylphosphatidylinositol (GPI) anchor (Turnbaugh et al., 2011).
PrPC has been shown to localize in lipid rafts (Campana et al., 2005; Taylor and Hooper, 2006). To determine whether Δ23–31 PrP is also present in lipid rafts, lysates of HEK293 cells stably expressing Δ23–31 or WT PrP were fractionated by centrifugation in OptiPrep density gradients. Both WT and Δ23–31 PrP were enriched at the interface between the 0 and 30% fractions, which corresponds to lipid raft domains, as demonstrated by the presence of the marker protein, flotillin-1 (Fig. 2A, lanes 3, 4). Low levels of both WT and Δ23–31 PrP were also detected in fractions at the bottom of the OptiPrep gradient (Fig. 2A, lanes 10–12), presumably representing a subpopulation of molecules that was not completely solubilized.
Figure 2.

Δ23–31 PrP is localized to detergent-resistant microdomains, is defective in endocytosis, and is converted into PrPSc in cultured cells. A, Cell lysates from HEK293 cells stably expressing either WT or Δ23–31 PrP were subjected to density gradient ultracentrifugation in an OptiPrep gradient. Fractions were analyzed by Western blotting using antibodies for PrP (6D11) and flotillin-1, a raft-resident protein. The majority of both flotillin-1 and PrP were found at the interface of the 0 and 30% OptiPrep fractions (lanes 2–4). A small amount of PrP, presumably derived from unlysed cells, can be found in the bottom fractions of the gradient (lanes 10–12). Molecular weight markers are shown in kilodaltons. B, N2a cells were transiently transfected with vector (i, iv), or with vector encoding WT PrP (ii, v) or Δ23–31 PrP (iii, vi). Cells were surface-stained with PrP antibody 3F4 on ice, and then incubated at 37°C to initiate endocytosis. Subsequently, cells were incubated in the absence (i–iii) or presence (iv–vi) of PIPLC. Cells were then fixed, permeabilized, and incubated with a fluorescently tagged secondary antibody (green) before DAPI staining (blue). Staining is observed for both WT and Δ23–31 PrP without PIPLC treatment (ii, iii), while only WT PrP (v), but not Δ23–31 PrP (vi), shows a punctate pattern of intracellular staining after PIPLC treatment. Scale bar, 25 μm. C, Chronically infected ScN2a cells were transiently transfected with vector, or with vector encoding WT (3F4) PrP or Δ23–31 (3F4) PrP. Cell lysates were incubated without (lanes 1–3) or with (lanes 4–6) PK before Western blotting with 3F4 antibody to detect transfected PrP. Both WT PrP (lane 5) and Δ23–31 PrP (lane 6) are converted into PK-resistant forms.
Collectively, these results show that Δ23–31 and WT PrPs have indistinguishable localization patterns in cultured cells and that the mutant protein, like its WT counterpart, is glycosylated and transits the secretory pathway to the cell surface, where it is GPI-anchored in lipid rafts.
Δ23–31 PrP displays decreased endocytosis
The N-terminal domain of PrPC has been previously identified as important for PrPC endocytosis via clathrin-coated pits (Shyng et al., 1995; Sunyach et al., 2003).
We therefore tested whether Δ23–31 PrP was defective in endocytosis. N2a cells expressing either WT or Δ23–31 PrP were incubated on ice with an anti-PrP antibody, and then warmed to 37°C to initiate endocytosis. After this, cells were incubated in the absence (Fig. 2Bi–iii) or presence (Fig. 2Biv–vi) of PIPLC, which cleaves off any PrP remaining on the cell surface. Cells were then fixed, permeabilized, and incubated with a fluorescently labeled secondary antibody. Both WT and Δ23–31 PrP-expressing cells showed staining in the absence of PIPLC (Fig. 2Bii,iii). After PIPLC treatment, cells expressing WT PrP displayed a punctate pattern of intracellular staining, corresponding to endocytic structures containing PrP (Fig. 2Bv). In contrast, Δ23–31 PrP cells lacked these structures (Fig. 2Bvi). These results indicate that Δ23–31 PrP is defective in endocytosis, confirming previous evidence regarding the role of residues 23–31 in this process.
Δ23–31 PrP converts into Δ23–31 PrPSc in cultured cells
To test the conversion capability of Δ23–31 PrP in cell culture, scrapie-infected N2a cells were transiently transfected with plasmids encoding epitope-tagged WT or Δ23–31 PrP. Once confluent, cells were collected, a portion of the cell lysate was digested with PK, and both digested and undigested lysates were analyzed by Western blotting using 3F4 antibody to detect epitope-tagged PrP. Δ23–31 PrP was converted into PK-resistant forms that comigrated with those generated from WT PrP (Fig. 2C, lanes 5, 6), indicating that deletion of the polybasic domain does not abolish the conversion of PrPC into PrPSc. However, we noted that, in proportion to the amount of undigested PrP, the deletion mutant generated less protease-resistant protein than did its WT counterpart.
Generation of transgenic mice expressing Δ23–31 PrP
To analyze the role of residues 23–31 in prion propagation and toxicity in vivo, we generated Tg mice expressing Δ23–31 PrP under the control of the PrP half-genomic promoter (Borchelt et al., 1996). Tg founders were bred to Prn-p0/0 mice on the C57BL/6J background, and the expression level for each Tg line was then quantitated by Western blotting. Three different Tg(Δ23–31) mouse lines were selected for further study, expressing approximately six times, four times, and one times the endogenous WT PrP level (Fig. 3A, lanes 3–5). These relative expression levels were confirmed by quantitative Western blotting and analysis by Storm and Odyssey imaging systems (data not shown). Δ23–31 PrP from each of the Tg lines displayed several glycoforms, with the diglycosylated form being predominant (Fig. 3A, lanes 1–5). After deglycosylation with PNGase F, all PrP forms collapsed into a single band migrating at ∼24 kDa, slightly smaller than the corresponding band for WT PrP in Tga20+/+ and nontransgenic C57BL/6J (referred to as non-Tg) mice (Fig. 3A, lanes 6–10).
Figure 3.

Expression and solubility of Δ23–31 PrP in transgenic mice. A, Equal volumes of protein from the brains of mice of the indicated genotypes were either untreated (lanes 1–5) or were treated with PNGase F (lanes 6–10) before blotting with anti-PrP antibody 6D11. Δ23–31 PrP is expressed at six times, four times, and one times in the three transgenic lines (lanes 3–5), compared with the level of WT PrP in a non-Tg mouse (lane 2). The asterisk denotes the endogenous C1 cleavage product. B, Brain lysates from Prn-p0/0 (lanes 1, 2), non-Tg (lanes 3, 4), Tg(Δ23–311×) (lanes 5, 6), and Tg(PG14) (lanes 7, 8) mice were ultracentrifuged to separate soluble (S) and insoluble (I) fractions. Like WT, Δ23–31 PrP is found only in the soluble fraction, while PG14 PrP, an aggregation-prone mutant, is partially insoluble.
We also examined the localization and solubility of Δ23–31 PrP from transgenic mice. Like WT, Δ23–31 PrP was expressed on the plasma membrane of cerebellar granular neurons (Turnbaugh et al., 2011). In addition, both WT and Δ23–31 PrPs were soluble in detergents (Fig. 3B, lanes 3–6), in contrast to an aggregation-prone mutant (PG14) that was partially detergent insoluble (Fig. 3B, lanes 7, 8). Together, these results show that Δ23–31 and WT PrPs have similar localization and biochemical properties in vivo.
Tg(Δ23–31) mice show prolonged survival after scrapie inoculation
Mice expressing Δ23–31 PrP at six times, four times, or one times on the Prn-p0/0 background showed no evidence of spontaneous disease and remained healthy for >600 d (data not shown). To test the role of residues 23–31 in prion conversion in vivo, we inoculated Tg(Δ23–311×), Tg(Δ23–314×), Tg(Δ23–316×) mice on the Prn-p0/0 background with the RML strain of scrapie and compared survival times as well as accumulation of PK-resistant PrP in the brains of these animals. As controls, we also inoculated three kinds of mice with WT PrP expression levels spanning those of mutant PrP in the TgΔ23–31 lines: non-Tg mice, which express endogenous PrP (1 times); and Tga20+/0 and Tga20+/+ mice, which express WT PrP from a transgene at 5 and 10 times, respectively (Shmerling et al., 1998).
