

Abstract
The exchange for deuterium of the C-6 protons of uridine 5′-monophosphate (UMP) and 5-fluorouridine 5′-monophosphate (F-UMP) catalyzed by yeast orotidine 5′-monophosphate decarboxylase (ScOMPDC) at pD 6.5 – 9.3 and 25 °C was monitored by 1H NMR spectroscopy. Deuterium exchange proceeds by proton transfer from C-6 of the bound nucleotide to the deprotonated side chain of Lys-93 to give the enzyme-bound vinyl carbanion. The pD-rate profiles for kcat give turnover numbers for deuterium exchange into enzyme-bound UMP and F-UMP of 1.2 × 10−5 and 0.041 s−1, respectively, so that the 5-fluoro substituent results in a 3400-fold increase in the first-order rate constant for deuterium exchange. The binding of UMP and F-UMP to ScOMPDC results in 0.5 and 1.4 unit decreases, respectively, in the pKa of the side chain of the catalytic base Lys-93, showing that these nucleotides bind preferentially to the deprotonated enzyme. We also report the first carbon acid pKas for proton transfer from C-6 of uridine (pKCH = 28.8) and 5-fluorouridine (pKCH = 25.1) in aqueous solution. The stabilizing effects of the 5-fluoro substituent on C-6 carbanion formation in solution (5 kcal/mol) and at ScOMPDC (6 kcal/mol) are similar. The binding of UMP and F-UMP to ScOMPDC results in a greater than 5 × 109-fold increase in the equilibrium constant for proton transfer from C-6 so that ScOMPDC stabilizes the bound vinyl carbanions, relative to the bound nucleotides, by at least 13 kcal/mol. The pD-rate profile for kcat/Km for deuterium exchange into F-UMP gives the intrinsic second-order rate constant for exchange catalyzed by the deprotonated enzyme as 2300 M−1 s−1. This was used to calculate a total rate acceleration for ScOMPDC-catalyzed deuterium exchange of 3 × 1010 M−1, which corresponds to a transition state stabilization for deuterium exchange of 14 kcal/mol. We conclude that a large portion of the total transition state stabilization for the decarboxylation of orotidine 5′-monophosphate can be accounted for by stabilization of the enzyme-bound vinyl carbanion intermediate of the stepwise reaction.
INTRODUCTION
Orotidine 5′-monophosphate decarboxylase (OMPDC) is an essential enzyme that catalyzes the decarboxylation of orotidine 5′-monophosphate (OMP) to give uridine 5′-monophosphate (UMP), a critical step in the de novo biosynthesis of pyrimidine nucleotides (Scheme 1). In mammals, OMPDC is found as part of the bifunctional uridine 5′-monophosphate synthase complex, but in many lower organisms it exists as a discrete protein.1 The enzyme is an obligate dimer and residues from both subunits contribute to the active site architecture.2,3 Remarkably, OMPDC requires no metal ions or other cofactors but yet it effects an enormous 1017-fold acceleration of the decarboxylation of enzyme-bound OMP.3,4 A significant fraction of the total transition state stabilization for decarboxylation of ca. 31 kcal/mol3 results from utilization of the 12 kcal/mol intrinsic binding energy of the 5′-phosphodianion group of OMP.5–8
Scheme 1.

The first X-ray crystal structures for OMPDC appeared in 2000 and, notably, they revealed the absence of suitably placed catalytic residues required for several of the previously proposed reaction mechanisms.9–12 This precipitated a renewal of the scientific debate about the mechanism and the origin of the enzymatic rate acceleration for decarboxylation.13–19 The simplest possibility is the direct decarboxylation of OMP by a stepwise mechanism through the unstable UMP vinyl carbanion intermediate that undergoes subsequent proton transfer from the enzyme to generate the product (Scheme 1). We have argued that this mechanism is mandated by the observation of product isotope effects of unity for the decarboxylation of OMP and 5-fluoroorotidine 5′-monophosphate (F-OMP) in a mixture of 50:50 (v/v) H2O/D2O, which show that there is no discrimination between H and D at the proton donor in the transition state for the product-determining step (Scheme 2).20,21 These results eliminated a concerted reaction mechanism for decarboxylation in which proton transfer from the enzyme provides electrophilic push to the loss of CO2.9,13
Scheme 2.

