

Abstract
HSP90 is a central player in the folding and maturation of many proteins. More than two hundred HSP90 clients have been identified by classical biochemical techniques including important signaling proteins with high relevance to human cancer pathways. HSP90 inhibition has thus become an attractive therapeutic concept and multiple molecules are currently in clinical trials. It is therefore of fundamental biological and medical importance to identify, ideally, all HSP90 clients and HSP90 regulated proteins. To this end, we have taken a global and a chemical proteomic approach in geldanamycin treated cancer cell lines using stable isotope labeling with amino acids in cell culture and quantitative mass spectrometry. We identified >6200 proteins in four different human cell lines and ∼1600 proteins showed significant regulation upon drug treatment. Gene ontology and pathway/network analysis revealed common and cell-type specific regulatory effects with strong connections to unfolded protein binding and protein kinase activity. Of the 288 identified protein kinases, 98 were downregulated upon geldanamycin treatment including >50 kinases not formerly known to be regulated by HSP90. Protein turn-over measurements using pulsed stable isotope labeling with amino acids in cell culture showed that protein down-regulation by HSP90 inhibition correlates with protein half-life in many cases. Protein kinases show significantly shorter half lives than other proteins highlighting both challenges and opportunities for HSP90 inhibition in cancer therapy. The proteomic responses of the HSP90 drugs geldanamycin and PU-H71 were highly similar suggesting that both drugs work by similar molecular mechanisms. Using HSP90 immunoprecipitation, we validated several kinases (AXL, DDR1, TRIO) and other signaling proteins (BIRC6, ISG15, FLII), as novel clients of HSP90. Taken together, our study broadly defines the cellular proteome response to HSP90 inhibition and provides a rich resource for further investigation relevant for the treatment of cancer.
The protein HSP90 is a evolutionary conserved molecular chaperone that is abundantly and ubiquitously expressed in cells from bacteria to man. In concert with multiple cochaperones and other accessory proteins, its primary function is to assist in the proper folding of proteins and thereby helps to maintain the structural and functional integrity of the proteome (proteostasis). Over the past 30 years, more than 200 such “client” proteins have been identified using classical biochemical and biophysical methods (1–3) More recently, genome wide screens in yeast suggest that 10–20% of the yeast proteome may be regulated by HSP90 (1, 4). Therefore, not surprisingly HSP90 clients span a very wide range of protein classes (kinases, nuclear receptors, transcription factors etc.) and biological functions (signal transduction, steroid signaling, DNA damage, protein trafficking, assembly of protein complexes, innate immunity to name a few) (1, 2, 5). Because many HSP90 clients are key nodes of biological networks, HSP90 not only exercises important functions in normal protein homeostasis, but also in disease. Many HSP90 clients are oncogenes (EGFR, c.KIT, BCR-ABL etc.) that drive a wide range of cancers and whose cells have often become “addicted” to HSP90 function (1). The disruption of HSP90 function by small molecule drugs has therefore become an attractive therapeutic strategy and about a dozen of HSP90 inhibitors are currently undergoing clinical trials in a number of tumor entities and indications (2, 5, 6). Geldanamycin is the founding member of a group of HSP90 inhibitors that target the ATP binding pocket of HSP90 and block the chaperone cycle, which on the one hand leads to transcription factor activation and subsequent gene expression changes (e.g. HSF1) (7, 8) and, on the other hand, to proteasome mediated degradation of HSP90 substrates (5, 9). Experience from clinical trials shows that the efficacy and toxicity of HSP90 targeted therapy varies greatly between tumors suggesting that the current repertoire of client proteins and our understanding of drug mechanism of action is incomplete (10). To predict an individual patient's responsiveness, it would thus be highly desirable to identify the complete set of HSP90 regulated proteins. Because HSP90 directly (e.g. by degradation) and indirectly (e.g. by induction of gene/protein expression) affects proteostasis, proteomic approaches are particularly attractive for studying e.g. the HSP90 interactome and the global effects of HSP90 inhibition on cellular systems. A number of proteomic approaches have been taken to explore the HSP90 regulated proteome including global proteome profiling using two-dimensional gels and mass spectrometry (11) as well as focused proteomic experiments utilizing immunoprecipitation of HSP90 complexes and chemical precipitation using immobilized HSP90 inhibitors (12). These studies have identified some important new HSP90 clients but generally fail to provide a global view of HSP90 regulated proteome because the attained proteomic depth was very limited and many HSP90 interactions are too transient or of too weak affinity to be purified by these methods. Very recently, a report on the global proteomic and phosphoproteomic response of HeLa cells to the HSP90 inhibitor 17-dimethylaminoethylo-17-demethoxygeldanamycin (17-DMAG) has appeared in the online version of Molecular and Cellular Proteomics (13) indicating that the cellular effects of HSP90 inhibition are much larger than previously anticipated.
In this study, we have profiled the global response of the proteomes and kinomes of the four cancer cell lines K562, Colo205, Cal27, and MDAMB231 to the HSP90 inhibitor geldanamycin. Using a combination of stable isotope labeling in cell culture (14), core proteome profiling(15), chemical precipitation of kinases(16), and quantitative mass spectrometry (17), we identified >6200 proteins of which ∼1600 proteins showed common as well as cell type specific regulation upon drug treatment. Bioinformatic analysis enabled a functional organization of this data into protein pathways, networks and complexes highlighting many known and novel aspects of HSP90 function. Protein turn-over measurements using pulsed stable isotope labeling with amino acids in cell culture (SILAC)1 (18, 19) showed that, for a significant number of proteins, the rate of HSP90 inhibition induced protein down-regulation correlates with protein half-life and that protein kinases have significantly shorter half lives than other proteins with potentially important implications for HSP90 inhibition in cancer therapy. A comparison of the effects of geldanamycin and the phase I drug PU-H71 (20) suggests that both molecules work by similar molecular mechanisms. Using HSP90 immunoprecipitation and pulldowns with immobilized geldanamycin, we validated several kinases (AXL, DDR1, TRIO) and other signaling proteins (BIRC6, ISG15, FLII), as novel bona fide clients of HSP90. Collectively, the data demonstrate the value of the global drug profiling approach and provides a rich resource for future investigation in HSP90 dependent biological processes.
EXPERIMENTAL PROCEDURES
SILAC Labeling and Cell Culture
CAL27 and MDAMB231 cells were cultured in Dulbecco's modified Eagle's medium (4.5 g/l glucose) medium, K562 and COLO205 cells were cultured in Roswell Park Memorial Institute 1640 medium. For SILAC labeling cells were grown in normal medium deficient in Arginine and Lysine (PAA, Pasching, Austria) supplemented with either stable isotope encoded heavy Arginine and Lysine (Euriso-top) or normal Arginine and Lysine for the light. SILAC medium was supplemented with 10% dialyzed fetal bovine serum (Invitrogen®, Invitrogen, Darmstadt) and 200 mm l-proline (Sigma-Aldrich, Germany). Cells were cultured in humidified air supplemented with 10% CO2 at 37 °C. Cells were seeded at a density of 105 cells and maintained in culture for 24 h prior to treatment with Geldanamycin or Dulbecco's modified Eagle's medium. Pulsed SILAC experiments were performed as described previously (18, 19). Briefly, cells were grown cells in “light” medium until exponential phase, subsequently cells were switched to “heavy” medium and harvested at three time points (6, 12, and 24 h). SF268 cells expressing DDR1 isoform b (21) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (PAA, Pasching, Austria) and 150 μg/ml of Hygromycin B (PAA).