Surprisingly, none of the nine Tg(Δ23–311×) mice displayed symptoms of disease at >400 d postinoculation (dpi) (Fig. 4, green line; Table 1). In contrast, non-Tg control mice became terminally ill at ∼160 dpi (Fig. 4, gray line). Both Tg(Δ23–314×) (Fig. 4, blue line) and Tg(Δ23–316×) mice (Fig. 4, red line) also showed a significant increase in life span, compared with Tga20+/0 and Tga20+/+ overexpressing controls (Fig. 4, dashed black and solid black lines, respectively). Interestingly, Tg(Δ23–314×) mice fell into two distinct groups, with one group surviving up to 166 dpi, and the second reaching terminal disease much later, between 336 and 427 dpi (Fig. 4, blue line; Table 1). Tg(Δ23–316×) mice had a more homogenous survival time, with all mice becoming terminally ill at an average of ∼130 dpi (Fig. 4, red line; Table 1). Tga20+/+ and Tga20+/0 control mice had much shorter survival times than any of the Tg(Δ23–31) mice, succumbing at 73 and 81 dpi on average, respectively (Fig. 4, solid black and dashed black lines; Table 1).
Figure 4.

Tg(Δ23–31) mice survive longer than control mice after scrapie inoculation. Survival times were monitored in RML-inoculated mice of the following genotypes: Tga20+/+ (solid black line); Tga20+/0 (dashed black line); non-Tg (gray line); Tg(Δ23–316×) (red line); Tg(Δ23–314×) (blue line); Tg(Δ23–311×) (green line). Each data point represents one animal. A statistical analysis of these data is shown in Table 1.
Table 1.
Tg(Δ23–31) mice have increased survival times after scrapie inoculation
| Host | PrP expression | Time to terminal illness (d) | No. of mice (affected/total) |
|---|---|---|---|
| Tga20+/+ | 10× | 72.5 ± 6 | 17/17 |
| Tga20+/0 | 5× | 81.1 ± 10.3 | 19/19 |
| Non-Tg | 1× | 159.3 ± 4.5 | 15/15 |
| Tg(Δ23–31) | 6× | 131.7 ± 13* | 10/10 |
| Tg(Δ23–31) | 4× | 243.6 ± 122.7* | 11/11 |
| Tg(Δ23–31) | 1× | N/A | 0/9 (5 mice, >500 d; 4 mice, >400 d) |
Mice of the indicated genotypes were intracerebrally inoculated with RML scrapie, and time to terminal illness was monitored. Survival times are significantly increased in both Tg(Δ23–316×) and Tg(Δ23–314×) mice, compared with either Tga20+/+ or Tga20+/0 mice. Tg(Δ23–311×) mice showed no signs of disease for >400 dpi.
The asterisks indicate statistically significant differences in survival compared with either Tga20+/+ or Tga20+/0 mice (*p < 0.01 by Student's t test).
These results show that deletion of PrP residues 23–31 dramatically extends the life span of Tg mice inoculated with RML prions, suggesting that these residues play an important role in the process of prion propagation and/or toxicity.
Terminally ill Tg(Δ23–31) mice accumulate low levels of PK-resistant PrP
To determine whether the increase in life span observed in RML-injected Tg(Δ23–31) mice was related to a decreased accumulation of PrPSc, brain homogenates from mice at 70 dpi and from terminally ill mice were digested with PK, and the amount of PK-resistant PrP was analyzed by Western blot (Figs. 5, 6). At 70 dpi, no PK-resistant PrP was detected in RML-injected Tg(Δ23–31) mice (Fig. 5, lanes 7–12), while WT-expressing controls accumulated low levels (Fig. 5, lanes 1–6). At the time of terminal disease, when control mice showed prominent accumulation of PrPSc, Tg(Δ23–31) mice had little or no PK-resistant PrP (Fig. 6A). In particular, asymptomatic Tg(Δ23–311×) mice, killed at ∼300 dpi, showed no detectable, PK-resistant PrP in their brains (Fig. 6A, lanes 15, 16), while, as expected, non-Tg mice had high levels (Fig. 6A, lanes 5, 6). In addition, the average amount of PK-resistant PrP found in Tg(Δ23–316×) and short-survival Tg(Δ23–314×) mouse brains was only 10 and 16% of the level of non-Tg mice (Fig. 6A, lanes 7–14), while Tga20+/+ and Tga20+/0 mice accumulated substantially more PK-resistant PrP (46 and 62% the level of non-Tg animals, respectively; Fig. 6A, lanes 1–4). Further evaluation of the brains of long-survival Tg(Δ23–314×) mice revealed that these animals accumulated higher levels of PK-resistant PrP (Fig. 6B, lanes 8–11) than short-survival mice of the same genotype (Fig. 6B, lanes 4–7). However, even the long-survival mice had only 51% of the amount of PK-resistant PrP found in the brains of terminally ill Tga20+/0 mice (Fig. 6B, lanes 1–2).
Figure 5.

Tg(Δ23–31) mice accumulate less PrPSc than controls at 70 dpi. Brain homogenates from RML-inoculated mice of the indicated genotypes at 70 dpi were treated without (top panel) or with (bottom panel) PK and were then subjected to Western blotting using anti-PrP antibody 6D11.
Figure 6.

Tg(Δ23–31) mice accumulate greatly reduced amounts of PrPSc. A, Brain homogenates from RML-inoculated mice of the indicated genotypes at the terminal stage were treated without (top panels) or with (bottom panels) PK and were then subjected to Western blotting using anti-PrP antibody 6D11. The average signal intensity for PrP in terminally ill mice of each genotype was quantitated (numbers below the lanes) and is reported as a percentage of the amount of PK-resistant PrP in non-Tg mice. Ages of terminal mice in lanes 1–16 are as follows (in dpi): 69, 71, 76, 86, 159, 159, 112, 140, 142, 146, 147, 150, 157, 168, 301, 304. Actin is shown as a loading control. B, Brain homogenates from terminally ill, RML-infected Tga20+/0 mice and from Tg(Δ23–314×) mice (short and long survivors) (+ lanes), and from age-matched, uninoculated control mice (− lanes) were treated with or without PK and Western blotted using anti-PrP antibody D18. Mice were killed at the following times (in dpi): 79, 86, 150, 152, 157, 168, 336, 399, 419, 427 (lanes 1, 2, 4–11, respectively). Uninoculated mice were killed at the following ages (in d): 82, 365 (lanes 3, 12, respectively). C, Brain homogenates from a terminally ill RML-inoculated Tga20+/0 mouse (86 dpi) and a terminally ill Tg(Δ23–316×) mouse (140 dpi) were digested with the indicated amounts of PK and subjected to Western blotting with 6D11 antibody to detect PrP. Both short (top) and long (bottom) exposures of the blot are shown.
To test the possibility that Tg(Δ23–31) mice accumulate a form of PrPSc that is protease sensitive, brains from terminally ill Tg(Δ23–316×) mice were digested with increasing concentrations of PK. At all concentrations of PK, the amount of PK-resistant PrP in the brain of Tg(Δ23–316×) mice was much lower than that of Tga20+/0 mice (Fig. 6C, top panels). However, a longer exposure of the Western blots (Fig. 6C, bottom panels) revealed that the small amount of PrPSc formed in Tg(Δ23–316×) mice was fully resistant to PK (up to 50 μg/ml). Collectively, these results demonstrate that RML-inoculated Tg(Δ23–31) mice accumulate much less PK-resistant PrP over the course of disease than mice expressing similar levels of the WT protein. However, the small amount of PrPSc present in the brains of Tg(Δ23–31) mice is still fully PK resistant, suggesting that this biochemical property of the RML scrapie strain is preserved when it is passaged into Tg(Δ23–31) mice.