We reported earlier that OMPDC from Saccharomyces cerevisiae (ScOMPDC) catalyzes the exchange for deuterium from solvent D2O of the C-6 proton of UMP to give UMP labeled with deuterium at C-6 (d-UMP, Scheme 2).22 These relatively slow reactions precluded extensive investigation of the kinetics and mechanism of OMPDC-catalyzed deuterium exchange into UMP. We reasoned that the addition of a 5-fluoro substituent to give 5-fluorouridine 5′-monophosphate (F-UMP) should result in stabilization of the negative charge at C-6 of the corresponding vinyl carbanion.23,24 Therefore, study of OMPDC-catalyzed deuterium exchange into F-UMP should be much more feasible than for UMP, and it would provide insight into the proton transfer reaction catalyzed by OMPDC. Our initial report of the OMPDC-catalyzed C-6 deuterium exchange reaction of F-UMP employed wildtype and the D70N and D70G mutants of OMPDC from Methanothermobacter thermautotrophicus.25
We present here a full report of our studies of the ScOMPDC-catalyzed C-6 deuterium exchange reaction of UMP,22 along with a complete set of data for the pD-dependence of the corresponding ScOMPDC-catalyzed C-6 deuterium exchange reaction of the more reactive substrate F-UMP. The results are consistent with a deuterium exchange reaction that is the formal reverse of the proton transfer “half reaction” that occurs during the decarboxylation of OMP, and they provide convincing support for our conclusion that the decarboxylation of OMP proceeds by a stepwise mechanism through the UMP vinyl carbanion intermediate (Scheme 2).20,21 We also report the first determination of reliable C-6 carbon acid pKas for a series of substituted uracils in aqueous solution, including the novel C-6 carbon acidities of the nucleosides uridine and 5-fluorouridine. A comparison of the data for the enzyme-catalyzed deuterium exchange reactions of UMP and F-UMP with those for proton transfer from uridine and 5-fluorouridine in aqueous solution shows that ScOMPDC stabilizes the bound UMP and F-UMP vinyl carbanions, relative to the bound nucleotides, by at least 13 kcal/mol. We conclude that a large portion of the rate acceleration for the decarboxylation of OMP can be accounted for by stabilization of the enzyme-bound vinyl carbanion intermediate of the stepwise reaction.
EXPERIMENTAL SECTION
Materials
Orotidine 5′-monophosphate was purchased from Sigma or was prepared by chemical or enzymatic methods using modifications of literature procedures.24,26,27 Uridine, uridine 5′-monophosphate disodium salt (99%, from yeast), 5-fluorouridine and 2-(N-morpholino)ethanesulfonic acid sodium salt (MES) were purchased from Sigma. 5-Fluoro-1,3-dimethyluracil, phosphorus(V) oxychloride and imidazole were purchased from Aldrich. Trimethyl phosphate, glycylglycine (> 99%) and 3-(N-morpholino)propanesulfonic acid (MOPS, ≥ 99.5%) were purchased from Fluka. 6-Azauridine 5′-monophosphate (free acid) was purchased from R. I. Chemical. Deuterium oxide (99.9% D), sodium deuteroxide (30% in D2O, 99.5% D) and deuterium chloride (35% w/w, 99.9% D) were purchased from Cambridge Isotope Laboratories. Bovine serum albumin (BSA) was from Roche and was dialyzed against 0.1 M NaCl in D2O. Trimethyl phosphate was dried over phosphorus pentoxide and distilled under reduced pressure. Imidazole was recrystallized from benzene. Water was from a Milli-Q Academic purification system. All other chemicals were reagent grade or better and were used without further purification.
5-Fluorouridine 5′-Monophosphate
5-Fluorouridine 5′-monophosphate was prepared as its triethylammonium salt from 5-fluorouridine using modifications of a literature procedure.28 Phosphorus(V) oxychloride (0.13 mL, 1.4 mmol) and 5-fluorouridine (0.20 g, 0.76 mmol) were dissolved in dry trimethyl phosphate (2 mL) at 0 °C. After 16 h the mixture was poured into cold water (100 mL), the solution was adjusted to pH 7 using 1 M NaOH, and water was added to give a total volume of 180 mL. The mixture was applied to a column of DEAE Sephadex A25 equilibrated with 50 mM triethylammonium bicarbonate and an elution gradient of 50 – 600 mM triethylammonium bicarbonate was applied. Fractions containing the desired product were pooled and the buffer was removed by lyophilization to give a 50% yield of 5-fluorouridine 5′-monophosphate as its triethylammonium salt. This material was shown to be free of inorganic phosphate by 31P NMR. 1H NMR (500 MHz, D2O, pD ≈ 7) δ7.83 (1H, 3JHF = 6.0 Hz, C6-H), 5.87 (1H, 3JHH = 5.0 Hz, 5JHF = 1.5 Hz, C1′-H), 4.21 (1H, m, C2′-H), 4.19 (1H, m, C3′-H), 4.08 (1H, m, C4′-H), 3.84 (2H, m, C5′-H). Chemical shifts are reported relative to HOD at 4.67 ppm.
Preparation of Solutions
Solution pH or pD was determined at 25 °C using an Orion Model 720A pH meter equipped with a Radiometer pHC4006-9 combination electrode that was standardized at 25 °C. Values of pD were obtained by adding 0.40 to the observed reading of the pH meter.29
The acidic protons of glycylglycine were exchanged for deuterium by dissolution in D2O followed by evaporation and drying under vacuum at 55 °C. Buffered solutions of glycylglycine or MOPS in D2O were prepared by dissolving the acidic form and NaCl (if needed) in D2O followed by the addition of a measured amount of NaOD to give the desired acid/base ratio at I = 0.1. Buffered solutions of imidazole or MES in D2O were prepared by dissolving the basic form and NaCl (if needed) in D2O followed by the addition of a measured amount of DCl to give the desired acid/base ratio at I = 0.1. Solutions in D2O were stored in a dessicator.
The concentration of OMP in stock solutions was determined from its absorbance in 0.1 M HCl at 267 nm using ε = 9430 M−1 cm−1.30 Stock UMP (100 mM) was prepared by dissolving the disodium salt in D2O to give a solution at pD 8.2 (I = 0.3) and its concentration was determined from the absorbance in 0.1 M HCl at 262 nm using ε = 10,000 M−1 cm−1.31 Stock solutions of F-UMP were prepared by dissolving the triethylammonium salt in D2O to give a solution at pD ≈ 8 and its concentration was determined from the absorbance in 0.1 M HCl at 270 nm using ε = 9160 M−1 cm−1 (see below). Stock 6-azauridine 5′-monophosphate (6-aza-UMP, 12 mM) was prepared by dissolving the free acid in D2O and the pD was adjusted to 7.9 by the addition of 4.3 M NaOD. Solutions of OMP, UMP, F-UMP and 6-aza-UMP were stored at −20 °C.
Extinction Coefficient of 5-Fluorouridine 5′-Monophosphate
A stock solution of F-UMP was prepared in D2O (pD ≈ 8) and was diluted 10-fold with 60 mM imidazole buffer (50% free base) in D2O at pD 7.6 and I = 0.1 (NaCl) to give a final concentration of 54 mM imidazole. The solution was analyzed by 1H NMR spectroscopy and the integrated areas of the signals due to the protons of F-UMP were compared with those due to the C-4 and C-5 protons of imidazole to give the concentration of F-UMP in the stock solution in D2O as 102 mM. The stock solution of F-UMP in D2O (102 mM) was then used to determine λmax = 270 nm and an extinction coefficient of ε = 9160 M−1 cm−1 for F-UMP in 0.1 M HCl.
pKa of the N-3 Hydron of 5-Fluorouridine 5′-Monophosphate in H2O and D2O (pKNL)
The acidity of the N-3 hydron of F-UMP in H2O and D2O was determined spectrophotometrically by monitoring the decrease in absorbance at 269 nm, Aobsd, that accompanies ionization at N-3. The absorbance of 163 μM F-UMP was determined as a function of pL in H2O or D2O at 25 °C and I = 0.1 (NaCl) using the following buffers: 0.1 M HCl, 0.01 M LCl, acetate (pH 4.7), MES (pD 6.4), imidazole (pD 7.0), MOPS (pL 7.1 – 7.4), glycylglycine (pL 7.6 – 9.4), glycine (pL 9.7 – 10.3), 0.01 M NaOL. The data were fit to eq 1, where Aacid and Abase are the absorbances at the acidic and basic extremes, to give the acidity constants KNL for ionization of F-UMP at N-3 in H2O and D2O.
| (1) |
Prepation of Yeast OMPDC in D2O
OMPDC from Saccharomyces cerevisiae (ScOMPDC) was prepared as described previously.32,33 The protein sequence differs from the published sequence for true wildtype yeast OMPDC by the following mutations: S2H,34 C155S,35 A160S and N267D.34 Except for the C155S mutation, the sequence is the same as that observed in the published crystal structure of wildtype yeast OMPDC.11 This C155S variant is more stable than, but kinetically and structurally essentially identical with, the Cys-155 enzyme.35 In this work, all experiments were conducted using this C155S variant.
Samples of overexpressed and purified ScOMPDC that had been stored at −80 °C were defrosted and extensively dialyzed at 4 °C against 10 mM MOPS (50% free base, pH 7.1) containing 100 mM NaCl in order to remove glycerol. This was followed by dialysis against several changes of buffer in D2O using a D-tube dialyzer (6 – 8 kDa MWCO, Novagen) placed inside a narrow vessel that was isolated from atmospheric moisture using parafilm. The buffers used for dialysis were: 62.5 or 125 mM glycylglycine, 80% free base (pD 9.3); 125 mM glycylglycine, 20% free base (pD 8.1); 100 mM MOPS, 50% free base or 60 mM imidazole, 50% free base (pD 7.6); 100 mM MOPS, 35% free base (pD 7.4); 60 or 125 mM imidazole, 20% free base (pD 7.0); 100 mM MES, 40% free base (pD 6.5). All buffers were in D2O at I = 0.1 (NaCl). The concentration of ScOMPDC in the stock solutions in D2O, determined by standard assay as described below, was 0.3 – 0.5 mM (9 – 15 mg/mL).
Enzyme Assays
The activity of ScOMPDC in the enzyme stock solutions in D2O and in the deuterium exchange reaction mixtures was determined by monitoring the decrease in absorbance at 279 nm accompanying the enzyme-catalyzed decarboxylation of OMP (Δε = −2400 M−1 cm−1 at 25 °C).2,7,22,32 Prior to assay, the stock solutions of ScOMPDC were diluted with 10 mM MOPS, 50% free base (pH 7.1) containing 100 mM NaCl and 0.4 mg/mL BSA to give a final concentration of ca. 20 μM ScOMPDC. Assays in a total volume of 1 mL were conducted in 10 mM MOPS, 50% free base (pH 7.1), at 25 °C and I = 0.105 (NaCl), with 40 – 50 μM OMP (25 – 30Km). The reaction was initiated by the addition of 1 SL of the diluted stock solution of ScOMPDC or an aliquot (up to 100 μL) of the deuterium exchange reaction mixture to give a final enzyme concentration of 20 – 40 nM, and the initial velocity of the ensuing decarboxylation of OMP was determined within 1 min. The concentration of ScOMPDC in the stock solution or in the deuterium exchange reaction mixture was then calculated from the observed value of Vmax (M s−1) using the relationship Vmax = kcat[E], with kcat = 15 s−1.2,7
NMR Analyses
1H NMR spectra at 500 MHz (8 – 128 transients) were acquired at 25 °C using a Varian Unity Inova 500 spectrometer with a sweep width of 6000 Hz, a 90° pulse angle, an acquisition time of 6 s and a total relaxation delay between pulses of 90 s (≥ 10T1) for the reactions of UMP or 30 s (≥ 9T1) for the reactions of F-UMP. Chemical shifts were referenced to HOD at 4.67 ppm. Values of T1 = 9 s and 7 s were determined for the C-5 and C-1′ protons, respectively, of [6-2H]-uridine 5′-monophosphate (d-UMP) in glycylglycine buffer at pD 9.4. Values of T1 = 1.5 s and 3.3 s were determined for the C-6 and C-1′ protons, respectively, of [6-1H]-5-fluorouridine 5′-monophosphate (h-F-UMP) in imidazole buffer at pD 7.6. Before determination of the integrated peak areas, the signals of interest were greatly expanded, accurately phased and subjected to a first-order drift correction.
Deuterium Exchange at C-6 of F-UMP Monitored by 1H NMR Spectroscopy
The C-6 deuterium exchange reactions of F-UMP catalyzed by ScOMPDC in D2O at pD 6.5 – 9.3 were followed directly by 1H NMR spectroscopy at 500 MHz. Reactions in a volume of 1 mL were initiated by the addition of an aliquot of F-UMP in D2O to a mixture of the appropriate buffer, NaCl, BSA and ScOMPDC in D2O to give final concentrations of 0.1 – 5 mM F-UMP, 0.4 mg/mL (0.04%) BSA (see below), and 0.2 – 40 μM ScOMPDC at I = 0.1 (NaCl). 700 μL of the reaction mixture was transferred to an NMR tube and the progress of deuterium exchange was monitored by 1H NMR spectroscopy at 25 °C for 2 – 8 h. The remaining portion of the reaction mixture was incubated at 25 °C and the activity of ScOMPDC was monitored by periodic standard assay as described above. After completion of the deuterium exchange reaction the enzyme was removed by ultrafiltration and the pD of the filtrate was recorded. There was no change in pD of the reaction mixture during these reactions at 25 °C which were followed for up to 8 h.
In all cases the concentration of enzyme used was chosen to accommodate the time needed to obtain at least 4 timepoints, with acquisition of an appropriate number of NMR transients, in one half-time for the deuterium exchange reaction. This required that lower concentrations of enzyme be used for the reactions at low concentrations of F-UMP for which more transients per timepoint were required for reasons of sensitivity. For the reactions of F-UMP in the presence of low concentrations of ScOMPDC (≥ 2 μM) in the absence of BSA there was up to a 25% decrease in the observed activity of ScOMPDC during the deuterium exchange reaction. However, the inclusion of 0.4 mg/mL (0.04%) BSA stabilized this loss of enzyme activity with time, which we attribute mainly to protein adsorption. There were also small decreases (up to 10%) in enzyme activity after extended reaction times in the presence of low concentrations of F-UMP (≤ Km) which we attribute to the instability of the dimeric form of unliganded ScOMPDC.2 A control experiment showed that the incubation of 10 μM ScOMPDC for 24 h at pD 9.4 (100 mM glycylglycine) and 25 °C resulted in a 15% loss in enzyme activity in the presence of 0.3 mM F-UMP (1.5Km) but no loss in enzyme activity in the presence of 2 mM F-UMP (10Km).
Deuterium Exchange at C-6 of UMP Monitored by 1H NMR Spectroscopy
The C-6 deuterium exchange reactions of UMP catalyzed by ScOMPDC in D2O at pD 7.0 – 9.3 were followed by 1H NMR spectroscopy at 500 MHz. Reactions in a volume of 1 – 4 mL were initiated by the addition of an aliquot of UMP in D2O to a mixture of the appropriate buffer, NaCl and ScOMPDC in D2O to give final concentrations of 2.5 – 10 mM UMP and 100 – 300 μM ScOMPDC at I = 0.1 (NaCl). The reaction mixture was divided into 1 mL portions that were placed in screw-cap centrifuge tubes equipped with O-rings and incubated in a water bath at 25 °C. At various times a tube was withdrawn and lightly centrifuged to sediment any particulate matter. An aliquot of the reaction mixture was removed and the enzyme-catalyzed deuterium exchange reaction was stopped by the addition of a 2 to 3-fold excess over enzyme of the tight-binding competitive inhibitor 6-aza-UMP (12 mM stock, pD 7.9). The enzyme-inhibitor complex, which was formed quantitatively, was removed by ultrafiltration using an Amicon Microcon device (10K MWCO) and the filtrate was transferred to an NMR tube. The extent of deuterium exchange was then determined by 1H NMR spectroscopy. After NMR analysis the sample was removed from the NMR tube and its pD was determined. The activity of ScOMPDC in the reaction mixture was monitored by periodic standard assay as described above. There was no significant loss of enzyme activity or change in pD of the reaction mixture during these reactions at 25 °C which were followed for up to 12 days.
RESULTS
Deuterium Exchange at C-6 of 5-Fluorouridine 5′-Monophosphate Catalyzed by Yeast OMPDC
The exchange for deuterium of the C-6 proton of [6-1H]-5-fluorouridine 5′-monophosphate (h-F-UMP) ([S]o = 0.1 – 5 mM) to give [6-2H]-5-fluorouridine 5′-monophosphate (d-F-UMP) catalyzed by OMPDC from Saccharomyces cerevisiae (ScOMPDC, 0.2 – 40 μM) in D2O at pD 6.5 – 9.3, 25 °C and I = 0.1 (NaCl) was followed directly by monitoring the disappearance of the C-6 proton by 1H NMR spectroscopy (Scheme 3).
Scheme 3.

Figure 1 shows partial 1H NMR spectra at 500 MHz in the regions of the C-6 proton (left) and C-1′ (anomeric) proton (right) obtained at various times during the exchange for deuterium of the C-6 proton of h-F-UMP ([S]o = 2.5 mM) catalyzed by ScOMPDC (9.2 μM) in D2O at pD 9.3 (100 mM glycylglycine, 80% free base), 25 °C and I = 0.1 (NaCl). The spectra on the left of Figure 1 show that deuterium exchange at C-6 results in the clean disappearance of the doublet at 7.874 ppm (3JHF = 6.2 Hz) due to the C-6 proton. By contrast, after 43% exchange of the C-6 proton, there is a complex appearance of the signals in the region of the C-1′ (anomeric) proton (Figure 1B, right). This results from overlap of the signals of roughly equal size due to the anomeric proton of h-F-UMP at 5.885 ppm (double doublet, 3JHH = 5.2 Hz, 5JHF = 1.9 Hz, Figure 1A) and that of d-F-UMP, which appears as an upfield-shifted (Δδ = 0.002 ppm) double doublet at 5.883 ppm (3JHH = 5.2 Hz, 5JHF = 1.9 Hz, Figure 1C). At late reaction times the signal in the anomeric region is again simplified and is due mainly to the upfield-shifted anomeric proton of the product d-F-UMP. A similar upfield shift of the broad doublet due to the anomeric proton of UMP as a result of deuterium incorporation at C-6 was noted in our earlier communication.22
Figure 1.