Drug Treatment and Harvesting
Geldanamycin (LC Laboratories, Woburn, MA) stock solution (20 mm) was prepared by dissolving it in dimethyl sulfoxide and used within 2 weeks. Cells were treated for 24 h with IC50 concentration of geldanamycin (GA) according the drug-response curve (5 μm concentration for K562, CAL27, and MDMBA231 and 10 μm concentration for COLO205, supplemental Fig. S1). The corresponding control groups were incubated with the same concentration of dimethyl sulfoxide (0.1%). Cells were washed with phosphate-buffered saline, 50 mm Tris/HCl pH 7.5, 5% Glycerol, 0.8% Nonidet P-40, and lysed with freshly added protease (SIGMAFAST, Sigma-Aldrich) and phosphatase inhibitors (Sigma-Aldrich, Munich, Germany). Homogenates were centrifuged at 6000 × g at 4 °C for 10 min followed by ultracentrifugation at 4 °C for 1h at 145,000 × g, supernatants were collected and aliquots were frozen in liquid nitrogen and stored at −80 °C until further use. Protein concentration in lysates was determined by the Bradford assay.
Sample Preparation for Full Proteome Analysis
One hundred micrograms of protein was reduced and alkylated by 10 mm dithiothreitol and 55 mm iodoacetamide before denaturing at 95 °C with NuPAGE® LDS Sample Buffer (Invitrogen, Darmstadt, Germany). Proteins were separated on a 4–12% NuPAGE gel (Invitrogen, Darmstadt, Germany) and gels were cut into 16 slices prior to in-gel trypsin digestion. In-gel trypsin digestion was performed according to standard procedures.
Kinobead Affinity Purification
Kinobead pulldowns were performed as described previously (16, 22). Briefly, cell lysates were diluted with equal volumes of 1× compound pulldown buffer (50 mm Tris/HCl pH 7.5, 5% glycerol, 1.5 mm MgCl2, 150 mm NaCl, 20 mm NaF, 1 mm sodium orthovanadate, 1 mm dithiothreitol, 5 mm calyculin A, and protease inhibitors). Lysates were further diluted if necessary to a final protein concentration of 5 mg/ml using 1 × compound pulldown buffer supplemented with 0.4% Nonidet P-40 followed by incubation with Kinobeads at 4 °C for 4 h. Subsequently, beads were washed with 1 × compound pulldown buffer and collected by centrifugation. Bound proteins were eluted with 2 × NuPAGE® LDS Sample Buffer (Invitrogen, Darmstadt, Germany) and eluates were reduced and alkylated by 10 mm dithiothreitol and 55 mm iodoacetamide. Samples were then run into a 4–12% NuPAGE gel (Invitrogen, Darmstadt, Germany) for about 1 cm to concentrate the sample prior to in-gel tryptic digestion. In-gel trypsin digestion was performed according to standard procedures.
HSP90 Immunoprecipitation and GA-NHS Affinity Purification
SILAC labeled CAL27 and MDAMB231 cells were lysed in 50 mm Tris/HCl pH 7.5, 5% Glycerol, 0.8% Nonidet P-40, 1.5 mm MgCl2, 150 mm NaCl, 1 mm Na3VO4, 25 mm NaF, and protease inhibitors (SIGMAFAST, Sigma-Aldrich). Homogenates were centrifuged at 6000 g at 4 °C for 10 min to remove cell debris. Cleared lysates were either incubated overnight at 4 °C with anti-HSP90 antibody (C20, SantaCruz) or the same source of normal IgG (mouse; Santa Cruz, Santa Cruz, CA) and both followed by incubating with protein A/G beads (SantaCruz) for another 4 h at 4 °C. After extensive washing, immunoprecipitates from heavy and light cells were boiled in LDS buffer, combined, and then separated on a NuPAGE Novex 4–12% Bis-Tris Mini Gel. Gel lanes containing separated immunocomplexes were cut into 12 slices and in-gel trypsin digestion was performed according to standard protocols. For GA-NHS affinity purification, the same cell lysate as immunoprecipitate were pre-incubated with either 25 μm Geldanamycin or 0.1% dimethyl sulfoxide for 1 h at 4 °C and Sepharose beads with the immobilized GA were subsequently added for another 1 h at 4 °C. The following steps were the same as immnoprecipitation mentioned above.
Liquid Chromatography Tandem MS (LC-MS/MS) Analysis
Nanoflow LC-MS/MS was performed by coupling an Eksigent nanoLC-Ultra 1D+ (Eksigent, Dublin, CA) to a LTQ-Orbitrap XL ETD (Thermo Scientific, Bremen, Germany). Tryptic peptides were dissolved in 20 μl 0.1% formic acid and 10 μl was injected for each analysis. Peptides were delivered to a trap column (100 μm i.d. × 2 cm, packed with 5 μm C18 resin, Reprosil PUR AQ, Dr. Maisch, Ammerbuch, Germany) at a flow rate of 5 μl/min in 100% buffer A (0.1% formic acid in HPLC grade water). After 10 min of loading and washing, peptides were transferred to an analytical column (75 μm × 40 cm C18 column Reprosil PUR AQ, 3 μm, Dr. Maisch, Ammerbuch, Germany) and separated using a 210 min gradient from 2% to 35% of buffer B (0.1% formic acid in acetonitrile) at 300 nL/minute flow rate. The LTQ-Orbitrap was operated in data dependent mode, automatically switching between MS and MS2. Full scan MS spectra were acquired in the Orbitrap at 60,000 resolution. Internal calibration was performed using the ion signal (Si(CH3)2O)6H+ at m/z 445.120025 present in ambient laboratory air. Tandem mass spectra were generated for up to eight peptide precursors in the linear ion trap for fragment by using collision-induced dissociation.
Peptide and Protein Quantification and Identification
Raw MS spectra were processed by Maxquant (version 1.1.1.25) for peak detection and quantification (23). MS/MS spectra was searched against the IPI human database human (version 3.68, 87,061 sequences) by Andromeda (24) search engine enabling contaminants and the reversed versions of all sequences with the following search parameters: Carbamidomethylation of cysteine residues as fixed modification and Acetyl (Protein N-term), Gln_pyro-Glu (N-Term Q), Glu_pyro-Glu(N-Term E), Oxidation (M), Phospho (ST), Phospho(Y) as variable modifications. Trypsin was specified as the proteolytic enzyme with up to 2 miss cleavages were allowed. The mass accuracy of the precursor ions was decided by the time-dependent recalibration algorithm of Maxquant, fragment ion mass tolerance was set to of 0.6 Da. The maximum false discovery rate for proteins and peptides was 0.01 and a minimum peptide length of six amino acids was required.
Statistical Analysis
Statistical analysis of quantified proteins was performed using R (version 2.12.1) (25). Raw protein abundance values were first normalized using Variance Stabilization Normalization (VSN) (26). VSN is able to stabilize the variance across the entire intensity range and addresses the error structure in the data. The application of VSN has previously been shown to be beneficial for MS-based quantification (27, 28). To investigate the data distribution and ensure the appropriate application of statistical tools, normal quantile-quantile plots were created for all protein intensities in each cell line. Variance stabilization normalization was applied to the data and variance-mean dependences were visually verified (supplemental Figs. S2 and S3). Differential expression of paired samples was assessed with a moderated linear model using the limma package (29) in Bioconductor (30). Differences in protein expression between “treated” and “control” samples were estimated with the least squares linear model fitting procedure and tested for differential expression with moderated Student's t-statistic via the empirical Bayesian statistics described in the limma package (29). We accepted or rejected the null hypothesis on the basis of p values computed for the omnibus B-statistic via limma at a specified significance level. p values were adjusted for multiple testing to control the false discover rate at 5%. For multiple testing adjustments, we calculated the false discover rate using the algorithm of Benjamini and Hochberg (31). p values, with appropriate multiple testing adjustment to control the false discovery rate at 5% allowed us to identify differentially expressed proteins.
GO Enrichment/Pathway Analysis/Complex Analysis
Classification and functional enrichment analysis of the identified proteins were performed using Database for Annotation, Visualization and Integrated Discovery (DAVID) Bioinformatics Database (32, 33) for the biological process (BP), molecular function (MF), and cellular component (CC). In order to make the functional categories more understandable, terms were clustered according to their functional similarity using REVIGO (34). Pathway membership of the identified proteins were analyzed by the Ingenuity Pathway Analysis (IPA) tool (Ingenuity Systems, Redwood City, CA, USA) for their functional significance and in the context of biological association networks. To investigate HSP90 targeting of many macromolecular complexes, we analyzed the list of significantly expressed proteins (adjusted p < 0.05) using Comprehensive Resource of Mammalian protein complexes (35), a database of manually curated and validated mammalian protein complexes.