Primary sites of PrPSc accumulation and spongiform degeneration are similar in RML-injected Tg(Δ23–31) and control mice
One possible explanation for the extended life span observed in PrPSc-infected Tg(Δ23–31) mice is that a new strain, with neuropathological properties different from the original inoculum, was generated after passaging RML prions into these mice. To address this possibility, the distribution of PrPSc and the presence of spongiform degeneration were assessed, comparing terminally ill, RML-infected Tg(Δ23–31) and control mice (Fig. 7). The distribution of PrPSc in brain sections was assessed by immunohistochemical staining, and the presence of spongiform degeneration was evaluated by hematoxylin and eosin staining. RML-inoculated Tga20+/+, Tga20+/0, and non-Tg control mice accumulated PrPSc primarily in the thalamus (Fig. 7A1–A3) and brainstem (Fig. 7B1–B3), and to a much lesser extent in the cortex (Fig. 7C1–C3) and other brain areas (data not shown). Spongiform degeneration in these animals was detected primarily in the brainstem (Fig. 7D1–D3). As expected, the amount of PrPSc detected in the different brain areas of RML-injected Tg(Δ23–314×) and Tg(Δ23–316×) mice was generally lower than that of controls. However, both of these mouse lines showed a pattern of PrPSc distribution identical with that of WT-expressing controls, with the thalamus (Fig. 7A4,A5) and brainstem (Fig. 7B4,B5) being the main sites of PrPSc accumulation, while almost no PrPSc was detected in the cortex (Fig. 7C4,C5) and other brain areas (data not shown). Moreover, spongiform degeneration was observed mainly in the brainstem of Tg(Δ23–314×) and Tg(Δ23–316×) mice (Fig. 7D4,D5). PrPSc deposition and spongiform change were comparable in Tg(Δ23–314×) mice with short and long survival times (data not shown). Importantly, no PrPSc staining or spongiosis was detected in brain sections from RML-injected, Tg(Δ23–311×) mice at 300 dpi (Fig. 7A6,B6,C6,D6), or in uninoculated Tga20+/+ mice (Fig. 7A7,B7,C7,D7) and Tg(Δ23–316×) mice (Fig. 7A8,B8,C8,D8).
Figure 7.
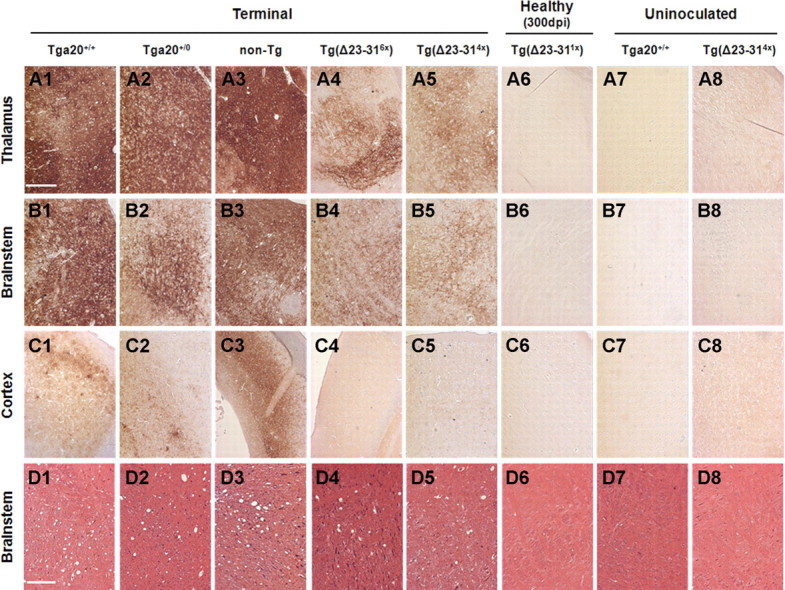
Tg(Δ23–31) and control mice display PrPSc accumulation and spongiform change in similar areas of the brain. A1–C8, Paraffin sections from mice of the indicated genotypes were stained for PrPSc. Representative staining of the thalamus (A1–A8), brainstem (B1–B8), and cortex (C1–C8) is shown. D1–D8, Paraffin sections from mice of the indicated genotypes were stained with hematoxylin and eosin. Representative sections of the brainstem are shown. Panels 1–6 (A–D) are from RML-inoculated mice, and panels 7 and 8 (A–D) are from uninoculated mice. Mice were killed at the following times (in dpi): 75 (A1, B1); 71 (C1); 75 (D1); 89 (A2, B2, C2); 82 (D2); 159 (A3, B3, C3); 150 (D3); 140 (A4, B4, C4); 112 (D4); 136 (A5, B5, C5); 152 (D5); 304 (A6, B6, C6, D6). Uninoculated mice were killed at the following ages (in d): 90 (A7, B7, C7, D7); 159 (A8, B8, C8, D8). Scale bars: A1–C8, 400 μm; D1–D8, 150 μm.
To confirm that the overall histopathological profile was similar in RML-inoculated Δ23–31 PrP and WT-expressing mice, we scored the extent of PrPSc deposition and spongiosis that occurred in each of six different brain regions (Table 2). All terminally ill Tga20+/+, Tga20+/0, Tg(Δ23–316×), and Tg(Δ23–314×) mice showed similar patterns of PrPSc deposition, with accumulation primarily in the thalamus and brainstem and the greatest amount of spongiosis in the brainstem. As expected based on previous work (Karapetyan et al., 2009), non-Tg mice showed more extensive PrPSc deposition throughout the brain, and higher levels of spongiform change in the cerebellum.
Table 2.
Quantitation of regional histopathology profiles for RML-inoculated mice
| Regional histopathology |
||||||
|---|---|---|---|---|---|---|
| Cortex | Hippocampus | Cerebellum | Brainstem | Thalamus | Striatum | |
| Tga20+/+ | ||||||
| Spongiosis (n = 3) | 0.33 ± 0.33 | 0.67 ± 0.67 | 1.0 ± 0 | 2.33 ± 0.33 | 1.0 ± 0 | 0.67 ± 0.33 |
| PrPSc (n = 3) | 1.0 ± 0 | 1.67 ± 0.33 | 1.33 ± 0.33 | 3.0 ± 0.58 | 2.67 ± 0.33 | 2.33 ± 0.33 |
| Tga20+/0 | ||||||
| Spongiosis (n = 3) | 0 ± 0 | 1.33 ± 0.33 | 1.33 ± 0.33 | 2.67 ± 0.33 | 1.67 ± 0.33 | 0.67 ± 0.33 |
| PrPSc (n = 3) | 1.0 ± 0 | 1.67 ± 0.33 | 1.33 ± 0.33 | 3.0 ± 0.58 | 2.67 ± 0.33 | 1.33 ± 0.33 |
| Non-Tg | ||||||
| Spongiosis (n = 4) | 1.0 ± 0.71 | 1.0 ± 0 | 3.25 ± 0.71 | 3.5 ± 0 | 1.0 ± 0 | 1.5 ± 0.35 |
| PrPSc (n = 3) | 2.33 ± 0.41 | 3.0 ± 0.82 | 3.67 ± 0.41 | 4.0 ± 0 | 4.0 ± 0 | 2.67 ± 0.82 |
| Tg(Δ23–31)6× | ||||||
| Spongiosis (n = 4) | 0.67 ± 0.33 | 0.33 ± 0.33 | 1.0 ± 0 | 2.0 ± 0.58 | 0.67 ± 0.67 | 0.67 ± 0.33 |
| PrPSc (n = 3) | 0.67 ± 0.67 | 0.67 ± 0.67 | 0 ± 0 | 2.33 ± 0.33 | 2.33 ± 0.33 | 1.0 ± 0.58 |
| Tg(Δ23–31)4× | ||||||
| Spongiosis (n = 7) | 0.14 ± 0.14 | 0.29 ± 0.18 | 1.0 ± 0.22 | 1.43 ± 0.20 | 1.0 ± 0.22 | 0.86 ± 0.26 |
| PrPSc (n = 7) | 0.29 ± 0.18 | 0 ± 0 | 0.14 ± 0.14 | 2.71 ± 0.29 | 2.29 ± 0.29 | 0.57 ± 0.30 |
| Tg(Δ23–31)1× 300 dpi | ||||||
| Spongiosis (n = 2) | 0 ± 0 | 0 ± 0 | 0 ± 0 | 1.5 ± 0.5 | 0.5 ± 0.5 | 0 ± 0 |
| PrPSc (n = 2) | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
Sagittal brain sections from mice of the indicated genotypes were stained for PrPSc, or with hematoxylin and eosin to reveal spongiosis. The relative amount of PrPSc deposition or spongiosis in the cortex, hippocampus, cerebellum, brainstem, thalamus, and striatum was scored on a scale from 0 to 4, as follows: 0, none; 1, slight; 2, moderate; 3, severe; 4, very severe. The average score ± SEM for each brain region is given, with the number of mice analyzed for each group shown in parentheses.