Partial 1H NMR spectra at 500 MHz in the regions of the C-6 proton (left) and C-1′ (anomeric) proton (right) obtained during exchange for deuterium of the C-6 proton of F-UMP ([S]o = 2.5 mM) catalyzed by ScOMPDC (9.2 μM) in D2O at pD 9.3 (100 mM glycylglycine, 80% free base), 25 °C and I = 0.1 (NaCl).
The progress of deuterium exchange into F-UMP was obtained from the integrated area of the doublet at 7.87 ppm due to the C-6 proton of h-F-UMP (AH), using the combined integrated areas of the signals due to the anomeric protons of both h-F-UMP and the product d-F-UMP at 5.88 ppm (AH+D) as an internal standard (Figure 1). Observed first-order rate constants for deuterium exchange were determined from the slopes of linear semi-logarithmic plots of reaction progress against time according to eq 2, covering 1 – 2 reaction half-times. Tables S1 – S6 of the Supporting Information give the values of kobsd (s−1) for exchange for deuterium of the C-6 proton of F-UMP in D2O at pD 6.45 – 9.33, 25 °C and I = 0.1 (NaCl) that were determined in these experiments.
| (2) |
Deuterium Exchange at C-6 of Uridine 5′-Monophosphate Catalyzed by Yeast OMPDC
The relatively slow exchange for deuterium of the C-6 proton of [6-1H]-uridine 5′-monophosphate (h-UMP) ([S]o = 2.5 – 10 mM) to give [6-2H]-uridine 5′-monophosphate (d-UMP) catalyzed by ScOMPDC (100 – 300 μM) in D2O at pD 7.0 – 9.3, 25 °C and I = 0.1 (NaCl) was followed by monitoring the appearance of the broad signal due to the C-5 proton of the product d-UMP by 1H NMR spectroscopy.22 The large coupling of the C-5 proton to the C-6 proton for h-UMP (3JHH = 8 Hz) is replaced by a small unresolved 3JHD coupling of the C-5 proton to deuterium at C-6 for d-UMP.22 The relatively high enzyme concentrations used in these experiments resulted in extremely broad 1H NMR signals for UMP so that the reactions could not be followed directly. At various times an aliquot of the reaction mixture was withdrawn and the reaction was stopped by the addition of a 2 to 3-fold excess over enzyme of the tight-binding competitive inhibitor 6-azauridine 5′-monophosphate (6-aza-UMP, Ki = 93 nM at pH 7.2).2,36 The enzyme•inhibitor complex, which was formed quantitatively, was removed by ultrafiltration and the filtrate was analyzed by 1H NMR spectroscopy.
The fractional extent of deuterium incorporation into UMP, f(d-UMP), was determined from analysis of the integrated areas of the signals due to the C-5 protons of h-UMP and the product d-UMP.22 Observed first-order rate constants for deuterium exchange were determined from the slopes of semi-logarithmic plots of the fraction of h-UMP remaining, f(h-UMP) = {1 - f(d-UMP)} against time, covering up to 30% reaction.22 The values of kobsd (s−1) for exchange for deuterium of the C-6 proton of UMP in D2O at pD 7.03 – 9.34, 25 °C and I = 0.1 (NaCl) were reported in the Supporting Information of the earlier communication.22 There was no detectable deuterium exchange into UMP (2 mM) after its incubation for 2 days in the presence of 260 μM ScOMPDC and 2 mM 6-aza-UMP at pD 9.3 (50 mM glycylglycine, 80% free base) at 25 °C. This shows that the observed enzyme-catalyzed deuterium exchange reactions occur at the active site of ScOMPDC.
The ScOMPDC-catalyzed deuterium exchange reactions of UMP at pD 7.6 – 9.3 were accompanied by up to 4% hydrolysis of the substrate 5′-phosphoryl group to give uridine. The very slow deuterium exchange reactions at pD 7.0 were accompanied by hydrolysis of both the 5′-phosphoryl group of UMP (up to 19%) and its glycosidic bond to give uracil (up to 24%). Control experiments conducted in the absence of ScOMPDC indicated that these side reactions are likely catalyzed by small contaminating enzyme activities present in our preparations of ScOMPDC. The rates of these hydrolysis reactions should be unaffected by the presence of deuterium at C-6 of UMP, so that they did not interfere with quantification of the extent of deuterium incorporation into the remaining UMP. However, at pD 7.0 in the presence of 2.5 mM UMP, the significant depletion of the pool of total UMP by these side reactions resulted in an apparent increase in the velocity of deuterium exchange with time (see kinetic analysis in the Discussion). In these cases, the observed rate constants for deuterium exchange were determined from the portion of the reaction that exhibited good first-order kinetics.22
Ionization of F-UMP and UMP at N-3
The pKa of the N-3 hydron of F-UMP in H2O and in D2O at 25 °C and I = 0.1 (NaCl) was determined spectrophotometrically by monitoring the absorbance of F-UMP at 269 nm as a function of pL. The data gave pKNH = 7.84 ± 0.03 in H2O and pKND = 8.37 ± 0.05 in D2O, at 25 °C and I = 0.1 (NaCl). The pKa of 7.84 for the N-3 proton of F-UMP is very similar to the reported pKas of 7.90 for the N-3 proton of 5-fluoro-2′-deoxyuridine 5′-monophosphate,2 and 7.67 for the N-3 proton of 5-fluoro-2′-deoxyuridine,37 in H2O at 25 °C and I = 0.1. This shows that there is little or no effect of a 2′-OH or a 5′-phosphoryl group on the pKa of the N-3 proton of simple uridine derivatives. The pKa of 9.3 reported for the N-3 proton of 2′-deoxyuridine in H2O at 25 °C and I = 0.1 (NaCl)37 and the 0.5 unit difference in the values of pKNH and pKND for F-UMP determined here can be used to estimate pKND = 9.8 for ionization of the N-3 hydron of UMP in D2O. Therefore, there is little dissociation of UMP at N-3 in the range of pD examined in our enzyme-catalyzed deuterium exchange reactions.
Nonenzymatic Deuterium Exchange at C-6 of Uridine, 5-Fluorouridine and 1,3-Dimethyl-5-Fluorouracil Catalyzed by Deuterioxide Ion in D2O
The exchange for deuterium of the C-6 protons of uridine, 5-fluorouridine and 1,3-dimethyl-5-fluorouracil at various temperatures and values of pD in D2O was followed by monitoring the disappearance of the C-6 proton of the substrate by 1H NMR spectroscopy, as described in the Supporting Information. The reactions of 5-fluorouridine in the presence of 1 M NaOD and of 1,3-dimethyl-5-fluorouracil at pD 10.2 were conducted at 25 °C. The slower reactions of uridine in the presence of 1 M NaOD and of 5-fluorouridine at pD 7.1 – 9.4 (I = 0.1) were conducted at 60 – 90 °C and rate constants for the reactions at 25 °C were obtained by extrapolation of Arrhenius plots (see the Supporting Information). Table S7 of the Supporting Information summarizes the observed second-order rate constants (kDO)obsd (M−1 s−1) for the deuterioxide-ion-catalyzed exchange reactions that were determined in these experiments. The data gave the following second-order rate constants kDO for deuterioxide-ion-catalyzed deuterium exchange at 25 °C (see Table S8 of the Supporting Information): 3.8 × 10−4 M−1 s−1 for uridine, 1.5 × 10−8 M−1 s−1 for uridine N-3 anion; 1.7 M−1 s−1 for 5-fluorouridine, 6.8 × 10−5 M−1 s−1 for 5-fluorouridine N-3 anion; 0.043 M−1 s−1 for 1,3-dimethyl-5-fluorouracil.
DISCUSSION
Kinetic Parameters for Enzyme-Catalyzed Deuterium Exchange into UMP and F-UMP
The velocity of enzyme-catalyzed deuterium exchange into h-UMP or h-F-UMP to give d-UMP or d-F-UMP (Scheme 3) is given by eq 3 derived for Scheme 4 (written for the reaction of UMP), where kex (s−1) is the turnover number for the E•h-UMP or E•h-F-UMP complex, [S]o is the total concentration of UMP or F-UMP irrespective of its isotopic composition, Kd is the binding constant for dissociation of the E•UMP or E•F-UMP complex, and [E] is the concentration of ScOMPDC.
Scheme 4.

| (3) |
| (4) |
| (5) |
Eq 4 relates the observed second-order rate constants for deuterium exchange, kobsd/[E] (M−1 s−1), to the kinetic parameters kex and Kd (Scheme 4), where kobsd is the observed first-order rate constant for deuterium exchange that was determined from the slope of a semi-logarithmic plot of reaction progress against time (see Experimental Section). This equation has two limits: (1) At [S]o ≫ Kd, the observed second-order rate constant for deuterium exchange is inversely proportional to [S]o, kobsd/[E] = kex/[S]o, where kex is the turnover number or the first-order rate constant for deuterium exchange into saturating enzyme-bound substrate. (2) At [S]o ≪ Kd, the observed second-order rate constant for deuterium exchange tends to the true second-order rate constant kex/Kd.
Figure 2 shows the dependence of kobsd/[E] (M−1 s−1) for deuterium exchange into F-UMP catalyzed by ScOMPDC (0.22 – 14 μM) on the total concentration of F-UMP in D2O at pD 8.15, 25 °C and I = 0.10 (NaCl). This observed second-order rate constant decreases as the concentration of F-UMP is increased, which is the expected behavior for an enzyme-catalyzed isotope exchange reaction for which the substrate and the product bind to the enzyme with essentially equal affinities (Kd, Scheme 4). The data were fit to eq 4 to give values of kex = 0.048 s−1 and Kd = 0.14 mM for deuterium exchange into F-UMP at pD 8.15.
Figure 2.
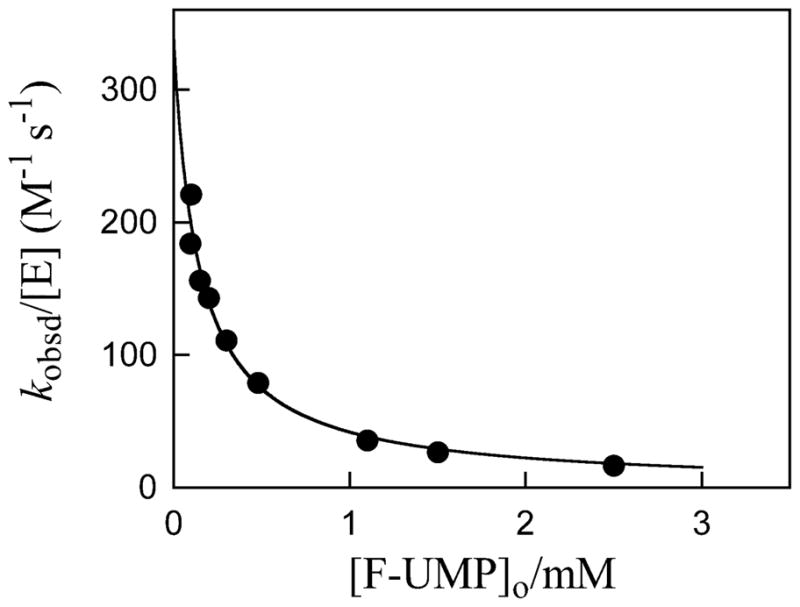
Dependence of the observed second-order rate constant kobsd/[E] (M−1 s−1) for exchange for deuterium of the C-6 proton of F-UMP catalyzed by ScOMPDC (0.22 – 14 μM) on the total concentration of F-UMP in D2O at pD 8.15, 25 °C and I = 0.10 (NaCl). The solid line shows the fit of the data to eq 4, which gives values of kex = 0.048 s−1 and Kd = 0.14 mM.
Eq 4 may be transformed to give eq 5 which describes the familiar hyperbolic dependence of enzymatic initial rate on total substrate concentration. Figure 3A shows the dependence of the quantity kobsd[F-UMP]o/[E] (s−1) for ScOMPDC-catalyzed deuterium exchange into F-UMP on the total concentration of F-UMP at pD 6.45 – 9.33, 25 °C and I = 0.1 (NaCl). The data were fit to eq 5 to give values of kex (s−1) for deuterium exchange into saturating enzyme-bound F-UMP, and Kd (M) for binding of F-UMP to ScOMPDC. Table 1 gives the values of kex (s−1), Kd (M) and the second-order rate constants kex/Kd (M−1 s−1) for enzyme-catalyzed deuterium exchange into F-UMP obtained from these fits.
Figure 3.