Immunoblot Analysis
Anti-DDR1 and α-tubulin antibodies were purchased from Santa Cruz. For immunoblot analysis, cells were washed with cold phosphate-buffered saline and lysed in RIPA buffer. Protein concentration was determined by the Bradford assay. Fifty micrograms of lysate was mixed with an equal volume of 2 × NuPAGE® LDS sample buffer containing 10 mm dithiothreitol and bloiled for 5 min at 95 °C. Proteins were subsequently separated by 4–12% NuPAGE gel and transferred onto to polyvinylidene difluoride membranes (Invitrogen, Darmstadt, Germany). Membranes were blocked for 1 h in blocking solution (2% bovine serum albumin in 1×Tris Buffered Saline, TBS, 20 mm Tris-HCl, pH 7.4, 150 mm NaCl, and 0.1% Tween-20) at room temperature and probed overnight at 4 °C with the respective primary antibody. Immunoreactivity was detected using IRDye® conjugated secondary antibody (LI-COR®, Nebraska) and visualized by Odyssey imaging system (LI-COR).
RESULTS
High Quality Map of HSP90 Regulated Proteins
The idea behind the experimental strategy taken (Fig. 1) was to compare the proteomes of cancer cell lines in response to HSP90 inhibition by the small molecule drug GA. Proteins that are significantly down-regulated upon drug treatment should represent HSP90 clients, interactors and more complex regulatory events whereas proteins up-regulated upon drug treatment should primarily represent the latter category. In practice, human cell lines were SILAC labeled, “light” cells were treated with geldanamycin (5–10 μm for 24 h, supplemental Fig. S1) and “heavy” cells were left untreated. Cell lysates were mixed 1:1 and subjected in parallel to protein separation by 1D-SDS-PAGE and to the enrichment of kinases using an affinity reagent called kinobeads (a set of unselective kinase inhibitors immobilized on Sepharose beads, (16, 22)). Following digestion into tryptic peptides, protein identification and differential quantification was performed by LC-MS/MS on an LTQ-Orbitrap XL mass spectrometer and using the software package MaxQuant. Follwowing the above strategy, we analyzed four human cancer cell lines from different origin in the human body (Cal27, tongue; Colo205, colon, MDAMB231, breast; K562, blood). Each analysis was performed in biological triplicates to enable statistical analysis within and across cell lines. Collectively, 6283 proteins including 288 protein kinases were identified (Figs. 2A and 2B, supplemental Fig. S4 and S5, supplemental Tables S1 and S2) at a false discovery rate of <1%. For all subsequent data analysis and bioinformatic analysis, we only used proteins that were quantified in all three biological replicates of a particular cell line. Using these rigorous criteria, more than. 1600 proteins including 117 protein kinases were significantly (p < 0.05) up- or down-regulated following geldanamycin treatement (Fig. 2C, supplemental Fig. S6, supplemental Tables S3 and S4). The mass spectra and extracted ion chromatograms shown in Fig. 2D give examples of three protein kinases that are down-regulated (left, DDR1), not regulated (middle, CK2A2), or up-regulated (right, AURKA). Depending on the cell line, between 20% (MDAMB231 cells) and 30% (K562 cells) of all proteins were significantly regulated and, in all cell lines, more proteins were down-regulated (52–65%) than up-regulated (36–48%; supplemental Fig. S7). This observation was far more pronounced for protein kinases for which 89–92% were down regulated upon drug treatment (supplemental Fig. S8).
Fig. 1.
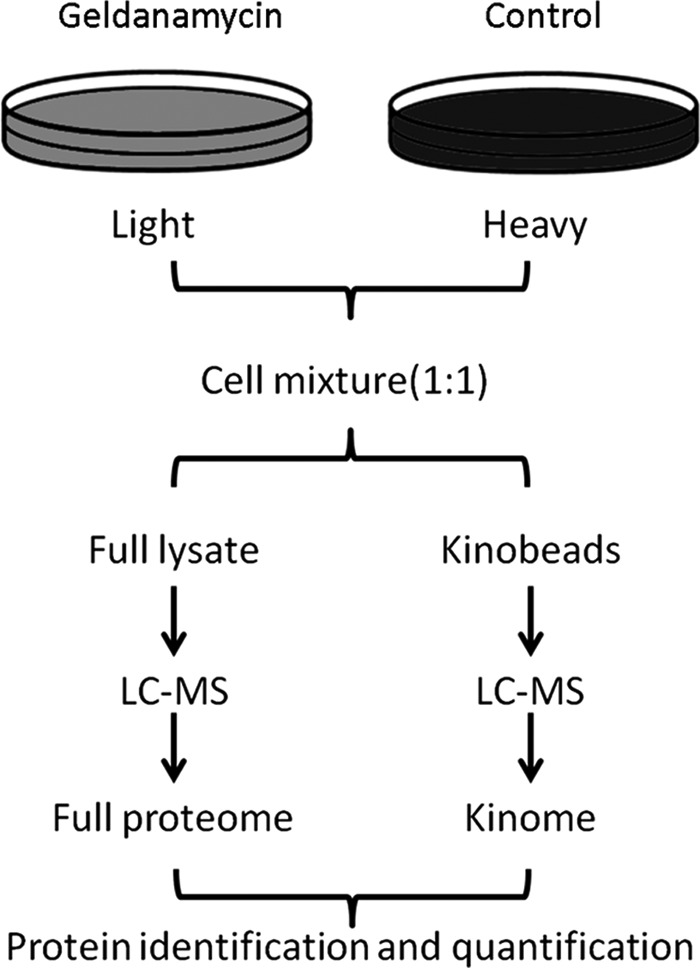
Experimental strategy for the identification of the HSP90 regulated proteome. Cells are grown in “light” and “heavy” SIALC medium. Light cells are treated with the HSP90 inhibitor geldanamycin. Treated and untreated cells are combined and full lysates are either separated by 1D gel electrophoresis (left branch) or first subjected to purification of kinases using kinobeads (right branch). Following trypsin digestion, both samples are analyzed by LC-MS/MS and proteins are subsequently identified and quantified. Each experiment was performed in biological triplicates.
Fig. 2.

Quantitative analysis of protein expression in four human cancer cell lines. A, protein identification summary from the four human cancer cell lines Cal 27 (headandneck), Colo205 (colon), MDAMB231 (breast) and K562 (blood). B, kinase identification summary for the same cell lines. C, volcano plot summarizing the protein quantification results in terms of the magnitude (log2foldchange >0 indicates down-regulation upon drug treatment) and significance (p value of <0.05 indicated by red line) of the observed protein expression changes. D, Example mass spectra (upper panel) and extracted ion chromatograms (lower panel) of drug induced down-regulated (left), not regulated (middle) and up-regulated (right) proteins. L and H denote peptide species containing either light or heavy amino acids.