In summary, the accumulation of PrPSc in the thalamus and brainstem, as well as the presence of spongiform degeneration of the brainstem, both typical of the RML strain, were indistinguishable between WT-expressing controls and Tg(Δ23–314×) or Tg(Δ23–316×) mice, although both of these neuropathological features were generally less severe in mice expressing the PrP mutant. These results argue against the possibility that the extended life span of RML-inoculated Tg(Δ23–31) mice is related to generation of a new prion strain displaying different properties than the original inoculum.
Deletion of residues 23–31 does not create a sequence barrier for prion propagation
Another possible explanation for the longer survival times observed in RML-infected Tg(Δ23–31) mice is the presence of a sequence mismatch between the original RML seed (which carries a WT PrP sequence) and the Δ23–31 PrP substrate. This mismatch could constitute a barrier for prion propagation and lead to suboptimal conversion of Δ23–31 PrP into Δ23–31 PrPSc. To test this possibility, we performed secondary passage experiments, inoculating Tg(Δ23–316×), Tga20+/+, and non-Tg hosts with brain homogenates from RML scrapie-infected Tg(Δ23–316×) mice (hereafter referred to as RMLΔ23–31). If a sequence mismatch were responsible for the prolonged survival times seen in the primary inoculation experiments, then secondary passage of RMLΔ23–31 into Tg(Δ23–316×) host mice should result in shorter survival. In fact, we observed no difference in the life span of Tg(Δ23–316×) mice infected with the original, WT RML inoculum compared with those inoculated with RMLΔ23–31 (Fig. 8A, red lines with circles and squares, respectively; selected data from Fig. 4 and Table 1 are reported again in Fig. 8 and Table 3 to allow direct comparison). Moreover, the survival times after inoculation of Tga20+/+ mice with WT RML and RMLΔ23–31 were almost identical (Fig. 8, black lines with circles and squares, respectively; Table 3). The only statistically significant difference was found between non-Tg mice inoculated with RML and RMLΔ23–31, with the latter surviving an additional 10 dpi, on average (Fig. 8, gray lines with circles and squares, respectively; Table 3). This small increase in life span may be attributable to the lower amount of PK-resistant PrPSc in the RMLΔ23–31 inoculum, compared with the WT RML inoculum (as shown in Fig. 6A).
Figure 8.

Secondary passage of RMLΔ23–31 scrapie into Tg(Δ23–31) mice does not shorten survival times. A, Survival times were monitored in mice of the following genotypes inoculated with RML or RMLΔ23–31: Tg(Δ23–316×) (red lines); Tga20+/+ (black lines); non-Tg (gray lines). The circles represent individual mice inoculated with RML, and the squares represent mice inoculated with RMLΔ23–31. A statistical analysis of these data is shown in Table 3. B, Brain homogenates from terminally ill mice of the indicated genotypes inoculated with RML (lanes 1–3) or RMLΔ23–31 (lanes 4–15) were treated without (top panel) or with (bottom panel) PK, and were then subjected to Western blotting using anti-PrP antibody 6D11. Actin is shown as a loading control.
Table 3.
Mice inoculated with RML or RMLΔ23–31 show similar survival times
| Host | Time to terminal (d) |
|
|---|---|---|
| RML | RMLΔ23–31 | |
| Tg(Δ23–316×) | 131.7 ± 13 (n = 10) | 133.6 ± 19.4 (n = 8) |
| Tga20+/+ | 72.5 ± 6.0 (n = 17) | 75.0 ± 6.1 (n = 9) |
| Non-Tg | 159.3 ± 4.5 (n = 15) | 170.0 ± 3.7 (n = 8)* |
Host mice of the indicated genotypes were intracerebrally inoculated with either RML (primary passage) or RMLΔ23–31 (secondary passage), and time to terminal disease was monitored. The number of mice is given in parentheses. Survival times in Tg(Δ23–316x) or Tga20+/+ mice is the same with both inocula.
Non-Tg mice inoculated with RMLΔ23–31 have a small, but statistically significant increase in survival time compared with mice inoculated with RML (*p < 0.01 by Student's t test).
Together, these data argue against the idea that deletion of residues 23–31 from PrPC creates a sequence barrier that affects propagation of the RML strain.
RMLΔ23–31 maintains the biological properties of the original inoculum
To test whether the biochemical and neuropathological properties of the original RML inoculum were maintained after passaging this strain into Tg(Δ23–316×) mice, we compared the amount and site of accumulation of PK-resistant PrP in mice infected with RML or with RMLΔ23–31. Brain homogenates from terminally ill Tg(Δ23–316×), Tga20+/+, or non-Tg mice inoculated with RML or with RMLΔ23–31 were incubated with PK and analyzed by Western blotting (Fig. 8B). We observed low or undetectable levels of PK-resistant PrP in the brains of Tg(Δ23–316×) mice inoculated with either RML (Fig. 8B, lanes 2, 3) or RMLΔ23–31 (Fig. 8B, lanes 4–7). Tga20+/+ and non-Tg mice consistently accumulated larger amounts of PK-resistant PrP, which was similar after infection with either RML (Fig. 8B, lane 1) (see also Fig. 6A, lanes 1, 2, and 5, 6) or RMLΔ23–31 (Fig. 8B, lanes 8–11 and 12–15).
We then assessed the distribution of PrPSc as well as the presence of spongiform degeneration in different brain areas from terminally ill Tg(Δ23–31) and control mice infected with RMLΔ23–31 (Fig. 9, Table 4). Only low levels of PrPSc were detected in RMLΔ23–31-infected Tg(Δ23–316×) mice, although, as for the first passage, the main sites of accumulation were the thalamus (Fig. 9C) and brainstem (Fig. 9F). The same two areas were also found to be the primary sites of PrPSc accumulation in RMLΔ23–31-infected Tga20+/+ (Fig. 9A,D) and non-Tg (Fig. 9B,E) mice. All the animals showed a similar pattern of spongiform degeneration, with the brainstem being the primary site affected (Fig. 9G–I).
Figure 9.

RML strain characteristics are preserved during secondary passage in Tg(Δ23–31) mice. Paraffin sections from terminally ill Tga20+/+, non-Tg, and Tg(Δ23–316×) mice inoculated with RMLΔ23–31 display staining for PrPSc in the thalamus (A–C) and the brainstem (D–F). Hematoxylin and eosin staining shows spongiosis in the brainstem of these animals (G–I). Mice were killed at the following times (in dpi): 72 (A, D), 171 (B, E), 132 (C, F), 68 (G), 171 (H), 144 (I). Scale bars: A–F, 400 μm; G–I, 150 μm.
Table 4.