A. Dependence of kobsd[F-UMP]o/[E] (s−1) for exchange for deuterium of the C-6 proton of F-UMP catalyzed by ScOMPDC on the total concentration of F-UMP in D2O at 25 °C and I = 0.1 (NaCl). The solid lines show the fit of the data to eq 5 to give the values of kex (s−1) and Kd (M) given in Table 1. Key: ●, Data at pD 6.45; ○ data at pD 7.03; ■, data at pD 7.37;□, data at pD 7.64; ▲, data at pD 9.33; △, data at pD 8.15. B. Dependence of kobsd[UMP]o/[E] for exchange for deuterium of the C-6 proton of UMP catalyzed by ScOMPDC on the total concentration of UMP in D2O at 25 °C and I = 0.1 (NaCl). Key: ■, Data at pD 7.03; □, data at pD 7.58; ○, data at pD 8.13; ●, data at pD 9.34.
Table 1.
Kinetic Parameters for Exchange for Deuterium of the C-6 Proton of 5-Fluorouridine 5′-Monophosphate Catalyzed by ScOMPDC in D2O at 25 °C and I = 0.1 (NaCl).a
| pD b | kex c s−1 | Kd d mM | kex/Kd e M−1 s−1 |
|---|---|---|---|
| 9.33 | 3.84 × 10−2 | 0.19 | 200 |
| 8.15 | 4.38 × 10−2 | 0.11 | 400 |
| 7.64 | 3.24 × 10−2 | 0.11 | 300 |
| 7.37 | 2.42 × 10−2 | 0.17 | 140 |
| 7.03 | 1.69 × 10−2 | 0.51 | 33 |
| 6.45 | 8.18 × 10−3 | 0.99 | 8.3 |
Observed first-order rate constants, kobsd (s−1), for deuterium exchange into F-UMP were determined by monitoring the disappearance of the C-6 proton of F-UMP by 1H NMR spectroscopy (Tables S1 – S6 of the Supporting Information).
Average value of pD for experiments at several values of [F-UMP]o.
Turnover number for deuterium exchange into enzyme-bound F-UMP, obtained from the nonlinear least squares fits of the values of kobsd to eq 5 (Figure 3A). The estimated error in these rate constants is ± 10%.
Dissociation constant for binding of F-UMP to ScOMPDC, obtained from the nonlinear least squares fits of the values of kobsd to eq 5 (Figure 3A). The estimated error in these binding constants is ± 20%.
Second-order rate constant for enzyme-catalyzed deuterium exchange into F-UMP, calculated as the ratio of the values of kex and Kd. The estimated error in these rate constants is ± 25%.
The observed second-order rate constants kobsd/[E] (M−1 s−1) for the deuterium exchange reactions of h-UMP to give d-UMP at [S]o = 2.5 – 10 mM are inversely proportional to the total concentration of UMP which shows that [S]o ≫ Kd under these conditions (eq 4). Figure 3B shows the dependence of the quantity kobsd[UMP]o/[E] for ScOMPDC-catalyzed deuterium exchange into UMP on the total concentration of UMP at pD 7.03 – 9.34, 25 °C and I = 0.1 (NaCl). Table 2 gives the first-order rate constants kex (s−1) for enzyme-catalyzed deuterium exchange into saturating enzyme-bound UMP that were determined as the average of the values of kobsd[UMP]o/[E] at each pD ([UMP]o ≫ Kd, eq 5).
Table 2.
Kinetic Parameters for Exchange for Deuterium of the C-6 Proton of Uridine 5′-Monophosphate Catalyzed by ScOMPDC in D2O at 25 °C and I = 0.1 (NaCl).a
Observed first-order rate constants, kobsd (s−1), for deuterium exchange into UMP were determined by monitoring the appearance of deuterium at the C-6 position by 1H NMR spectroscopy [Ref. 22].
Average value of pD for experiments at several values of [UMP]o.
pD-Rate Profiles for Deuterium Exchange at C-6 of F-UMP and UMP
Figure 4 shows the pD-rate profile for the second-order rate constant kex/Kd (M−1 s−1) for ScOMPDC-catalyzed deuterium exchange at C-6 of F-UMP in D2O, constructed using the data in Table 1. This pD-rate profile is bell-shaped and the slope of the ascending limb at low pD approaches 2.0, which shows that the second-order rate constant for enzyme-catalyzed deuterium exchange into F-UMP is dependent on the deprotonation of two groups at the free enzyme/substrate. We assign the downward break at high pD to ionization of the phosphodianion form of F-UMP at N-3 to give the substrate trianion (KSD, Scheme 5), for which we have determined pKSD = 8.37 in D2O under the experimental conditions for deuterium exchange (see Results).
Figure 4.

pD-Rate profile for the second-order rate constant kex/Kd (M−1 s−1) for exchange for deuterium of the C-6 proton of F-UMP catalyzed by ScOMPDC in D2O at 25 °C and I = 0.1 (NaCl). The solid line shows the fit of the data to eq 6 with the values of pKSD2 = 6.8 and pKSD = 8.37 to give pKE = 8.5 ± 0.2 for deprotonation of the essential residue at the free enzyme and (kex)max/Kd = 2300 ± 700 M−1 s−1 as the intrinsic second-order rate constant for the deuterium exchange reaction.
Scheme 5.

The pH-rate profile for the second-order rate constant kcat/Km (M−1 s−1) for decarboxylation of the natural substrate OMP by ScOMPDC in H2O depends on the deprotonation of a group with an apparent pKa of 6.1 which is very similar to the pKa of 6.38 for the phosphoryl group of OMP determined by titration under the same conditions (25 °C, I = 0.1).2 The X-ray crystal structure of ScOMPDC liganded with the potent inhibitor 6-hydroxyuridine 5′-monophosphate (BMP) shows hydrogen bonds between the ligand phosphoryl group and the side chains of Gln-215, Tyr-217 and Arg-235, and the backbone NHs of Arg-235 and Gly-234 (see Figure 6). This strongly implies that the enzyme binds and turns over the phosphodianion form of the substrate.8,11,32 Therefore, a very reasonable assignment for the apparent pKa of 6.1 in the pH-rate profile for kcat/Km for decarboxylation of OMP is the second ionization of the phosphoryl group of OMP to give the substrate phosphodianion.2 By analogy, we assign the two ionizations at the free substrate/enzyme evident in the ascending limb at low pD in the pD-rate profile for kex/Kd for deuterium exchange into F-UMP (Figure 4) as ionization of the substrate phosphoryl monoanion (SD2−, Scheme 5) to give the phosphoryl dianion (SD2−), and the deprotonation of a group at the free enzyme to give the basic form of the enzyme.
Figure 6.

Schematic showing the contacts between yeast ScOMPDC and bound 6-hydroxyuridine 5′-monophosphate constructed using X-ray crystallographic data (PDB entry 1DQX). The phosphodianion binding motif consists of the side chains of Gln-215 and Tyr-217 in the mobile loop (loop 7), the side chain of Arg-235, and the backbone NHs of Gly-234 and Arg-235. The completely conserved “catalytic motif” (DxKxxD) consists of the side chains of Asp-91, Lys-93 and Asp-96′ of the second subunit.
The pKas of 6.26 and 6.25 for the phosphoryl groups of UMP and ribose 5′-monophosphate, respectively, determined by titration at 25 °C and I = 0.1,2 are essentially identical, which shows that the ionization of this group is relatively insensitive to the presence of the pyrimidine ring. Therefore, we estimate pKSD2 = 6.8 for ionization of the phosphoryl group of F-UMP in D2O at I = 0.1 (Scheme 5, X = F), based on the pKa of 6.3 for the phosphoryl group of UMP in H2O at I = 0.1,2 and the 0.5 unit higher pKa of phosphate monoanion in D2O than in H2O.38,39
Scheme 6 shows our proposed kinetic model for the ScOMPDC-catalyzed deuterium exchange reactions of F-UMP and UMP that is consistent with the observed bell-shaped pD-rate profile for kex/Kd for F-UMP (Figure 4). In this model the ionization of the substrate phosphoryl monoanion to give the phosphodianion (KSD2) is required for binding and reaction of the substrate, and the deprotonation of an acidic residue at the enzyme (KE) is required to generate a Brønsted base for the proton abstraction from C-6 of the substrate, a necessary step for deuterium exchange. Further ionization of the substrate at N-3 (pKSD = 8.37) to give the F-UMP trianion results in a form of the substrate that is unable to bind and react, as evidenced by the downward break in the pD-rate profile at high pD (Figure 4). The values of kex/Kd for deuterium exchange into F-UMP were fit to eq 6, derived for Scheme 6, with the values of pKSD2 = 6.8 and pKSD = 8.37 to give pKE = 8.5 ± 0.2 for deprotonation of the essential residue at the free enzyme. The fit also gives (kex)max/Kd = 2300 ± 700 M−1 s−1 as the intrinsic second-order rate constant for the deuterium exchange reaction of the phosphodianion form of F-UMP (SD2−) catalyzed by the deprotonated enzyme (E). This intrinsic rate constant is ca. 6-fold larger than the largest experimental second-order rate constant for the deuterium exchange reaction, observed at pD 8.15 (Table 1).
Scheme 6.

| (6) |
| (7) |
Figure 5 shows the pD-rate profiles for the first-order rate constants kex (s−1) for the ScOMPDC-catalyzed deuterium exchange at C-6 of enzyme-bound F-UMP and UMP in D2O, constructed using the data in Tables 1 and 2. As required by the model in Scheme 6, these profiles exhibit a dependence on the ionization state of only the enzyme-substrate complex ED+•SD2−, and they are consistent with the deprotonation of an essential residue at this complex to generate a Brønsted base for proton abstraction from C-6 of the substrate (KES, Scheme 6). The values of kex for F-UMP and UMP were fit to eq 7, derived for Scheme 6, to give pKES = 7.1 ± 0.1 and 8.0 ± 0.1 for deprotonation of the essential residue at the ED+•F-UMP and ED+•UMP complexes, respectively. The data also give the intrinsic first-order rate constants (turnover numbers) for deuterium exchange into saturating F-UMP and UMP bound to the deprotonated enzyme as (kex)max = 0.041 s−1 and 1.2 × 10−5 s−1, respectively. Therefore, the addition of a 5-fluoro substituent results in a 3400-fold increase in the rate constant for deuterium exchange into saturating enzyme-bound UMP at the complex with the basic form of the enzyme (E•SD2−, Scheme 6).
Figure 5.