HSP90 Regulated Proteome in Different Cell Lines
In order to organize the extensive list of HSP90 regulated proteins, we applied several layers of bioinformatic analysis. First, gene ontology (GO) analysis revealed that the ∼1600 regulated proteins across all four cell lines span a range of molecular functions and biological processes (Fig. 3A, supplemental Figs. S9–S13, supplemental Table S5). Expectedly, the most significant GO categories of the regulated proteins common to all four cell lines were unfolded protein binding (for up-regulated proteins) and kinase activity (for down-regulated proteins) thus validating the proteomic data given the well documented effect of HSP90 inhibition on proteins from these classes. Other common categories include among others “cell cycle” and “response to DNA damage.” For regulated proteins that are specific for a single cell line, the significant GO-categories are more diverse (supplemental Fig. S13, supplemental Table S5) likely representing the different biological backgrounds from which these cells are derived. For example, K562 cells are derived from progenitors of red blood cells and the GO-category “oxygen transport” only come up for this cell line. We next performed molecular pathway analysis in order to explore which cellular signaling pathways are affected by HSP90 inhibition. Again, expectedly, HSP90 inhibition affected multiple pathways and with strong differences between cell lines (Fig. 3B, supplemental Table S6). For example, the protein ubiquitination pathway is similarly up-regulated in all four cell lines reflecting the induction of degradation of HSP90 clients on HSP90 inhibition. Clathrin-mediated endocytosis signaling is a feature of many receptor-regulated processes and is increased in all solid cancer lines but not in the blood cell line K562. PPARα/RXRα activation is also implicated in several cancers and is increased in all cell lines but the colon line Colo205. For the down-regulated pathways, similar responses in e.g. the ERK/MAPK pathway are observed in all cell lines, which is not surprising given that many signaling events converge on this cascade. Ephrin receptor signaling is strongly down-regulated in all three solid tumor lines but not in the leukemia cell line likely because the latter cells do not require much capacity for cell-cell communication (a process in which Ephrin receptors are involved). Finally, down-regulation of mTOR signaling is much more pronounced in the colon cancer line Colo205 and the breast cancer line MDAMB231 compared with the other two cell lines. Collectively, the pathway analysis suggests that significant similarities and differences in the cellular response to HSP90 inhibition indeed do exist. Similarities and differences in the general signaling capacities of the different cell lines can also be observed when placing regulated proteins into molecular networks derived from the literature (supplemental Figs. S14 and S15, supplemental Table S7). Regulated networked proteins are often involved in the cell cycle, cell death, cellular growth, and proliferation, DNA replication, recombination, and repair. The identified networks around the tumor suppressor p53 from the breast cancer line MDAMB231 and the transcription factor complex NFkB from the head and neck line Cal27 are typical examples (supplemental Figs. S14 and S15). Given the large number of regulatory proteins in these networks, it is not hard to imagine that HSP90 inhibition will exert pleiotropic effects on cancer (and normal) cells. HSP90 itself engages in many protein complexes and our data recapitulates much of the known binary interactions including many (mostly down-regulated) kinases (Fig. 3C, supplemental Fig. S16). Interestingly, by querying the Comprehensive Resource of Mammalian protein complex database (35), we also found that members of protein complexes sometimes appear to be coregulated by HSP90 inhibition (supplemental Table S8). For example, core members of the kinase maturation complex 1 (HSP90a and b, CDC37, HSP70) were up-regulated upon drug treatment. We also detected six out of the seven members of the Arp2/3 complex (a cellular complex responsible for actin filament nucleation and branching) and all were up-regulated upon geldanamycin treatment. Examples for down-regulated protein complexes included the DNA-PK-Ku-eIF2-NF90-NF45 complex (DNA repair) the Nop56p-associated preribosomal ribonucleoprotein complex (ribosome biogenesis), the spliceosome (mRNA splicing), and the MNK1-eIF4F complex (protein translation initiation).
Fig. 3.

Bioinformatic analysis of the HSP90 regulated proteome part I. A, Results of GO term enrichment analysis of all four cell lines (colored). B, Example results of Ingenuity Pathway Analysis (IPA) across the geldanamycin regulated proteomes of the four cell lines analyzed. The histogram bars indicate the degree (significance) of overrepresentation of a particular pathway in a particular cell line. C, Binary protein-protein interaction map of HSP90alpha and HSP90beta extracted by IPA from the list of geldanamycin regulated proteins. Orange edges and nodes indicate down-regulation, black ones indicate up-regulation upon drug treatment (see supplemental Fig. S16 for a high resolution map).
In order to investigate if the observed similarities and differences of HSP90 inhibition in different cell lines are mainly caused by differences in protein expression (or p value cutoff), we performed an “all against all” comparison of proteins quantified in the four different cell lines. When comparing the SILAC ratios of proteins upon HSP90 inhibition of Cal27 and K562 cells (Fig. 4A, supplemental Fig. S17 for all other combinations), many proteins with similar expression levels (as measured by MS intensity, inset of Fig. 4A) also show similar behavior in both cell lines (e.g. HSP70, DDR1, YES). There is also a significant number of regulated proteins that are exclusively expressed in one of the cell lines (e.g. BTK, FRK) and there are proteins that show different response to HSP90 inhibition despite similar overall expression (e.g. AURA, PLK1). Because fold change measurement are often not reliable measures of significance, we also compared the cell lines by the p values of protein changes on drug treatment and obtained a similar overall picture (Fig. 4B, supplemental Fig. S18 for all other combinations). Although AURA is significantly changed in Cal27 cells, it is completely insignificant in K562 cells suggesting that the response of proteins to HSP90 inhibition does include a cell-type specific component. As mentioned previously, the GO analysis also revealed a very strong overrepresentation of protein kinases in the list of regulated proteins, ∼90% of which are down-regulated by geldanamycin treatment. Mapping all identified protein kinases onto the phylogenetic kinome tree (Fig. 4C) shows that the majority of kinases are actually unaffected (blue marks) at the drug concentration and during the time of treatment used in this study. Drug regulated kinases were found in all the major kinase classes (red, yellow, and green marks) but some branches of the TK and AGC classes are particularly rich in regulated kinases. We identified a lot of kinases previously reported to be HSP90 clients (yellow marks) but our data also adds a further 51 down-regulated kinases not previously implicated in HSP90 regulation (red marks). Remarkably, and somewhat unexpectedly, there are 15 up-regulated kinases (green marks) including AURA, PERK, and AXL (see discussion section).
Fig. 4.

Bioinformatic analysis of the HSP90 regulated proteome part II. A, Comparison of the cell lines Cal27 and K562 for the magnitude of protein regulation in response to HSP90 inhibition by geldanamycin (expressed as Log (fold change,2)). Proteins marked in orange show statistically significant regulation (p < 0.05), proteins marked in blue do not. B, same data as in (A) but here the significance of protein regulation is plotted (expressed as -Log (p value,10)). Proteins marked in blue show now significant response to HSP90 inhibition. Proteins marked in violet are significantly regulated in both cell lines. Proteins marked in green or red are significantly regulated in one of the two cell lines. Analogous plots for all cell line comparisons are shown in supplemental Figs. S17 and S18. C, Phylogenetic tree showing all protein kinases identified in this study. Kinases unaffected by HSP90 inhibition are marked in blue, up-regulated kinases in green, down-regulated known HSP90 clients in yellow, and down-regulated novel kinases in red (according to Piscard's list).
HSP90 Inhibition and Protein Turn Over
In order to validate the results obtained above using quantitative mass spectrometry, we repeated the geldanamycin treatment of cells and used Western blotting to follow the quantities of a number of proteins over the course of 24 h (Fig. 5A). The Western blot data for all 10 proteins was congruent with the MS data showing strong reduction of EPHA2, BTK (both known HSP90 clients), and DDR1, moderate reduction of ERK1 and BCAR1 and strong induction of AURA and AXL. STAT3 protein levels were not affected by HSP90 inhibition but phosphorylation of STAT3 was completely abolished within 3 h of drug treatment. These results show that the MS data can recapitulate known HSP90 clients and be interpreted in quantitative terms. The WB time course also showed that some proteins are much more rapidly removed from cells upon drug treatment than others. We note that the proteomic profiling and WB experiments cannot measure HSP90 inhibition induced client degradation directly but instead measure the effect of HSP90 inhibition on the global quantity of proteins in a cell. Because HSP90 inhibition should lead to the rapid degradation of newly synthesized HSP90 clients, we reasoned that the observed differences in kinetics of the loss of proteins from cells may be related to the individual protein turnover rate. We therefore conducted pulsed SILAC experiments without drug treatment in all four cell lines and enriched kinases using kinobeads (Fig. 5B), which allowed the determination of protein half lives from the kinetics of stable isotope incorporation into proteins (Fig. 5C, supplemental Tables S9 and S10). For example, a half-life of 7 h was determined for the protein kinase DDR1 (32 h for EGFR; Fig. 5C), which closely follows the time required to remove 50% of DDR1 from cells upon geldanamycin treatment (Fig. 5A). To generalize this analysis, we plotted the determined protein turn-over of kinases against the level of kinase down-regulation by HSP90 inhibition (24 h pulsed SILAC versus 24 h GA treatment; Fig. 5D). The plot shows that protein turn over and GA induced protein removal from cells correlate for many proteins (blue area, e.g. DDR1, EPHA2, EGFR, AKT) suggesting that only a small pool of these proteins is in complex with HSP90 at any one time so that the rate of drug induced removal from cells is primarily determined by the rate of their normal turn over. For proteins with different behavior (e.g. BTK, CSNK1D), more complex scenarios must be discussed (see discussion section). Interestingly, the determined half-lives of proteins ranged from a few to more than a hundred hours (Fig. 5E, supplemental Tables S9 and S10) and that protein kinases have generally much shorter half-lives than other protein classes suggesting that therapeutic HSP90 inhibition will have faster effect on kinase signaling pathways than other cellular activities.