Quantitation of regional histopathology profiles for RMLΔ23–31-inoculated mice
| Regional histopathology |
||||||
|---|---|---|---|---|---|---|
| Cortex | Hippocampus | Cerebellum | Brainstem | Thalamus | Striatum | |
| Tga20+/+ | ||||||
| Spongiosis (n = 4) | 0.25 ± 0.25 | 1.5 ± 0.87 | 1.0 ± 0.41 | 2.25 ± 0.25 | 0.5 ± 0.25 | 0.25 ± 0.25 |
| PrPSc (n = 4) | 1.25 ± 0.25 | 1.5 ± 0.5 | 2.0 ± 0.41 | 3.25 ± 0.25 | 3.75 ± 0.25 | 1.0 ± 0.41 |
| Non-Tg | ||||||
| Spongiosis (n = 4) | 1.25 ± 0.25 | 1.25 ± 0.25 | 3.75 ± 0.25 | 3.5 ± 0.29 | 1.25 ± 0.48 | 1.25 ± 0.63 |
| PrPSc (n = 4) | 3.5 ± 0.29 | 3.5 ± 0.29 | 3.25 ± 0.25 | 3.0 ± 0 | 4.0 ± 0 | 2.25 ± 0.25 |
| Tg(Δ23–31)6× | ||||||
| Spongiosis (n = 4) | 0 ± 0 | 0.5 ± 0.29 | 0.75 ± 0.25 | 2.25 ± 0.25 | 0.75 ± 0.48 | 1.0 ± 0.41 |
| PrPSc (n = 3) | 0.33 ± 0.33 | 1.0 ± 0 | 0.67 ± 0.33 | 2.67 ± 0.33 | 2.67 ± 0.88 | 1.0 ± 0.58 |
Sagittal brain sections from mice of the indicated genotypes were stained for PrPSc, or with hematoxylin and eosin to reveal spongiosis. The relative amount of PrPSc deposition or spongiosis in various brain areas was scored as described in Table 2. The average score ± SEM for each brain region is given, with the number of mice analyzed for each group shown in parentheses.
To further analyze the extent and location of PrPSc deposition and spongiosis in the brains of RMLΔ23–31-infected mice, we quantified each of these pathologies in the cortex, hippocampus, cerebellum, brainstem, thalamus, and striatum of terminally ill mice of each genotype (Table 4). The highest levels of PrPSc were localized in the brainstem and thalamus of Tga20+/+ and Tg(Δ23–316×) mice, and the most severe spongiform degeneration was observed in the brainstem. As expected, non-Tg mice showed a wider extent of PrPSc deposition, as well as increased spongiosis in the cerebellum.
These results demonstrate that both the biochemical and neuropathological properties of the RML inoculum are unaltered after passaging this strain into Δ23–31 PrP-expressing mice.
Deletion of residues 23–31 makes PrP an inefficient substrate, but does not compromise its seeding ability
To gain additional insights into the role of residues 23–31 in prion conversion, we undertook experiments using an in vitro conversion system to complement our in vivo studies in mice. Protein misfolding cyclic amplification (PMCA) was used to test whether the original RML inoculum was able to seed the misfolding of WT or Δ23–31 PrP. Brain homogenates from either Tga20+/0 or Tg(Δ23–316×) mice were used as substrates for the reaction. The RML inoculum efficiently seeded the misfolding of full-length PrP (Fig. 10A, top right panel) but not Δ23–31 PrP (Fig. 10A, bottom right panel). These results indicated that, as observed in vivo, PrP molecules deleted for residues 23–31 are inefficiently converted into PrPSc.
Figure 10.

Deletion of residues 23–31 makes PrP an inefficient substrate, but does not compromise its seeding ability. PMCA reactions were run using the indicated seeds and substrates. Experiments were performed at least three times, and a representative example is shown. For each experiment, a nonamplified input sample is shown on the left, and an amplified sample on the right. The lanes represent reactions run with serial sixfold dilutions of the seed. All samples were digested with PK before Western blotting. A, RML from a terminally ill, non-Tg mouse was used to seed conversion of WT PrP (from a Tga20+/0 mouse) and Δ23–31 PrP [from a Tg(Δ23–316×) mouse]. B, RMLΔ23–31 [from a terminally ill, RML-inoculated Tg(Δ23–316×) mouse] or RML (from a terminally ill Tga20+/0 mouse) was used to seed conversion of WT PrP from a non-Tg mouse. The amounts of the two seeds were equalized, based on Western blotting for PK-resistant PrP. C, Brain homogenate from a healthy Tg(Δ23–311×) mouse at 300 d after inoculation with RML was used to seed conversion of WT PrP from a Tga20+/+ mouse.
Next, we compared the seeding activity of RML scrapie passaged into Tga20+/0 or Tg(Δ23–316×) mice. To allow direct comparison of the seeding activity of the two inocula, similar amounts of PK-resistant RML or RMLΔ23–31 were used to seed the misfolding of WT PrP derived from non-Tg brain homogenates (Fig. 10B, left panels). Consistent with our secondary passage experiments in Tga20+/+ mice (Fig. 8), we found that RML and RMLΔ23–31 seeds were equally capable of inducing the conversion of full-length PrP (Fig. 10B, right panels), indicating that the deletion of residues 23–31 does not affect the seeding activity of PrPSc.
These in vitro results recapitulated the observations made in vivo, demonstrating that deletion of residues 23–31 from PrPC impairs its conversion into PrPSc. However, once conversion is established, the resulting RMLΔ23–31 molecules show the same seeding activity as the original inoculum.
PMCA detects infectivity in healthy Tg(Δ23–311×) mice
As described above, RML-infected Tg(Δ23–311×) mice did not show any clinical or neuropathological signs of disease for >400 dpi. To test whether the brains of these mice accumulated subclinical amounts of infectious PrPSc, we took advantage of the high sensitivity of the PMCA reaction. Surprisingly, we found that brain homogenates from healthy, RML-infected Tg(Δ23–311×) mice at 300 dpi were able to seed the misfolding of WT PrP derived from Tga20+/+ mice (Fig. 10C, right panel) in the absence of any detectable PK-resistant seed (Fig. 10C, left panel). Additionally, when brain homogenates from the same Tg(Δ23–311×) mice were used to inoculate Tga20+/+ indicator mice, these mice developed prion disease (data not shown).
These results suggest that Tg(Δ23–311×) mice accumulated either very low levels of PK-resistant PrPSc or a PK-sensitive, nonpathogenic form of PrPSc that was only detectable using high-sensitivity techniques such as passage into PrP-overexpressing mice or PMCA.
Deletion of residues 23–31 compromises binding of PrPC to PrPSc
To test whether inefficient conversion of Δ23–31 PrP is a consequence of decreased binding to PrPSc, we performed immunoprecipitation experiments comparing the amount of PrPSc pulled down by WT or Δ23–28 PrP molecules (the latter, like Δ23–31 PrP, harbors a deletion of the polybasic domain). Recombinant (rec), myc-tagged WT or Δ23–28 PrP was incubated with brain homogenates from RML-infected, non-Tg mice. An anti-myc antibody was then used to immunoprecipitate the PrPC–PrPSc complexes, followed by digestion with PK to detect PrPSc. Detection of myc-tagged PrP in absence of PK treatment confirmed that both recWT and recΔ23–28 proteins were efficiently immunoprecipitated in presence or absence of PrPSc (Fig. 11A, top panel, lanes 2–5). Importantly, we found that recΔ23–28 PrP was much less efficient in pulling down PrPSc when compared with the recWT PrP control (Fig. 11A, bottom panel, compare lanes 2, 4). Averaging the results of three independent experiments, the polybasic domain mutant pulled down 41.8% (SEM, ±18.0) of the PrPSc that was pulled down by WT PrP. No signal was detected when the immunoprecipitation was performed in absence of RML brain homogenate, recPrP, or anti-myc antibody (Fig. 11A, bottom panel, lanes 3, 5, 6, 7). These results indicate that the impaired conversion of Δ23–31 PrP is likely due to defective binding to PrPSc seeds.
Figure 11.