pD-Rate profiles for the first-order rate constants kex (s−1) for exchange for deuterium of the C-6 proton of enzyme-bound F-UMP and UMP catalyzed by ScOMPDC in D2O at 25 °C and I = 0.1 (NaCl). The solid lines show the fits of the data to eq 7. A. Data for F-UMP which give pKES = 7.1 ± 0.1 and (kex)max = 0.041 s−1. B. Data for UMP which give pKES = 8.0 ± 0.1 and (kex)max = 1.2 × 10−5 s−1.
Identity of the Catalytic Base for Deuterium Exchange
The first X-ray crystal structures of OMPDC appeared in 2000 and they strongly implicated the protonated side chain of Lys-93 (numbering for the yeast enzyme) as the catalytic Brønsted acid needed to deliver the proton incorporated at C-6 during the decarboxylation of OMP to give UMP (Scheme 2).9–13 Figure 6, based on the X-ray crystal structure of ScOMPDC complexed with BMP,11 shows that Lys-93 at ScOMPDC is part of the strictly conserved DxKxxD “catalytic motif” that is found in all OMPDCs,40 the integrity of which is essential for robust decarboxylase activity.25,41 The hydrogen bond between the ammonium group of Lys-93 and the anionic oxygen at C-6 of BMP, an analog of the putative carbanion intermediate,36 is consistent with its proposed role as a Brønsted acid in the decarboxylation of OMP.9–12 This is in accord with the results of earlier mutagenesis studies of yeast OMPDC that showed that Lys-93 is essential for robust enzymatic activity. The inactive K93C mutant was rescued by covalent modification of the Cys-93 side chain by 2-bromoethylamine, which restores a primary amino group to the side chain at position 93.42 More recently, the OMPDCs from M. thermautotrophicus (MtOMPDC), Plasmodium falciparum (PfOMPDC) and the OMPDC domain of the bifunctional human UMP synthase (HsOMPDC)43 have been shown to undergo covalent modification by nucleophilic attack of the side chains of Lys-72, Lys-138 and Lys-314, respectively, at C-6 of bound 6-iodouridine 5′-monophosphate (6-I-UMP) and 6-azidouridine 5′-monophosphate (6-N3-UMP), resulting in displacement of the good leaving groups iodide and azide ions (Scheme 7).44–48 These lysine residues correspond to Lys-93 of the enzyme from yeast. The results of these structural and chemical studies strongly suggest that Lys-93 at the yeast enzyme functions not only to provide the proton incorporated at C-6 during the decarboxylation of OMP, but also as a Brønsted base in the reverse proton transfer “half reaction” that occurs as the first step of the C-6 deuterium exchange reactions of UMP and F-UMP (Scheme 2).
Scheme 7.

Figure 7 shows the X-ray crystal structures of wildtype HsOMPDC liganded by UMP and the D312N (D91N at ScOMPDC) mutant liganded by F-UMP. There is good superimposition of the side chains of the residues in the active sites of these enzymes and for simplicity Figure 7 depicts side chains of only the wildtype enzyme. Both UMP and F-UMP bind in the same manner observed for BMP at the yeast enzyme (Figure 6),11 with the pyrimidine ring in the syn conformation and C-6 directed towards Lys-314. The pyrimidine rings of UMP and F-UMP are “sandwiched” between the hydrophobic side chains of Pro-417 (Pro-202 at ScOMPDC) and Ile-318′ (Ile-97′ at ScOMPDC) of the second subunit. The fluorine of F-UMP projects into a hydrophobic pocket lined by the side chains of Ile-368, Met-371 and Ile-401 (Leu-150, Leu-153 and Ile-183 at ScOMPDC).46 The hydrophobic pocket thought to be occupied by the molecule of CO2 generated during the decarboxylation of OMP lies on the opposite side of the pyrimidine ring from the essential lysine, and is lined by the side chains of Phe-310, Ile-401 and Ile-448 (Phe-89, Ile-183 and Ile-232 at ScOMPDC).46 This pocket is occupied by a molecule of water in HsOMPDC liganded with UMP (Figure 7) and the same “conserved’ water molecule is observed at OMPDCs liganded with BMP.49 Lys-314 is positioned close to C-6 of UMP and F-UMP with an Nζ-C6 distance of 3.06 Å in the UMP liganded structure. We therefore propose that the catalytic Brønsted base for the observed deuterium exchange reactions of F-UMP and UMP catalyzed by ScOMPDC is the deprotonated form of Lys-93.
Figure 7.

X-ray crystal structures of wildtype human OMPDC liganded by UMP (cyan, PDB entry 2QCD) and the D312N mutant liganded by F-UMP (cornflower blue, PDB entry 2QCF). For simplicity, side chains of only the wildtype enzyme are shown. A molecule of water (purple) is observed bound in the hydrophobic pocket adjacent to C-6 in the UMP-liganded structure but this water is absent in the F-UMP-liganded structure.
Figure 8 shows the X-ray crystal structures of wildtype HsOMPDC liganded by UMP and the D312N mutant liganded by the substrate OMP. UMP and OMP bind in the same manner but the carboxylate group at C-6 of OMP is distorted out of the plane of the pyrimidine ring by 36 degrees,46,50 and there is no bound water molecule observed in the hydrophobic pocket. Similar out of plane distortions have been observed for the cyano group of 6-cyanouridine 5′-monophosphate and the functional groups at C-6 of other substituted pyrimidine nucleotides bound to OMPDC.47,48,51 Nζ of Lys-314 lies within hydrogen bonding distance of the carboxylate group of OMP with an Nζ-O7 distance of 2.54 Å. The close correspondence of these structures supports the proposal that the protonated side chain of Lys-93 at the yeast enzyme acts as a catalytic acid in the decarboxylation of OMP and that its conjugate base is the catalytic base in the C-6 deuterium exchange reactions of UMP and F-UMP.
Figure 8.

X-ray crystal structures of wildtype human OMPDC liganded by UMP (cyan, PDB entry 2QCD) and the D312N mutant liganded by the substrate OMP (green, PDB entry 2QCL). The carboxylate group at C-6 of OMP is distorted out of the plane of the pyrimidine ring by 36 degrees. Nζ of Lys-314 lies within hydrogen bonding distance of the carboxylate group of OMP with an Nζ-O7 distance of 2.54 Å.
The pH-rate profile for kcat/Km for decarboxylation of OMP by ScOMPDC in H2O is consistent with the requirement that a residue with a pKa of 7.7 be in the acidic form, and this pKa can be assigned to the side chain of Lys-93.2,26 The pKas of amine bases are ca. 0.6 unit higher in D2O than in H2O38 so that the catalytic acid is expected to have a pKa of around 8.3 in D2O, which is very similar to pKE = 8.5 for deprotonation of the essential group at the free enzyme determined from the pD-rate profile for kex/Kd for the deuterium exchange reaction of F-UMP in D2O (Figure 4). The close identity of the pKas at the free enzyme of the essential acid for decarboxylation of OMP and of the conjugate acid of the essential base for the deuterium exchange reaction of F-UMP supports our proposal that this base is the deprotonated form of Lys-93. Table 3 summarizes the pKas in D2O of the essential catalytic side chain at free ScOMPDC (pKE) and those at the enzyme liganded by OMP, UMP and F-UMP (pKES), determined from the pL-rate profiles for decarboxylation of OMP2 and for the C-6 deuterium exchange reactions of UMP and F-UMP. The value of pKE = 8.5 in D2O for the side chain of Lys-93 at free ScOMPDC is ca. 2.5 units lower than the expected intrinsic pKa of 11.0 for this residue in D2O.52 This depressed pKa is intriguing because the protonated side chain of Lys-93 appears to be involved in hydrogen bonds with the carboxylate side chains of both Asp-91 and Asp-96′ of the second subunit in the DxKxxD motif (Figure 6). The relatively low pKa of the ammonium group of Lys-93 has been noted previously,3,42 but the structural data offer no strong clues as to its origin. However, we note here that the 2.3 unit variation in the pKa of the ammonium group of Lys-93 at ScOMPDC complexed with OMP, UMP and F-UMP (Table 3) shows that the pKa of this residue is highly sensitive to its local environment. The downward perturbation in the pKa of Lys-93 may result in part from its proximity to the array of hydrophobic residues that surrounds the pyrimidine base (Figure 6), which should result in destabilization of the cationic protonated form.
Table 3.
Apparent pKa of the Essential Residue at Free ScOMPDC and at the Enzyme Liganded by OMP, UMP and F-UMP in D2O at 25 °C and I = 0.1 (NaCl).
pKa of the essential group at the free enzyme (Scheme 6).
pKa of the essential group at the E•S complex (Scheme 6).
Determined from the pH-rate profiles of kcat/Km (for pKE) and kcat (for pKES) for decarboxylation of OMP in H2O [Ref. 2]. The pKas in D2O were estimated from the values in H2O and the 0.6 unit higher pKas of ammonium ions in D2O than in H2O [Ref. 38].
Determined from the pD-rate profile for kex/Kd for deuterium exchange into F-UMP (Figure 4).
Determined from the pD-rate profile for kex for deuterium exchange into UMP (Figure 5B).
Determined from the pD-rate profile for kex for deuterium exchange into F-UMP (Figure 5A).
Intrinsic Affinity of OMPDC for UMP and F-UMP
The pD-rate profiles for kex for the deuterium exchange reactions of UMP and F-UMP (Figure 5) show that the pKa of the conjugate acid of the essential base is lowered from pKE = 8.5 for the free enzyme to pKES = 8.0 and 7.1 for the E•UMP and E•F-UMP complexes, respectively (Table 3). These results are consistent with a neutral rather than an anionic base because the presence of the hydrophobic pyrimidine ring at the liganded enzyme would be expected to destabilize a neighboring negative charge and result in an increase rather than a decrease in the basicity of the catalytic base. The relatively large 1.4 unit decrease in the pKa of the side chain of Lys-93 upon the binding of F-UMP (Table 3) is consistent with the destabilization of positive charge at Nζ of protonated Lys-93 by the combined effects of the electron-withdrawing 5-fluoro substituent and the hydrophobic pyrimidine ring of F-UMP (Figure 7). Scheme 6 includes the thermodynamic cycle that relates the binding of F-UMP and UMP to the protonated (Kd′) and deprotonated enzymes (Kd) to the pKas of the essential group at the free (pKE) and liganded (pKES) enzymes. The relationship Kd′/Kd = KES/KE and the difference (pKE - pKES) = 1.4 gives Kd′/Kd = 25 as the ratio of the dissociation constants for F-UMP from the protonated and the deprotonated enzyme, respectively. Therefore, F-UMP binds 25-fold more tightly to the deprotonated form of ScOMPDC, E, than it does to the form of the enzyme that dominates at neutral pD, ED+. Similarly, the difference (pKE - pKES) = 0.5 for UMP (Table 3) shows that UMP binds 3-fold more tightly to E than to ED+.
The intrinsic second-order rate constant for ScOMPDC-catalyzed deuterium exchange into F-UMP, (kex)max/Kd = 2300 M−1 s−1, can be combined with the intrinsic first-order rate constant (kex)max = 0.041 s−1 to give Kd = 20 μM as the intrinsic affinity of the deprotonated enzyme for the phosphodianion form of F-UMP in D2O. This intrinsic affinity is 6-fold greater than the largest experimental affinity observed at pD 7.6 – 8.2 (Table 1). The observed affinities, (Kd)obsd, of ScOMPDC for 2′-deoxyuridine 5′-monophosphate and 2′-deoxy-5-fluorouridine 5′-monophosphate of 170 μM and 250 μM, respectively, at pH 7.2 in H2O show that the addition of a 5-fluoro substituent results in only a small ca. 1.5-fold change in the observed affinity of ScOMPDC for the 2′-deoxypyyrimidine nucleotide in H2O at pH 7.2.2 These values of (Kd)obsd at pH 7.2 can be combined with eq 8, derived for Scheme 6 (in H2O), to give relative intrinsic affinities Kd of the deprotonated form of ScOMPDC for the phosphodianion forms of UMP and F-UMP of 1.5:1. This suggests that the intrinsic affinity of ScOMPDC for UMP in D2O is essentially the same as Kd = 20 μM determined for F-UMP. Therefore, we conclude that the larger decrease in the affinity of the enzyme for F-UMP than for UMP upon protonation of the catalytic base results largely from a specific destabilizing interaction of the electron-withdrawing fluorine at C-5 of bound F-UMP with the protonated enzyme.
| (8) |
By contrast with F-UMP and UMP, the binding of OMP to ScOMPDC results in a ca. 1.1 unit increase in the pKa of 8.3 in D2O for Lys-93 at the free enzyme, as evidenced by the downward break at pH 8.8 in the pH-rate profile for kcat for decarboxylation of OMP,2 which corresponds to a pKa of 9.4 in D2O (Table 3). This increase in basicity of the side chain of Lys-93 upon the binding of OMP is consistent with electrostatic stabilization of the positive charge at Nζ by its proximity to the negative charge of the carboxylate group at C-6 of bound OMP (Figure 8).
Mechanism of Deuterium Exchange: Formation of a Vinyl Carbanion at OMPDC
We reported earlier that the decarboxylation of OMP catalyzed by wildtype and several mutant ScOMPDCs in a mixed solvent of 50:50 (v/v) H2O/D2O results in the formation of an equimolar mixture of the products h-UMP and d-UMP labeled with H and D at C-6, respectively, so that the product deuterium isotope effect (PIE) on decarboxylation is unity (Scheme 2).20,21 Furthermore, the values of ΦNL3+ ≈ 1.0 for H/D fractionation between L2O and R-NL3+ show that the primary kinetic solvent isotope effect (KIE) is essentially equal to the PIE.21,53 Therefore, there is no discrimination between H and D at the proton donor in the transition state for the rate-determining step of the decarboxylation of OMP. The absence of any significant KIE shows that the rate-limiting step for enzyme-catalyzed decarboxylation of OMP is strongly uncoupled from the proton transfer step. It eliminates the possibility of a concerted mechanism for decarboxylation of OMP9,13 in which proton transfer from Lys-93 to C-6 provides electrophilic push to the loss of CO2. We concluded that the OMPDC-catalyzed decarboxylation of OMP proceeds by the stepwise mechanism through the UMP vinyl carbanion intermediate shown in Scheme 8, in which the transition state for the loss of CO2 from OMP to give the bound UMP carbanion is insensitive to the isotopic composition of the adjacent ζ-NL3+ group of Lys-93.20,21 The PIE of unity requires that hydron transfer to the bound vinyl carbanion be much faster than the possible rotation of the terminal CH2-NL3+ bond of the side chain of the proton donor Lys-93, which exchanges the positions of its NL3+ hydrons (kp ≫ krot). Therefore, the yields of h-UMP and d-UMP from the decarboxylation of OMP in an H2O/D2O mixture are determined by the initial isotopic enrichment of the ζ-NL3+ group of Lys-93 (governed by the fractionation factor ΦNL3+ ≈ 1) at the E•OMP Michaelis complex.20,21 The observation of a small inverse PIE of 0.93 for the decarboxylation of F-OMP catalyzed by wildtype ScOMPDC shows that the expected decrease in the chemical reactivity of the carbanion intermediate as a result of the 5-fluoro substituent does not result in any large change in the inequality kp ≫ krot.21 This small inverse PIE is consistent with a KIE of unity for hydron transfer to the enzyme-bound F-UMP vinyl carbanion intermediate, along with a small increase in the fractionation factor for the ζ-NL3+ group of Lys-93 at the E•F-OMP Michaelis complex to ΦNL3+ ≈ 1.1.21
Scheme 8.