Fig. 5.

Kinetics of geldanamycin induced proteins removal from cancer cells. A, This Western blot time course analysis of cells treated with geldanamycin shows that proteins are removed from cells at different rates. B, Schematic representation of a pulsed SILAC experiment. Cells are labeled with heavy amino acids for six, twelve of 24 h. Light and heavy samples for each time point were combined, kinases enriched by kinobeads and all proteins were analyzed by MS. Newly synthesized proteins are detected as heavy peptides (red) whereas old proteins are detected as light peptides (blue). C, Example mass spectra (upper panel) used for protein half-life determinations of the receptor tyrosine kinase DDR1 and additional examples for other proteins (lower panel). D, Correlation plot of kinase protein turnover (24h pulsed SILAC) and levels of kinase regulation induced by geldanamycin (24h treatment). Kinases in the blue zone show good correlation between turn over and drug treatment suggesting that newly synthesized proteins are rapidly degraded upon drug treatment and that the measurable pool of cellular kinases is removed from cells at the rate of normal turn over. Cellular levels of kinases in the gray zone are regulated in a more complex fashion. E, Comparison of protein half lives by different protein classes. Kinases have significantly shorter half lives than other protein classes.
Pharmacological Intervention with HSP90 Regulated Pathways
Geldanamycin is a very effective HSP90 inhibitor but also a quite toxic compound. Therefore, other HSP90 inhibitors have been developed with better drug-like properties. We hence asked the question if there are strong differences in specificity between geldanamycin and the fairly novel HSP90 inhibitor PU-H71 that is currently in phase I clinical trials. To effect this, we repeated the kinobead profiling experiment using both drugs (Fig. 6A). The GA and PU data sets show a very high degree of correlation (R2>0.9) suggesting that the mechanism by which these drugs inhibit HSP90 is similar. We next asked if global HSP90 inhibition by GA can be meaningfully combined with more targeted therapeutic approaches in cancers that are primarily driven by a specific oncogene (exemplified here by the head and neck cancer line Cal27 that is growth dependent on EGFR (36)). HSP90 inhibitors are quite toxic so reducing their dose would be clinically beneficial. Treating Cal27 cells with the highly selective EGFR inhibitor lapatinib or GA showed that a combination of the two drugs (fixed dose of 25 nm GA) is more effective than either drug alone (Fig. 6B) suggesting that such combination treatments might be effective (see also discussion). The molecular pathway analysis presented above (Fig. 3B) showed that mTOR signaling in Cal27 cells was strongly negatively affected by HSP90 inhibition, which would potentially represent yet another avenue for treating these cancer cells. To validate this hypothesis we used the potent dual PI3K/mTOR inhibitor PF-04691502, which very effectively killed Cal27 cells (and more so than the rather unspecific pan-SRC family inhibitor dasatinib). Given that the cells are growth dependent on EGFR and respond to lapatinib, this data suggests that EGFR signals into the PI3K/mTOR pathway however in an AKT independent fashion because the selective AKT inhibitor MK-2262 failed to kill the same cells (Fig. 6C). This experiment showed that the proteomic profiling data and subsequent bioinformatic analysis actually provide meaningful information that can be biologically and mechanistically interpreted.
Fig. 6.

Pharmacological intervention with HSP90 regulated processes. A, comparison of kinase expression regulated by the two ATP binding site HSP90 inhibitors geldanamycin and PU-H71 in Cal27 cells. The strong correlation suggests that both drugs work by mechanisms leading to a very similar outcome. B, Combination of HSP90 inhibition with targeted kinase inhibition. Cal27 cells are growth dependent on the receptor tyrosine kinase EGFR. Combined treatment of Cal27 cells with geldanamycin and the EGFR inhibitor lapatinib kills cells more effectively than either drug alone. C, Targeting the PI3K/mTOR pathway in Cal27 cells. HSP90 inhibition strongly affects the PI3K/mTOR pathway in Cal27 cells (see Fig. 3B). The dual PI3K/mTOR inhibitor PF-04691502 kills Cal27 cells in an AKT independent fashion (indicated by the inability of the AKT inhibitor MK-2262 to kill the cells) and with similar efficacy as the unspecific kinase inhibitor dasatinib.
Preliminary Validation of Novel HSP90 Regulated Proteins
Our global proteome profiling data does not distinguish between HSP90 regulated proteins, HSP90 client proteins, or HSP90 interactors. We therefore performed a series of affinity purification experiments to identify proteins that physically interact with HSP90. Immunoprecipitation using HSP90 antibodies and DDR1 Western blot detection showed that DDR1 is an interactor of HSP90 (and CDC37; Fig. 7A). Together with the strong regulation of DDR1 in response to geldanamycin treatment (Figs. 4A and 5A) we conclude that DDR1 is also a HSP90 client. Further HSP90 co-IP experiments using SILAC labeled Cal27 and MDAMB231 cells together with quantitative mass spectrometry (in biological triplicate) revealed ∼50 significantly enriched proteins including known and potential novel HSP90 interactors (Fig. 7B, supplemental Tables S11 and S12). As a complementary approach to HSP90 IPs, we additionally performed pull-down experiments using immobilized geldanamycin from SILAC labeled MDAMB231 cells (in biological triplicate). Again, the quantitative MS analysis identified about 40 significantly drug-enriched proteins including several known HSP90 interactors (supplemental Tables S13 and S14). Finally, we compared the obtained list of physical HSP90 interactors with the 1600 geldanamycin regulated proteins in order to examine which of these proteins may qualify as HSP90 clients. About 50 such proteins were identified that are currently our best validated set of novel HSP90 client proteins (Fig. 7C, supplemental Table S15).
Fig. 7.

HSP90 interactome. A, Co-immunoprecipitation and Western blot analysis reveals that DDR1 is a physical interactor of the HSP90/CDC37 complex. B, Results of HSP90 co-immunoprecipitation experiments and quantitative MS analysis from SILAC labeled Cal27 and MDAMB231 cells. The analysis in biological triplicates allowed for the identification of ∼50 known and novel high confidence HSP90 interactors. C, Graphical representation of the HSP90 interactome as obtained in this study. HSP90 isoforms are shown in the center, surrounded by cochaperones in the next layer, kinases in the next layer and other proteins of diverse functions in the outer layer.