The N-terminal, polybasic domain mediates binding between PrPC and PrPSc. A, Myc-tagged WT or Δ23–28 recombinant PrPs were incubated in the presence or absence of RML-infected brain homogenate before immunoprecipitation with anti-myc antibody 9E10 (+ lanes) or naked beads (− lane). A portion of each sample was then incubated in the absence of PK, and Western blots probed with anti-myc antibody 4A6 (top panel). The remainder of each sample was digested with PK, and Western blots probed with anti-PrP antibody D18 (bottom panel). A sample of the RML used as input was is shown in lane 1 after PK digestion. Δ23–28 PrP pulled down far less PK-resistant PrP than WT PrP (compare lanes 2, 4). Blots shown are representative of at least three independent experiments. B, Model for the role of residues 23–31 in generation of PrPSc. Top panel, When residues 23–31 (represented by yellow plus signs) are present, PrPC (blue) binds strongly to PrPSc (red), leading to efficient conversion. Bottom panel, When these amino acids are deleted, as in Δ23–31 PrP, the PrPC–PrPSc interaction is less favorable, leading to reduced conversion. In this case, PrPSc is still generated but relies upon binding of PrPSc to PrPC at secondary sites. The red explosion shape represents the PK-resistant core of PrPSc. The 23–31 region of PrPSc is not involved in binding to PrPC.
Discussion
In the present study, we have demonstrated that PrPSc formation depends on a critical 9 aa domain at the N terminus of PrP (residues 23–31). Deletion of these residues severely compromised the ability of PrPC to serve as a substrate for conversion into PrPSc, and Tg mice expressing Δ23–31 PrP showed dramatically increased survival after scrapie inoculation, accumulating reduced amounts of PrPSc in their brains. Our biochemical assays indicate that this phenomenon is a consequence of a reduced ability of PrPC substrate molecules missing the N-terminal, polybasic domain to bind to PrPSc seeds, demonstrating that this region is required for the initial steps of formation of the PrPC–PrPSc complex.
Tg(Δ23–31) mice display reduced susceptibility to prion disease
Tg(Δ23–311×) mice expressing the mutant protein at physiological levels did not exhibit clinical signs of scrapie infection for >400 dpi, nor did they accumulate detectable amounts of protease-resistant PrP. Mice expressing higher levels of Δ23–31 PrP (four times and six times) eventually succumbed to disease, but with dramatically prolonged incubation times and reduced levels of PrPSc compared with mice expressing equivalent levels of WT PrP.
Remarkably, brain homogenates from clinically healthy Tg(Δ23–311×) mice at 300 dpi were able to transmit disease to Tga20+/+ indicator mice, and to seed the misfolding of WT PrP in PMCA reactions. Thus, the brains of these animals contained infectious prions. These results are consistent with studies indicating that accumulation of infectivity precedes development of clinical disease and can be uncoupled from the presence of protease-resistant PrPSc (Büeler et al., 1994; Chiesa and Harris, 2001; Sandberg et al., 2011).
Interestingly, we observed a differential attack rate after scrapie inoculation of Tg(Δ23–314×) mice: one group of animals reached terminal disease at ∼160 dpi, and the remainder at ∼400 dpi. This suggests the operation of a stochastic process related to inefficient generation of PrPSc, such that critical levels of toxic PrP forms are reached in subsets of animals at different times. Thus, mice that overexpress the Δ23–31 PrP substrate at even higher levels (six times) have more uniform, but still significantly prolonged survival times, while those expressing lower, physiological levels (one times) are completely resistant to clinical disease for >400 dpi.
We addressed several possible explanations for the decreased susceptibility of Tg(Δ23–31) mice to scrapie prions, and their reduced accumulation of PrPSc. Since sequence compatibility is a well known factor that influences the generation of PrPSc (Priola et al., 1994; Prusiner, 1998; Priola, 1999; Raymond et al., 2000), a mismatch between the sequence of the WT PrPSc seed and the Δ23–31 PrPC substrate might impede PrPSc formation. If this were the case, however, incubation and survival times would become shorter upon secondary passage of RMLΔ23–31 into Tg(Δ23–316×) mice. In contrast, we observed identical survival times in Tg(Δ23–316×) mice inoculated with either RML or RMLΔ23–31, indicating that deletion of residues 23–31 does not create a sequence barrier for prion propagation. Additionally, PrPSc accumulated in the same areas of the brain in Tg(Δ23–314×), Tg(Δ23–316×), and WT PrP-expressing control mice. Thus, the characteristics of the RML strain were unaltered upon passage into Tg(Δ23–314×) and Tg(Δ23–316×) mice.
Another possibility was that deletion of residues 23–31 altered the cellular trafficking or localization of PrPC, thereby affecting production of PrPSc. Consistent with the role of residues 23–31 in endocytic trafficking (Shyng et al., 1995; Sunyach et al., 2003), we observed impaired internalization of Δ23–31 PrP in transfected cells. However, we found that Δ23–31 PrP molecules were inefficiently converted into PrPSc in the PMCA assay, as well as in vivo. In addition, recombinant Δ23–28 PrP had a decreased affinity for PrPSc in binding assays. Together, our results argue that residues 23–31 affect production of PrPSc independent of effects on cellular trafficking or localization.
The polybasic domain forms an essential part of the PrPC–PrPSc binding site
Δ23–31 PrP supported generation of PrPSc in PMCA reactions, scrapie-infected N2a cells, and scrapie-inoculated Tg(Δ23–314×) and Tg(Δ23–316×) mice. In all cases, however, conversion of Δ23–31 PrP was less efficient than conversion of WT PrP, demonstrating that deletion of the polybasic domain significantly impairs the efficiency of PrPC as a substrate for conversion into PrPSc.
What is the molecular mechanism responsible for the resistance of Δ23–31 PrPC to conversion? Several lines of evidence suggest that propagation of PrPSc involves two mechanistically distinct steps: binding of the PrPSc seed to PrPC substrate followed by a conformational change that results in PrPSc formation (DebBurman et al., 1997; Horiuchi and Caughey, 1999). Our study suggests that the polybasic domain of PrPC forms a critical part of the binding surface with PrPSc in the initial step of the conversion process (Fig. 11B, top). In the absence of these residues, conversion can still occur, presumably via binding of PrPSc to more C-terminal domains of PrPC, but this interaction is much less efficient (Fig. 11B, bottom). Our results leave open the possibility that residues 23–31, in addition to forming a binding site for PrPSc, might also mediate interaction with cofactor molecules that influence the conversion reaction, such as polyanions or lipids (Deleault et al., 2007; Wang et al., 2010).
While deletion of residues 23–31 impaired the ability of PrPC to serve as a substrate for generation of PrPSc, this region played no detectable role in the ability of PrPSc to serve as a seed for converting WT PrP. Secondary passage of brain homogenates from scrapie-inoculated Tg(Δ23–316×) mice into Tga20+/+ mice produced disease with incubation times and strain properties that were indistinguishable from those obtained after inoculation with WT RML. These observations are consistent with previous findings showing that proteinase K-digested PrPSc molecules that are missing part of the N terminus (up to residue 90) retain full infectivity (Prusiner et al., 1983, 1984; Bolton et al., 1985). Strikingly, the same efficiency of conversion was observed when equal amounts of brain-extracted RML or RMLΔ23–31 were used to seed the misfolding of WT PrP in the PMCA reaction.
Together, our results demonstrate that the binding between PrPC and PrPSc is asymmetric: the polybasic domain of PrPC constitutes a critical docking site for PrPSc, but once the two molecules come together, conformational conversion is templated by residues in the protease-resistant core of PrPSc independent of the polybasic region. Thus, the convertibility of PrPC and the infectivity of PrPSc are governed by different domains of the polypeptide chain.
Several other studies have suggested a role for N-terminal regions in the formation of PrPSc, although our study significantly extends previous work by pinpointing a 9 aa segment in PrPC that is essential for PrPSc binding and conversion both in vitro and in transgenic mice. Consistent with the results presented here, mice expressing PrP with a larger deletion (Δ23–88) that includes the 23–31 region displayed a prolonged incubation time after scrapie inoculation, although the animals eventually produced PrPSc and succumbed to illness (Supattapone et al., 2001). Studies using motif-grafted antibodies identified residues 23–33 as one of three domains involved in binding of PrPC to PrPSc (Moroncini et al., 2004; Solforosi et al., 2007), and a peptide encompassing PrP residues 19–30 was found to bind to PrPSc in plasma (Lau et al., 2007). Mutation of positively charged residues within the 23–33 region (Abalos et al., 2008), or deletion of residues 23–88 (Rogers et al., 1993), did not prevent formation of PrPSc in neuroblastoma cells, although deletion of PrPC residues 23–28 decreased binding to PrPSc and impaired conversion in PMCA reactions (Miller et al., 2011). Thus, the factors controlling conversion in neuroblastoma cells appear to differ from those operative in transgenic mice or in PMCA reactions.