The ScOMPDC-catalyzed deuterium exchange reactions of UMP and F-UMP examined here show that deuterium exchange into saturating enzyme-bound F-UMP [(kex)max = 0.041 s−1] is 3400-fold faster than exchange into enzyme-bound UMP [(kex)max = 1.2 × 10−5 s−1] so that there is a large 4.8 kcal/mol stabilization of the transition state for deuterium exchange by the electron-withdrawing 5-fluoro substituent at F-UMP. This is consistent with the formation of a carbanion intermediate at which the negative charge is stabilized by interaction with the electron-withdrawing 5-fluoro substituent, and a deuterium exchange reaction that is the reverse of the proton transfer “half reaction” involved in the physiological decarboxylation of OMP.
Scheme 9 shows our proposed mechanism for the OMPDC-catalyzed deuterium exchange reactions of UMP and F-UMP.22 Proton transfer from C-6 of UMP and F-UMP to the neutral side chain of Lys-93 (k-p) results in an enzyme-bound vinyl carbanion intermediate that can be rapidly reprotonated via “internal return” of the abstracted hydron to regenerate the unlabeled nucleotide (kp ≫ krot). This reversible chemical step is followed by the rare rate-limiting rotation of the terminal CH2-ND2H+ bond of the side chain of Lys-93 in to a “reactive position” for delivery of a deuteron to C-6 (krot).
Scheme 9.

The rate constant for CH2-NL3+ bond rotation in water is expected to be similar to the value of kreorg = 1011 s−1 estimated for reorganization that exchanges the relative positions of -O and -S at a thiobenzoate•carbocation ion pair.54 However, the side chain of protonated Lys-93 is “anchored” by hydrogen bonding to the carboxylate groups of Asp-91 and Asp-96′ in a “double salt bridge” (Figure 6), which is expected to result in a substantial barrier to rotation of the terminal CH2-NL3+ bond of this side chain at the ScOMPDC•carbanion complex. The strength of the stabilizing interactions in salt bridges buried in the interior of proteins is 3 –5 kcal/mol55,56 so that we estimate this barrier, which should involve the breaking of hydrogen bonds between protonated Lys-93 and both Asp-91 and Asp-96′, to be at least 8 kcal/mol. This gives an upper limit on the rate constant for bond rotation of krot ≤ 107 s−1.
Carbon Acidity of Substituted Uracils in Aqueous Solution
Prior to the current work, a critical unknown was the effect of a 5′-phosphoribosyl group at N-1 and/or a 5-fluoro substituent on the C-6 carbon acidity of uracil derivatives in aqueous solution (Scheme 10). Therefore, we have determined second-order rate constants kDO (M−1 s−1) for the DO−-catalyzed deuterium exchange reactions of uridine, 5-fluorouridine and 1,3-dimethyl-5-fluorouracil in D2O at a variety of temperatures and pD (see Results and Supporting Information). The exchange reactions of uridine and 5-fluorouridine in the presence of 1 M NaOD provided kDO (M−1 s−1) for deuterium exchange into their N-3 anions, while the exchange reaction of 5-fluorouridine at pD 7.1 provided kDO for deuterium exchange into the neutral nucleoside (pKND = 8.4, this work). The value of kDO for deuterium exchange into neutral uridine was calculated from kDO determined for its N-3 anion, with the assumption that ionization of uridine at N-3 results in the same 2.5 × 104-fold decrease in reactivity of the C-6 proton that is observed upon ionization of 5-fluorouridine at N-3 (Table S8 of the Supporting Information).
Scheme 10.
The values of kDO for the substituted uracils (Table S8 of the Supporting Information) were used to calculate the second-order rate constants kHO (M−1 s−1) for deprotonation of these carbon acids by hydroxide ion in H2O given in Table 4, using a secondary solvent isotope effect of kDO/kHO = 2.4.57–59 The carbon acid pKas were then calculated using eq 9, derived for Scheme 11, with pKw = 14 for H2O and kHOH = 1011 s−1 for the reverse protonation of localized unstable carbanions by solvent water that is limited by the rotation of a molecule of water into a reactive position.57–60
Table 4.
Kinetic and Thermodynamic Acidity of the C-6 Protons of N-1, N-3 and C-5 Substituted Uracils in Water at 25 °C (Schemes 10 and 11).
| Substrate | X | R1 | R2 | kHO a M−1 s−1 | pKCH b |
|---|---|---|---|---|---|
| Uridine | H | Ribosyl | H | 1.6 × 10−4 | 28.8 ± 1 |
| Uridine N-3 Anion | H | Ribosyl | Anion | 6.3 × 10−9 | 33.2 ± 1 |
| 1,3-Dimethyluracil | H | Me | Me | 2 × 10−6 c | 30.7 ± 1 |
| 5-Fluorouridine | F | Ribosyl | H | 0.71 | 25.1 ± 0.5 |
| 5-Fluorouridine N-3 Anion | F | Ribosyl | Anion | 2.8 × 10−5 | 29.6 ± 0.5 |
| 1,3-Dimethyl-5-fluorouracil | F | Me | Me | 1.8 × 10−2 | 26.7 ± 0.5 |
Second-order rate constant for proton transfer from C-6 to hydroxide ion in H2O, calculated from the value of kDO for deuterium exchange (Table S8 of the Supporting Information) using a secondary solvent isotope effect of kDO/kHO = 2.4 (see text).
Carbon acid pKa for ionization of the substrate at C-6 in H2O, calculated from kHO (M−1 s−1) for deprotonation of the carbon acid by hydroxide ion and kHOH = 1011 s−1 for the reverse protonation of the vinyl carbanion by solvent water, using eq 9.
Calculated from data for the competing C-6 deuterium exchange and hydrolysis reactions of the substrate in 1 M NaOD [Ref. 61].
Scheme 11.
| (9) |
Table 4 summarizes the C-6 carbon acidities, pKCH (Scheme 10), of the model nucleosides uridine and 5-fluorouridine in water, along with those of their N-3 anions and the corresponding 1,3-dimethyluracils. The value of pKCH = 30.7 for the C-6 proton of 1,3-dimethyluracil (Scheme 10, X = H, R1 = R2 = Me), obtained from rate constants for its competing exchange and hydrolysis reactions in 1 M NaOD at 25 °C,61 is lower than the earlier reported value of 34 ± 2 determined from the temperature dependence of deuterium exchange in acetate buffers at 175 – 217 °C.62 There is a good linear correlation (not shown) with a slope of ρI = −8.0 of the carbon acid pKas of 3-R-4-methylthiazolium cations (Scheme 12),63 “normal” carbon acids that undergo ionization to generate localized carbanions,59,64,65 with the inductive substituent constant σI for the 3-substituent, and, by analogy, a similar free energy relationship is expected for the carbon acid pKas of N-1 substituted uracils. The value of σI = −0.05 for the methyl group66 and the estimated value of σI = 0.14 for the ribosyl group67 can be combined with ρI = −8.0 to estimate a decrease in C-6 carbon acidity of 1.5 pK units upon the change from a methyl to a ribosyl group at N-1 of substituted uracils (R1, Scheme 12). This is consistent with the 1.6 unit difference in the values of pKCH for 1,3-dimethyl-5-fluorouracil and 5-fluorouridine determined here (Table 4).
Scheme 12.