DISCUSSION
We have studied the global effects the HSP90 inhibitor geldanamycin on the proteomes of four different cancer cells. This not only allowed us broadly to define the HSP90 regulated proteome but also enabled the identification of common as well as cell-type specific drug effects which will serve the community as a valuable starting point for further biological investigation. We identified >6300 proteins including almost 300 protein kinases and rigorous statistical analysis of the quantitative data showed that the expression levels of a rather unexpectedly high number of ∼1600 proteins was affected by the drug. Work in yeast has suggested that up to 10–20% of all proteins may be regulated by HSP90 (1, 4) and we find that this is also broadly the case in human cancer cells. Bioinformatic classification of the proteomic data faithfully recapitulated the well-known major effects of HSP90 inhibition on the up-regulation of the protein folding machinery and the strong down-regulation of kinase activity in the cell. Interestingly, bioinformatics also revealed similarities and differences between cancer cell types on the levels of signaling pathways, protein networks, and individual proteins, which has broad implications for the mode of action of the drug in these cells. This is likely also of clinical relevance because the efficacy of HSP90 inhibition varies a lot between tumor entities (10). In fact, up- as well as down-regulation of proteins in response to drug treatment could both be inhibiting (desired) and promoting (undesired) cancer cell survival and proliferation. Reduction in kinase activity will generally lead to cell death in cancer cells but the observed up-regulation of the receptor tyrosine kinase AXL may do the opposite (37). At the same time, we find that the tumor suppressor p53 is down-regulated in MDAMB231 cells, which might have adverse effects particularly if the drug is systemically used over extended periods of time (38). Likewise, the up-regulation of the chaperone machinery of the cell is probably not a desirable effect because cancer cell death may be delayed or require higher drug doses (39). Another noteworthy result of the global data analysis concerns the down-regulation of protein synthesis effected by the down-regulation of proteins all the way from ribosome biogenesis, mRNA splicing, and translation initiation. Very recently, a conceptually similar report on the global proteomic response of HeLa cells to the HSP90 inhibitor 17-DMAG has appeared in the online version of Molecular and Cellular Proteomics (13). Despite the many differences in experimental detail (different drug and dose, different cell line etc.; supplemental Fig. S19) both studies identified ∼500 commonly regulated proteins (supplemental Fig. S20) that represent the two major expected global effects of HSP90 inhibition on the cellular proteome (supplemental Fig. S21). Notably, the unfolded protein response is increasing and kinase associated processes are decreasing suggesting that the experimental strategy for the identification of the HSP90 regulated proteome is valid. There are further similarities e.g. the highlighted DNA damage response although this is more pronounced in HeLa cells, which are a commonly used model system to study DNA damage. The comparison of the four cell lines investigated in this study also revealed numerous differences and, not surprisingly, the Sharma study adds further complementary data most likely representing the different biologies of the different cell lines.
The very deep coverage of the cancer kinome (almost 300) obtained in this study allowed us to identify more than 50 protein kinases as potential novel targets of HSP90 inhibition. Although 90% of all kinases are down-regulated in response to the drug, 19 kinases showed the opposite behavior. Among these are a significant number of kinases involved in the cell cycle (AURA, PBK, PKAb, NEK2, ERK3, TTk, PKR). One of the effects of HSP90 inhibition is arresting cells in the cell cycle (G2/M), which may either be mediated by these kinases or result in their accumulation during the drug treatment (40, 41). Two of the kinases are involved in stress responses (OSR1, MST3), which may explain why they are up-regulated along with other components of the cellular stress response. We serendipitously discovered that some kinases were removed from cells much faster than others upon drug treatment, which led us to investigate this phenomenon in more detail. The proteomic approach employed in this study measures the global pool of a kinase (or other protein) in a cell. Hence, we cannot directly measure the pool of protein associated with HSP90 and the observed quantitative changes are a mixture of HSP90 inhibition induced degradation and other cellular activities such as ordinary protein turn-over, induction of protein expression etc. To address the influence of the former, we conducted pulsed SILAC experiments to determine the turn-over of individual proteins (mainly kinases, Fig. 5). For many of these, GA response and turnover correlate, which is consistent with an interpretation in which HSP90 inhibition leads to the rapid degradation of newly synthesized protein and the cellular pool of such proteins is depleted by their normal turn-over. For proteins that do not show this behavior, more complex scenarios must be discussed. The case in which proteins that are removed from cells by GA treatment at a faster rate than their normal protein turn-over may suggest that much of the cellular pool of the proteins is in complex with HSP90 (possibly indicating slow kinase maturation). In the reverse scenario, the regulated protein may be a HSP90 client but its expression is also induced by the drug treatment, which would result in an apparent slower removal from cells. Yet other proteins may simply accumulate in GA treated cells because the cells arrest in the cell cycle. Future experiments will have to aim at dissecting the different factors more clearly e.g. by separating synthesis and degradation rates using HSP90 inhibition in conjunction with translation or proteasomal inhibition. Interestingly, kinases turned out to have in general much shorter half-lives (about a factor 2) than other proteins, a fact that we believe has not been observed before. Given that kinase down-regulation is a major consequence of HSP90 inhibition, these differences in pharmacokinetics on individual targets may have several therapeutically relevant implications. First, it is likely that blocking kinase signaling cascades is the first and possibly major contributor to drug efficacy. Second, tumors that are driven by kinase of rapid turn-over rates (say AKT EPHA2, or DDR1, Fig. 5A) are likely to respond more quickly to the drug than tumors driven by longer lived proteins (e.g. EGRF). Third, rapid (or prolonged) removal of certain kinases may be undesirable. For example, BTK, which is an indispensable kinase for B lymphocyte development, differentiation, and signaling, will be very rapidly removed upon HSP90 targeted therapy and may therefore lead to a loss of function of immune cells of the innate as well as adaptive immune system (42, 43).
The observation that a large number of human proteins with diverse molecular functions appear to be affected by geldanamycin begs the question if different HSP90 inhibitors would have different target profiles possibly leading to different cellular outcomes. Our preliminary comparison of geldanamycin and the inhibitor PU-H71 show no drastic differences in their kinase profiles (Fig. 6). This may not be surprising as both drugs target the ATP binding site of HSP90 and thus their primary mechanism of cellular action should be similar. However, Moulick et al. recently showed by using immobilized HSP90 inhibitors (including PU-H71) that different inhibitors can favor distinct HSP90 client and co-chaperone complexes (44). This apparent discrepancy can be resolved by the fact that our kinobeads experiments purify kinases from cells (with or without interacting HSP90 and complex members) and thus monitor the outcome of the HSP90 inhibition by the drug rather than capturing the physical interaction. At least in principle, it should be possible to drug different parts of the HSP90 protein (2, 5). Once such molecules become more widely available, proteome-wide profiling experiments such as the ones described here should help to define their specificities. HSP90 inhibition is also being considered in the context of combination therapies in order to suppress resistance formation by “oncogene switching” (2, 45) or in order to extend the effective treatment time of targeted drugs such as kinase inhibitors (46). Our preliminary data using combinations of geldanamycin and the EGFR inhibitor lapatinib (Fig. 6) confirm earlier work that the drug combination is more effective than either drug alone but more work is required to establish if this concept can be generalized (47). A further pharmacologically interesting outcome of this proteomic study is the observation that certain signaling pathways (e.g. PI3K/mTOR) are more affected by HSP90 inhibition in some cancer cells than in others (Fig. 3). This may, on the one hand, merely reflect the overall activity of these pathways in the respective cell line but, on the other hand, may also highlight alternative therapeutic intervention points exemplified by the AKT independent killing of Cal27 cells by the dual PI3K/mTOR inhibitor PF-04691502 (Fig. 6). Alternatively, the discovery of highly active pathways in cancer cells using HSP90 drugs may also provide a means to sensitize cells for more targeted drugs.