Residues 23–31 as a therapeutic target
Our results suggest that the N-terminal, polybasic region of PrPC may represent a target for therapeutic compounds that prevent the formation of PrPSc. Indeed, sulfated glycosaminoglycans, which bind to this region, have been shown to reduce accumulation of PrPSc and prolong the life span of scrapie-infected mice (Doh-ura et al., 2004). We have recently demonstrated that residues 23–31 control the neurotoxicity associated with certain deleted forms of PrP, as well as the ability of WT PrP to suppress these effects (Solomon et al., 2011; Turnbaugh et al., 2011; Westergard et al., 2011). Thus, this region may also be important for PrP interactions with cellular machinery that regulates neuronal death and survival.
Interestingly, evidence suggests that the polybasic domain of PrPC may also play a role in other neurodegenerative disorders. Residues 23–28 were identified as one of the two binding sites on PrPC for Aβ oligomers (Chen et al., 2010), which some studies have shown to deliver a synaptotoxic signal via PrPC (Laurén et al., 2009; Chung et al., 2010; Gimbel et al., 2010; Barry et al., 2011; Freir et al., 2011). Another study showed that deletion of PrP residues 27–89 prevented the toxic effects of several other β-sheet-rich oligomers in addition to Aβ, implying an even broader role for this region (Resenberger et al., 2011). Therefore, molecules capable of binding to the N-terminal, polybasic region of PrPC may function as potent inhibitors of the neurotoxicity associated not only with prions but also with other misfolded proteins.
Footnotes
This work was supported by NIH Grants NS040975 and NS052526 (D.A.H.) and NS046478 and NS055875 (S.S.). J.A.T. was supported by NIH Predoctoral and Postdoctoral Training Grant in the Biochemistry of Aging 5T32AG000115-25, and M.B.M. was supported by Kirschstein MD/PhD National Research Service Award Fellowship F30 NS064637. We thank Cheryl Adles, Su Deng, and Jorge De Castro for mouse colony maintenance and genotyping. We acknowledge Mike Green for generation of the ScN2a.3 cell line, and Rick Kascak for providing antibodies 6D11 and 3F4. The hybridoma cell line used to purify antibody D18 was provided by Dennis Burton.
References
- Abalos GC, Cruite JT, Bellon A, Hemmers S, Akagi J, Mastrianni JA, Williamson RA, Solforosi L. Identifying key components of the PrPC-PrPSc replicative interface. J Biol Chem. 2008;283:34021–34028. doi: 10.1074/jbc.M804475200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry AE, Klyubin I, McDonald JM, Mably AJ, Farrell MA, Scott M, Walsh DM, Rowan MJ. Alzheimer's disease brain-derived amyloid-β-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J Neurosci. 2011;31:7259–7263. doi: 10.1523/JNEUROSCI.6500-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JE, Gentleman SM, Ironside JW, McCardle L, Lantos PL, Doey L, Lowe J, Fergusson J, Luthert P, McQuaid S, Allen IV. Prion protein immunocytochemistry—UK five centre consensus report. Neuropathol Appl Neurobiol. 1997;23:26–35. [PubMed] [Google Scholar]
- Bolton DC, Meyer RK, Prusiner SB. Scrapie PrP 27–30 is a sialoglycoprotein. J Virol. 1985;53:596–606. doi: 10.1128/jvi.53.2.596-606.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13:159–163. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- Büeler H, Raeber A, Sailer A, Fischer M, Aguzzi A, Weissmann C. High prion and PrPSc levels but delayed onset of disease in scrapie-inoculated mice heterozygous for a disrupted PrP gene. Mol Med. 1994;1:19–30. [PMC free article] [PubMed] [Google Scholar]
- Campana V, Sarnataro D, Zurzolo C. The highways and byways of prion protein trafficking. Trends Cell Biol. 2005;15:102–111. doi: 10.1016/j.tcb.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Chen S, Yadav SP, Surewicz WK. Interaction between human prion protein and amyloid-β (Aβ) oligomers. J Biol Chem. 2010;285:26377–26383. doi: 10.1074/jbc.M110.145516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiesa R, Harris DA. Prion diseases: what is the neurotoxic molecule? Neurobiol Dis. 2001;8:743–763. doi: 10.1006/nbdi.2001.0433. [DOI] [PubMed] [Google Scholar]
- Chiesa R, Piccardo P, Ghetti B, Harris DA. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron. 1998;21:1339–1351. doi: 10.1016/s0896-6273(00)80653-4. [DOI] [PubMed] [Google Scholar]
- Chung E, Ji Y, Sun Y, Kascsak RJ, Kascsak RB, Mehta PD, Strittmatter SM, Wisniewski T. Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer's disease model mouse. BMC Neurosci. 2010;11:130. doi: 10.1186/1471-2202-11-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci. 2001;24:519–550. doi: 10.1146/annurev.neuro.24.1.519. [DOI] [PubMed] [Google Scholar]
- DebBurman SK, Raymond GJ, Caughey B, Lindquist S. Chaperone-supervised conversion of prion protein to its protease-resistant form. Proc Natl Acad Sci U S A. 1997;94:13938–13943. doi: 10.1073/pnas.94.25.13938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A. 2007;104:9741–9746. doi: 10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doh-ura K, Ishikawa K, Murakami-Kubo I, Sasaki K, Mohri S, Race R, Iwaki T. Treatment of transmissible spongiform encephalopathy by intraventricular drug infusion in animal models. J Virol. 2004;78:4999–5006. doi: 10.1128/JVI.78.10.4999-5006.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996;15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, Asante EA, Farrow MA, Sessions RB, Saibil HR, Clarke AR, Rowan MJ, Walsh DM, Collinge J. Interaction between prion protein and toxic amyloid beta assemblies can be therapeutically targeted at multiple sites. Nat Commun. 2011;2:336. doi: 10.1038/ncomms1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Laurén J, Gimbel ZA, Strittmatter SM. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci. 2010;30:6367–6374. doi: 10.1523/JNEUROSCI.0395-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi M, Caughey B. Specific binding of normal prion protein to the scrapie form via a localized domain initiates its conversion to the protease-resistant state. EMBO J. 1999;18:3193–3203. doi: 10.1093/emboj/18.12.3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karapetyan YE, Saá P, Mahal SP, Sferrazza GF, Sherman A, Salès N, Weissmann C, Lasmézas CI. Prion strain discrimination based on rapid in vivo amplification and analysis by the cell panel assay. PLoS One. 2009;4:e5730. doi: 10.1371/journal.pone.0005730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT, Caughey B. Cell-free formation of protease-resistant prion protein. Nature. 1994;370:471–474. doi: 10.1038/370471a0. [DOI] [PubMed] [Google Scholar]