The data in Table 4 show that the addition of a 5-fluoro substituent to substituted uracils results in an increase in the C-6 carbon acidity of 3.6 – 4.0 pK units. By contrast, the addition of a 5-fluoro substituent results in decreases of 1.5, 1.6 and 1.7 – 1.9 units in the pKa of the N-3 proton of uracil,37 2′-deoxyuridine,37 and UMP,68 respectively. These substituent effects are consistent with a fall-off factor of ca. 2 for the presence of the additional carbon atom between the 5-fluoro substituent and the center of negative charge at substituted uracil N-3 anions compared with their C-6 carbanions.57
Stabilization of the UMP and F-UMP Vinyl Carbanions by ScOMPDC
Figure 9 shows free energy profiles for the enzyme-catalyzed deuterium exchange reactions of UMP and F-UMP through the respective C-6 vinyl carbanion intermediates. These profiles were constructed according to the mechanism shown in Scheme 9, with the observed 3400-fold faster deuterium exchange into enzyme-bound F-UMP [(kex)max = 0.041 s−1] than UMP [(kex)max = 1.2 × 10−5 s−1] at high pD (Figure 5), and a barrier to rotation of the terminal CH2-NL3+ bond of the side chain of Lys-93 of 8 kcal/mol (krot = 107 s−1, see above). These profiles refer to the exchange reactions of the substrate phosphodianions (SD2−) catalyzed by ScOMPDC at which Lys-93 is deprotonated (END2). The essentially identical values of Kd = 20 μM for binding of F-UMP and UMP to the deprotonated enzyme correspond to a free energy of binding to form the Michaelis complex of 6.4 kcal/mol. Proton abstraction from C-6 (k-p) leads to the formation of the initial enzyme-carbanion complex which can undergo rapid internal return by reprotonation (kp) or the occasional, rate-limiting, rotation of the side-chain of Lys-93 into a position to deliver a deuteron to C-6 (krot), with an estimated barrier to rotation of at least 8 kcal/mol (see above). Once this rotational event occurs there is rapid and irreversible transfer of a deuteron to C-6 to generate the deuterium exchange product, kp ≫ krot, as evidenced by the PIEs of unity determined for the decarboxylation of OMP in 50:50 (v/v) H2O/D2O.20,21
Figure 9.

Free energy profiles for the enzyme-catalyzed deuterium exchange reactions of the phosphodianion forms of UMP and F-UMP (SD2−) through the respective C-6 vinyl carbanion intermediates. These profiles were constructed according to the mechanism shown in Scheme 9, with the observed 3400-fold faster deuterium exchange into enzyme-bound F-UMP than UMP and a barrier to rotation of the terminal CH2-NL3+ bond of the side chain of Lys-93 of 8 kcal/mol (see text).
The formation of vinyl carbanions derived from UMP and F-UMP as intermediates of their C-6 deuterium exchange reactions (Scheme 9 and Figure 9) allows equilibrium constants (Keq)enz for proton transfer from C-6 of enzyme-bound UMP and F-UMP to the side chain of Lys-93 (Scheme 13A) to be obtained from the first-order rate constants (kex)max for deuterium exchange into the enzyme-bound nucleotides, according to eqs 10 and 11. The values of (kex)max = 1.2 × 10−5 and 0.041 s−1 for deuterium exchange into enzyme-bound UMP and F-UMP, respectively, and krot = 107 s−1 were substituted into eq 11 to give (dimensionless) equilibrium constants for proton transfer from C-6 of the bound nucleotide to ScOMPDC of (Keq)enz = 1.2 × 10−12 and 4.1 × 10−9, respectively, so that there is a large 4.8 kcal/mol stabilizing effect of the 5-F substituent on this proton transfer equilibrium (Figure 9). The values of pKES = 8.0 and 7.1 for the side chain of Lys-93 at ScOMPDC complexed with UMP and F-UMP, respectively (Table 3), show that there is an offsetting 1.2 kcal/mol destabilizing effect of the 5-fluoro substituent on equilibrium proton transfer to Nζ of Lys-93, as a result of interactions of the electron-withdrawing fluorine at C-5 of bound F-UMP with the positively charged ammonium group (vide infra). Therefore, the 5-fluoro substituent at F-UMP results in an overall decrease in the thermodynamic barrier for proton transfer from C-6 of 6.0 kcal/mol, which corresponds to a 4.4 unit increase in the carbon acidity of the bound nucleotide. This is similar to the 3.7 pK unit difference in the C-6 carbon acidities of the model nucleosides uridine (pKCH = 28.8 ± 1) and 5-fluorouridine (pKCH = 25.1 ± 0.5) in aqueous solution (Table 4).
Scheme 13.