It should be noted that the catalog of HSP90 regulated proteins reported here should not be mistaken for a list of HSP90 clients. However, it is likely that HSP90 clients and physical interactors are significantly represented in the catalog. In order to identify novel interaction partners, we used HSP90 immunoprecipitation and pull-down experiments employing immobilized geldanamycin. Although classical experiments, they are somewhat complicated by the fact that the affinity of most clients to HSP90 is not very strong (12) and they may therefore be difficult to retrieve within the time frame of the biochemical experiment. Nevertheless, the HSP90 IPs purified more than 30 known HSP90 interaction partners including many of the cochaperones and client kinases (Fig. 7). In addition, the experiments highlight about 20 significant potential novel interactors. Again, these include a number of kinases (AXL, TRIO) but also GTPases and activators (ARHGAP29, SMAP1, XPOT), metabolite transporters (ASCT2, SLC3A2, GLUT1) as well as other (signaling) proteins such as the apoptosis related proteins BIRC6. It is beyond the scope of this manuscript to discuss in detail how these proteins relate to HSP90 and cancer, but a few interesting observations can be made. The protein BAG2 inhibits the chaperone activity of HSP70/HSC70 by promoting substrate release (48). Whether or not BAG2 also binds to HSP90 or is co-purified with HSC70 is currently not known. EDRF1 is a transcription factor involved in erythroid differentiation (49) but its relationship to HSP90 has not been described. FLII is a coactivator in transcriptional activation by hormone-activated nuclear receptors and is involved in estrogen hormone signaling, which is particularly relevant for breast cancer (50). ISG15 is a ubiquitin-like protein that is conjugated to several intracellular target proteins including EIF2AK2, which was found in the same IP experiment. ISG15 may thus be a ubiquitin donor in HSP90 mediated protein degradation. Finally, BIRC6 is a protein that contains a BIR (baculoviral inhibition of apoptosis protein repeat) domain and a UBCc (ubiquitin-conjugating enzyme E2, catalytic) domain and functions as an apoptosis inhibitor by promoting the degradation of apoptotic proteins by ubiquitination (51). Although many HSP90 cochaperones have been identified that often confer substrate specificity, the number of ubiquitin ligases that form part of HSP90 complexes has only recently begun to expand from the established E3-ligase CHIP (44). It is therefore tempting to speculate that BIRC6 may be part of the HSP90 ubiquitination system. Recent literature also suggests that BIRC6 is a potential therapeutic target in colon cancer stem cells, which warrants further research on this protein (52). Around 50 of the proteins that were identified in HSP90 complexes (purified by HSP90 IP or immobilized GA) are also on the list of regulated proteins of the global proteome profiling experiments. These proteins represent our currently best validated novel HSP90 clients and include several eukaryotic translation initiation factors, the ubiquitin-like protein ISG15, the protein kinases ARAF and AXL, the transcriptional coactivator FLII, and the HSP70 inhibitor BAG2.
Conclusions and Outlook
The results of this study show that the systematic identification of HSP90 regulated proteins from cancer cells by quantitative proteomics provides valid data that includes novel biological information and from which multiple starting points for further research in the area can be defined. On the technical level, one logical extension of this work would be to repeat the geldanamycin profiling experiment in the presence and absence of heat stress or proteasomal and translation inhibitors, which may provide a sharper distinction between direct effects of HSP90 inhibition and more distantly related cellular responses to drug treatment. Profiling of further cell lines from other tumor entities, profiling tumor models in rodents and comparing the effects of different HSP90 drugs on the proteome are further attractive lines of future inquiry as these may provide clinically valuable insights into pharmacokinetic details that may be used to stratify tumor entities for HSP90 targeted therapy or to predict the response of an individual's tumor to such treatment.
Acknowledgments
ZW is grateful to the China Scholarship Council (CSC) and the Faculty Graduate Center Weihenstephan of the TUM Graduate School for supporting his studies in Germany. We thank Karl Kramer and Konrad Mueller for the provision of the cell line expressing DDR1, Matthias Selbach for fruitful discussions on the design of the pulsed SILAC experiments and Simone Lemeer for critically reading the manuscript.
Footnotes
 This article contains supplemental Figs. S1 to S21 and Tables S1 to S15.
This article contains supplemental Figs. S1 to S21 and Tables S1 to S15.
1 The abbreviations used are:
- SILAC
- stable isotope labeling with amino acids in cell culture
- PBS
- phosphate buffered saline
- REVIGO
- Reduce and Visualize Gene Ontology
- VSN
- variance stabilization normalization.
REFERENCES
- 1. Taipale M., Jarosz D. F., Lindquist S. (2010) HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 11, 515–528 [DOI] [PubMed] [Google Scholar]
- 2. Trepel J., Mollapour M., Giaccone G., Neckers L. (2010) Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 10, 537–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Picard D. HSP90 interactors. http://www.picard.ch/downloads/Hsp90interactors.pdf
- 4. Zhao R., Houry W. A. (2007) Molecular interaction network of the Hsp90 chaperone system. Adv. Exp. Med. Biol. 594, 27–36 [DOI] [PubMed] [Google Scholar]
- 5. Whitesell L., Lindquist S. L. (2005) HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 5, 761–772 [DOI] [PubMed] [Google Scholar]
- 6. Hao H., Naomoto Y., Bao X., Watanabe N., Sakurama K., Noma K., Motoki T., Tomono Y., Fukazawa T., Shirakawa Y., Yamatsuji T., Matsuoka J., Takaoka M. (2010) HSP90 and its inhibitors. Oncol. Rep. 23, 1483–1492 [DOI] [PubMed] [Google Scholar]
- 7. Zou J., Guo Y., Guettouche T., Smith D. F., Voellmy R. (1998) Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94, 471–480 [DOI] [PubMed] [Google Scholar]
- 8. Kim H. R., Kang H. S., Kim H. D. (1999) Geldanamycin induces heat shock protein expression through activation of HSF1 in K562 erythroleukemic cells. IUBMB Life 48, 429–433 [DOI] [PubMed] [Google Scholar]
- 9. Gray P. J., Jr., Prince T., Cheng J., Stevenson M. A., Calderwood S. K. (2008) Targeting the oncogene and kinome chaperone CDC37. Nat. Rev. Cancer 8, 491–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bohonowych J. E., Gopal U., Isaacs J. S. (2010) Hsp90 as a gatekeeper of tumor angiogenesis: clinical promise and potential pitfalls. J. Oncol. 2010, 412985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hartson S. D., Matts R. L. (2011) Approaches for defining the Hsp90-dependent proteome. Biochim. Biophys. Acta. 1823, 656–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tsaytler P. A., Krijgsveld J., Goerdayal S. S., Rüdiger S., Egmond M. R. (2009) Novel Hsp90 partners discovered using complementary proteomic approaches. Cell Stress Chaperones 14, 629–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sharma K., Vabulas R. M., Macek B., Pinkert S., Cox J., Mann M., Hartl F. U. (2011) Quantitative proteomics reveals that Hsp90 inhibition preferentially targets kinases and the DNA damage response. Mol. Cell. Proteomics 11, M111.014654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 15. Schirle M., Heurtier M. A., Kuster B. (2003) Profiling core proteomes of human cell lines by one-dimensional PAGE and liquid chromatography-tandem mass spectrometry. Mol. Cell. Proteomics 2, 1297–1305 [DOI] [PubMed] [Google Scholar]
- 16. Bantscheff M., Eberhard D., Abraham Y., Bastuck S., Boesche M., Hobson S., Mathieson T., Perrin J., Raida M., Rau C., Reader V., Sweetman G., Bauer A., Bouwmeester T., Hopf C., Kruse U., Neubauer G., Ramsden N., Rick J., Kuster B., Drewes G. (2007) Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 25, 1035–1044 [DOI] [PubMed] [Google Scholar]
- 17. Bantscheff M., Schirle M., Sweetman G., Rick J., Kuster B. (2007) Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 389, 1017–1031 [DOI] [PubMed] [Google Scholar]