- Lau AL, Yam AY, Michelitsch MM, Wang X, Gao C, Goodson RJ, Shimizu R, Timoteo G, Hall J, Medina-Selby A, Coit D, McCoin C, Phelps B, Wu P, Hu C, Chien D, Peretz D. Characterization of prion protein (PrP)-derived peptides that discriminate full-length PrPSc from PrPC. Proc Natl Acad Sci U S A. 2007;104:11551–11556. doi: 10.1073/pnas.0704260104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J. 2007;26:548–558. doi: 10.1038/sj.emboj.7601507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MB, Geoghegan JC, Supattapone S. Dissociation of infectivity from seeding ability in prions with alternate docking mechanism. PLoS Pathog. 2011;7:e1002128. doi: 10.1371/journal.ppat.1002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroncini G, Kanu N, Solforosi L, Abalos G, Telling GC, Head M, Ironside J, Brockes JP, Burton DR, Williamson RA. Motif-grafted antibodies containing the replicative interface of cellular PrP are specific for PrPSc. Proc Natl Acad Sci U S A. 2004;101:10404–10409. doi: 10.1073/pnas.0403522101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissey MP, Shakhnovich EI. Evidence for the role of PrPC helix 1 in the hydrophilic seeding of prion aggregates. Proc Natl Acad Sci U S A. 1999;96:11293–11298. doi: 10.1073/pnas.96.20.11293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norstrom EM, Mastrianni JA. The charge structure of helix 1 in the prion protein regulates conversion to pathogenic PrPSc. J Virol. 2006;80:8521–8529. doi: 10.1128/JVI.00366-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunziante M, Gilch S, Schätzl HM. Essential role of the prion protein N terminus in subcellular trafficking and half-life of cellular prion protein. J Biol Chem. 2003;278:3726–3734. doi: 10.1074/jbc.M206313200. [DOI] [PubMed] [Google Scholar]
- Pan T, Wong BS, Liu T, Li R, Petersen RB, Sy MS. Cell-surface prion protein interacts with glycosaminoglycans. Biochem J. 2002;368:81–90. doi: 10.1042/BJ20020773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasupuleti M, Roupe M, Rydengård V, Surewicz K, Surewicz WK, Chalupka A, Malmsten M, Sörensen OE, Schmidtchen A. Antimicrobial activity of human prion protein is mediated by its N-terminal region. PLoS One. 2009;4:e7358. doi: 10.1371/journal.pone.0007358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauly PC, Harris DA. Copper stimulates endocytosis of the prion protein. J Biol Chem. 1998;273:33107–33110. doi: 10.1074/jbc.273.50.33107. [DOI] [PubMed] [Google Scholar]
- Peretz D, Williamson RA, Matsunaga Y, Serban H, Pinilla C, Bastidas RB, Rozenshteyn R, James TL, Houghten RA, Cohen FE, Prusiner SB, Burton DR. A conformational transition at the N terminus of the prion protein features in formation of the scrapie isoform. J Mol Biol. 1997;273:614–622. doi: 10.1006/jmbi.1997.1328. [DOI] [PubMed] [Google Scholar]
- Priola SA. Prion protein and species barriers in the transmissible spongiform encephalopathies. Biomed Pharmacother. 1999;53:27–33. doi: 10.1016/s0753-3322(99)80057-2. [DOI] [PubMed] [Google Scholar]
- Priola SA, Caughey B, Race RE, Chesebro B. Heterologous PrP molecules interfere with accumulation of protease-resistant PrP in scrapie-infected murine neuroblastoma cells. J Virol. 1994;68:4873–4878. doi: 10.1128/jvi.68.8.4873-4878.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB, McKinley MP, Bowman KA, Bolton DC, Bendheim PE, Groth DF, Glenner GG. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell. 1983;35:349–358. doi: 10.1016/0092-8674(83)90168-x. [DOI] [PubMed] [Google Scholar]
- Prusiner SB, Groth DF, Bolton DC, Kent SB, Hood LE. Purification and structural properties of a major scrapie prion protein. Cell. 1984;38:127–134. doi: 10.1016/0092-8674(84)90533-6. [DOI] [PubMed] [Google Scholar]
- Raymond GJ, Bossers A, Raymond LD, O'Rourke KI, McHolland LE, Bryant PK, 3rd, Miller MW, Williams ES, Smits M, Caughey B. Evidence of a molecular barrier limiting susceptibility of humans, cattle and sheep to chronic wasting disease. EMBO J. 2000;19:4425–4430. doi: 10.1093/emboj/19.17.4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resenberger UK, Harmeier A, Woerner AC, Goodman JL, Müller V, Krishnan R, Vabulas RM, Kretzschmar HA, Lindquist S, Hartl FU, Multhaup G, Winklhofer KF, Tatzelt J. The cellular prion protein mediates neurotoxic signalling of beta-sheet-rich conformers independent of prion replication. EMBO J. 2011;30:2057–2070. doi: 10.1038/emboj.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers M, Yehiely F, Scott M, Prusiner SB. Conversion of truncated and elongated prion proteins into the scrapie isoform in cultured cells. Proc Natl Acad Sci U S A. 1993;90:3182–3186. doi: 10.1073/pnas.90.8.3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg MK, Al-Doujaily H, Sharps B, Clarke AR, Collinge J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature. 2011;470:540–542. doi: 10.1038/nature09768. [DOI] [PubMed] [Google Scholar]
- Shmerling D, Hegyi I, Fischer M, Blättler T, Brandner S, Götz J, Rülicke T, Flechsig E, Cozzio A, von Mering C, Hangartner C, Aguzzi A, Weissmann C. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell. 1998;93:203–214. doi: 10.1016/s0092-8674(00)81572-x. [DOI] [PubMed] [Google Scholar]
- Shyng SL, Moulder KL, Lesko A, Harris DA. The N-terminal domain of a glycolipid-anchored prion protein is essential for its endocytosis via clathrin-coated pits. J Biol Chem. 1995;270:14793–14800. doi: 10.1074/jbc.270.24.14793. [DOI] [PubMed] [Google Scholar]
- Solforosi L, Bellon A, Schaller M, Cruite JT, Abalos GC, Williamson RA. Toward molecular dissection of PrPC–PrPSc interactions. J Biol Chem. 2007;282:7465–7471. doi: 10.1074/jbc.M610051200. [DOI] [PubMed] [Google Scholar]
- Solomon IH, Khatri N, Biasini E, Massignan T, Huettner JE, Harris DA. An N-terminal polybasic domain and cell surface localization are required for mutant prion protein toxicity. J Biol Chem. 2011;286:14724–14736. doi: 10.1074/jbc.M110.214973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunyach C, Jen A, Deng J, Fitzgerald KT, Frobert Y, Grassi J, McCaffrey MW, Morris R. The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J. 2003;22:3591–3601. doi: 10.1093/emboj/cdg344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supattapone S, Muramoto T, Legname G, Mehlhorn I, Cohen FE, DeArmond SJ, Prusiner SB, Scott MR. Identification of two prion protein regions that modify scrapie incubation time. J Virol. 2001;75:1408–1413. doi: 10.1128/JVI.75.3.1408-1413.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubner LM, Bienkiewicz EA, Copié V, Caughey B. Structure of the flexible amino-terminal domain of prion protein bound to a sulfated glycan. J Mol Biol. 2010;395:475–490. doi: 10.1016/j.jmb.2009.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DR, Hooper NM. The prion protein and lipid rafts. Mol Membr Biol. 2006;23:89–99. doi: 10.1080/09687860500449994. [DOI] [PubMed] [Google Scholar]
- Turnbaugh JA, Westergard L, Unterberger U, Biasini E, Harris DA. The N-terminal, polybasic region is critical for prion protein neuroprotective activity. PLoS One. 2011;6:e25675. doi: 10.1371/journal.pone.0025675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadia JS, Schaller M, Williamson RA, Dowdy SF. Pathologic prion protein infects cells by lipid-raft dependent macropinocytosis. PLoS One. 2008;3:e3314. doi: 10.1371/journal.pone.0003314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science. 2010;327:1132–1135. doi: 10.1126/science.1183748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner RG, Hundt C, Weiss S, Turnbull JE. Identification of the heparan sulfate binding sites in the cellular prion protein. J Biol Chem. 2002;277:18421–18430. doi: 10.1074/jbc.M110406200. [DOI] [PubMed] [Google Scholar]
- Weissmann C. The state of the prion. Nat Rev Microbiol. 2004;2:861–871. doi: 10.1038/nrmicro1025. [DOI] [PubMed] [Google Scholar]
- Westergard L, Turnbaugh JA, Harris DA. A nine amino acid domain is essential for mutant prion protein toxicity. J Neurosci. 2011;31:14005–14017. doi: 10.1523/JNEUROSCI.1243-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AR, Enever P, Tayebi M, Mushens R, Linehan J, Brandner S, Anstee D, Collinge J, Hawke S. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature. 2003;422:80–83. doi: 10.1038/nature01457. [DOI] [PubMed] [Google Scholar]