| (10) |
| (11) |
| (12) |
Similarly, overall equilibrium constants for proton transfer from C-6 of UMP and F-UMP to a primary amine base of pKRNH3 = 7 in water (Scheme 13B) can be calculated using eq 12, with the values of pKCH for the model nucleosides uridine and 5-fluorouridine from Table 4, as (= 2 × 10−22 and 8 × 10−19, respectively. Therefore, the binding of UMP or F-UMP to Keq)aq ScOMPDC results in a ~5 × 109-fold increase in the equilibrium constant for proton transfer from C-6 to an amine base, which corresponds to a thermodynamic stabilization of the enzyme-bound UMP and F-UMP vinyl carbanions, relative to the enzyme-nucleotide complexes, of 13 kcal/mol. A critical unknown is the magnitude of the barrier to rotation of the terminal CH2-NL3+ bond of the side chain of Lys-93, which is hydrogen-bonded to the anionic side chains of Asp-91 and Asp-96′ at the enzyme•carbanion complex (Figure 6). We have estimated this barrier as ≥ 8 kcal/mol, so that 13 kcal/mol is a lower limit on the stabilization of the bound UMP and F-UMP vinyl carbanions resulting from their interactions with ScOMPDC.
Enzymatic Rate Accelerations for Decarboxylation and Deuterium Exchange
The formation of vinyl carbanions as intermediates in the C-6 deuterium exchange reactions of UMP and F-UMP catalyzed by ScOMPDC provides convincing evidence for the decarboxylation of OMP and F-OMP by a stepwise mechanism through the same carbanions. The total rate acceleration for the decarboxylation of OMP by free ScOMPDC can be calculated as 4 × 1022 M−1, from the ratio of kcat/Km = 1.1 × 107 M−1 s−1 for decarboxylation at pH 7.1,7 which is the maximum in the pH-rate profile for decarboxylation,2 and ko = 2.8 × 10−16 s−1 for the nonenzymatic decarboxylation of OMP in aqueous solution.4 This corresponds to a total stabilization of the transition state for decarboxylation of OMP of 31 kcal/mol.3 It is more difficult to establish the rate acceleration for the enzyme-catalyzed deuterium exchange reactions of UMP and F-UMP, because the nonenzymatic proton transfer reactions of these normal carbon acids in solution proceed by proton transfer to lyoxide ion so that the observed rate constant depends strongly on pL. This notwithstanding, the observed rate constant for C-6 deprotonation of 5-fluorouridine (a model for F-UMP) by hydroxide ion in water at pH 7.1 can be calculated as ko = kHO[HO−] = 9 × 10−8 s−1 (Table 4). The intrinsic second-order rate constant for ScOMPDC-catalyzed deuterium exchange into F-UMP catalyzed by the deprotonated enzyme is (kex)max/Kd = 2300 M−1 s−1. The ratio of these rate constants gives the enzymatic rate acceleration for deuterium exchange as 3 × 1010 M−1, which corresponds to a transition state stabilization of 14 kcal/mol. Together with the ≥ 13 kcal/mol stabilization of the bound UMP and F-UMP vinyl carbanions by interactions with the enzyme estimated above, this shows that a large portion of the enzymatic rate acceleration for the decarboxylation of OMP results from stabilization of the bound carbanion intermediate by its interactions with ScOMPDC.
These data suggest that the total transition state stabilization for decarboxylation of OMP via the UMP vinyl carbanion intermediate exceeds that for its formation by proton transfer from C-6 of UMP to the enzyme by ca. 17 kcal/mol. We suggest several factors that may contribute to this apparent greater proficiency of ScOMPDC for decarboxylation than deuterium exchange.
(1) The difference in the transition state stabilization for decarboxylation compared with deuterium exchange would be lowered to 14 kcal/mol if there is uncertainty in the rate constant for the nonenzymatic decarboxylation of OMP such that this reaction is ca. 100-fold faster than the current best estimate.4
(2) The decarboxylation of OMP by ScOMPDC is limited in part by the barrier to the chemical step of the loss of CO2 to give the carbanion intermediate.2,26,33,69 By contrast, the rate-limiting step for enzyme-catalyzed deuterium exchange is the physical step of rotation of the terminal CH2-NHD2+ bond of the side chain of Lys-93 into a “reactive position”, with an estimated barrier of ≥ 8 kcal/mol (krot, Figure 9). Therefore, the energy of the transition state for the chemical proton transfer step (k-p) will be lower than that for the overall deuterium exchange reaction by an amount RTln(kp/krot) so that the rate acceleration for deuterium exchange underestimates the rate acceleration for formation of the bound vinyl carbanion from UMP by this amount.
(3) The initial product of the decarboxylation of OMP is the ternary complex of OMPDC with UMP and a molecule of CO2 bound in the hydrophobic pocket below C-6 of UMP (Figure 7). If the presence of CO2 in the hydrophobic pocket activates the enzyme for catalysis of proton transfer, then the true reactivity of the enzyme towards proton transfer from C-6 of bound UMP and F-UMP could be higher than that observed in the deuterium exchange reactions described here.
(4) The transition state for decarboxylation of OMP may be stabilized by favorable “solvation” of the departing CO2 molecule by the development of stabilizing interactions with the hydrophobic side chains of Phe-89, Ile-183 and Ile-232 that line the CO2 binding pocket at yeast OMPDC (Phe-310, Ile-401 and Ile-448 at HsOMPDC, Figure 7). These interactions, which are not present in the ground state of the free enzyme and OMP, would result in selective stabilization of the transition state for formation of the UMP carbanion intermediate by decarboxylation that is not available for the transition state for its formation by proton transfer from C-6 of UMP.
Finally, we note that ground state destabilization by the introduction of “strain” at the protein and/or electrostatic “stress” at the substrate in the OMPDC•OMP Michaelis complex may contribute to an increase in kcat for the decarboxylation of OMP, and, possibly, proton transfer from C-6 of UMP and F-UMP.12,17,70 However, the overall rate accelerations for decarboxylation and exchange calculated here from the rate constants kcat/Km are unaffected by such effects because they refer to the barrier for conversion of the free enzyme and substrate in solution to the enzyme-bound transition state.71
Supplementary Material
Acknowledgments
We acknowledge the National Institutes of Health Grant GM39754 (to J.P.R.), Grant GM65155 (to J.A.G.) and Grant GM095419 (to W.W.) for generous support of this work.
Footnotes
Experimental details of the C-6 deuterium exchange reactions of uridine, 5-fluorouridine and 1,3-dimethyl-5-fluorouracil in D2O monitored by 1H NMR spectroscopy. Tables S1 – S6: Observed first-order rate constants for the ScOMPDC- catalyzed C-6 deuterium exchange reactions of F-UMP at pD 6.45 – 9.33. Tables S7 and S8: Second-order rate constants for the C-6 deuterium exchange reactions of uridine, 5-fluorouridine, their N-3 anions, and 1,3-dimethyl-5-fluorouracil catalyzed by DO−. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Yablonski MJ, Pasek DA, Han BD, Jones ME, Traut TW. J Biol Chem. 1996;271:10704–10708. doi: 10.1074/jbc.271.18.10704. [DOI] [PubMed] [Google Scholar]
- 2.Porter DJT, Short SA. Biochemistry. 2000;39:11788–11800. doi: 10.1021/bi001199v. [DOI] [PubMed] [Google Scholar]
- 3.Miller BG, Wolfenden R. Annu Rev Biochem. 2002;71:847–885. doi: 10.1146/annurev.biochem.71.110601.135446. [DOI] [PubMed] [Google Scholar]
- 4.Radzicka A, Wolfenden R. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- 5.Amyes TL, Richard JP, Tait JJ. J Am Chem Soc. 2005;127:15708–15709. doi: 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- 6.Morrow JR, Amyes TL, Richard JP. Acc Chem Res. 2008;41:539–548. doi: 10.1021/ar7002013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toth K, Amyes TL, Wood BM, Chan KK, Gerlt JA, Richard JP. Biochemistry. 2009;48:8006–8013. doi: 10.1021/bi901064k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amyes TL, Ming SA, Goldman LM, Wood BM, Desai BJ, Gerlt JA, Richard JP. Biochemistry. 2012;51:4630–4632. doi: 10.1021/bi300585e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Appleby TC, Kinsland C, Begley TP, Ealick SE. Proc Natl Acad Sci U S A. 2000;97:2005–2010. doi: 10.1073/pnas.259441296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harris P, Poulsen J-CN, Jensen KF, Larsen S. Biochemistry. 2000;39:4217–4224. doi: 10.1021/bi992952r. [DOI] [PubMed] [Google Scholar]
- 11.Miller BG, Hassell AM, Wolfenden R, Milburn MV, Short SA. Proc Natl Acad Sci U S A. 2000;97:2011–2016. doi: 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu N, Mo Y, Gao J, Pai EF. Proc Natl Acad Sci U S A. 2000;97:2017–2022. doi: 10.1073/pnas.050417797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Begley TP, Appleby TC, Ealick SE. Curr Opin Struct Biol. 2000;10:711–718. doi: 10.1016/s0959-440x(00)00148-2. [DOI] [PubMed] [Google Scholar]
- 14.Warshel A, Strajbl M, Villa J, Florian J. Biochemistry. 2000;39:14728–14738. doi: 10.1021/bi000987h. [DOI] [PubMed] [Google Scholar]
- 15.Houk KN, Lee JK, Tantillo DJ, Bahmanyar S, Hietbrink BN. ChemBioChem. 2001;2:113–118. doi: 10.1002/1439-7633(20010202)2:2<113::AID-CBIC113>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 16.Warshel A, Florian J, Strajbl M, Villa J. ChemBioChem. 2001;2:109–111. doi: 10.1002/1439-7633(20010202)2:2<109::AID-CBIC109>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 17.Gao J. Curr Opin Struct Biol. 2003;13:184–192. doi: 10.1016/s0959-440x(03)00041-1. [DOI] [PubMed] [Google Scholar]
- 18.Begley TP, Ealick SE. Curr Opin Chem Biol. 2004;8:508–515. doi: 10.1016/j.cbpa.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 19.Callahan BP, Miller BG. Bioorg Chem. 2007;35:465–469. doi: 10.1016/j.bioorg.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 20.Toth K, Amyes TL, Wood BM, Chan K, Gerlt JA, Richard JP. J Am Chem Soc. 2007;129:12946–12947. doi: 10.1021/ja076222f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toth K, Amyes TL, Wood BM, Chan K, Gerlt JA, Richard JP. J Am Chem Soc. 2010;132:7018–7024. doi: 10.1021/ja102408k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amyes TL, Wood BM, Chan K, Gerlt JA, Richard JP. J Am Chem Soc. 2008;130:1574–1575. doi: 10.1021/ja710384t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shostak K, Jones ME. Biochemistry. 1992;31:12155–12161. doi: 10.1021/bi00163a026. [DOI] [PubMed] [Google Scholar]
- 24.Van Vleet JL, Reinhardt LA, Miller BG, Sievers A, Cleland WW. Biochemistry. 2008;47:798–803. doi: 10.1021/bi701664n. [DOI] [PubMed] [Google Scholar]
- 25.Chan KK, Wood BM, Fedorov AA, Fedorov EV, Imker HJ, Amyes TL, Richard JP, Almo SC, Gerlt JA. Biochemistry. 2009;48:5518–5531. doi: 10.1021/bi900623r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smiley JA, Paneth P, O’Leary MH, Bell JB, Jones ME. Biochemistry. 1991;30:6216–6223. doi: 10.1021/bi00239a020. [DOI] [PubMed] [Google Scholar]
- 27.Miller BG, Butterfoss GL, Short SA, Wolfenden R. Biochemistry. 2001;40:6227–6232. doi: 10.1021/bi0028993. [DOI] [PubMed] [Google Scholar]
- 28.Fujita K-i, Matsukawa A, Shibata K, Tanaka T, Taniguchi M, Oi S. Carb Res. 1994;265:299–302. doi: 10.1016/0008-6215(94)00231-2. [DOI] [PubMed] [Google Scholar]
- 29.Glasoe PK, Long FA. J Phys Chem. 1960;64:188–190. [Google Scholar]
- 30.Moffatt JG. J Am Chem Soc. 1963;85:1118–1123. [Google Scholar]
- 31.Dawson RMC, Elliott DC, Elliott WH, Jones KM. Data for Biochemical Research. 3. Clarendon Press; Oxford: 1986. [Google Scholar]
- 32.Barnett SA, Amyes TL, Wood BM, Gerlt JA, Richard JP. Biochemistry. 2008;47:7785–7787. doi: 10.1021/bi800939k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wood BM, Chan KK, Amyes TL, Richard JP, Gerlt JA. Biochemistry. 2009;48:5510–5517. doi: 10.1021/bi9006226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller BG, Smiley JA, Short SA, Wolfenden R. J Biol Chem. 1999;274:23841–23843. doi: 10.1074/jbc.274.34.23841. [DOI] [PubMed] [Google Scholar]
- 35.Sievers A, Wolfenden R. Bioorg Chem. 2005;33:45–52. doi: 10.1016/j.bioorg.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 36.Levine HL, Brody RS, Westheimer FH. Biochemistry. 1980;19:4993–4999. doi: 10.1021/bi00563a010. [DOI] [PubMed] [Google Scholar]
- 37.Privat EJ, Sowers LC. Mut Res. 1996;354:151–156. doi: 10.1016/0027-5107(96)00005-x. [DOI] [PubMed] [Google Scholar]
- 38.Laughton PM, Robertson RE. In: Solute-Solvent Interactions. Coetzee JF, Ritchie CD, editors. Vol. 1. Marcel Dekker; New York: 1969. pp. 399–538. [Google Scholar]
- 39.Schowen KB, Schowen RL. In: Methods in Enzymology. Purich DL, editor. Vol. 87. Academic Press; New York: 1982. pp. 551–606. [PubMed] [Google Scholar]
- 40.Traut TW, Temple BRS. J Biol Chem. 2000;275:28675–28681. doi: 10.1074/jbc.M003468200. [DOI] [PubMed] [Google Scholar]
- 41.Miller BG, Snider MJ, Wolfenden R, Short SA. J Biol Chem. 2001;276:15174–15176. doi: 10.1074/jbc.M011429200. [DOI] [PubMed] [Google Scholar]
- 42.Smiley JA, Jones ME. Biochemistry. 1992;31:12162–12168. doi: 10.1021/bi00163a027. [DOI] [PubMed] [Google Scholar]
- 43.Wittmann JG, Rudolph MG. Acta Crystallogr, Sect D Biol Crystallogr. 2007;D63:744–749. doi: 10.1107/S0907444907016605. [DOI] [PubMed] [Google Scholar]
- 44.Bello AM, Poduch E, Fujihashi M, Amani M, Li Y, Crandall I, Hui R, Lee PI, Kain KC, Pai EF, Kotra LP. J Med Chem. 2007;50:915–921. doi: 10.1021/jm060827p. [DOI] [PubMed] [Google Scholar]
- 45.Bello AM, Poduch E, Liu Y, Wei L, Crandall I, Wang X, Dyanand C, Kain KC, Pai EF, Kotra LP. J Med Chem. 2008;51:439–448. doi: 10.1021/jm7010673. [DOI] [PubMed] [Google Scholar]
- 46.Wittmann JG, Heinrich D, Gasow K, Frey A, Diederichsen U, Rudolph MG. Structure. 2008;16:82–92. doi: 10.1016/j.str.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 47.Fujihashi M, Wei L, Kotra LP, Pai EF. J Mol Biol. 2009;387:1199–1210. doi: 10.1016/j.jmb.2009.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heinrich D, Diederichsen U, Rudolph MG. Chem Eur J. 2009;15:6619–6625. doi: 10.1002/chem.200900397. [DOI] [PubMed] [Google Scholar]
- 49.Hu H, Boone A, Yang W. J Am Chem Soc. 2008;130:14493–14503. doi: 10.1021/ja801202j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu N, Gillon W, Pai EF. Biochemistry. 2002;41:4002–4011. doi: 10.1021/bi015758p. [DOI] [PubMed] [Google Scholar]
- 51.Callahan BP, Bell AF, Tonge PJ, Wolfenden R. Bioorg Chem. 2006;34:59–65. doi: 10.1016/j.bioorg.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 52.Harris TK, Turner GJ. IUBMB Life. 2002;53:85–98. doi: 10.1080/15216540211468. [DOI] [PubMed] [Google Scholar]
- 53.Tsang W-Y, Richard JP. J Am Chem Soc. 2007;129:10330–10331. doi: 10.1021/ja073679g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Richard JP, Tsuji Y. J Am Chem Soc. 2000;122:3963–3964. [Google Scholar]
- 55.Anderson DE, Becktel WJ, Dahlquist FW. Biochemistry. 1990;29:2403–2408. doi: 10.1021/bi00461a025. [DOI] [PubMed] [Google Scholar]
- 56.Tissot AC, Vuilleumier S, Fersht AR. Biochemistry. 1996;35:6786–6794. doi: 10.1021/bi952930e. [DOI] [PubMed] [Google Scholar]
- 57.Richard JP, Williams G, Gao J. J Am Chem Soc. 1999;121:715–726. [Google Scholar]
- 58.Richard JP, Williams G, O’Donoghue AC, Amyes TL. J Am Chem Soc. 2002;124:2957–2968. doi: 10.1021/ja0125321. [DOI] [PubMed] [Google Scholar]
- 59.Amyes TL, Diver ST, Richard JP, Rivas FM, Toth K. J Am Chem Soc. 2004;126:4366–4374. doi: 10.1021/ja039890j. [DOI] [PubMed] [Google Scholar]
- 60.Richard JP, Amyes TL, Toteva MM. Acc Chem Res. 2001;34:981–988. doi: 10.1021/ar0000556. [DOI] [PubMed] [Google Scholar]
- 61.Wong FM, Capule CC, Wu W. Org Lett. 2006;8:6019–6022. doi: 10.1021/ol0624981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sievers A, Wolfenden R. J Am Chem Soc. 2002;124:13986–13987. doi: 10.1021/ja021073g. [DOI] [PubMed] [Google Scholar]
- 63.Washabaugh MW, Jencks WP. Biochemistry. 1988;27:5044–5053. doi: 10.1021/bi00414a015. [DOI] [PubMed] [Google Scholar]
- 64.Kresge AJ. Acc Chem Res. 1975;8:354–360. [Google Scholar]
- 65.Washabaugh MW, Jencks WP. J Am Chem Soc. 1989;111:683–692. [Google Scholar]
- 66.Charton M. Prog Phys Org Chem. 1981;13:119–251. [Google Scholar]
- 67.Estimated as the sum of the σI values of 0.11 for the CH2OMe group and 0.03 for the CH2CHOH group [Ref. 66].
- 68.The literature values of pKNH for UMP of 9.7 [Ref. 2] and 9.5 [ Jencks WP, Regenstein J. In: Handbook of Biochemistry and Molecular Biology (Physical and Chemical Data) 3. Fasman GD, editor. Vol. 1. CRC Press; Cleveland, OH: 1976. pp. 305–351.] are 1.7 – 1.9 units higher than pKNH = 7.84 for F-UMP determined in this work.
- 69.Ehrlich JI, Hwang C-C, Cook PF, Blanchard JS. J Am Chem Soc. 1999;121:6966–6967. [Google Scholar]
- 70.Fersht A. Structure and Mechanisms in Protein Science. W. H. Freeman & Co; New York: 1999. p. 373. [Google Scholar]
- 71.Jencks WP. Adv Enzymol Relat Areas Mol Biol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.