- 18. Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 19. Schwanhäusser B., Gossen M., Dittmar G., Selbach M. (2009) Global analysis of cellular protein translation by pulsed SILAC. Proteomics 9, 205–209 [DOI] [PubMed] [Google Scholar]
- 20. Immormino R. M., Kang Y., Chiosis G., Gewirth D. T. (2006) Structural and quantum chemical studies of 8-aryl-sulfanyl adenine class Hsp90 inhibitors. J. Med. Chem. 49, 4953–4960 [DOI] [PubMed] [Google Scholar]
- 21. Lemeer S., Bluwstein A., Wu Z., Leberfinger J., Muller K., Kramer K., Kuster B. (2011) Phosphotyrosine mediated protein interactions of the discoidin domain receptor 1. J. Proteomics [DOI] [PubMed] [Google Scholar]
- 22. Wu Z., Doondeea J. B., Gholami A. M., Janning M. C., Lemeer S., Kramer K., Eccles S. A., Gollin S. M., Grenman R., Walch A., Feller S. M., Kuster B. (2011) Quantitative chemical proteomics reveals new potential drug targets in head and neck cancer. Mol. Cell. Proteomics 10, M111.011635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 24. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 25. Team R. D. C. (2011) R: A language and environment for statistical computing. R Foundation for Statistical Computing,. Vienna, Austria., ISBN 3-900051-07-0, URL ≤http://www.R-project.org/>. [Google Scholar]
- 26. Huber W., von Heydebreck A., Sültmann H., Poustka A., Vingron M. (2002) Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 18 Suppl 1, S96–104 [DOI] [PubMed] [Google Scholar]
- 27. Wu Z., Doondeea J. B., Moghaddas Gholami A., Janning M. C., Lemeer S., Kramer K., Eccles S. A., Gollin S. M., Grenman R., Walch A., Feller S. M., Kuster B. (2011) Quantitative chemical proteomics reveals new potential drug targets in head and neck cancer. Mol. Cell. Proteomics [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Karp N. A., Huber W., Sadowski P. G., Charles P. D., Hester S. V., Lilley K. S. (2010) Addressing accuracy and precision issues in iTRAQ quantitation. Mol. Cell. Proteomics 9, 1885–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smyth G. K. (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, Article3 [DOI] [PubMed] [Google Scholar]
- 30. Gentleman R. C., Carey V. J., Bates D. M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., Hornik K., Hothorn T., Huber W., Iacus S., Irizarry R., Leisch F., Li C., Maechler M., Rossini A. J., Sawitzki G., Smith C., Smyth G., Tierney L., Yang J. Y., Zhang J. (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Benjamini Y., Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 [Google Scholar]
- 32. Huang da W., Sherman B. T., Tan Q., Kir J., Liu D., Bryant D., Guo Y., Stephens R., Baseler M. W., Lane H. C., Lempicki R. A. (2007) DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 35, W169–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 34. Supek F., Bošnjak M., Škunca N., Šmuc T. (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6, e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ruepp A., Brauner B., Dunger-Kaltenbach I., Frishman G., Montrone C., Stransky M., Waegele B., Schmidt T., Doudieu O. N., Stümpflen V., Mewes H. W. (2008) CORUM: the comprehensive resource of mammalian protein complexes. Nucleic Acids Res. 36, D646–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Magné N., Fischel J. L., Dubreuil A., Formento P., Poupon M. F., Laurent-Puig P., Milano G. (2002) Influence of epidermal growth factor receptor (EGFR), p53 and intrinsic MAP kinase pathway status of tumour cells on the antiproliferative effect of ZD1839 (“Iressa”). Br. J. Cancer 86, 1518–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Linger R. M., Keating A. K., Earp H. S., Graham D. K. (2010) Taking aim at Mer and Axl receptor tyrosine kinases as novel therapeutic targets in solid tumors. Expert Opin. Ther. Targets 14, 1073–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Price J. T., Quinn J. M., Sims N. A., Vieusseux J., Waldeck K., Docherty S. E., Myers D., Nakamura A., Waltham M. C., Gillespie M. T., Thompson E. W. (2005) The heat shock protein 90 inhibitor, 17-allylamino-17-demethoxygeldanamycin, enhances osteoclast formation and potentiates bone metastasis of a human breast cancer cell line. Cancer Res. 65, 4929–4938 [DOI] [PubMed] [Google Scholar]
- 39. Guo F., Rocha K., Bali P., Pranpat M., Fiskus W., Boyapalle S., Kumaraswamy S., Balasis M., Greedy B., Armitage E. S., Lawrence N., Bhalla K. (2005) Abrogation of heat shock protein 70 induction as a strategy to increase antileukemia activity of heat shock protein 90 inhibitor 17-allylamino-demethoxy geldanamycin. Cancer Res. 65, 10536–10544 [DOI] [PubMed] [Google Scholar]
- 40. Kim H. R., Lee C. H., Choi Y. H., Kang H. S., Kim H. D. (1999) Geldanamycin induces cell cycle arrest in K562 erythroleukemic cells. IUBMB Life 48, 425–428 [DOI] [PubMed] [Google Scholar]
- 41. Jiang S., Katayama H., Wang J., Li S. A., Hong Y., Radvanyi L., Li J. J., Sen S. (2010) Estrogen-induced aurora kinase-A (AURKA) gene expression is activated by GATA-3 in estrogen receptor-positive breast cancer cells. Horm. Cancer 1, 11–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shinozaki F., Minami M., Chiba T., Suzuki M., Yoshimatsu K., Ichikawa Y., Terasawa K., Emori Y., Matsumoto K., Kurosaki T., Nakai A., Tanaka K., Minami Y. (2006) Depletion of hsp90beta induces multiple defects in B cell receptor signaling. J. Biol. Chem. 281, 16361–16369 [DOI] [PubMed] [Google Scholar]
- 43. Yorgin P. D., Hartson S. D., Fellah A. M., Scroggins B. T., Huang W., Katsanis E., Couchman J. M., Matts R. L., Whitesell L. (2000) Effects of geldanamycin, a heat-shock protein 90-binding agent, on T cell function and T cell nonreceptor protein tyrosine kinases. J. Immunol. 164, 2915–2923 [DOI] [PubMed] [Google Scholar]
- 44. Moulick K., Ahn J. H., Zong H., Rodina A., Cerchietti L., Gomes DaGama E. M., Caldas-Lopes E., Beebe K., Perna F., Hatzi K., Vu L. P., Zhao X., Zatorska D., Taldone T., Smith-Jones P., Alpaugh M., Gross S. S., Pillarsetty N., Ku T., Lewis J. S., Larson S. M., Levine R., Erdjument-Bromage H., Guzman M. L., Nimer S. D., Melnick A., Neckers L., Chiosis G. (2011) Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat. Chem. Biol. 7, 818–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shimamura T., Shapiro G. I. (2008) Heat shock protein 90 inhibition in lung cancer. J. Thorac. Oncol. 3, S152–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tauchi T., Okabe S., Ashihara E., Kimura S., Maekawa T., Ohyashiki K. (2011) Combined effects of novel heat shock protein 90 inhibitor NVP-AUY922 and nilotinib in a random mutagenesis screen. Oncogene 30, 2789–2797 [DOI] [PubMed] [Google Scholar]
- 47. LaBonte M. J., Wilson P. M., Fazzone W., Russell J., Louie S. G., El-Khoueiry A., Lenz H. J., Ladner R. D. (2011) The dual EGFR/HER2 inhibitor lapatinib synergistically enhances the antitumor activity of the histone deacetylase inhibitor panobinostat in colorectal cancer models. Cancer Res. 71, 3635–3648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Takayama S., Xie Z., Reed J. C. (1999) An evolutionarily conserved family of Hsp70/Hsc70 molecular chaperone regulators. J. Biol. Chem. 274, 781–786 [DOI] [PubMed] [Google Scholar]
- 49. Wang D., Li Y., Shen B. (2002) A novel erythroid differentiation related gene EDRF1 upregulating globin gene expression in HEL cells. Chin. Med. J. 115, 1701–1705 [PubMed] [Google Scholar]
- 50. Sakurai T., Kondoh N., Arai M., Hamada J., Yamada T., Kihara-Negishi F., Izawa T., Ohno H., Yamamoto M., Oikawa T. (2007) Functional roles of Fli-1, a member of the Ets family of transcription factors, in human breast malignancy. Cancer Sci. 98, 1775–1784 [DOI] [PubMed] [Google Scholar]
- 51. Hauser H. P., Bardroff M., Pyrowolakis G., Jentsch S. (1998) A giant ubiquitin-conjugating enzyme related to IAP apoptosis inhibitors. J. Cell Biol. 141, 1415–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Van Houdt W. J., Emmink B. L., Pham T. V., Piersma S. R., Verheem A., Vries R. G., Fratantoni S. A., Pronk A., Clevers H., Borel Rinkes I. H., Jimenez C. R., Kranenburg O. (2011) Comparative Proteomics of Colon Cancer Stem Cells and Differentiated Tumor Cells Identifies BIRC6 as a Potential Therapeutic Target. Mol. Cell. Proteomics 10, M111.011353 [DOI] [PMC free article] [PubMed] [Google Scholar]